

ABSTRACT
Inhibitors of DNA methyltransferases (DNMTis) or histone deacetylases (HDACis) are epigenetic drugs which are investigated since decades. Several have been approved and are applied in the treatment of hematopoietic and lymphatic malignancies, although their mode of action has not been fully understood. Two recent findings improved mechanistic insights: i) activation of human endogenous retroviral elements (HERVs) with concomitant synthesis of double-stranded RNAs (dsRNAs), and ii) massive activation of promoters from long terminal repeats (LTRs) which originated from past HERV invasions. These dsRNAs activate an antiviral response pathway followed by apoptosis. LTR promoter activation leads to synthesis of non-annotated transcripts potentially encoding novel or cryptic proteins. Here, we discuss the current knowledge of the molecular effects exerted by epigenetic drugs with a focus on DNMTis and HDACis. We highlight the role in LTR activation and provide novel data from both in vitro and in vivo epigenetic drug treatment.
KEYWORDS: Epigenetic therapy, endogenous retroviral elements, viral mimicry
Introduction
Cancer is driven by a combination of genetic and epigenetic alterations, which together modulate gene expression patterns. This cooperativity of genetic and epigenetic events is best exemplified by recent genome sequencing efforts of international consortia identifying mutations in proteins that control and regulate the epigenome [1,2]. These proteins read, write, or modify a complex interplay of epigenetic processes involving chromatin remodeling, histone modifications, DNA methylation, and RNA-mediated targeting [3–6]. In healthy cells, epigenetic modifications regulate tissue-specific and developmental processes as for example the inactivation of one X-chromosome in a female cell [7,8], regulation of the expression of imprinted genes through allele-specific modification of imprinting control regions [9], tissue-specific cell differentiation, or age-related processes [10,11]. Furthermore, epigenetic marks are involved in the silencing of retrotransposable elements in the genome [12,13]. Recent genome-wide profiling highlights the complexity of epigenomic patterns and their alterations in cancer.
DNA methylation
Methylation of cytosine is the most abundant DNA modification leading to 5-methylcytosine, which occurs predominantly in CpG dinucleotides and has been shown to affect transcriptional regulation [14]. Members of the highly conserved family of DNA methyltransferases (DNMTs) catalyze DNA methylation throughout the cell cycle [15]. DNMT1 maintains DNA methylation patterns, and DNMT3A and DNMT3B methylate DNA de novo. The majority (70–80%) of the 28 million CpG dinucleotides are generally methylated, only less than 3 million CpG sites, mostly located in CpG-rich regions designated CpG islands (CGIs), are largely unmethylated [16]. In line with the importance of DNA methylation in developmental processes, the majority of dynamic CpG sites are located within cis-regulatory elements.
Histone modifications
Besides chemical modification of DNA, a multitude of covalent post-translational histone modifications regulate chromatin function [17]. A complex regulatory system of proteins, referred to as ‘writers’, ‘readers’, and ‘erasers’ of histone marks [2,18] regulates the numerous histone modifications. Methylation, phosphorylation, and acetylation of the basic histone tails are catalyzed by enzymes, which share evolutionarily conserved domains. These post-translational modifications alter the physical properties of histone tails and affect the interactions of histones with DNA, non-histone proteins, and between each other [19]. For example, acetylation of lysine residues reduces the positive charge of the histone tails and consequently the interaction with the negatively charged backbone of the DNA double helix leading to a decondensation of chromatin [20]. Histone deacetylases (HDACs) are an important class of ‘erasers’ that antagonize histone acetyltransferases (HATs) by removing acetylation from histones and non-histone proteins [21]. Eighteen HDAC proteins are encoded in the human genome which are categorized into four classes (I, IIa, IIb, III, and IV) based on their sequence similarity to yeast counterparts and dependence on Zn2+ (class I, II, and IV) or the need of a NAD+ cofactor for enzymatic activity (class III). Loss of many of the HDACs results in embryonic lethality or strong developmental defects. Such drastic phenotypes reflect the important role of HDACs in proper down regulation of genes [22].
Transcription regulation
The regulation of transcriptional activity is a complex interplay of different mechanisms defining the ‘epigenetic code’. For example, DNA methylation of CGIs in promoter regions has been correlated with transcriptional repression [23]. Similarly, histone 3 lysine 9 di- and trimethylation (H3K9me2/3) mark repressed, while histone 3 lysine 4 mono- and trimethylation (H3K4me1/3) mark transcriptionally active sites [24,25]. However, causes and consequences of transcriptional activation or repression remain debated, since most knowledge is based on correlative studies. Recently, several studies showed causative roles for both DNA methylation and histone modifications by targeting epigenetic factors using the CRISPR-dCas9 technique [26]. Targeting of DNMT3A to promoter regions caused hypermethylation and consequently transcriptional repression of several genes [27,28]. In line with this observation, recruitment of ten-eleven translocation (TET) enzymes, which are involved in DNA demethylation, activated gene expression [29]. In contrast, catalytically inactive DNMT3A was not able to alter transcriptional activity at these sites [27]. Accordingly, removal of H3K4 methylation and H3K27 acetylation from enhancers repressed proximal transcription [30], whereas introduction of H3K27 acetylation at promoters enhanced transcriptional activity [31].
Epigenetic drug treatment
In contrast to genetic mutations, epigenetic modifications are potentially reversible and, hence, are attractive targets for cancer prevention and treatment (Figure 1). First epigenetic drugs have been developed decades ago and have been tested in in vitro (cell culture) and in vivo (mouse models, clinical trials) assays [32]. A multitude of small-molecule inhibitors directed against distinct epigenomic components or single regulators has since then been developed and tested: Two DNMT inhibitors (DNMTis), 5-azacytidine (Azacitidine, Vidaza®) and 5-aza-2'-deoxycytidine (Decitabine (DAC), Dacogen®), with considerable response rates and survival benefits for patients with MDS and AML; as well as five histone deacetylase inhibitors (HDACis), Vorinostat (SAHA, Zolinza®), Romidepsin (Istodax®), Belinostat (Beleodaq®), Tucidinostat (chidamide, Epidaza®) and Panobinostat (Farydak®). Four of these HDACis received FDA approval and one, Tucidinostat, has been approved in China. The first four HDACis are used for the treatment of peripheral T-cell lymphomas, the fifth, Panobinostat, for the combinatorial treatment of myeloma [33–35]. In March 2014, another pan-HDACi, Pracinostat (SB939), has been granted Orphan Drug status for the treatment of acute myelocytic leukemia (AML) and T-cell lymphoma. In 2016, Pracinostat received therapy designation by the FDA in combination with azacitidine for the treatment of patients with newly diagnosed acute myeloid leukemia (AML) who are older than 75 years of age or unfit for intensive chemotherapy. In clinical phase II and III studies, Pracinostat is currently being investigated in combinatorial treatment with Azacytidine (in MDS and AML patients) or with the JAK-inhibitor Ruxolitinib (in patients with Myelofibrosis). Current activities focus on the development of novel small molecule inhibitors directed against regulators of the epigenome other than DNMTs and HDACs to further improve the efficiency and specificity of therapies [36]. Under examination are, for example, the pharmacological inhibition of lysine acetyltransferases (KATs, e.g. Tip60) [37] or of BET family bromodomain proteins, which recognize acetylation of lysine residues [38]. BET inhibition has already entered clinical phase I trials [39]. Additionally, polypharmacological agents have recently been developed such as dual kinase/BET inhibitors, dual HDAC/BET inhibitors and agents that degrade BET family proteins, e.g. proteolysis-targeting chimeras (PROTACs) [40]. Other epigenetic targets are the histone 3 lysine 79 (H3K79) methyltransferase DOT1L, the catalytic subunit of Polycomb repressive complex 2 (PRC2) EZH2, or the histone demethylase LSD1 [41]. Combinatorial treatment with an LSD1-inhibitor and DNMT1- or HDAC-inhibitor showed synergistic effects in gene reactivation and increased the therapeutic efficacy of the DNMT1- and HDAC-inhibitors [42]. In contrast to their treatment successes of hematologic malignancies, epigenetic agents have not shown significant efficacy as monotherapy against solid tumors. However, recent studies of solid tumor treatment demonstrated that epigenetic drugs have favorable modifier effects when combined with other epigenetic agents, chemotherapy, hormonal therapy, or immunotherapy [43–46]. Future studies will show whether combinatorial treatments are pivotal in using epigenetic drugs.
Figure 1.
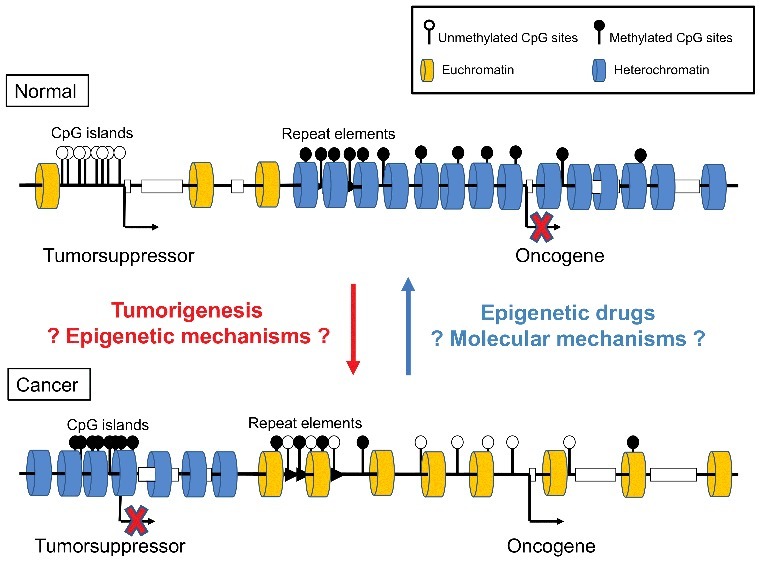
Simplified models for epigenetic regulation
Gene promoters are usually CpG-rich and frequently associated with CpG islands. In normal cells (upper), the promoter of a tumor suppressor gene is unmethylated and the chromatin in an active, euchromatic state. In cancer cells (lower), the promoter of a tumor suppressor gene is methylated and in an inactive, heterochromatic state. In contrast, an oncogene's promoter is methylated and inactive in normal cells and unmethylated and active in cancer cells. Repetitive elements are usually embedded in densely packed (heterochromatic) chromatin in normal cells and in more loosely packed chromatin in cancer cells. Epigenetic mechanisms involved in tumor transformation and chromatin remodeling following epigenetic drug treatment are currently under intensive investigation.
DNMT-inhibitors
Silencing of tumor suppressor genes by promoter hypermethylation is a common feature of tumors [47]. Inhibition of the maintenance DNA methyltransferase DNMT1 leads to DNA demethylation upon DNA replication and can thus rescue the expression of tumor suppressor genes and cell cycle regulators [48,49]. Due to their therapeutic potential, many DNMT inhibitors (DNMTis) have been developed and used for the treatment of hematopoietic malignancies [33]. Treatment of cells with Azacitidine or Decitabine leads to their incorporation into the newly synthesized DNA strands of the dividing cells, where they covalently bind and irreversibly inhibit DNMT1 [50]. Both Azacitidine and Decitabine have been used extensively in the treatment of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) with several phase II/III trials demonstrating an increased overall survival rate and improved quality of life for the patients [33,51]. Zebularine and Guadecitabine are additional nucleoside-analog DNMTis, which are currently under investigation for their therapeutic potential [33,52].
HDAC-inhibitors
HDACs deacetylate histones but also other proteins such as transcription factors or proteins involved in DNA repair [53]. Deacetylation of lysine residues in the histone tails is associated with condensed chromatin and, thus, with repression of transcription. Human HDACs comprise 18 members categorized into four groups [54]. The observation of HDAC overexpression in diverse malignancies led to the assumption that HDACs may act as oncogenes [53]. Therefore, HDACs became a novel target for therapeutic approaches, and various inhibitors were developed. These compounds comprise a set of five different classes: benzamides, hydroxamic acids, fatty acids (short chain), sirtuin inhibitors and cyclic tetrapeptides. HDAC inhibitors (HDACis) can selectively target a specific HDAC isoenzyme (“isoenzyme-selective inhibitor”) or can act on some but not all HDAC enzymes (“pan-inhibitor”) [54]. For our experiments, Pracinostat (SB939), a small-molecule panHDACi based on hydroxamic acid, which is binding all HDAC isozymes with similar affinity with the exception of HDAC6 and 7 has been used. Generally, antitumor effects were increased by combined administration of HDACis with other drugs. For instance, HDACis have been combined with the DNMTis Azacitidine (Aza) [54], or Decitabine [55–57], with agents increasing the abundance of reactive oxygen species [58] or with proteasomal inhibitors [59].
Although the precise mode of action of HDACis remains elusive, some of them have been successfully applied in the clinic and, hence, been approved by health authorities like the US Food and Drug Administration (FDA). So far, it is known, that HDACi treatment leads to cell cycle arrest (G1 or G2 phase) and apoptosis of cancer cells [54,60]. Moreover, autophagy and angiogenesis is reduced and modulation of the immune response has been observed. Cyclin dependent kinase inhibitor p21 (CDKN1A) expression increases after HDACi treatment, and p21 inhibits complex formation of cyclins and cyclin dependent kinases [61]. Extrinsic as well as intrinsic apoptotic pathways are triggered by HDAC inhibition [54].
Bromodomain inhibitors
Bromodomains are conservative modular domains which play crucial roles of readers for recognizing acetyl-lysine binding proteins on histone tails, recruiting complex to act on promoter region and drive downstream gene transcription [62]. Thus developing antagonists targeting BET bromodomain may contribute to potent epigenetic therapies [63,64]. The mammalian BET family protein is comprised of four members including ubiquitously expressed BRD2, BRD3, BRD4 and germ cells restricted BRDT. Among the BET family, BRD4 has been found to play a diverse role in the regulation of cell cycle progression and thereby responsible for the development of a variety of cancers [65]. BRD4 usually binds with the acetylated histones in chromatin and with positive transcription factor b (p-TEF-b) [66]. JQ1 is a thiendizaepine-based first small inhibitor molecule designed against Bromodomain (BRD4) which could bind to acetyl-lysine recognition motifs or the bromodomain and extraterminal (BET) family of bromodomain proteins. Since the first research reports revealed that JQ1 could competitively bind to bromodomain and replace position of oncoprotein from chromatin in 2010 [67], increasing studies provided evidence about pharmacological inhibition of JQ1 to be potential therapeutic treatments in several different disease models. To be more specific, JQ1 could disrupt androgen recruitment through targeting BET bromodomain, decreasing cell proliferation of castration-resistant prostate cancer cells [38]. JQ1 as a BET bromodomain inhibitor could also downregulate MYC expression to reduce cell viability of medulloblastoma which may cause malignant brain tumors in children [68]. Other studies reported about the potential synergetic effect of BET inhibitors with some other epigenetic drugs, especially in lymphomas treatment [69]. JQ1 produced the significant synergetic effect with rapamycin to inhibit the growth and survival of Osteosarcoma cells [70].
The synthesis of small molecules to target bromodomain is still continuing, and several novel inhibitors have already shown the potency to suppress growth of cancer cells [71]. Moreover, few of BET bromodomain inhibitors entered clinical trials, showing inhibition in several hematological malignancies from patients and effect on coronary atherosclerosis [72,73].
Activation of human endogenous retroviral elements by DNMT inhibition
Recent investigations of low-dose DNMTi treatment in cancer cell lines identified a novel molecular mechanism of action. Two publications on DNMTi (5-azacytidine and 5-aza-2´-deoxycytidine) described the generation of double-stranded RNA (dsRNA) originating from codogenic and non-codogenic human endogenous retroviral elements (HERVs) [74,75]. The dsRNAs triggered the activation of the dsRNA-targeting viral response pathway mediated by MAD5/MAVS/IRF7 followed by interferon response and apoptosis. Moreover, a high anti-viral gene expression signature correlated with durable response in melanoma patients that received immune checkpoint therapy [74]. Delayed gene expression changes in many IRF7 target genes and no correlation with DNA methylation changes suggested that these events are secondary to hypomethylating agent-mediated effects.
Combined treatment with chemotherapy or immune checkpoint inhibitors
HDACi- and DNMTi-treatment have shown their efficacy when combined with chemotherapeutic drugs [76]. DNMTi treatment, for example, decreased platinum resistance of cancer cells [77], and clinical phase I trials proved that Decitabine in combination with carboplatin could lower chemotherapy resistance in recurrent platinum-resistant ovarian cancer [78]. Other preclinical studies [60,79] and clinical trials [80] also revealed HDACis as chemosensitizers as they increased sensitivity of cancer cells under chemotherapeutic treatments.
Since HDACi and DNMTi were demonstrated to be able to affect the immune system, the combination of epigenetic drugs and immune checkpoint inhibitors was considered to be a promising cancer therapy [46,81]. Several preclinical studies indicate practicability of co-treatment with HDACi [82,83] or DNMTi [84,85] and immune checkpoint inhibitors such as anti-PD-1 and anti-CTLA-4 antibodies in both in vitro and in vivo experiments. Moreover, many clinical trials are being conducted to evaluate synergistic activity under HDACi or DNMTi combined with immunotherapies in different types of cancer. Clinical phase I/II trials are performed to assess efficacy of combination with Vorinostat and Pembrolizumab (Keytruda®; human IgG4 anti-PD-1 monoclonal antibody) in patients with non-small cell lung cancer, advanced renal cell carcinoma or hormone-resistant breast cancer. The co-treatment of Decitabine and Nivolumab (Opdivo®; human IgG4 anti-PD-1 monoclonal antibody) or Pembrolizumab will also be evaluated in patients with AML, metastatic VRC, or NSCLC in clinical phase II trials [46].
Transposable elements
Transposable elements (TEs) are mobile genomic segments which represent roughly 50% of the human genome [86]. During evolution, TEs have accumulated many mutations which disabled the vast majority to transpose. The intermediates of mobilization define two major TE classes: DNA transposons constitute about 3% of the human genome [86] and transpose in an RNA-independent manner, while retrotransposons require a reverse-transcribed RNA intermediate [87].
Retrotransposons are subdivided into those with and those without long terminal repeats (LTRs). In humans, non-LTR retrotransposons are in the majority and some are still active [88]. HERVs, which make up about 8% of the human genome, are retrotransposons consisting of the genes gag, pol, and env which are flanked by two LTR elements [89]. The LTRs have promoter function required for proviral transcription [90,91]. Loss of internal proviral sequences through homologous recombination between the LTRs results in the formation of solitary LTRs that are unable to retrotranspose [89] and are with about 650,000 copies (90%) in the majority [92]. Transposition of the remainder can have a detrimental effect on genome integrity of the host cell and, is therefore prevented by epigenetic surveillance mechanisms including DNA methylation and repressive histone modifications [93].
Epigenetic regulation of retroviral elements
Most LTRs are heavily methylated in somatic cells [94,95], and it has been proposed that DNA methylation has primarily evolved to silence HERVs and other TEs [96]. Loss of DNA methylation results in LTR expression in somatic cells [97–99]. How LTR activity is contained has been extensively studied in embryonic mouse cells. Additional to DNA methylation, the interplay of up to several hundred proteins may be required during early embryonic development, when the genome is largely unmethylated [100,101]. Many of these proteins are involved in chromatin remodeling or contribute to diverse post-translational modifications of histones [102]. The methyltransferase SETDB1, for example, catalyzes the addition of methyl groups to H3K9 and is required for LTR silencing in mouse embryonic stem cells (ESCs) but not in mouse embryonic fibroblasts [100]. Many LTRs are covered with H3K9me3 in mouse ESCs, while LTRs in differentiated cells are largely devoid of this histone mark [103]. The TRIM28 (also known as KAP1) co-repressor recruits SETDB1 to certain ERV superfamilies and is also essential for LTR silencing [104]. Other histone modifying enzymes such as the histone demethylase KDM1A (also known as LSD1) and the NURD HDAC complex have also been shown to interact with TRIM28 [105]. In line with these findings, HDACis activate the expression of latent exogenous retroviruses after host cell integration, suggesting that histone deacetylation is involved in retroviral silencing [106]. Despite the evolution of distinct epigenetic silencing mechanisms, different families of LTRs are coordinately expressed in a developmental and tissue-specific manner [107,108] and often serve as alternative promoters or cis-regulatory elements [92,109–112]. Given the high sequence homology between members of the same LTR subfamily, they are also particularly suitable for the evolution of transcriptional networks that require the orchestrated expression of stage- or stimulus-specific genes [113,114].
Results
Activation of HERV promoters by DNMT-and HDAC-inhibitors
In order to better understand the molecular events upon epigenetic drug treatment, we profiled in the lung cancer cell line NCI-H1299 genome-wide transcription start site activities using cap-analysis of gene expression (CAGE)[115] as well as associated changes in DNA methylation (whole-genome bisulfite sequencing (WGBS)) and chromatin (chromatin immunoprecipitation sequencing (ChIP-Seq)) following treatment with DNMTi (Decitabine), HDACi (Pracinostat, SB939) or both. Our unprecedented CAGE study revealed that all three drug combinations largely induced de novo transcription from previously uncharacterized transcription start sites (TSSs) rather than expression from canonical promoters [116]. We observed activation of more than 2000 treatment-induced non-annotated TSSs (TINATs), all of them not being annotated in common genome databases before. The strongest effects were found with the combinatorial treatment (Decitabine plus SB939). While DNMTi treatment-dependent transcriptional activation coincided with DNA hypomethylation and gain of activating histone marks, HDACi treatment specifically induced a subset of TINATs in association with H2AK9ac, H3K14ac, and H3K23ac. Strikingly, the vast majority of these newly identified TSSs originated in LTRs, especially in LTR12C elements from the HERV9 family. Moreover, newly induced transcripts contained open reading frames either encoding for known, truncated, chimeric, or truncated plus chimeric proteins. New transcripts which initiated within introns of protein-coding genes were frequently spliced with downstream exons; many such transcripts contained a truncated open reading frame, potentially encoding novel immunogenic proteins owing to their predicted property to bind to MHC class I molecules and, thus, to represent novel surface antigens. The up-regulation of immune system-related genes following DNMTi treatment has been connected in previous studies with the transcription of codogenic HERVs and the formation of dsRNA [74,75,85]. Using our own CAGE dataset, we were able to detect the reactivation of double-stranded ERV RNAs as well as the induction of Aza-induced viral defense genes.
LTR12C activation by epigenetic drugs is a general but drug-specific phenomenon
We extended DNMTi and HDACi treatment to four more cancer cell lines, the leukemic lines HL60, K562, Raji, and Jurkat, and profiled genome-wide TSS activities by nano cap-analysis of gene expression (nanoCAGE) [117]. HERVs were activated in all four leukemic cell lines, and again, in a synergistic mode upon combinatorial treatment with Decitabine (DAC) plus SB939. Moreover, cryptic transcripts initiating in LTR12C elements were the most prominent (Figure 2).
Figure 2.
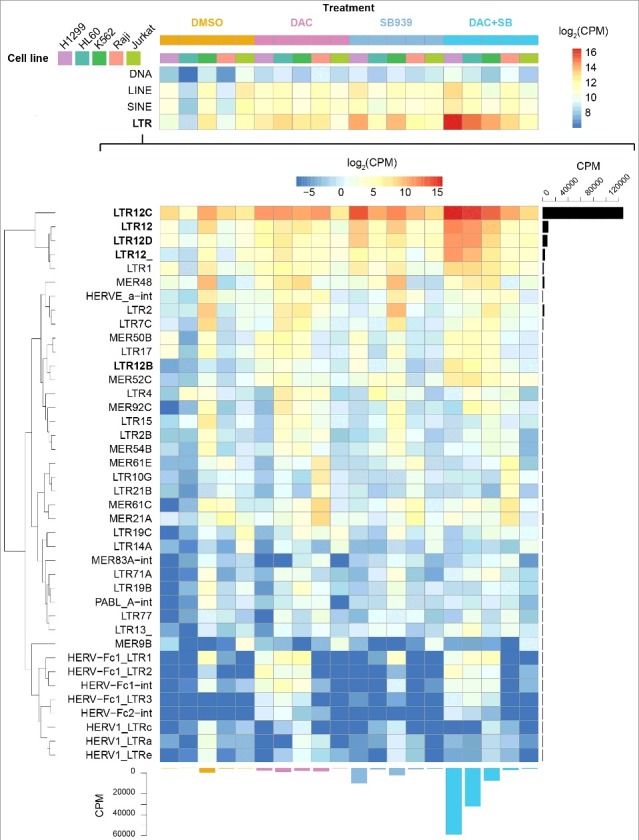
Expression analysis of transposable elements upon epigenetic drug treatment in various cell lines by nano-CAGE sequencing
The log2 of transposable elements (TEs) expression of five cell line models (NCI-H1299, HL60, K562, Raji, Jurkat) upon epigenetic drug treatment (SB939 (SB), Decitabine (DAC), combinatorial treatment (DAC+SB), and control (DMSO)) is depicted in summary for the different TE families (upper part of heatmap) as well as in detail for the different LTR subgroups (lower part of heatmap). CPM, counts per million.
To examine if LTR12C activation upon epigenetic drug treatment also occurs in vivo, we analyzed human peripheral blood mononuclear cells (PBMNCs) from three patients treated with the pan-HDACi Vorinostat (for study details refer to Suppl. Table 1). Cells were isolated from peripheral blood samples which were collected at 0h, 0.5h, 1h, 1.5h, 2h, 4h, 6h and 8h after drug administration. In all three patients, activation of LTR12C elements could be observed, the patients differing only in the activation kinetics (Figure 3). We therefore conclude that activation of cryptic transcripts upon DNMTi and HDACi treatment is a general rather than a tissue specific phenomenon, also occurring in human patients. In patient 1 and 2, the LTR12C expression was analyzed after one week of Vorinostat administration, whereas patient 3 had already taken Vorinostat 5 months continuously and has reached the maximum tolerated dose before the LTR12C expression was investigated. Therefore, one could assume that for patient 3 the LTR12C expression level was already higher than for the other two patients resulting in a lower relative increase of LTR12C expression during the investigated time points.
Figure 3.
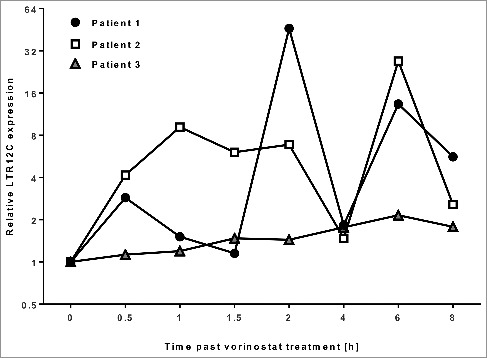
LTR12C expression in peripheral blood mononuclear cells (PBMNCs) upon Vorinostat (suberoylanilide hydroxamic acid, SAHA) treatment
Time course of LTR12C expression (normalized to housekeepers and relative to untreated samples) in three patients treated with Vorinostat. LTR12C transcription was induced upon Vorinostat intake in all three patients in an individual manner.
We examined additional types of inhibitors, such as those targeting chromatin-reading bromodomains, HATs, or histone methyl transferases (Table 1), for their capability to induce LTR12C expression in NCI-H1299 cells. Only the bromodomain inhibitor JQ1 increased LTR12C expression (Figure 4), suggesting that HERV silencing is exerted by specific epigenetic mechanisms.
Table 1.
Compounds used to test for LTR12C activation in H1299 cells.
| Drug | Target | MW | Recommended conc. | Used conc. |
|---|---|---|---|---|
| JQ1 | BRD4 | 456.99 | 500 nM | 250 nM |
| PU139* | HAT | 246.26 | 20 μM | 10 μM |
| GSK2699537* | LSD1 | 232.37 | 1 μM | 1 μM |
| UNC0638 | G9a | 509.73 | 500 nM | 500 nM |
| SGC0946 | Dot1L | 618.57 | 10 nM | 10 nM |
| GSK343 | EZH2 | 541.69 | 500 nM | 500 nM |
| TW36* | Spindlin1 | 350.48 | 50 μM | 25 μM |
Epigenetic compounds (Drug); specific target (Target); molecular weight (MW); recommended and used concentration for single drug treatment of NCI-H1299 cells; *[126–128].
Figure 4.
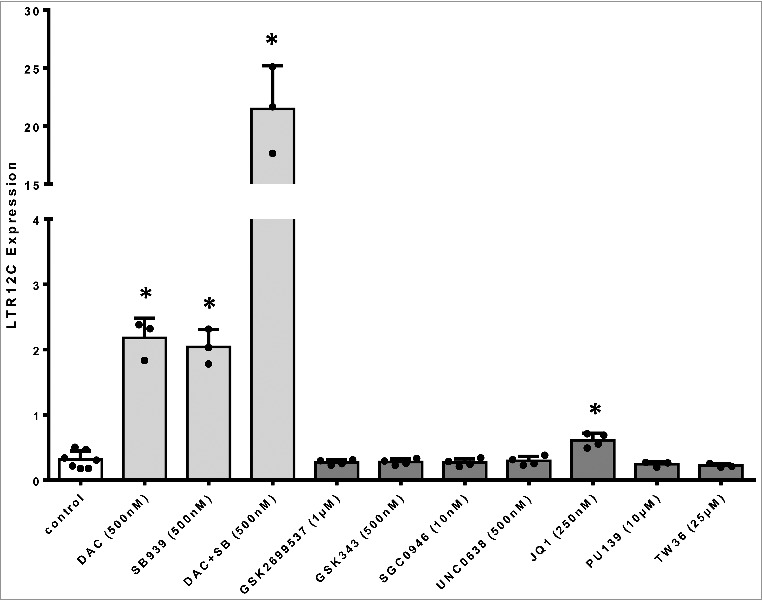
LTR12C expression following epigenetic drug treatment
LTR12C expression upon epigenetic drug treatment of NCI-H1299 cells. Light grey bars depict LTR12C expression (normalized to housekeepers) following treatment with DNMTi (DAC), HDACi (SB939) or both. Dark gray bars depict LTR12C expression (normalized to housekeepers) following treatment with compounds inhibiting other epigenetic targets (see Table 1. DNMTi and HDACi only treatment both induce LTR12C transcription, and a double treatment leads to a synergistic effect. Among the further compounds, only the bromodomain inhibitor JQ1 induces LTR12C transcription.
In conclusion, the activation of LTR12C retroviral elements can be found upon treatment with DNMT- and HDAC-inhibitors as well as by using JQ1-inhibitors and is a detectable phenomenon in different human cancer cell lines as well as in healthy human cells.
Discussion and Perspectives
Clinical relevance of ERV activation
The impact of epigenetic aberrations on carcinogenesis motivated the development of therapeutic strategies making use of drugs targeting the epigenome [118]. However, their mode of action and especially the global cellular effects of epigenetic drug treatment are only sparsely described. Recent publications highlight the effects of DNMTi and/or HDACi on human endogenous retroviral elements (HERVs) [74,75,81], or truncated ERVs of the LTR12C subgroup [116]. In two studies [74,75], treatment of cell lines using a DNMTi led to the induction of dsRNA which, in turn, induced an interferon-based viral mimicry response. Using the combinatorial treatment of 5-Azacytidine with HDACis, Topper and colleagues could achieve anti-tumor responses in non-small-cell lung cancer cell lines mediated by the induction of ERV9-1 accompanied by suppression of MYC signaling and an enhancement of the immune signaling due to upregulation of CCL5 secretion [81]. These findings suggest the potential for combining epigenetic drug treatment with immune checkpoint blockade. We demonstrated in our study [116] that DNMTi and HDACi treatment induces cryptic transcripts (TINATs) which are initiated from solitary LTR12C elements. Here we demonstrate that this induction is not only present in human cell lines (Figure 2) but also in normal PBMNCs from patients treated with Vorinostat (Figure 3). Of particular interest in the clinical setting is our observation that, at least in in silico analysis, TINATs encode peptides with immunomodulatory potential [116]. Polysomal fractionation analysis revealed in about one third of TINAT cases translational activity which underscores the possibility of truncated protein versions. To verify the existence of these predicted truncated peptides, proteomics studies are required.
Many open questions remain, e.g., whether TINAT induction also occurs in tumor cells of patients treated with epigenetic drugs. If so, patients may be stratified according to treatment response or outcome depending on presence or absence of TINATs and the occurrence of a higher mutational burden as an additional factor, as it has been described by Chiappinelli et al. [74] Possible immunomodulatory consequences of TINAT activation might serve as a prognostic marker for patients treated with epigenetic drugs.
Molecular mechanisms of HERV activation
The inhibition of specific HDACs was shown to be sufficient for HERV9-derived LTR12 activation [119]. Similarly, the induction of TINATs can occur upon HDACi treatment alone, in the absence of DNMTi. We observed that a subset of TINATs is induced upon HDACi treatment in association with the acetylation of H2AK9, H3K14, and H3K23, histone moieties different from the classical promoter- or enhancer-modulating sites of modification. The acetylation of these “non-canonical” histone sites might contribute to the synergistic effect of DNMTi plus HDACi double treatment which has already been described by others [120]. The elucidation of the mechanism and the identification of the participating factors for TINAT induction are urgent tasks to be resolved. Proteomic studies already pointed into a direction different from histone modifications but to proteins as targets of acetylation which are involved in the regulation of numerous cellular processes such as chromatin remodeling, cell cycle, splicing, nuclear transport, and actin nucleation [121,122].
TINAT induction by the BRD4 inhibitor JQ1
The repertoire of epigenetic drugs also includes inhibitors of bromodomain-containing chromatin readers like BRD4 which function in cell cycle progression and contribute to malignancies. When we tested diverse epigenetic drugs, also the BRD4-inhibitor JQ1 induced TINAT expression in NCI-H1299 cells (Figure 4). JQ1 acts as an acetyl-histone mimetic and prevents interactions between bromodomain proteins, acetylated histones, and transcription factors [67]. Consequently, JQ1 activates or represses genes and transposable elements, but it also enables reactivation of HIV-1 transcription in latently infected human T cells [123]. JQ1's mode of action in TINAT induction still remains to be clarified.
Concluding remarks
Treatment of cell lines with epigenetic drugs targeting diverse classes of epigenetic modifiers, including DNMTs, HDACs, and BET protein family members, can activate endogenous retroviral elements and massively induce novel transcripts of unknown function. Now the exciting task is to decipher if and how the diverse types of epigenetic drugs converge in their mode of action to specifically activate HERVs and to verify the existence of predicted truncated peptides resulting from splicing events of activated HERVs. And most important, clinical studies are needed to clarify the consequences of HERV activation upon epigenetic drug treatment in patients and to evaluate their immunomodulatory potential and their function as prognostic markers.
Materials and Methods
Cell culture and epigenetic drug treatment
NCI-H1299 (CRL-5803, ATCC), HL60 (ACC-3, DSMZ), K562 (ACC-10, DSMZ), RAJI (ACC-319, DSMZ), JURKAT (ACC-282, DSMZ) cells were grown in RPMI 1640 supplemented with 10% FCS and 1% PenStrep. Cell line authenticity and purity was confirmed using the Multiplex Cell Authentication and Cell Contamination Test by Multiplexion. Cells were treated with 500 nM (250 nM for HL60, 5 nM for RAJI, 10 nM for JURKAT) DAC, 500 nM SB939 (100nM for RAJI, 200nM for JURKAT), or 500 nM DAC (250 nM for HL60, 5 nM for RAJI, 10 nM for JURKAT) + 500 nM SB939 (100nM for RAJI, 200nM for JURKAT) for 72, 18, or 54 + 18 h, respectively, and compound-containing media was refreshed every 24 h.
Single treatment of epigenetic drugs from other different target classes (Table 1) was applied once at the beginning of the 72-hour treatment period, using a final concentration of 250 nM for JQ1, 10 µM for PU139, 1µM for GSK2699537, 500 nM for UNC0638, 10 nM for SGC0946, 500 nM for GSK343 and 25 µM for TW36, respectively. Recommended concentrations were taken from the literature for published compounds or from preliminary cytotoxicity tests on leukemic cells (data not shown). Due to cytotoxicity reasons in NCI-H1299, the recommended dosage for JQ1, PU139, and TW36 was reduced by 50%. After 72 hours, cells were harvested, RNA isolated, cDNA synthesized and LTR12C expression determined by qRT-PCR analysis.
Vorinostat treatment of study patients
All 3 patients gave consent to participation and were included in the study “Phase I/II intra-patient dose escalation study of Vorinostat in children with relapsed solid tumor, lymphoma or leukemia” (NCT-2007-11-02-1004; for study details refer to Suppl. Tabl 1). Blood was collected under continuous Vorinostat treatment once daily, during pharmakokinetic sampling collected before (0 min) and 30 min, 60 min, 90 min, 120 min, 4h, 6h, and 8h after oral Vorinostat intake (Suppl. Table 1). Using density-gradient centrifugation technique (see below), peripheral blood mononuclear cells (PBMNCs) were separated, RNA isolated, cDNA synthesized and LTR12C expression determined by qRT-PCR analysis.
Nano cap analysis of gene expression (nanoCAGE) sequencing
NanoCAGE was performed in one experiment on five treated cell line models (NCI-H1299, HL-60, K562, RAJI, JURKAT) by DNA form, Japan. Libraries were sequenced in paired-end mode on a HiSeq 2000 V4 system (125bp, paired-end) by the DKFZ Genomics and Proteomics Core facility. Resulting reads were quality trimmed using fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and aligned against the reference genome (hg19) using HISAT2 [124]. Only the first in pair read of uniquely mapped reads was retained to generate coverage tracks. Read counts for repeat classes and subfamilies were normalized to the library size using edgeR [125].
Isolation of peripheral mononuclear cells from peripheral blood samples
For qRT-PCR expression analysis, PBMNCs were isolated by Lymphoprep (Lymphoprep, 250ml, #01-63-12-002A, LOT 12GLS14, Fresenius Kabi Norge AS for Axis Shield PoC AS, Oslo, Norway) density-gradient centrifugation according to the manufacturer's protocol.
qRT-PCR expression analysis
RNA was transcribed to cDNA using random hexamers and Superscript III Reverse Transcriptase (Invitrogen) according to the manufacturer's instructions. Unless stated otherwise, expression analysis was performed on the Roche Lightcycler 480 system and target-gene expression was normalized to the housekeeping genes GAPDH, and HPRT1 (primer sequences in Supplementary Table 2).
Supplementary Material
Funding Statement
Deutsche Forschungsgemeinschaft [grant numbers SPP1463, FOR2674, W1461/4-1, Oe542/2-1, CRC992]; Deutsche Kinderkrebsstiftung [grant number DKS2009.16]. Funding details: the project was funded in part by DFG Priority Program SPP1463 and FOR 2674, German Cancer Research Consortium (DKTK), German Center for Lung Diseases (DZL). OW (W1461/4-1) and IO (Oe542/2-1) are funded by the Deutsche Forschungsge meinschaft (DFG). Investigator initiated Vorinostat phase I/II trial was supported by a grant from the Deutsche Kinderkrebsstiftung to OW (DKS 2009.16). MJ thanks the DFG (Deutsche Forschungsgemeinschaft, Project A04 within CRC992 Medical Epigenetics) for funding.
Acknowledgments
We thank M. Bähr, M. Helf, O. Mücke, K. Klimo and the Genomics and Proteomics Core Facility at the German Cancer Research Center for their excellent technical support and expertise.
Disclosures
ChristophPlass: Advisory board BioMedX
Olaf Witt: Scientific Advisory Board Novartis: Clinical Development of BRAF/MEKi in pediatric gliomas; Scientific Advisory Board Astra Zeneca: Clinical Development of MEKi in pediatric low grade gliomas/NF1.
References
- [1].You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. PMID:22789535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Plass C, Pfister SM, Lindroth AM, et al. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14:765–780. doi: 10.1038/nrg3554. PMID:24105274 [DOI] [PubMed] [Google Scholar]
- [3].Esteller M. Epigenetics in cancer. The New Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. PMID:18337604 [DOI] [PubMed] [Google Scholar]
- [4].Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. PMID:21941284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. PMID:22770212 [DOI] [PubMed] [Google Scholar]
- [6].Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. PMID:23239728 [DOI] [PubMed] [Google Scholar]
- [7].Brockdorff N. Chromosome silencing mechanisms in X-chromosome inactivation: unknown unknowns. Dev (Camb, Engl). 2011;138:5057–5065. doi: 10.1242/dev.065276. PMID:22069184 [DOI] [PubMed] [Google Scholar]
- [8].Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12:542–553. doi: 10.1038/nrg3035. PMID:21765457 [DOI] [PubMed] [Google Scholar]
- [9].Adalsteinsson BT, Ferguson-Smith AC. Epigenetic control of the genome-lessons from genomic imprinting. Genes. 2014;5:635–655. doi: 10.3390/genes5030635. PMID:25257202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. PMID:21663790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thiagarajan RD, Morey R, Laurent LC. The epigenome in pluripotency and differentiation. Epigenomics. 2014;6:121–137. doi: 10.2217/epi.13.80. PMID:24579950 [DOI] [PubMed] [Google Scholar]
- [12].Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci: CMLS. 2008;65:3329–3347. doi: 10.1007/s00018-008-8494-3. PMID:18818875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Leung DC, Lorincz MC. Silencing of endogenous retroviruses: when and why do histone marks predominate? Trends Biochem Sci. 2012;37:127–133. doi: 10.1016/j.tibs.2011.11.006. PMID:22178137 [DOI] [PubMed] [Google Scholar]
- [14].Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. PMID:22641018 [DOI] [PubMed] [Google Scholar]
- [15].Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. PMID:15952895 [DOI] [PubMed] [Google Scholar]
- [16].Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. PMID:23400093 [DOI] [PubMed] [Google Scholar]
- [17].Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. PMID:19234478 [DOI] [PubMed] [Google Scholar]
- [18].Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. PMID:11498575 [DOI] [PubMed] [Google Scholar]
- [19].Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. PMID:21321607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Workman JL, Kingston RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. PMID:9759497 [DOI] [PubMed] [Google Scholar]
- [21].Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res: MCR. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. PMID:17951399 [DOI] [PubMed] [Google Scholar]
- [22].Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. PMID:19065135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16 (Spec No 1):R50–R59. doi: 10.1093/hmg/ddm018. PMID:17613547 [DOI] [PubMed] [Google Scholar]
- [24].Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. PMID:20813257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. PMID:21116306 [DOI] [PubMed] [Google Scholar]
- [26].Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. PMID:23849981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vojta A, Dobrinic P, Tadic V, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. doi: 10.1093/nar/gkw159. PMID:26969735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McDonald JI, Celik H, Rois LE, et al. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol Open. 2016;5:866–874. doi: 10.1242/bio.019067. PMID:27170255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen H, Kazemier HG, de Groote ML, et al. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2014;42:1563–1574. doi: 10.1093/nar/gkt1019. PMID:24194590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mendenhall EM, Williamson KE, Reyon D, et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013;31:1133–1136. doi: 10.1038/nbt.2701. PMID:24013198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hilton IB, D'Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. PMID:25849900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sorm F, Vesely J. Effect of 5-aza-2'-deoxycytidine against leukemic and hemopoietic tissues in AKR mice. Neoplasma. 1968;15:339–343. PMID:5684460 [PubMed] [Google Scholar]
- [33].Navada SC, Steinmann J, Lubbert M, et al. Clinical development of demethylating agents in hematology. J Clin Invest. 2014;124:40–46. doi: 10.1172/JCI69739. PMID:24382388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. PMID:24382387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Moreau P. How I treat myeloma with new agents. Blood. 2017;130(13):1507–1513. doi: 10.1182/blood-2017-05-743203. PMID:28747306 [DOI] [PubMed] [Google Scholar]
- [36].Federation AJ, Bradner JE, Meissner A. The use of small molecules in somatic-cell reprogramming. Trend Cell Biol. 2014;24:179–187. doi: 10.1016/j.tcb.2013.09.011. PMID:24183602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Farria A, Li W, Dent SY. KATs in cancer: functions and therapies. Oncogene. 2015;34(38):4901–4913. doi: 10.1038/onc.2014.453. PMID:25659580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Asangani IA, Dommeti VL, Wang X, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. PMID:24759320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Albrecht BK, Gehling VS, Hewitt MC, et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J Med Chem. 2016;59:1330–1339. doi: 10.1021/acs.jmedchem.5b01882. PMID:26815195 [DOI] [PubMed] [Google Scholar]
- [40].Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol: Off J Eur Soc Med Oncol. 2017;28:1776–1787. doi: 10.1093/annonc/mdx157. PMID:28838216 [DOI] [PubMed] [Google Scholar]
- [41].Morera L, Lubbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenet. 2016;8:57. doi: 10.1186/s13148-016-0223-4. PMID:27222667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zahnow CA, Topper M, Stone M, et al. Inhibitors of DNA methylation, histone deacetylation, and histone demethylation: a perfect combination for cancer therapy. Adv Cancer Res. 2016;130:55–111. doi: 10.1016/bs.acr.2016.01.007. PMID:27037751 [DOI] [PubMed] [Google Scholar]
- [43].Juo YY, Gong XJ, Mishra A, et al. Epigenetic therapy for solid tumors: from bench science to clinical trials. Epigenomics. 2015;7:215–235. doi: 10.2217/epi.14.73. PMID:25942532 [DOI] [PubMed] [Google Scholar]
- [44].Schiffmann I, Greve G, Jung M, et al. Epigenetic therapy approaches in non-small cell lung cancer: update and perspectives. Epigenetics. 2016;11:858–870. doi: 10.1080/15592294.2016.1237345. PMID:27846368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Duruisseaux M, Esteller M. Lung cancer epigenetics: from knowledge to applications. Semin Cancer Biol. 2017. Sep 14. pii: S1044-579X(17)30166-9. doi: 10.1016/j.semcancer.2017.09.005. PMID:28919484 [DOI] [PubMed] [Google Scholar]
- [46].Mazzone R, Zwergel C, Mai A, et al. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigeneti. 2017;9:59. doi: 10.1186/s13148-017-0358-y. PMID:28572863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Weisenberger DJ. Characterizing DNA methylation alterations from the cancer genome atlas. J Clin Invest. 2014;124:17–23. doi: 10.1172/JCI69740. PMID:24382385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2 (Suppl 1):S4–S11. doi: 10.1038/ncponc0354. PMID:16341240 [DOI] [PubMed] [Google Scholar]
- [49].Daskalakis M, Nguyen TT, Nguyen C, et al. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2'-deoxycytidine (decitabine) treatment. Blood. 2002;100:2957–2964. doi: 10.1182/blood.V100.8.2957. PMID:12351408 [DOI] [PubMed] [Google Scholar]
- [50].Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticanc Res. 2013;33:2989–2996. PMID:23898051 [PubMed] [Google Scholar]
- [51].Lubbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol: Off J Am Soc Clin Oncol. 2011;29:1987–1996. doi: 10.1200/JCO.2010.30.9245. PMID:21483003 [DOI] [PubMed] [Google Scholar]
- [52].Issa JJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16:1099–1110. doi: 10.1016/S1470-2045(15)00038-8. PMID:26296954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Brit J Cancer. 2016;114:605–611. doi: 10.1038/bjc.2016.36. PMID:26908329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Eckschlager T, Plch J, Stiborova M, et al. Histone deacetylase inhibitors as anticancer drugs. Int J Mole Sci. 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Walton TJ, Li G, Seth R, et al. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate. 2008;68:210–222. doi: 10.1002/pros.20673. PMID:18092350 [DOI] [PubMed] [Google Scholar]
- [56].Cecconi D, Donadelli M, Dalla Pozza E, et al. Synergistic effect of trichostatin A and 5-aza-2'-deoxycytidine on growth inhibition of pancreatic endocrine tumour cell lines: a proteomic study. Proteomics. 2009;9:1952–1966. doi: 10.1002/pmic.200701089. PMID:19294695 [DOI] [PubMed] [Google Scholar]
- [57].Chen MY, Liao WS, Lu Z, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer. 2011;117:4424–4438. doi: 10.1002/cncr.26073. PMID:21491416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hu Y, Lu W, Chen G, et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound beta-phenylethyl isothiocyanate. Blood. 2010;116:2732–2741. doi: 10.1182/blood-2009-11-256354. PMID:20566897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Nawrocki ST, Carew JS, Pino MS, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–3781. doi: 10.1158/0008-5472.CAN-05-2961. PMID:16585204 [DOI] [PubMed] [Google Scholar]
- [60].Tang F, Choy E, Tu C, et al. Therapeutic applications of histone deacetylase inhibitors in sarcoma. Cancer Treat Rev. 2017;59:33–45. doi: 10.1016/j.ctrv.2017.06.006. PMID:28732326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367–1374. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- [62].Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mole Cell Biol. 2017;18:246–262. doi: 10.1038/nrm.2016.143. PMID:28053347 [DOI] [PubMed] [Google Scholar]
- [63].Zaware N, Zhou MM. Chemical modulators for epigenome reader domains as emerging epigenetic therapies for cancer and inflammation. Curr Opin Chem Biol. 2017;39:116–125. doi: 10.1016/j.cbpa.2017.06.012. PMID:28689146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ramadoss M, Mahadevan V. Targeting the cancer epigenome: synergistic therapy with bromodomain inhibitors. Drug Disc Today. 2017; PMID:28943305 [DOI] [PubMed] [Google Scholar]
- [65].Xu Y, Vakoc CR. Targeting cancer cells with BET bromodomain inhibitors. CSH Perspect Med. 2017; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Fu LL, Tian M, Li X, et al. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015;6:5501–5516. doi: 10.18632/oncotarget.3551. PMID:25849938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. PMID:20871596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bandopadhayay P, Bergthold G, Nguyen B, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20:912–925. doi: 10.1158/1078-0432.CCR-13-2281. PMID:24297863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol. 2015;6:128–141. doi: 10.1177/2040620715576662. PMID:26137204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lee DH, Qi J, Bradner JE, et al. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int J Cancer J Int Du Cancer. 2015;136:2055–2064. doi: 10.1002/ijc.29269. PMID:25307878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chaidos A, Caputo V, Gouvedenou K, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014;123:697–705. doi: 10.1182/blood-2013-01-478420. PMID:24335499 [DOI] [PubMed] [Google Scholar]
- [72].Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–e195. doi: 10.1016/S2352-3026(15)00247-1. PMID:27063977 [DOI] [PubMed] [Google Scholar]
- [73].Nicholls SJ, Puri R, Wolski K, et al. Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: results of the phase 2b, randomized, double-blind, multicenter, ASSURE trial. Am J Cardiovas Drugs: Drugs, Devices, Other Intervent. 2016;16:55–65. doi: 10.1007/s40256-015-0146-z. PMID:26385396 [DOI] [PubMed] [Google Scholar]
- [74].Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. PMID:26317466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Roulois D, Loo Yau H, Singhania R, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. PMID:26317465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630–641. doi: 10.1038/nrg.2016.93. PMID:27629931 [DOI] [PubMed] [Google Scholar]
- [77].Li Y, Hu W, Shen DY, et al. Azacitidine enhances sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis. Am J Obstet Gynecol. 2009;200:177:e1–e9. [DOI] [PubMed] [Google Scholar]
- [78].Fang F, Balch C, Schilder J, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116:4043–4053. doi: 10.1002/cncr.25204. PMID:20564122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Diyabalanage HV, Granda ML, Hooker JM. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett. 2013;329:1–8. doi: 10.1016/j.canlet.2012.09.018. PMID:23032720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hu B, Younes A, Westin JR, et al. Phase-I and randomized phase-II trial of panobinostat in combination with ICE (ifosfamide, carboplatin, etoposide) in relapsed or refractory classical Hodgkin lymphoma. Leuk Lymph. 2017:1–8. [DOI] [PubMed] [Google Scholar]
- [81].Topper MJ, Vaz M, Chiappinelli KB, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171:1284-300:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kim K, Skora AD, Li Z, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Nat Acad Sci USA. 2014;111:11774–11779. doi: 10.1073/pnas.1410626111. PMID:25071169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zheng H, Zhao W, Yan C, et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res: Off J Am Assoc Cancer Res. 2016;22:4119–4132. doi: 10.1158/1078-0432.CCR-15-2584. PMID:26964571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Coral S, Sigalotti L, Altomonte M, et al. 5-aza-2'-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res: Off J Am Assoc Cancer Res. 2002;8:2690–2695. PMID:12171902 [PubMed] [Google Scholar]
- [85].Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. PMID:24583822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. PMID:11237011 [DOI] [PubMed] [Google Scholar]
- [87].Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9:411–412. doi: 10.1038/nrg2165-c1. PMID:18421312 [DOI] [PubMed] [Google Scholar]
- [88].Han JS. Non-long terminal repeat (non-LTR) retrotransposons: mechanisms, recent developments, and unanswered questions. Mobile DNA. 2010;1:15. doi: 10.1186/1759-8753-1-15. PMID:20462415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10(6):395–406. doi: 10.1038/nrmicro2783. PMID:22565131 [DOI] [PubMed] [Google Scholar]
- [90].Yu HL, Zhao ZK, Zhu F. The role of human endogenous retroviral long terminal repeat sequences in human cancer (Review). Int J Mol Med. 2013;32:755–762. doi: 10.3892/ijmm.2013.1460. PMID:23900638 [DOI] [PubMed] [Google Scholar]
- [91].Suntsova M, Garazha A, Ivanova A, et al. Molecular functions of human endogenous retroviruses in health and disease. Cell Mole Life Sci: CMLS. 2015;72:3653–3675. doi: 10.1007/s00018-015-1947-6. PMID:26082181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Friedli M, Trono D. The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol. 2015;31:429–451. doi: 10.1146/annurev-cellbio-100814-125514. PMID:26393776 [DOI] [PubMed] [Google Scholar]
- [93].Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. PMID:17363976 [DOI] [PubMed] [Google Scholar]
- [94].Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Smith ZD, Chan MM, Humm KC, et al. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. PMID:25079558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/S0168-9525(97)01181-5. PMID:9260521 [DOI] [PubMed] [Google Scholar]
- [97].Groudine M, Eisenman R, Weintraub H. Chromatin structure of endogenous retroviral genes and activation by an inhibitor of DNA methylation. Nature. 1981;292:311–317. doi: 10.1038/292311a0. PMID:6166864 [DOI] [PubMed] [Google Scholar]
- [98].Leonova KI, Brodsky L, Lipchick B, et al. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Nat Acad Sci. 2012;110:E89–E98. doi: 10.1073/pnas.1216922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. PMID:9771701 [DOI] [PubMed] [Google Scholar]
- [100].Matsui T, Leung D, Miyashita H, et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. PMID:20164836 [DOI] [PubMed] [Google Scholar]
- [101].Karimi Mohammad M, Goyal P, Maksakova Irina A, et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. PMID:21624812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yang Bin X, El Farran Chadi A, Guo Hong C, et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell. 2015;163:230–245. doi: 10.1016/j.cell.2015.08.037. PMID:26365490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. PMID:17603471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rowe HM, Jakobsson J, Mesnard D, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–240. doi: 10.1038/nature08674. PMID:20075919 [DOI] [PubMed] [Google Scholar]
- [105].Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet. 2012;13:283–296. doi: 10.1038/nrg3199. PMID:22421730 [DOI] [PubMed] [Google Scholar]
- [106].Archin NM, Liberty AL, Kashuba AD, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. PMID:22837004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Göke J, Lu X, Chan Y-S, et al. Dynamic Transcription of Distinct Classes of Endogenous Retroviral Elements Marks Specific Populations of Early Human Embryonic Cells. Cell Stem Cell. 2015;16:135–141. doi: 10.1016/j.stem.2015.01.005. PMID:25658370 [DOI] [PubMed] [Google Scholar]
- [108].Pavlicev M, Hiratsuka K, Swaggart KA, et al. Detecting Endogenous Retrovirus-Driven Tissue-Specific Gene Transcription. Genome Biol Evol. 2015;7:1082–1097. doi: 10.1093/gbe/evv049. PMID:25767249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Buzdin A, Kovalskaya-Alexandrova E, Gogvadze E, et al. At Least 50% of Human-Specific HERV-K (HML-2) Long Terminal Repeats Serve In Vivo as Active Promoters for Host Nonrepetitive DNA Transcription. J Virol. 2006;80:10752–10762. doi: 10.1128/JVI.00871-06. PMID:17041225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Fort A, Hashimoto K, Yamada D, et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet. 2014;46:558–566. doi: 10.1038/ng.2965. PMID:24777452 [DOI] [PubMed] [Google Scholar]
- [111].Wolf G, Yang P, Füchtbauer AC, et al. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 2015;29:538–554. doi: 10.1101/gad.252767.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Veselovska L, Smallwood SA, Saadeh H, et al. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol. 2015;16:209. doi: 10.1186/s13059-015-0769-z. PMID:26408185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351:1083–1087. doi: 10.1126/science.aad5497. PMID:26941318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Macfarlan TS, Gifford WD, Driscoll S, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63. doi: 10.1038/nature11244. PMID:22722858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Takahashi H, Lassmann T, Murata M, et al. 5' end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat Protoc. 2012;7:542–561. doi: 10.1038/nprot.2012.005. PMID:22362160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Brocks D, Schmidt CR, Daskalakis M, et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49:1052–1060. doi: 10.1038/ng.3889. PMID:28604729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Salimullah M, Sakai M, Plessy C, et al. NanoCAGE: a high-resolution technique to discover and interrogate cell transcriptomes. Cold Spring Harbor Protoc. 2011;2011:pdb prot5559. doi: 10.1101/pdb.prot5559. PMID:21205859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Shortt J, Ott CJ, Johnstone RW, et al. A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer. 2017;17:160–183. doi: 10.1038/nrc.2016.148. PMID:28228643 [DOI] [PubMed] [Google Scholar]
- [119].Kronung SK, Beyer U, Chiaramonte ML, et al. LTR12 promoter activation in a broad range of human tumor cells by HDAC inhibition. Oncotarget. 2016;7:33484–33497. doi: 10.18632/oncotarget.9255. PMID:27172897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Cameron EE, Bachman KE, Myohanen S, et al. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. PMID:9916800 [DOI] [PubMed] [Google Scholar]
- [121].Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. PMID:19608861 [DOI] [PubMed] [Google Scholar]
- [122].Choudhary C, Weinert BT, Nishida Y, et al. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. PMID:25053359 [DOI] [PubMed] [Google Scholar]
- [123].Banerjee C, Archin N, Michaels D, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukocyte Biol. 2012;92:1147–1154. doi: 10.1189/jlb.0312165. PMID:22802445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Meth. 2015;12:357–360. doi: 10.1038/nmeth.3317. PMID:25751142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Chen Y, Lun AT, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research. 2016;5:1438. PMID:27508061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gajer JM, Furdas SD, Grunder A, et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis. 2015;4:e137. doi: 10.1038/oncsis.2014.51. PMID:25664930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Mohammad HP, Smitheman KN, Kamat CD, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28:57–69. doi: 10.1016/j.ccell.2015.06.002. PMID:26175415 [DOI] [PubMed] [Google Scholar]
- [128].Robaa D, Wagner T, Luise C, et al. Identification and structure-activity relationship studies of small-molecule inhibitors of the methyllysine reader protein spindlin1. Chem Med Chem. 2016;11:2327–2338. doi: 10.1002/cmdc.201600362. PMID:27634332 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.