

Cardiovascular disease (CVD) is the leading cause of mortality worldwide and increases significantly as we age. Gaining a better understanding of how molecular pathways related to aging can influence atherosclerosis and cardiovascular function is therefore an important research goal. Rare, monogenic disorders can provide powerful new insights into common pathologies such as CVD, pointing to heretofore unexpected proteins as key drivers of disease. Such is the case for prelamin A and the premature aging disease progeria.
The nuclear lamina is a meshwork of intermediate filament proteins on the inner aspect of the nuclear membrane whose building blocks in somatic cells are lamins A, B1, B2 and C. Mutations in the lamin A/C gene (LMNA), which encodes the lamin A precursor, prelamin A, and lamin C, can cause a variety of diseases referred to as the laminopathies.1 The most dramatic of these is Hutchinson-Gilford progeria syndrome (HGPS), also called progeria. HGPS arises from a de novo autosomal dominant mutation in a portion of LMNA encoding prelamin A.2,3 Children with HGPS develop several features of accelerated aging, including growth retardation, thin skin, hair loss, joint ailments, lipodystrophy and, most significantly, aggressive early onset atherosclerosis and CVD. These children invariably die from myocardial infarctions or strokes in the second decade of life. Their blood lipid profiles are normal, yet autopsies have revealed atherosclerotic plaques similar to those seen in elderly individuals.4 Other notable features in the large vessels of children with HGPS are dramatic vascular smooth muscle cell loss and thickened, fibrotic adventia. In this issue of Circulation, Hamczyk et al5 report that loss of vascular smooth muscle cells plays a key role in the acceleration of atherosclerosis in a mouse model of HGPS.
Defects in the posttranslational processing pathway of prelamin A lead to HGPS and related progeroid disorders.6 Prelamin A has a carboxyl-terminal cysteine-aliphatic-aliphatic-any amino acid (CAAX) motif that is a signal for the initiation of a series of processing reactions. These reactions include farnesylation of the cysteine residue of the CAAX motif, cleavage of the last 3 residues and carboxyl methylation of the farnesylcysteine (Figure 1A, Steps 1–3). While most CAAX proteins rely on farnesylation for their biological function, prelamin A undergoes a final processing step mediated by the zinc metalloprotease ZMPSTE24 that catalyzes the cleavage of the last 15 amino acids, including its farnesylated cysteine, to produce mature, unfarnesylated lamin A (Figure 1A, Step 4). Genetic mutations that diminish or block this final proteolytic cleavage result in the accumulation of permanently farnesylated forms of prelamin A, which in turn promote disease (Figure 1B). Mutations in ZMPSTE24 cause mandibuloacral dysplasia (MAD), atypical progeria or restrictive dermopathy (RD), with disease severity correlating with residual enzyme activity.7 A Point mutation in one LMNA allele abolishing the ZMPSTE24 recognition site in prelamin A (L647R) causes a mild progeroid disorder with features of MAD.8 The mutation in LMNA that underlies HGPS (G608G) activates a cryptic RNA splice donor site, leading to an internal deletion of 50 amino acids from prelamin A, including the ZMPSTE24 recognition site. This truncated, permanently farnesylated prelamin A variant is called progerin. Many lines of evidence indicate it is the dose-dependent presence of farnesylated prelamin A variants in HGPS, RD and MAD and not the lack of lamin A that causes progeroid disorders.1,6,7
Figure 1.
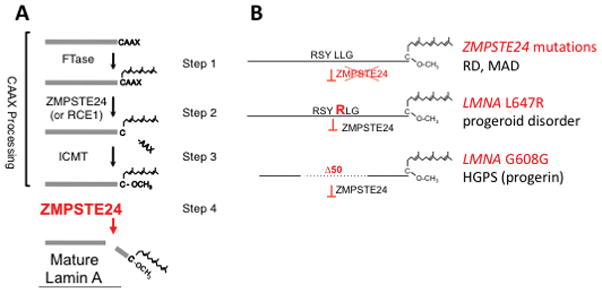
Prelamin A processing and diseases caused by mutations in genes altering it. A, Prelamin A processing pathway. In Step 1, protein farnesyltransferase (FTase) catalyzes the addition of a farnesyl lipid moiety to the cysteine residue of the CAAX motif (CSIM in prelamin A). In Step 2, the terminal three amino acids (SIM) are cleaved in a reaction catalyzed by zinc metalloproteinase STE24 (ZMPSTE24) or Ras converting CAAX endopeptidase 1 (RCE1) (Figure 1A, step 2). In Step 3, isoprenylcysteine carboxyl methyltransferase (ICMT) catalyzes the carboxymethylation of the farnesylcysteine. In Step 4, ZMPSTE24 recognizes the farnesylated protein and catalyzes the cleavage of the last 15 amino acids of prelamin A, including its farnesylated cysteine, resulting in the production of mature, unfarnesylated lamin A. B, Defective prelamin A processing results in expression of permanently farnesylated forms of prelamin A and progeroid diseases. Recessive mutations in ZMPSTE24 cause RD and MAD (top); a dominant mutation LMNA L647R disrupts prelamin A cleavage causing a progeroid disorder (middle); dominant G608G (or rarely G608S) mutations in LMNA generate a prelamin A variant with an internal deletion of 50 amino acids and cause HGPS; the truncated prelamin A variant is called progerin.
Investigators have generated several mouse models to study prelamin A processing and disease. These include mice with germline deletion of Zmpste24, which accumulate prelamin A and develop systemic features of progeria.9,10 and mice with a mutant Lmna allele (Δ50) that express progerin, and also exhibits systemic progeroid features.11 Neither of these mice develop the profound CVD seen in children with HGPS, possibly because they die too early from other problems. Nonetheless, Loren Fong and Stephen Young used these strains in pioneering research to show that treatment with protein farnesyltransferase inhibitors ameliorate the progeroid phenotypes, establishing the critical role of permanently farnesylated prelamin A variants in pathogenesis.11,12
More recent mouse models of HGPS in which Lmna splicing is altered exhibit some but not all of the vascular pathology of the disease. Transgenic mice with a bacterial artificial chromosome containing human LMNA engineered to contain the G608G mutation lack most systemic features of progeria but exhibit loss of smooth muscle cells and collagen deposition in the media of the aorta.13 Mice with a homozygous Lmna G609G mutation, which corresponds to the human LMNA G608G mutation that causes altered RNA splicing, similarly have loss of vascular smooth muscle cells in the aortic arch, as well as systemic features of progeria.14 Another strain of mice, LmnaG609G/G609G, develop near-complete loss of smooth muscle cells in the media of the ascending aorta as well as advanced adventitial fibrosis by four months of age.15 Taken together, it appears that the predominant common pathological characteristics of the large vessels in children with HGPS and mouse models of the disease are marked loss of vascular smooth muscle cells and arterial adventitial thickening and fibrosis, more severe than that typically observed in the atherosclerosis of physiological aging4
In contrast to humans with HGPS, where early onset aggressive atherosclerosis occurs, none of the mouse progeria models above with defects in prelamin A processing develop all of the features of atherosclerosis, particularly accumulation of lipids and lipid-laden macrophages within the intima of the vessel wall. This difference is likely because the mouse, as a species, is highly resistant to atherosclerosis. Only mice with genetic defects in lipid metabolism, such as Apoe−/− mice deficient in apolipoprotein E or Ldlr−/− mice deficient in low-density lipoprotein receptor, develop atherosclerosis.16 Hamczyk et al5 now report that male (female mice were not used in their studies) Apoe−/−LmnaG609/G609 mice with a progeria phenotype develop atherosclerosis, which includes medial lipid deposition and low-density lipoprotein retention, along with adventitial thickening and loss of smooth muscle cells within the media. This alone is perhaps not a surprising discovery in that these mice have the combined pathology seen in Apoe−/− and homozygous progerin-expressing LmnaG609G/G609G mice. However, Apoe−/−LmnaG609/G609 mice have a higher atherosclerotic burden (reported in the aortic arch and thoracic aorta) than Apoe−/−Lmna+/+ mice, indicating that the vascular changes induced by progerin accelerates the atherosclerosis in Apoe−/− mice. Mice fed either a chow or high-fat diet show similar pathological features but were more pronounced for the latter. However, Apoe−/−LmnaG609G/G609G mice had the same short lifespan as Apoe+/+LmnaG609G/G609G, demonstrating that accelerated atherosclerosis does not hasten death and that LmnaG609G/G609G mice apparently die from other systemic progeroid defects caused by the ubiquitous expression of progerin. This is in sharp contrast to children with HGPS, with only a heterozygous LMNA mutation, who also express progerin ubiquitously but generally succumb to myocardial infarction and stroke resulting from atherosclerosis.
To circumvent systemic effects of ubiquitous progerin overexpression, Hamczyk et al5 also generated a mouse with vascular smooth muscle cell-restricted progerin expression. To do so, they started with mice having a conditional mutant Lmna G609G allele.14 By crossing these mice to an appropriate Cre driver line and then to Apoe−/− mice, the authors created strains expressing progerin selectively in vascular smooth muscle cells. Satisfyingly, the Apoe−/−LmnaG609/G609 mice exhibited accelerated atherosclerosis independent of other systemic progeroid symptoms and died prematurely, likely due to the sequelae of atherosclerosis. The atheromata in these mice had features of vulnerable plaque and some data, although not quite conclusive, suggested that they suffered from myocardial infarctions. Thus, these mice may provide a useful preclinical model to study atherosclerosis-associated premature death in children with progeria and possibly more generally. Mice generated by similar methods expressing progerin selectively in macrophages rather than smooth muscle cells did not have accelerated atherosclerosis. Furthermore, mice expressing progerin in vascular smooth muscle cells on the Apoe+/+ background also had loss of these cells and aortic adventitial thickening but did not develop atherosclerosis and had a lifespan similar to that of Apoe+/+ mice. Thus, combining the Apoe−/− and HGPS (LmnaG609G/G609G) mouse models and restricting progerin expression to vascular smooth muscle cells were the critical innovations in this study that allowed these investigators to develop a novel model for studying the role of vascular smooth muscle cells in driving atherosclerosis.
It is important to point out that the results of Hamczyk et al5 are reminiscent of past research showing that chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis in Apoe−/− mice.17 This suggests that most or all of the effects of progerin on atherosclerosis observed in the current experiments are a result of the previously-described vascular smooth muscle cell loss in mice expressing this prelamin A variant. But understanding precisely how progerin promotes vascular smooth muscle cell loss remains to be determined and may provide an important link between lamin A processing and the atherosclerosis occurring in progeria.
HGPS is a monogenic disease with progeroid defects beyond just early-onset CVD. Do studies on mouse models of this extraordinarily rare syndrome with features of accelerated aging provide any information relevant to the extraordinarily common atherosclerosis seen with physiological aging? Sporadic use of the same cryptic splice site in LMNA that is activated in HGPS leads to very low levels of progerin expression in all individuals.18 One study has suggested that low levels of progerin can be detected in coronary arteries of individuals without HGPS and that the quantity increases with age.4 Similarly, prelamin A is normally expressed, albeit transiently, in nearly all cells and perturbations of ZMPSTE24-catalyzed processing could lead to its accumulation. Indeed it has been reported that unprocessed prelamin A accumulates in vascular smooth muscle cells in human arteries and atherosclerotic lesions of old individuals and that vascular smooth muscle cells passaged in vitro accumulate prelamin A due to decreased expression or activity of ZMPSTE24.19 Hence, one could hypothesize that farnesylated prelamin A or its farnesylated variant progerin may function in atherosclerosis of physiological aging, perhaps by causing vascular smooth muscle cell loss in a species – humans – that is not as resistant to atherosclerosis as mice. Should further research prove this hypothesis true, treatment with drugs that target farnesylated prelamin A or progerin, such as those used in genetically modified mice with alterations in prelamin A processing or in children with progeria,10,11,13,14, may someday be used to help prevent atherosclerosis that occurs with physiological aging.
Footnotes
DISCLOSURES
Dr Worman and Dr Michaelis are principal investigators on a pending NIH grant (R21AG058032) entitled “Role of permanently farnesylated prelamin in the cardiovascular disease of aging.” Dr Michaelis is funded for progeria studies by NIH grant R01GM041223.
Contributor Information
Howard J Worman, Department of Medicine and Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY.
Susan Michaelis, Department of Cell Biology, The Johns Hopkins University School of Medicine, Baltimore, MD.
References
- 1.Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119:1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Lévy N. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamczyk MR, Villa-Bellosta R, Gonzalo P, Andrés-Manzano MJ, Nogales P, Bentzon JF, López-Otín C, Andrés V. Vascular smooth muscle-specific progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation. 2018 doi: 10.1161/CIRCULATIONAHA.117.030856. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michaelis S, Hrycyna CA. Biochemistry. A protease for the ages. Science. 2013;339:1529–1530. doi: 10.1126/science.1236764. [DOI] [PubMed] [Google Scholar]
- 7.Barrowman J, Wiley PA, Hudon-Miller SE, Hrycyna CA, Michaelis S. Human ZMPSTE24 disease mutations: residual proteolytic activity correlates with disease severity. Hum Mol Genet. 2012;21:4084–4093. doi: 10.1093/hmg/dds233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Lichter-Konecki U, Anyane-Yeboa K, Shaw JE, Lu JT, Östlund C, Shin JY, Clark LN, Gundersen GG, Nagy PL, Worman HJ. A mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder. J Cell Sci. 2016;129:1975–1980. doi: 10.1242/jcs.187302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergo MO, Gavino B, Ross J, Schmidt WK, Hong C, Kendall LV, Mohr A, Meta M, Genant H, Jiang Y, Wisner ER, Van Bruggen N, Carano RA, Michaelis S, Griffey SM, Young SG. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodríguez F, Tryggvason K, López-Otín C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 11.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 13.Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:3250–3255. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osorio FG, Navarro CL, Cadiñanos J, López-Mejía IC, Quirós PM, Bartoli C, Rivera J, Tazi J, Guzmán G, Varela I, Depetris D, de Carlos F, Cobo J, Andrés V, De Sandre-Giovannoli A, Freije JM, Lévy N, López-Otín C. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra107. doi: 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- 15.Lee JM, Nobumori C, Tu Y, Choi C, Yang SH, Jung HJ, Vickers TA, Rigo F, Bennett CF, Young SG, Fong LG. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016;126:1592–1602. doi: 10.1172/JCI85908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- 17.Clarke MC, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, Bennett MR. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529–1538. doi: 10.1161/CIRCRESAHA.108.175976. [DOI] [PubMed] [Google Scholar]
- 18.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200–2210. doi: 10.1161/CIRCULATIONAHA.109.902056. [DOI] [PubMed] [Google Scholar]