

SUMMARY
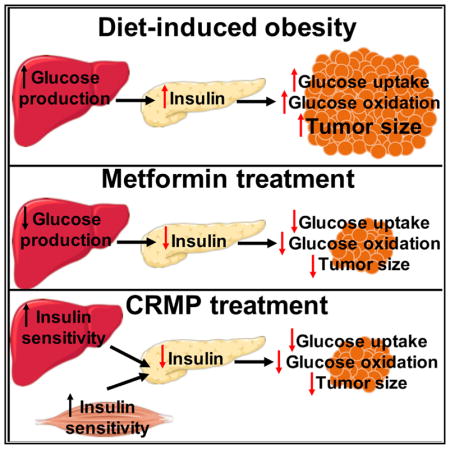
Obesity is associated with colon cancer pathogenesis, but the underlying mechanism is actively debated. Here, we confirm that diet-induced obesity promotes tumor growth in two murine colon cancer models and show that this effect is reversed by an orally administered controlled-release mitochondrial protonophore (CRMP) that acts as a liver-specific uncoupler of oxidative phosphorylation. This agent lowered circulating insulin, and the reduction of tumor growth was abrogated by an insulin infusion raising plasma insulin to the level of high-fat-fed mice. We also demonstrate that hyperinsulinemia increases glucose uptake and oxidation in vivo in tumors and that CRMP reverses these effects. This study provides evidence that perturbations of whole-organism energy balance or hepatic energy metabolism can influence neoplastic growth. Furthermore, the data show that glucose uptake and utilization by cancers in vivo are not necessarily constitutively high but rather may vary according to the hormonal milieu.
In Brief
Wang et al. demonstrate that diet-induced hyperinsulinemia increases colon adenocarcinoma tumor glucose uptake and oxidation in mice. They further demonstrate that reversal of hyperinsulinemia by a liver-specific mitochondrial protonophore is sufficient to reverse the obesity-induced acceleration of tumor growth.
INTRODUCTION
It has been predicted that by 2050, most Americans will be overweight or obese (Wang et al., 2008) and one in three Americans will have type 2 diabetes (T2D) (Boyle et al., 2001). Obesity and type 2 diabetes are positively associated with prevalence and poor prognosis of many cancers in both men and women, with a recent study by the Centers for Disease Control and Prevention finding 40% of new diagnoses of cancer associated with overweight or obesity (Steele et al., 2017). Thus, the obesity epidemic has worrisome implications for cancer epidemiology.
The mechanisms underlying the influence of obesity on cancer are an area of active debate and may differ from tumor type to tumor type. Colon cancer is one of approximately one dozen types of cancer that have been associated with obesity (Bardou et al., 2013; Dong et al., 2017; Fardet et al., 2017; Øines et al., 2017). Hyperinsulinemia has been proposed as a mechanism by which obesity and diabetes may lead to increased colon cancer risk (Cohen and LeRoith, 2012; Pollak, 2012; Wolpin et al., 2009), perhaps through activation of insulin-like growth factor-I (IGF-I) signaling by insulin-induced suppression of IGF binding protein-1 expression and/or through increased tumor angiogenesis. However, because insulin has profound effects on metabolism in many tissues, it is plausible that insulin also modulates substrate metabolism in cancer, specifically by increasing glucose uptake and oxidation in a manner that contributes to tumor progression. This hypothesis is consistent with the widespread expression of the insulin receptor by transformed cells (Abbruzzese et al., 2015; Klil-Drori et al., 2017; Mattarocci et al., 2009; Pollak, 2012), but it is not consistent with the commonly held view that glucose uptake by neoplastic tissue is constitutively high and uninfluenced by hormones that regulate metabolism in normal tissues.
Treatment with metformin (the most commonly prescribed antidiabetic drug worldwide) or phenformin (a more potent biguanide) slows in vivo growth of adenocarcinomas (Algire et al., 2010, 2011; Shackelford et al., 2013; Tomimoto et al., 2008). There is clear clinical evidence that even low-dose oral metformin reduces colon polyp formation (Higurashi et al., 2016), although evidence that patients with diabetes treated with metformin have reduced cancer burden is controversial (Farmer et al., 2017; Nie et al., 2016). It is unclear whether the clinical and experimental antineoplastic actions of metformin are attributable to systemic effects, such as reduction of circulating insulin levels; to direct effects on neoplastic cells; or to both mechanisms.
Thus, a causative mechanistic link from hyperinsulinemia per se, to tumor glucose utilization, to tumor growth remains to be conclusively established in colon cancer, partly because methodologic limitations have prevented assessment of tumor glucose oxidation in awake animals. To that end, we show here that hyperinsulinemia drives tumor glucose uptake and oxidation in awake MC38 colon adenocarcinoma tumor-bearing mice, as well as in ApcMin+/− mice, a model of familial adenomatous polyposis, and that two interventions that reverse hyperinsulinemia abrogate the effect of a high-fat diet (HFD) to increase glucose uptake and oxidation, thereby slowing tumor growth. These agents, a controlled-release mitochondrial protonophore (Perry et al., 2015b) and metformin, an inhibitor of gluconeogenesis, act by different mechanisms, but both agents’ ability to slow tumor growth depended on reduction of hyperinsulinemia. These data highlight insulin-lowering agents as candidates deserving investigation as therapeutic strategies for colon cancer.
RESULTS
Controlled-Release Mitochondrial Protonophore Lowers Fasting Glucose and Insulin
To test the potential therapeutic effect of a controlled-release formulation of 2,4-dinitrophenol (DNP), which acts as a liver-targeted mitochondrial uncoupler (Perry et al., 2015b), MC38 tumor-bearing mice were provided ad lib access to controlled-release mitochondrial protonophore containing HFD. Plasma DNP concentrations were maintained in a therapeutic range (5–15 μM) throughout the day, whereas tumor DNP concentrations were negligible (~0.15 μM) (Figures 1A and 1B). Although HFD increased fasting plasma glucose and insulin concentrations and whole-body glucose turnover, each of these effects was reversed by treatment with controlled-release mitochondrial protonophore, which lowered plasma glucose and insulin concentrations and whole-body glucose turnover to levels measured in chow-fed controls (Figures 1C–1E). Although HFD increased both food intake and body weight, controlled-release mitochondrial protonophore had no impact on either parameter. Insulin treatment increased plasma glucagon and epinephrine concentrations, likely as a counterregulatory mechanism against hypoglycemia, but did not alter corticosterone (Figure S1). To examine the mechanistic significance of HFD-induced hyperinsulinemia on tumor growth in HFD tumor-bearing mice, we infused controlled-release mitochondrial protonophore-treated HFD mice with insulin by subcutaneous pellet to raise plasma insulin concentrations to those measured in HFD controls. This intervention tripled plasma insulin concentrations without any significant effect on plasma glucose (Figures 1C–1E). Consistent with previous data (Perry et al., 2015a), HFD-fed mice exhibited increased non-esterified fatty acid (NEFA) concentrations, but plasma NEFA were further increased by controlled-release mitochondrial protonophore and normalized with insulin replacement in controlled-release mitochondrial protonophore-treated mice (Figure 1F). Finally, whereas triglyceride content was increased with high-fat feeding in both liver and skeletal muscle, controlled-release mitochondrial protonophore reversed these alterations, independent of plasma insulin concentrations (Figures 1G and 1H).
Figure 1. Liver-Targeted Mitochondrial Uncoupling with a Controlled-Release Mitochondrial Protonophore Reverses Fasting Hyperglycemia and Hyperinsulinemia in HFD MC38 Tumor-Bearing Mice.

(A) Plasma DNP concentration in tumor-bearing mice given ad lib access to food containing controlled-release mitochondrial protonophore.
(B) Plasma, liver, and tumor DNP concentrations measured in a separate group of ad lib-fed mice at 10 a.m.
(C and D) Fasting plasma glucose (C) and insulin concentrations (D).
(E) Endogenous glucose production.
(F) Plasma non-esterified fatty acid concentrations.
(G and H) Liver (G) and quadriceps (H) triglyceride content
In (A) and (B), n = 4. In (C)–(H), n = 7 HFD mice and 8 in all other groups. Data are the mean ± SEM, with comparisons by ANOVA with Bonferroni’s multiple comparisons test. See also Figure S1.
Controlled-Release Mitochondrial Protonophore Slows Tumor Growth by Suppressing Hyperinsulinemia and Consequently Reducing Tumor Glucose Uptake and Oxidation
Next, we sought to determine the effect of HFD and controlled-release mitochondrial protonophore on tumor growth. Consistent with previous data (Algire et al., 2010; Nimri et al., 2015; VanSaun et al., 2009; Yakar et al., 2006), high-fat feeding accelerated MC38 tumor growth. This effect was abrogated by treatment of HFD mice with controlled-release mitochondrial protonophore but restored when controlled-release mitochondrial protonophore-treated HFD mice were infused with insulin (Figures 1D and 2A). To examine the mechanism by which controlled-release mitochondrial protonophore slowed tumor growth, we first sought to determine whether controlled-release mitochondrial protonophore suppressed tumor growth directly. Incubating MC38 cells in the DNP concentrations measured in tumors (0.15 μM) or in plasma (11 μM) had no effect on cell division in vitro (Figure 2B). We then assessed the effect of alterations in diet and insulin sensitivity on glucose metabolism. High-fat feeding promoted [14C] 2-deoxyglucose glucose uptake in tumors and increased glucose oxidation, as indicated by the ratio of flux through pyruvate dehydrogenase (VPDH), which controls the entry of glucose carbons into the tricarboxylic acid cycle, to citrate synthase flux (VCS) (Figures 2C and 2D). However, reversing hyperinsulinemia with controlled-release mitochondrial protonophore treatment reversed these increases in glucose uptake and VPDH/VCS, whereas restoring plasma insulin concentrations increased both parameters to levels measured in HFD controls. In contrast, although plasma acetate concentrations and whole-body acetate turnover increased with high-fat feeding, tumor growth did not correlate with acetate turnover (Figures 2E and 2F) or uptake (Figure 2G). Finally, we observed no difference in tumor acetate metabolism, indicated by an unchanged ratio of tricarboxylic acid (TCA) cycle intermediate enrichment to plasma acetate enrichment (Figure 2H).
Figure 2. Controlled-Release Mitochondrial Protonophore Slows MC38 Tumor Growth and Limits Tumor Glucose Uptake and Oxidation due to Reversal of Hyperinsulinemia.

(A) Tumor size.
(B) In vitro proliferation of cells incubated in media containing DNP. Data are the mean ± SEM of 6 replicate wells for each condition, each counted in duplicate. The two conditions were compared by the two-tailed unpaired Student’s t test.
(C and D) Tumor glucose uptake (C) and oxidation (VPDH/VCS) (D).
(E and F) Plasma acetate concentrations (E) and whole-body acetate turnover (F).
(G) Ratio of tumor to plasma acetate enrichment in mice infused with [1-13C] acetate.
(H) Ratio of tumor TCA cycle intermediate enrichment to plasma acetate enrichment in mice infused with [1-13C] acetate.
In (A) and (C)–(H), n = 7 HFD mice and 8 in all other groups. In all panels, data are the mean ± SEM with comparisons by ANOVA with Bonferroni’s multiple comparisons test unless otherwise stated.
Controlled-Release Mitochondrial Protonophore Reduces Polyp Number and Area in ApcMin+/− Mice
To confirm the potential utility of mitochondrial uncoupling as a therapeutic strategy against obesity-associated colon cancer, we studied male ApcMin heterozygous mice. Similar to MC38 tumor-bearing mice, ApcMin+/− mice fed HFD exhibited increases in plasma glucose, insulin, and NEFA concentrations, as well as whole-body glucose turnover on a HFD, associated with increased caloric intake and weight gain (Figures 3A and 3B; Figures S2A–S2D). Despite not altering either food intake or weight gain compared to the HFD group, controlled-release mitochondrial protonophore treatment normalized plasma glucose and insulin concentrations and glucose turnover associated with a reversal of diet-induced increases in ectopic lipid accumulation, despite an increase in plasma NEFA concentrations likely secondary to reversal of hyperinsulinemia (Figures 3A and 3B; Figures S2A–S2E). To examine the physiologic impact of hyperinsulinemia in this model, we also infused a group of controlled-release mitochondrial protonophore-treated HFD ApcMin+/− mice with insulin chronically by subcutaneous pellet and found that this intervention lowered plasma glucose and endogenous glucose turnover within the physiologic range but did not lead to severe hypoglycemia or any alterations in ectopic lipid content (Figures 3A and 3B; Figures S2A–S2G). Next, we examined the impact of diet-induced obesity and hyperinsulinemia on ApcMin+/− colon polyp appearance and growth. HFD increased the number and size of polyps in the small intestine, as well as the total weight of the small intestine, but these alterations were reversed by controlled-release mitochondrial protonophore. However, restoring hyperinsulinemia in controlled-release mitochondrial protonophore-treated mice increased colon polyp number and size to those measured in HFD controls (Figures 3C–3E; Figures S2H and S2I). Tumor glucose uptake and oxidation correlated with tumor size: both [14C] 2-deoxyglucose uptake and VPDH/VCS were increased in ApcMin+/− tumors of HFD mice. However, this effect was reversed by lowering plasma glucose and insulin concentrations to those measured in chow-fed mice using controlled-release mitochondrial protonophore and restored when insulin concentrations were replaced in HFD-controlled-release mitochondrial protonophore-treated mice (Figures 3F and 3G).
Figure 3. Controlled-Release Mitochondrial Protonophore Slows Tumor Growth in High-Fat-Fed ApcMin+/− Mice, a Murine Model of Familial Adenomatous Polyposis, by Reversing Hyperinsulinemia.

(A and B) Fasting plasma glucose (A) and insulin concentrations (B).
(C) Representative images of small intestine stained for β-catenin.
(D and E) Quantification of total polyp number (D) and area (E).
(F and G) Tumor glucose uptake (F) and oxidation (VPDH/VCS) (G).
In (A)–(E), n = 5 per group. In (F) and (G), n = 4–5 per group. In all panels, data are the mean ± SEM and groups were compared by ANOVA with Bonferroni’s multiple comparisons test. See also Figure S2.
Reductions in Fasting Plasma Glucose and Insulin Associated with Suppression of Tumor Glucose Uptake and Oxidation Explain Metformin’s Tumor-Suppressive Effect
Next, we aimed to determine the molecular mechanism by which metformin slows MC38 tumor growth and to assess its potential reliance on reversal of hyperinsulinemia. To that end, we treated HFD mice with metformin in drinking water for four weeks. In contrast to controlled-release mitochondrial protonophore treatment, metformin treatment led to significant metformin accumulation in tumors (~70 μM) and plasma (~180 μM) (Figure 4A). Metformin did not affect food intake, body weight, or ectopic lipid content, but not surprisingly, chronic treatment with metformin lowered fasting plasma glucose and insulin concentrations and glucose turnover but raised NEFA concentrations, presumably secondary to reductions in plasma insulin (Figures 4B–4D; Figures S3A–S3E). Reductions in fasting hyperglycemia and hyperinsulinemia with metformin treatment were associated with a marked slowing of tumor growth, reducing tumor size by 80% compared to HFD control mice after four weeks of treatment, without affecting tumor growth in vitro (Figures 4E and 4F). To assess the potential impact of reductions in plasma insulin concentrations on tumor growth and metabolism, we also studied a group of metformin-treated HFD mice infused with insulin to restore plasma insulin concentrations to the concentrations measured in HFD controls and found that this intervention did not alter food intake, body weight, or ectopic lipid content but did lower plasma glucose and NEFA concentrations (Figure 4B; Figures S3A–S3E). Although metformin did not affect plasma glucagon, catecholamine, or corticosterone concentrations, insulin replacement increased both glucagon and catecholamine concentrations (Figures S3F–S3I), presumably as a compensatory mechanism to avoid dangerous hypoglycemia. Most importantly, insulin replacement fully abrogated the protective effect of metformin on tumor growth, increasing tumor growth rates to those measured in HFD controls (Figure 4E). Similar to the suppressive effect of controlled-release mitochondrial protonophore on tumor fluxes, this was insulin dependent: metformin suppressed tumor glucose uptake and oxidation (VPDH/VCS), but both changes were reversed with replacement insulin (Figures 4G and 4H). In contrast, metformin had no effect on plasma acetate concentrations, whole-body acetate turnover, or tumor acetate uptake or metabolism (Figures S3J–S3M), implicating metformin’s effect to lower insulin as the likely explanation for its antitumor effect in our model.
Figure 4. Metformin Slows Tumor Growth in HFD Mice by Reducing Tumor Glucose Uptake and Oxidation Secondary to Hyperinsulinemia.

(A) Plasma, liver, and tumor metformin concentrations.
(B and C) Fasting plasma glucose (B) and insulin concentrations (C).
(D) Endogenous glucose turnover.
(E) Tumor size.
(F) In vitro proliferation of MC38 cells incubated in media containing metformin.
(G and H) Tumor glucose uptake (G) and oxidation (VPDH/VCS) (H).
In (A), n = 6. In (B)–(H), data are the mean ± SEM of n = 8 per group, with comparisons by ANOVA with Bonferroni’s multiple comparisons test. See also Figure S3.
DISCUSSION
Obesity and type 2 diabetes are well-known pathogenic factors promoting several cancers, including colorectal cancer. Hyperinsulinemia, which occurs frequently with obesity and type 2 diabetes and universally with insulin resistance, has been proposed as a potential mediator of the increased risk of colon cancer in obese individuals (Kim, 1998; Pollak, 2012; Schoen et al., 1999). Consistent with this hypothesis, chronic insulin sensitization with exercise reduces tumor burden in ApcMin+/− mice (Baltgalvis et al., 2008; McClellan et al., 2014), while metformin has a similar impact in both ApcMin+/− tumor-bearing mice (Tomimoto et al., 2008) and MC38 tumor-bearing mice (Algire et al., 2010, 2011; Shackelford et al., 2013), suggesting that systemic metabolic alterations may have therapeutic promise in colon cancer. Unfortunately, the independent roles of hyperinsulinemia and hyperglycemia are difficult to probe in human subjects in vivo, because hyperinsulinemia typically occurs in the setting of hyperglycemia in insulin-resistant and diabetic individuals. Several studies have raised the possibility of a detrimental effect of exogenous insulin therapy on colon cancer risk in type 2 diabetes patients (Chung et al., 2008; Rosato et al., 2016; Yang et al., 2004; Yin et al., 2014). However, such retrospective pharmaco-epidemiologic data are controversial because of the difficulty in defining an appropriate control group, as doing so would require withholding insulin from patients for whom it is medically indicated. However, strong data indicate that obesity adversely effects the burden of many cancers, and the possibility that hyperinsulinemia is mechanistically involved deserves clarification. To that end, we performed in vivo flux studies to simultaneously examine the impact of diet-induced insulin resistance and hyperinsulinemia on both uptake and oxidation of glucose in colon adenocarcinomas in mice. The striking tumor-promoting effect of high-fat feeding was associated with increases in tumor glucose uptake and oxidation and was associated with both hyperinsulinemia and hyperglycemia in two genetically divergent models of colon cancer, ApcMin+/− and MC38 tumor-bearing C57BL/6J mice (Figures 1C, 1D, 2A, 2C, 2D, and 3A–3H). However, reversing hyperinsulinemia with a controlled-release mitochondrial protonophore or with metformin reversed the tumor-promoting effect of HFD. These data suggest that hyperinsulinemia and/or hyperglycemia may stimulate tumor growth but do not differentiate between the two. In the case of metformin, a direct inhibitory effect of the drug on the tumor is not ruled out, but this possibility is excluded for the liver-specific protonophore, because it does not accumulate in neoplastic tissue. Both agents acted in a weight-independent manner. These data do not argue against an effect of obesity on tumor growth—to the contrary, diet-induced obesity was associated with a greater tumor burden in HFD mice—but instead suggest that interventions targeting metabolic dysregulation may be effective without altering appetite or body weight, two considerations of great importance in cancer patients.
Although many previous studies have shown that metformin slows tumor growth in vitro at millimolar concentrations, therapeutic plasma metformin concentrations are in the 50–100 μM range. When we incubated MC38 cells in metformin at the concentration (70 μM) measured in tumors of mice treated with an oral metformin dose that lowered plasma glucose and insulin concentrations and inhibited tumor growth, we found no effect on proliferation in vitro (Figure 4F). These data show that, while suprapharmacologic concentrations of metformin are capable of slowing tumor growth in vitro, the concentrations of metformin measured in vivo in animals treated with typical doses of metformin do not have any direct effect on tumor growth. However, because tumor microenvironment differs substantially from tissue culture media in concentrations of molecules, such as glucose and serine, that influence sensitivity to metformin, the possibility that micromolar concentrations of metformin directly affect tumor growth cannot be excluded. In contrast to metformin, DNP did not accumulate significantly in tumors (0.15 μM, compared to plasma concentrations of 11 μM), and there is no evidence that this concentration of DNP uncouples oxidative phosphorylation (Perry et al., 2015b) or inhibits tumor cell proliferation in vitro (Figure 2B). The higher concentration of DNP measured in plasma (11 μM) also had no impact on tumor growth in vitro. Thus, in vivo tumor growth inhibition by this agent likely reflects indirect actions.
To examine the impact of hyperinsulinemia per se on tumor growth, we treated groups of controlled-release mitochondrial protonophore- and metformin-treated HFD mice with chronic administration of insulin by subcutaneous pellet. This intervention restored both MC38 and ApcMin+/− tumor size to that measured in control HFD mice, suggesting that hyperinsulinemia alone, without hyperglycemia, is sufficient to mediate the tumor-promoting effect of obesity. These data are consistent with a previous study demonstrating that exogenous insulin treatment promotes MC38 tumor growth in vivo (Hvid et al., 2012) and directly demonstrate that the tumor growth inhibitory effects of a liver-specific mitochondrial uncoupling agent require reduction of insulin levels.
It is likely that hyperinsulinemia, and resulting increases in glucose uptake and oxidation, is just one of multiple factors promoting tumor pathogenesis. Studies have implicated numerous factors, including 5′ adenosine monophosphate-activated protein kinase (AMPK), reactive oxygen species, inflammatory cytokines, adiponectin, leptin, and insulin-like growth factor-1 (IGF-1), in colon cancer pathogenesis. Several of these alternative mechanisms may be associated with hyperinsulinemia—for instance, chronic inflammation is associated with insulin resistance, although the direction of causality has been under debate—and may, in addition to insulin’s ability to promote tumor glucose uptake, be one of multiple mechanisms by which obesity promotes tumor growth. However, restoring hyperinsulinemia promotes tumor growth in controlled-release mitochondrial protonophore- or metformin-treated mice to rates similar to those measured in untreated HFD controls, which argues that hyperinsulinemia is the major factor responsible for the observed increases in MC38 and ApcMin tumor growth in obese mice. The link between hyperinsulinemia and tumor growth has been explored previously, including in a classic study in which Kalaany and Sabatini (2009) demonstrate that dietary restriction reduces and insulin promotes growth of several tumor types only in tumors that do not exhibit constitutive phosphatidylinositol-3-kinase activation. However, to our knowledge, the impact of hyperinsulinemia on tumor glucose metabolism, and resulting alterations in tumor growth, has not been explored mechanistically in vivo in colon tumors. These findings could provide such a mechanism. We demonstrate here that obesity-related hyperinsulinemia is a key contributory factor to MC38 and ApcMin+/− adenocarcinoma tumor growth by increasing glucose uptake and oxidation, demonstrating that these processes are not constitutively active in MC38 or ApcMin+/− intestinal tumors. These data support hyperinsulinemia as the primary mediator of the adverse effect of obesity on neoplasia, at least in these cancer models, and suggest that insulin-lowering agents may be attractive therapeutic targets against obesity-associated tumors.
EXPERIMENTAL PROCEDURES
Cell Lines
MC38 cells were maintained in DMEM (5.6 mM glucose) containing 10% fetal bovine serum (FBS), 2 mM glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM HEPES buffer, and broad-spectrum antibiotics (penicillin and streptomycin). They were incubated at 37°C and trypsinized and split 1:10 twice weekly.
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee of Yale University. In the MC38 tumor studies, 8-week-old male C57BL/6J mice (Jackson Laboratory) were used for all experiments and were randomly assigned to treatment groups. They were housed in the Yale Animal Resource Center on a 12 hr light/dark cycle. Mice were injected with MC38 colon adenocarcinoma cells (5 × 105 cells, injected into the right flank) and on the day of injection were placed on regular chow (Harlan Teklad no. 2018S), HFD (Research Diets D12492), or the same HFD containing controlled-release mitochondrial protonophore (7 mg controlled-release mitochondrial protonophore per gram of diet, with a daily dose of ~5 mg active DNP per kilogram of body weight per day). The dietary or pharmacologic interventions were initiated on the day of tumor implantation to avoid confounding effects of treatment on tumor implantation. Mice given replacement insulin were fed HFD containing controlled-release mitochondrial protonophore as described earlier and were implanted on the day of tumor injection with three subcutaneous insulin pellets (LinShin). Mice treated with metformin were given ad lib access to metformin-containing drinking water (1 mg/mL) beginning the day of tumor injection. In all MC38 studies, tumors were measured weekly using calipers. Food intake was measured 3 weeks after tumor injection and treatment initiation by weighing the food in each mouse’s cage daily for 3 days. Mice were individually housed during this three-day period.
For the ApcMin+/− studies, C57BL/6J-ApcMin heterozygous male mice were obtained from Jackson Laboratory at 7 weeks of age. After one week of acclimation, they were randomly divided into treatment groups, placed on diets, and implanted with insulin pellets as described earlier. Body weight was measured weekly, and food intake was measured during week 3 of treatment.
One week before all in vivo studies, mice underwent surgery under general isoflurane anesthesia to place a catheter in the jugular vein for in vivo flux studies. All studies were performed after an overnight fast.
In Vivo Flux Studies
Mice used for VPDH/VCS flux studies and measurement of endogenous glucose turnover were infused with [U-13C6] glucose (1 mg/[kg-min] after a 5 min 3× prime) for 120 min. At 120 min, a blood sample was taken by tail massage for measurement of glucose turnover. [13C6] glucose enrichment was measured as described later, and glucose turnover was calculated using Equation 1:
| (Equation 1) |
where APE denotes the atom percent enrichment measured by gas chromatography-mass spectrometry (GC-MS). While the [13C6] glucose infusion continued, mice were injected with a bolus of [14C] 2-deoxyglucose (10 μCi), and blood samples were taken 5, 15, 25, 35, 45, and 55 min later. Mice were then euthanized by intravenous (i.v.) pentobarbital, and MC38 tumors were freeze-clamped in tongs pre-chilled in liquid N2, while colon polyps in ApcMin+/− were visualized and excised with dissection scissors before freeze-clamping. Based on the area under the curve of the plasma [14C] 2-de-oxyglucose-specific activity measured using a scintillation counter, we calculated tumor glucose uptake as previously described (Youn and Buchanan, 1993). Based on the assumptions described previously (Shulman et al., 1987), we measured calculated VPDH/VCS as the ratio of [4,5-13C2] glutamate/[13C3] alanine, with glutamate enrichment measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and alanine enrichment by GC-MS as described in the following section.
In a separate group of MC38 tumor-bearing mice, a primed-continuous 120 min infusion of [1-13C] acetate (prime of 60 μmol/[kg-min] × 5 min and then a continuous infusion rate of 20 μmol/[kg-min]) was administered. Plasma acetate enrichment was measured by GC-MS, and acetate turnover was calculated using Equation 1. Tumor enrichment was also measured by GC-MS as described later, and the ratio of tumor to plasma enrichment was calculated.
Biochemical Analysis
Plasma glucose was measured using the YSI Biochemistry Analyzer; plasma insulin and glucagon were measured by radioimmunoassay by the Yale Diabetes Research Center; plasma epinephrine, norepinephrine, and corticosterone were measured by ELISA (Abnova, Abnova, and Abcam, respectively), and plasma NEFA was measured using the Wako NEFA-HR(2) enzymatic method. To measure plasma glucose enrichment, 10 μL of plasma taken after 120 min of [1,2,3,4,5,6-13C6] glucose infusion was deprotonized using equal volumes of barium sulfate and zinc hydroxide, dried, and derivatized using 1:1 acetic anhydride:pyridine. GC-MS (chemical ionization [CI] mode, m/z 331 [m0], 332 [m+1], 333 [m+2], …, 337 [m+6]) was used to determine the 13C6 glucose enrichment. Plasma acetate concentration and enrichment were also measured by GC-MS (electron ionization [EI] mode) (Perry et al., 2016).
Tissue Analysis
Tissue triglyceride content was measured enzymatically using the method of Bligh and Dyer (1959) and the Sekisui triglyceride reagent. Plasma and tissue DNP (Perry et al., 2015b) and metformin (Madiraju et al., 2014) concentrations were measured by LC-MS/MS. To measure tumor [4,5-13C2] glutamate enrichment, ~25–50 mg tissue was homogenized in 10× volume 50% methanol and filtered through a Nanosep filter, and LC-MS/MS (AbSCIEX 6500 QTRAP with a Shimadzu ultrafast liquid chromatography system, negative ion mode) was used to monitor the relevant ion pairs: [m0] C4-5 glutamate, 146/41, and [m+2] C4-5 glutamate, 148/48. The same extraction was used to measure tumor [m+1] glutamate, glutamine, citrate, and malate enrichment (ion pairs: [m0] glutamate and glutamine 146/128, [m1] glutamate and glutamine 147/ 129, [m0] citrate, 191/173, [m+1] citrate, 192/174, [m0] malate, 133/115, [m1] malate, 134/116) in mice infused with [1-13C] acetate. GC-MS was used to measure [13C3] alanine enrichment as we have described (Perry et al., 2016).
In Vitro Studies
MC38 cells were plated in 24 well plates (105 cells per well) and incubated in the standard culture media described in the Cell Lines section or the same media containing 0.15 or 11 μM DNP or 70 or 180 μM metformin. 48 hr later, the cells were trypsinized and counted using a hemocytometer by a blinded investigator (6 wells per condition, with two aliquots per well counted and averaged).
Histologic Analysis
Sections were fixed in 10% neutral buffered formalin and transferred the next day to 70% ethanol. Sections were cut and stained for β-catenin (Biocare Medical, CM406) by the Yale Research Histology core. 100× fields were taken of the tumor samples using an Olympus BX41 microscope with an Insight 2 camera running SPOT Advanced 5.3 (SPOT Imaging). β-catenin positive polyp number and area were measured in all ApcMin+/− intestines by an investigator blinded as to group allocation.
Statistical Analysis
Data were analyzed using GraphPad Prism v.7.0. Data are presented as the mean ± SEM of the numbers indicated in the figure legends. The two-tailed Student’s t test (paired or unpaired, as specified in the figure legends) was used to perform comparisons of two groups, while ANOVA with Bonferroni’s multiple comparisons test was employed to compare three or more groups. Differences with a p value of less than 0.05 were considered significant.
Supplementary Material
Highlights.
Hyperinsulinemia in HFD mice increases glucose uptake and oxidation in tumors
Metformin and CRMP slow colon tumors in two mouse models by reversing hyperinsulinemia
Reducing tumor glucose oxidation may slow obesity-associated cancers
Acknowledgments
The authors thank Dr. Marie-José Blouin for her advice and Xiaoxian Ma, Wanling Zhu, Gina Butrico, Maria Batsu, and Amos Brooks for their technical contributions to this study. Clipart representing pancreas, muscle, and liver were used under a Creative Commons License from Servier Medical Art. This study was funded by grants from the U.S. Public Health Service (K99 CA-215315 [to R.J.P.], R01 DK-113984 [to G.I.S.], R01 DK-40936 [to G.I.S.], P30 DK-059635, and UL1TR000142) and from the Canadian Institutes of Health Research (to M.N.P.).
Footnotes
AUTHOR CONTRIBUTIONS
M.N.P., G.I.S., and R.J.P. designed the study and wrote the manuscript. All authors edited and approved the manuscript. Y.W., A.R.N., W.E.D., C.J.P., X.-M.Z., A.R.-C., and R.J.P. performed experiments and analyzed data.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Supplemental Information includes three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.008.
References
- Abbruzzese C, Diodoro MG, Sperduti I, Mileo AM, Pattaro G, De Salvo L, Cosimelli M, Perrotti N, Paggi MG. Detection of phosphorylated insulin receptor in colorectal adenoma and adenocarcinoma: implications for prognosis and clinical outcome. J Cell Physiol. 2015;230:562–567. doi: 10.1002/jcp.24733. [DOI] [PubMed] [Google Scholar]
- Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17:351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- Algire C, Amrein L, Bazile M, David S, Zakikhani M, Pollak M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011;30:1174–1182. doi: 10.1038/onc.2010.483. [DOI] [PubMed] [Google Scholar]
- Baltgalvis KA, Berger FG, Peña MM, Davis JM, Carson JA. Effect of exercise on biological pathways in ApcMin/+ mouse intestinal polyps. J Appl Physiol (1985) 2008;104:1137–1143. doi: 10.1152/japplphysiol.00955.2007. [DOI] [PubMed] [Google Scholar]
- Bardou M, Barkun AN, Martel M. Obesity and colorectal cancer. Gut. 2013;62:933–947. doi: 10.1136/gutjnl-2013-304701. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Boyle JP, Honeycutt AA, Narayan KM, Hoerger TJ, Geiss LS, Chen H, Thompson TJ. Projection of diabetes burden through 2050: impact of changing demography and disease prevalence in the U.S. Diabetes Care. 2001;24:1936–1940. doi: 10.2337/diacare.24.11.1936. [DOI] [PubMed] [Google Scholar]
- Chung YW, Han DS, Park KH, Eun CS, Yoo KS, Park CK. Insulin therapy and colorectal adenoma risk among patients with type 2 diabetes mellitus: a case-control study in Korea. Dis Colon Rectum. 2008;51:593–597. doi: 10.1007/s10350-007-9184-1. [DOI] [PubMed] [Google Scholar]
- Cohen DH, LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer. 2012;19:F27–F45. doi: 10.1530/ERC-11-0374. [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhou J, Zhu Y, Luo L, He T, Hu H, Liu H, Zhang Y, Luo D, Xu S, et al. Abdominal obesity and colorectal cancer risk: systematic review and meta-analysis of prospective studies. Biosci Rep. 2017;37:1–12. doi: 10.1042/BSR20170945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardet A, Druesne-Pecollo N, Touvier M, Latino-Martel P. Do alcoholic beverages, obesity and other nutritional factors modify the risk of familial colorectal cancer? A systematic review. Crit Rev Oncol Hematol. 2017;119:94–112. doi: 10.1016/j.critrevonc.2017.09.001. [DOI] [PubMed] [Google Scholar]
- Farmer RE, Ford D, Forbes HJ, Chaturvedi N, Kaplan R, Smeeth L, Bhaskaran K. Metformin and cancer in type 2 diabetes: a systematic review and comprehensive bias evaluation. Int J Epidemiol. 2017;46:745. doi: 10.1093/ije/dyx046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, Uchiyama T, Taniguchi L, Hata Y, Uchiyama S, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016;17:475–483. doi: 10.1016/S1470-2045(15)00565-3. [DOI] [PubMed] [Google Scholar]
- Hvid H, Fendt SM, Blouin MJ, Birman E, Voisin G, Svendsen AM, Frank R, Vander Heiden MG, Stephanopoulos G, Hansen BF, Pollak M. Stimulation of MC38 tumor growth by insulin analog X10 involves the serine synthesis pathway. Endocr Relat Cancer. 2012;19:557–574. doi: 10.1530/ERC-12-0125. [DOI] [PubMed] [Google Scholar]
- Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009;458:725–731. doi: 10.1038/nature07782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI. Diet, lifestyle, and colorectal cancer: is hyperinsulinemia the missing link? Nutr Rev. 1998;56:275–279. doi: 10.1111/j.1753-4887.1998.tb01765.x. [DOI] [PubMed] [Google Scholar]
- Klil-Drori AJ, Azoulay L, Pollak MN. Cancer, obesity, diabetes, and antidiabetic drugs: is the fog clearing? Nat Rev Clin Oncol. 2017;14:85–99. doi: 10.1038/nrclinonc.2016.120. [DOI] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. Met-formin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattarocci S, Abbruzzese C, Mileo AM, Visca P, Antoniani B, Alessandrini G, Facciolo F, Felsani A, Radulescu RT, Paggi MG. Intracellular presence of insulin and its phosphorylated receptor in non-small cell lung cancer. J Cell Physiol. 2009;221:766–770. doi: 10.1002/jcp.21916. [DOI] [PubMed] [Google Scholar]
- McClellan JL, Steiner JL, Day SD, Enos RT, Davis MJ, Singh UP, Murphy EA. Exercise effects on polyp burden and immune markers in the ApcMin/+ mouse model of intestinal tumorigenesis. Int J Oncol. 2014;45:861–868. doi: 10.3892/ijo.2014.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Zhu H, Gu M. Reduced colorectal cancer incidence in type 2 diabetic patients treated with metformin: a meta-analysis. Pharm Biol. 2016;54:2636–2642. doi: 10.1080/13880209.2016.1176057. [DOI] [PubMed] [Google Scholar]
- Nimri L, Saadi J, Peri I, Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity to altered metabolism in mice colon carcinogenesis. Oncotarget. 2015;6:38195–38209. doi: 10.18632/oncotarget.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Øines M, Helsingen LM, Bretthauer M, Emilsson L. Epidemiology and risk factors of colorectal polyps. Best Pract Res Clin Gastroenterol. 2017;31:419–424. doi: 10.1016/j.bpg.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015a;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steato-hepatitis in rats. Science. 2015b;347:1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Pe-tersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- Rosato V, Tavani A, Gracia-Lavedan E, Guinó E, Castaño-Vinyals G, Vil-lanueva CM, Kogevinas M, Polesel J, Serraino D, Pisa FE, et al. Type 2 Diabetes, Antidiabetic Medications, and Colorectal Cancer Risk: Two Case-Control Studies from Italy and Spain. Front Oncol. 2016;6:210. doi: 10.3389/fonc.2016.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen RE, Tangen CM, Kuller LH, Burke GL, Cushman M, Tracy RP, Dobs A, Savage PJ. Increased blood glucose and insulin, body size, and incident colorectal cancer. J Natl Cancer Inst. 1999;91:1147–1154. doi: 10.1093/jnci/91.13.1147. [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, Shaw RJ. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI, Rossetti L, Rothman DL, Blair JB, Smith D. Quantitative analysis of glycogen repletion by nuclear magnetic resonance spectroscopy in the conscious rat. J Clin Invest. 1987;80:387–393. doi: 10.1172/JCI113084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele CB, Thomas CC, Henley SJ, Massetti GM, Galuska DA, Agurs-Collins T, Puckett M, Richardson LC. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity - United States, 2005–2014. MMWR Morb Mortal Wkly Rep. 2017;66:1052–1058. doi: 10.15585/mmwr.mm6639e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, Takahashi H, Nakajima N, Nagashima Y, Wada K, Nakagama H, Nakajima A. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008;99:2136–2141. doi: 10.1111/j.1349-7006.2008.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSaun MN, Lee IK, Washington MK, Matrisian L, Gorden DL. High fat diet induced hepatic steatosis establishes a permissive micro-environment for colorectal metastases and promotes primary dysplasia in a murine model. Am J Pathol. 2009;175:355–364. doi: 10.2353/ajpath.2009.080703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. Will all Americans become overweight or obese? estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring) 2008;16:2323–2330. doi: 10.1038/oby.2008.351. [DOI] [PubMed] [Google Scholar]
- Wolpin BM, Meyerhardt JA, Chan AT, Ng K, Chan JA, Wu K, Pollak MN, Giovannucci EL, Fuchs CS. Insulin, the insulin-like growth factor axis, and mortality in patients with nonmetastatic colorectal cancer. J Clin Oncol. 2009;27:176–185. doi: 10.1200/JCO.2008.17.9945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Nunez NP, Pennisi P, Brodt P, Sun H, Fallavollita L, Zhao H, Scavo L, Novosyadlyy R, Kurshan N, et al. Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology. 2006;147:5826–5834. doi: 10.1210/en.2006-0311. [DOI] [PubMed] [Google Scholar]
- Yang YX, Hennessy S, Lewis JD. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients. Gastroenterology. 2004;127:1044–1050. doi: 10.1053/j.gastro.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Yin S, Bai H, Jing D. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients: a systemic review and meta-analysis. Diagn Pathol. 2014;9:91. doi: 10.1186/1746-1596-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JH, Buchanan TA. Fasting does not impair insulin-stimulated glucose uptake but alters intracellular glucose metabolism in conscious rats. Diabetes. 1993;42:757–763. doi: 10.2337/diab.42.5.757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.