

Abstract
The establishment of more effective treatments that can circumvent chemoresistance in Multiple Myeloma (MM) is a priority. Although bortezomib (BTZ) is one of the most potent proteasome inhibitors available, still possesses limitations related to dose limiting side effects. Several strategies have been developed to improve the delivery of chemotherapies to MM by targeting different moieties expressed on MM cells to nanoparticle delivery systems (NPs), which have failed mainly due to their heterogeneous expression on these cells. Our goal was to test CD38 targeted chitosan NPs as novel targeting moiety for MM to improve the potency and efficacy of BTZ in MM cells and reduce the side effects in healthy tissue. We have showed preferential BTZ release in tumor-microenvironment, specific binding to MM cells, and an improved drug cellular uptake through BTZ diffusion from the surface and endocytosed NPs, which translated in enhanced proteasome inhibition and robust cytotoxic effect on MM cells when BTZ was administered through anti-CD38 chitosan NPs. Furthermore, the anti-CD38 chitosan NPs specifically delivered therapeutic agents to MM cells improving therapeutic efficacy and reducing side effects in vivo. The anti-CD38 chitosan NPs showed low toxicity profile allowing enhancement of proteasome-inhibitory activity and specificity of BTZ by endocytosis-mediated uptake of CD38 representing a promising therapy in MM.
Keywords: CD38, Bortezomib, endocytosis, Nanoparticles, Multiple Myeloma
Graphical abstract
INTRODUCTION
Multiple Myeloma (MM) is the second most common hematologic malignancy, accounting for >10,000 deaths annually in the USA [1, 2]. Despite development of novel drugs which improved the treatment outcomes, MM remains incurable with relatively low response rates especially in the relapsed patient population [3-5]. Current methods of MM therapy include proteasome inhibitors, immunomodulatory drugs, and monoclonal antibodies [5, 6]. Bortezomib (BTZ), the first proteasome inhibitor used in the treatment of MM, is a specific and reversible inhibitor of the proteasome, which is responsible for the degradation of intracellular proteins and contribute to the maintenance of protein homeostasis and clearance of misfolded and/or unfolded and cytotoxic proteins [7, 8]. However, BTZ induces a variety of systemic toxicities when administered to patients including vomiting, diarrhea, nausea, or constipation, lack of strength, fatigue, as well as, thrombocytopenia and peripheral neuropathy (PN) [9, 10]. In general, PN and thrombocytopenia occurs in approximately 30% of patients and are the most problematic dose limiting toxicities [9]. PN is characterized by a burning sensation, paresthesias, numbness and/or neuropathic pain, and there is a recommended dose modification for bortezomib-related neuropathy and/or neuropathic pain by the National Cancer Institute. Dose limiting thrombocytopenia is transient and reversible, and it can be generally managed with platelet transfusions without reducing or omitting doses [9, 11].
Therefore, there is an urgent need for development of a more specific delivery of BTZ to decrease side effects in the normal tissues and increase its potency in the MM cells. BTZ has been previously loaded into nanoparticle delivery systems (NPs). Liposomes loaded with BTZ have shown to be successful to in vitro proteasome inhibition, induction of apoptosis, as well as reduced body weight loss in vivo [12]. However, this liposomal formulation showed reduced cytotoxic effects in MM cells compared to free BTZ, attributed to the kinetics of BTZ release from their NPs. Moreover, these particles were relying on passive targeting to the tumor through enhanced permeability and retention (EPR) effect, which is the tendency for NPs and other molecular agents to conglomerate more in tumor versus normal tissue [13-15]. But in addition to the unspecific nature of the EPR effect, large tumors may not display the EPR effect, thus causing for fewer NPs to accumulate in the central areas [16]. Therefore, a more specific and active targeting approach is needed.
In this paper, we loaded BTZ in crosslinked chitosan NPs conjugated with anti-CD38 monoclonal antibodies. We hypothesize that the specific targeting with anti-CD38 chitosan NPs will improve the potency and efficacy of BTZ in MM cells and reduce the side effects in healthy tissue.
MATERIALS AND METHODS
Materials & Reagents
Unless stated otherwise, all materials were purchased from Sigma (St. Louis, MO, USA). Chitosan low molecular weight, acetic acid 99.7%, and sodium tripolyphosphate (TPP) were used for the preparation of the chitosan NPs. Alexa Fluor 633 (AF633) was purchased from Life Technologies (Carlsbad, CA, USA). Streptavidin conjugation kit (abcam, Cambridge, MA, USA), Mix-n-Stain Biotin labeling kit, and human anti-CD38 (Clone 240742, R&D systems, Minneapolis, MN, USA) were used to prepared targeted anti-CD38 NPs.
Cells
The MM cell lines MM.1S, H929, RPMI8226, and U266 were purchased from ATCC, OPM1 and MM1s-GFP-Luc were a kind gift from Dr. Irene Ghobrial (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA). MSP-1 cell line was developed in our lab and used as myeloma-derived stromal cell line for co-cultures [17]. Bone Marrow mononuclear cells (BM MNCs) from 5 different patient samples were purchased from Allcells (Alameda, CA). Peripheral blood mononuclear cells (PB MNCs) were isolated from pheresis leukopaks from the Siteman Cancer Center, Washington University in Saint Louis. 1× RBC Lysis Buffer was added to whole blood, gently vortex and incubated at room temperature, protected from light, for 10-15 minutes. Cell were washed and cultured in RPMI completed media (RPMI-1640 media (Corning CellGro, Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS, Gibco, Life technologies, Grand island, NY), 2 mmol/l of L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Corning CellGro)). Primary CD138+ cells were isolated from BM aspirates of MM patients from the Siteman Cancer Center, Washington University in Saint Louis, by magnetic-bead sorting, as previously described [18]. Informed consent was obtained from all patients with an approval from the Washington University Medical School IRB committee and in accord with the Declaration of Helsinki. All cells were cultured at 37°C, 5% CO2; MM cells in RPMI completed media, and stromal cells in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning CellGro) supplemented with 20% FBS, L-glutamine, penicillin, and streptomycin.
Preparation of chitosan NPs
Chitosan NPs were obtained by ionic crosslinking of a 2 mg/ml chitosan solution dissolved in 2.1% acetic acid with TPP solution (0.25 – 1 mg/ml). Chitosan solution was added drop-wise with a 30G needle to a TPP solution and kept with constant medium stirring for 15 minutes (Ratio 5:1 in volume). After NPs stabilization, the NPs were ultracentrifuged at 40.000g for 30 minutes at 10°C (Rotor 25.50, Avanti J-E, Beckman Coulter, Indianapolis, IN, USA). AF633-labeled chitosan polymer was elaborated for in vivo, confocal, and flow cytometry studies and used for preparation of AF633 chitosan NPs. Chitosan NPs were conjugated with streptavidin using streptavidin-conjugation kit, according to the manufacturer’s instructions (100μg streptavidin per 10mg of NPs). Anti-CD38 monoclonal antibody was labeled with biotin using biotin labeling kit according to manufacturer’s instructions (50μg anti-CD38 antibody in biotin kit for 20-50μg). Then, the streptavidin-chitosan NPs (100mg/ml) were mixed with the biotin-anti-CD38 (200μg/ml) overnight at 4°C to obtain anti-CD38 targeted chitosan NPs. Moreover, chitosan NPs from the same batch without conjugation to streptavidin and biotin-antibody were used as non-targeted NPs. All NPs were ultracentrifuged at 40.000g for 30 minutes at 10°C for purification before were dispersed in double distilled water (DDW) for characterization of size and stability and dispersed in RPMI completed media with 10% FBS for all experiments with cells.
Characterization of size and stability of anti-CD38 chitosan NPs
The effect of TPP concentration (0.25 – 1 mg/ml), BTZ loading (intermediate (50 μM) and high-dose (1 mM)) and storage time (over a month) on particle size and ζ-potential (the potential difference between the dispersion medium and the surface of the nanoparticle) were determined by dynamic light scattering (DLS) using a Malvern Zetasizer Nano ZS (ZEN3600) (Malvern, Herrenberg, Germany) with a 633-nm He-Ne laser operating at a 173° angle. Malvern’s Dispersion Technology Software version 6.01 was used to collect and analyze the data. Chitosan NPs crosslinked with 0.25mg/ml TPP concentration were further characterized by Transmission Electron Microscopy (TEM, FEI Tecnai G2 Spirit TEM at 120 KV) where a drop of the sample was placed on 200 mesh carbon film coated copper grid and the sample was then air dried for imaging.
HPLC Assay for Detection of Bortezomib
Bortezomib was analyzed using high performance liquid chromatography (HPLC, Agilent 1100 series, Santa Clara, CA) with a reverse phase C-18 column (Agilent Zorbax Eclipse XDB C18). A 50% acetonitrile solution in water containing 0.1% trifluoroacetic acid was used as the mobile phase at a flow rate of 1 mL/min. A calibration curve was formed by plotting the area under curve (AUC) of the BTZ HPLC peak (at retention time = 2 minutes, λ = 270 nm) for a concentration range of 0 to 1.5 mM. A linear correlation for the curve was obtained a R2=0.9995, with a limit of detection of approximately 25 μM.
Bortezomib encapsulation efficiency in anti-CD38 chitosan NPs
The effect of intermediate (50 μM) and high-dose (1 mM) BTZ on the encapsulation efficiency of BTZ in NPs was assessed by the aforementioned HPLC assay. 1h BTZ incubation at 4°C either in 10 mg/ml NPs already crosslinked in PBS solution or incorporated of BTZ into TPP solution before crosslinking was tested. BTZ solutions (50 μM and 1mM) were used as controls for each study group. After ultracentrifugation, supernatants were collected from each condition and AUC values were used in the calibration curve to determine BTZ concentration. The encapsulation efficiency (EE) was calculated as follows: %EE = 100 − [(Concentration BTZ-NPs supernatant/Concentration BTZ Control) *100].
In vitro drug release from anti-CD38 chitosan NPs in normal and tumor environments
In vitro release studies were assessed by the aforementioned HPLC assay (1h BTZ incubation at 4°C in 10 mg/ml NPs solution in DDW solution). BTZ loaded nanoparticles were included in dialysis bags at 4°C with middle mixing in DDW in a light protected environment for 72 hours. BTZ was added to the DDW solution that contains BTZ non-loaded nanoparticles as total release control, and also BTZ non-loaded nanoparticles were included in DDW as blank. The DDW solution with the nanoparticles was sampled at different time points and the samples were analyzed by HPLC. The % release (%R) was calculated as follows: %R = (Nanoparticles – Blank) / (Total – Blank) *100.
In addition, MM cell lines (MM.1S, H929 and RPMI) and normal mononuclear cells (PB MNCs) were cultured in RPMI media with 10% FBS for 72 hours, conditioned media samples were centrifuged and supernatants were obtained. The pH of the media samples was tested and divided into two vials. The first vial was used as is (conditioned media), while the pH in the other vial was adjusted back to the pH of media (8.4) (conditioned media-Corrected pH), with non-MM cultured used as a control. Chitosan NPs loaded with doxorubicin (model fluorescent drug) were incubated in the different media samples (non-cultured media, normal mononuclear cells, and MM-media cultured with and without adjustment of the pH) for 24 h at 37°C. Samples were ultracentrifuged at 40.000g for 30 minutes at 10°C, supernatant extracted, and analyzed by fluorescence plate reader (Exc 480nm/Em 580nm).
Swelling of anti-CD38 chitosan NPs in normal and tumor environments
The swelling properties of anti-CD38 chitosan NPS was evaluated. Pre-weighed anti-CD38 chitosan NPs were immersed in excess of two swelling medium, non-conditioned media and MM cells conditioned media. At various time intervals, NPs were ultra-centrifuged, the swelling medium was removed and NPs weighed. Results were calculated according to the following equation: Q = (Ws – Wd) /Wd. Here, Q is the swelling ratio, Ws is the weight in the swollen state and Wd is the weight in the dried state.
Characterization of the kinetics of the binding/uptake of the anti-CD38 to MM cells
First, we detected the expression of CD38 in three MM cell lines (MM.1S, H929, and RPMI8226) with the monoclonal antibody APC-anti-CD38 (incubation at 4°C for 1 h) and analyzed the expression by detection of the mean fluorescent intensity (MFI) of APC in the MM cells by flow cytometry.
Then, MM cell lines were treated with the anti-CD38 chitosan NPs pre-labeled with AF633 (1mg/ml) for increasing times (0, 30 minutes, 2, 4, 6, 16 and 24 h), then cells were washed and analyzed for the MFI of AF633 by flow cytometry (to ascertain quantitatively the total binding/uptake). In addition, the cells were imaged using FV1000 confocal microscope with an XLUMPLFLN 20XW/1.0 immersion objective lens (Olympus, PA, USA), with excitation of 633 nm and the emission filter of 650 long pass, to determine the sub-cellular distribution of the nanoparticle.
To characterize the kinetics of potential dissociation, MM cells were treated with the AF633 anti-CD38 chitosan NPs for 2hrs, then excess particles were washed from the cells, and the cells were put back to culture and tested for the fluorescence intensity at 0, 2, 6, 12 and 24 h after the washing by flow cytometry.
Characterization of specificity of the binding/uptake of the anti-CD38 chitosan NPs to MM and normal cells in vivo
MM cell lines (n=5, MM.1S, H929, RPMI8226, and U266), primary CD138+ MM cells isolated from MM patients (n=5), normal mononuclear cells isolated from the peripheral blood of normal subjects (n=5), and normal plasma cells isolated from the BM of normal subjects (n=5) were incubated with the targeted AF633 anti-CD38 chitosan NPs or with AF633 non-targeted chitosan NPs for 2 h. In addition, non-labeled non-targeted chitosan NPs were used as control. Cells were washed and analyzed for the MFI of AF633 by flow cytometry.
To further confirm the mechanism of binding/uptake of the targeted and non-targeted particles, MM cell lines (MM1s, H929, and RPMI8226) were treated with or without free unlabeled anti-CD38 monoclonal antibody (for blocking the epitopes), and then incubated with the targeted and the non-targeted NPs for 2 h, washed, and analyzed by flow cytometry.
Characterization of specificity and biodistribution of anti-CD38 NPs in vivo
Approval for these studies was obtained from the Ethical Committee for Animal Experiments at Washington University in St. Louis Medical School. MM1s-GFP-Luc cells were injected into 20 female, 7-week old SCID mice (Taconic Farms, Hudson, NY) intravenously (i.v.) at the concentration of 2 × 106 cells per mouse and tumor progression was confirmed using bioluminescent imaging (BLI) at 4 weeks post cell injection. Radiance was measured by BLI in photons/sec within a region of interest (ROI) covering full body. Mice were then randomized to 4 groups of 5 mice each, and treated with (a) vehicle control, (b) free deactivated AF633 (5 mg/kg), (c) non-targeted NPs with a dye content equivalent to 5 mg/kg, (d) anti-CD38 NPs loaded with a dye content equivalent to 5 mg/kg. The AF633 that we use for labelling the chitosan contains a succinimidyl ester group. This group may react with amine groups when injected i.v and change its biodistribution. We therefore, deactivate this group in the AF633 (free deactivated AF633) with NH3SO4 before injection. Mice were sacrificed 24 h post-injection of each treatment and organs were harvested (femurs (BM), blood, heart, kidney, liver, lung, and spleen). Organs were homogenized with a Tissue Homogenizer (Omni TH), collected cells were filtered through a 35μm strainer flow cap and analyzed by flow cytometer. MM cells (GFP+ cells) were detected and analyzed for anti-CD38 chitosan NPs uptake as MFI of AF633.
The effect of bortezomib-loaded anti-CD38 chitosan NPs on cell viability of MM cells and normal MNCs in vitro
MM cell lines (MM.1S, H929, and RPMI8826) and PB MNCs were cultured with vehicle (control), BTZ as a free drug (5 nM), empty NPs, non-targeted NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 NPs loaded with BTZ equivalent amounts to 5 nM for 48 h. We also tested toxicity at 48 h after a 2 h pulse exposure to the same conditions and determine the effect on cell viability. We also tested increasing concentrations of anti-CD38 antibody (0.5 – 5 μg/ml) to rule out an effect of CD38 on MM cells for 48 h. Cell viability was assessed using MTT solution followed by absorbance reading at 570 nm using a spectrophotometer as previously described [19]. Briefly, the MTT solution was added to the cells 48 h after starting the treatment, and 2 - 4 h later the stop solution was added.
In addition, cell proliferation assay of MM.1S cells co-culture with or without MM-derived MSP-1 stromal cell line with vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h was analyzed. Furthermore, the effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM on proliferation of MM cells cultured in normoxic (21% O2) or hypoxic (1% O2) conditions for 48 h was measured.
MM.1S (pre-labeled with Invitrogen cell tracers DiO (10 μg/ml) for 1 h) were cultured in relevant three-dimensional tissue engineered bone marrow (3DTEBM) cultures through crosslinking of fibrinogen as previously described [20-24]. The effect of vehicle (control), BTZ as a free drug (10 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 10 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 10 nM for 48 h was analyzed by flow cytometry. Higher BTZ doses (10 nM) are used in the 3DTEBM based on the known drug resistance of the cells in 3DTEBM cultures [20]. Cell proliferation assays were performed by digestion of 3DTEBM cultures with type I collagenase (25 mg/ml for 2 - 3 h at 37°C), followed by flow cytometry analysis using MACSQuant Analyzer (Miltenyi Biotec), and the data was analyzed using FlowJo program v10 (Ashland, OR). Moreover, 3DTEBM cultures treated with AF633 anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 10 nM were imaged to determine NPs distribution by confocal microscopy after 24 h incubation.
The effect of bortezomib loaded anti-CD38 chitosan NPs on Cell Cycle of MM cells
H929 cells (1 × 106 cell/ml) were cultured with vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h, and cell cycle was analyzed as previously described [25]. Briefly, cells were washed, fixed with 70% ethanol, and washed again with PBS. RNA was degraded by incubation in RNAase for 30 minutes at 37°C, and the DNA was stained with PI solution for 10 minutes, then cells were analyzed by flow cytometry.
The effect of bortezomib loaded anti-CD38 chitosan NPs on apoptosis of MM cells
H929, U266 and RPMI cells (1 × 106 cell/ml) were cultured with vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h, and apoptosis was analyzed as previously described [26]. Cells were washed and resuspended in 1× Annexin binding buffer, incubated with Annexin V (5μl of BD Pharmingen FITC Annexin V #556420 100 test) for 15 minutes followed by staining with PI (5 μl of 50μg/ml) for extra 15 minutes, 1× binding buffer was added, and the cells were analyzed with flow cytometry.
The effect of bortezomib loaded anti-CD38 chitosan NPs on proliferation, cell cycle and apoptosis cell signaling in MM cells
To test the effect of BTZ as a free drug or encapsulated in chitosan NPs on proliferation, cell cycle, and apoptosis signaling, H929 cells were treated with vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 6 h (survival) and 12 h (cell cycle and apoptosis). Cells were washed and lysed with 1× Phenylmethylsulfonyl fluoride (PMSF) for 15 minutes. Protein concentration in the cell lysates was normalized and 50 μg of protein was loaded per lane. Electrophoresis was performed using NuPAGE 4% - 12% Bis-Tris gels (Novex, Life Technologies, Grand Island, NY) and transferred to a nitrocellulose membrane using iBlot (Invitrogen, Life Technologies). Membranes were blocked with 5% non-fat milk in Tris-Buffered Saline/Tween20 (TBST) buffer and incubated with primary antibodies overnight at 4°C for proliferation signaling with pAKT and pERK1/2; for cell cycle with pRb; and for apoptosis with cleaved Caspase-3 and cleaved PARP. α-Tubulin was used as a loading control. The membranes were washed with TBST for 30 minutes, incubated for 1hr at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibody, washed, and developed using Novex ECL Chemiluminescent Substrate Reagent Kit (Invitrogen). Antibodies were purchased from Cell Signaling Technology (Danvers, MA). Phospho-Erk1/2 (Thr202/Tyr204) (D13.14.4E) XP® (rabbit mAb #4370), phospho-Akt (Ser473) (D9E) XP® (rabbit mAb #4060), pRb (Ser807/811) (rabbit mAb #9308), cleaved-Caspase-3 (Asp175) (5A1E) (rabbit mAb #9664), cleaved-PARP (Asp214) (D64E10) (rabbit mAb #5625), and α-Tubulin (11H10) (rabbit mAb #2125) were used at a dilution of 1:1000.
The effect of bortezomib loading in anti-CD38 chitosan NPs on proteasome inhibition activity.
Cell extracts were prepared in NP-40 lysis buffer from control and treated cells (BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 NPs loaded with BTZ equivalent amounts to 5 nM) retrieved after 2h treatment with BTZ. Proteasome activity was determined using the AMC-tagged peptide substrate in a Proteasome Activity Assay Kit (abcam, Cambridge, MA, USA) according to the manufacturer’s protocol. Fluorescence was determined in a SpectraMax i3 Multi-Mode Detection Platform (Molecular Devices, Sunnyvale, CA, USA) and proteasome activity was expressed as the amount of proteasome which generates 1.0nmol of AMC per minute at 37°C.
The effect of bortezomib loading in anti-CD38 chitosan NPs on intracellular delivery of bortezomib by detection of boron cellular accumulation using ICP-OES
MM.1S cells (40 × 106 cell/ml) were cultured with vehicle (control), BTZ as a free drug (100 nM), non-targeted chitosan NPs loaded with an equivalent BTZ amount (100 nM), and anti-CD38 chitosan NPs loaded with an equivalent BTZ amount (100 nM) for 1.5 h. After treatment the cells were spun down, washed, and the cell pellet was digested in concentrated nitric acid (HNO3). After diluting samples with deionized water to a final 5% HNO3 content (v/v), samples were microwave digested in a sealed tube (MARS 6 Microwave Digestion System, CEM, Matthews, NC) and then analyzed by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES, Optima 7300DV series, Perkin Elmer, Waltham, MA). Samples were analyzed for boron content (λem = 249.677 nm) against a calibration curve of boron standards of 0, 0.625, 1.25, 2.5, 5, 10 and 20 parts per billion (ppb) in 5% HNO3 prepared from a 10 parts per million boron standard solution (Inorganic Ventures, Christiansburg, VA) in 5% HNO3. Scandium was used as an internal standard throughout ICP-OES analysis.
Investigation of the anti-CD38 chitosan NPs uptake mechanism
Endocytosis is critical to the uptake of NPs in order to allow toxic effects in cells. MM cells were plated on glass bottom 8 well chambers and allowed to adhere overnight. CellLight Early Endosomes-GFP BacMam 2.0 reagent (targeting rab5) was added to result in a final concentration of 30 particles per cells and allowed to incubate with the cells for 18–24 h. Cells were washed twice with PBS and incubated with AF633 anti-CD38 chitosan NPs for 24 h. Cells were again washed twice with PBS and allowed to stay in phenol red free medium for imaging live under culture conditions of 37 °C and 5% CO2 at specified time points using FV1000 confocal microscope.
To understand the process of internalization of anti-CD38 chitosan NPs, MM cells were pre-treated with inhibitors of macropinocytosis and phagocytosis (cytochalasin D), clathrin-mediated endocytosis (chlorpromazine), caveolae-mediated endocytosis (nystatin), or late endocytosis (EGA), prior to exposure to anti-CD38 chitosan NPs. All the inhibitors were tested on survival of MM cells to determine a concentration that does not induce cell death by MTT. MM cells (1 × 106 cell/ml) were pre-incubated at 37°C with no inhibitor (Ctrl), cytochalasin D 1μM, chlorpromazine 2μM, nystatin 0.5μg/ml, and EGA 2.5μM or at 4°C as control of the active uptake for 30 min. Cells were washed with PBS and incubated with AF633 anti-CD38 chitosan NPs for 2 h. Then, cells were washed and analyzed by flow cytometry for the fluorescence intensity of AF633.
Proteasome activity inhibition was also measured in combination with inhibitors of macropinocytosis and phagocytosis (cytochalasin D), clathrin-mediated endocytosis (chlorpromazine), caveolae-mediated endocytosis (nystatin), or late endocytosis (EGA), prior to exposure to anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM. MM cells were pre-incubated at 37°C with no inhibitor (Ctrl), cytochalasin D 1μM, chlorpromazine 2μM, nystatin 0.5μg/ml, and EGA 2.5μM for 30 min. Cells were washed with PBS and incubated with anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM, then proteasome Activity Assay Kit was followed as in the previous experiment.
The effect of bortezomib-loaded anti-CD38 NPs on MM tumor progression in vivo.
MM1s-GFP-Luc cells were injected into forty female, 7-week old SCID mice i.v. at the concentration of 2 × 106 cells per mouse and tumor progression was evaluated using BLI. After 4 weeks, tumor bearing mice were randomized into 4 groups of 10 animals each and treated with: (i) vehicle, (ii) BTZ as a free drug (1 mg/kg once a week), (iii) non-targeted NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week), (iv) and anti-CD38 NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week). Tumor progression was followed in the 4 groups twice a week (for 5 weeks) by BLI; moreover the survival, weight, and general health of the animals was followed. In addition, 3 mice were taken from each group 3 weeks after the beginning of the treatment, and specimens of the BM, blood smear, spleen, liver, kidney, brain, spinal-cord and intestine were fixed and pathologically evaluated for treatment efficacy and histological tissue damage. In order to determine how many mice per 4 different treatment groups (Control plus 3 conditions groups) we utilized a Tukey-Cramer multiple comparison test for independent means. Utilizing a multiple comparison alpha adjustment for the 6 tests and assuming a 100% difference with a standard deviation of 25% we have over 95% power (alpha = .025) to detect this difference, therefore we would need 10 mice per group to sufficiently power this aim.
Statistical analysis
Measurements were made in triplicate for each group. The in vitro data were expressed as means ± standard deviation. Results were analyzed using student t-test for statistical significance and were considered significantly different for p value less than 0.05. The in vivo data were analyzed using student t-test or ANOVA for statistical significance, and results are depicted as mean ± S.E.M. Variation within each group was equally variant and similar between the groups that were statistically compared. Values were considered significantly different for p value less than 0.05.
RESULTS
Characterization of anti-CD38 chitosan NPs
Chitosan NPs were prepared using ionotropic gelation technique in which the crosslinking reaction involves ionic interactions between the positively charged amino groups of chitosan and the negatively charged phosphate groups of TPP resulting in polymeric NPs (Fig. 1.A). Targeting with anti-CD38 antibody was obtained by conjugation of chitosan NPs with streptavidin and followed by incubation with biotinylated-anti-CD38 antibody (Fig. 1.B).
Fig. 1:
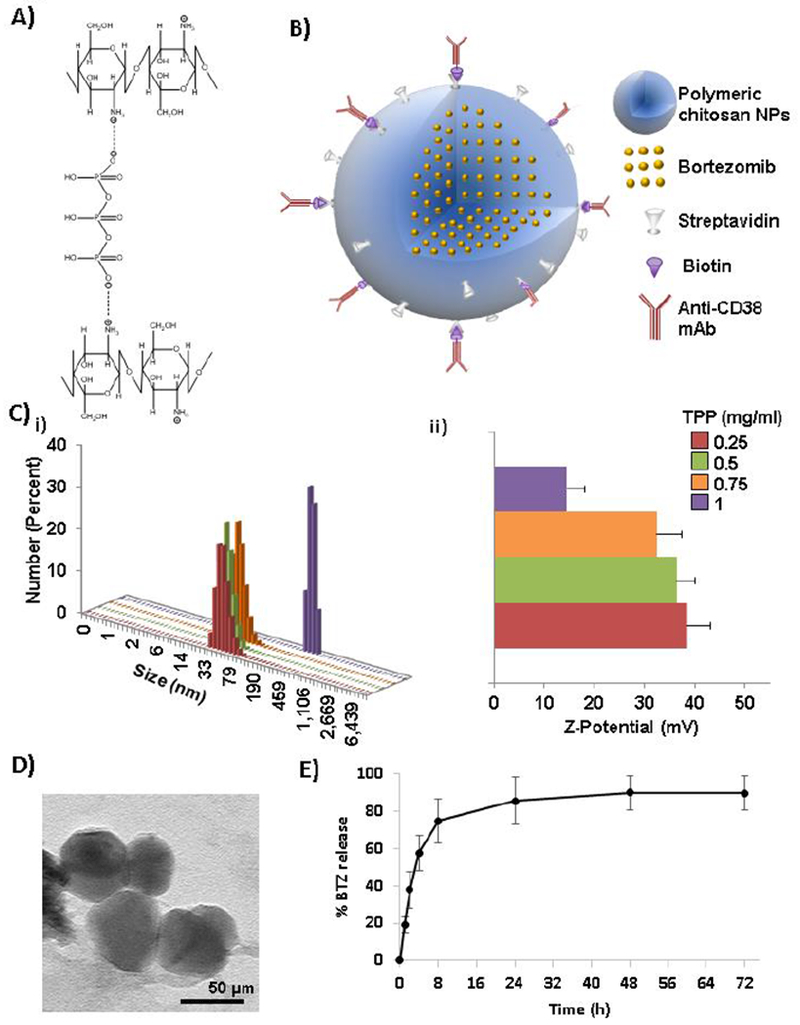
Characterization of anti-CD38 chitosan NPs. A) Crosslinking reaction showing ionotropic gelation of chitosan with TPP anions (ChemDraw Professional 15.1 was used for the chemical drawings). B) Scheme representing anti-CD38 chitosan NPs loaded with bortezomib by conjugation of polymeric chitosan NPs with streptavidin and further coupling with biotinylated anti-CD38 monoclonal antibody. C) Effect of TPP crosslinker (0.25 – 1 mg/ml) on i) size and ii) stability of anti-CD38 chitosan NPs. D) TEM image of chitosan NPs showing 50μm size. E) In vitro BTZ release from anti-CD38 chitosan measured by HPLC after 72 h (n=3).
We investigated the effect of the concentration of the crosslinker (TPP) on the size and the stability of the resulting NPs. The particle size and ζ-potential were determined by DLS. We found that increasing the concentration of the crosslinker increased the size of the particle (Fig. 1.Ci) and it decreased the stability of the particles (Fig. 1.Cii). A concentration of 0.25 mg/ml led to the highest ζ-potential, while 1 mg/ml reduced it dramatically to (15.1mV) a level less than the theoretical value needed to produce stable particles (30mV). These results showed that the crosslinking using 0.25 mg/ml maintained the highest stability of the nanoparticle with ζ-potential of 53.3 mV and the size of the particles obtained was 50 ± 11 nm, which was further confirmed by TEM analysis (Fig. 1.D). We used these conditions for all the further studies.
For evaluating the encapsulation efficiency of BTZ, a reverse phase HPLC assay was developed. BTZ had a retention time of 2 min (Fig. S1.A) and illustrated a linear dynamic range in the concentration range between 0 and 1.5 mM with a R2=0.9995 (Fig. S1.B). Anti-CD38 chitosan NPs were incubated for 1 h with intermediate (50 μM) or high-dose (1 mM) BTZ. Encapsulation efficiency was calculated using the AUC values for BTZ-loaded chitosan NPs. The encapsulation efficiency of intermediate and high-dose BTZ was 10.57 ± 4.90 and 9.67 ± 2.45 when incorporated after NPs crosslinking, and 85.31 ± 6.24 and 83.32 ± 5.22 when incorporated previous crosslinking in the crosslinker solution, respectively. In addition, the effect of BTZ loading inside of anti-CD38 chitosan NPs on size and stability was measured and we found that neither size (Fig. S1.C.i) nor ζ-potential (Fig. S1.C.ii) were affected by intermediate or high BTZ loading. We further tested the size and the stability of the particles as a function of storage time to optimize their stability for biological use, and we found that the size of empty and BTZ loaded NPs did not change over a month when particles were kept at 4°C (Fig. S1.D.i), and the stability (Fig. S1.D.ii) was not changed in the same period of time.
In vitro drug release from anti-CD38 chitosan NPs to different environments
The release of BTZ was analyzed in BTZ-loaded nanoparticles over a 72 h period. Fig. 1.E shows that BTZ was mainly released in the first 8 h with 75% of BTZ release. After that we could see that at 24 h the released reached a plateau that is maintained until 72 h into a DDW solution. We further hypothesize that the acidic tumor environment induces a rapid swelling of the chitosan NPs due to the free amine groups in the chitosan and faster release of the drug compared to neutral pH in the regular media (representing blood and normal tissues). First, pH of MM cell conditioned media was tested at different time points. We found that 72 h of MM cell culture generate acidic tumor microenvironment close to the pH tumor environment cited in the literature around 6.5-7.1 [27-29]. The pH of the conditioned media was measured after 72 h of culture and found a significant decreased in pH in MM-conditioned media (6.94 ± 0.23) compared to normal PB MNCS-conditioned media (8.38 ± 0.01) and non-conditioned media (8.6 ± 0.05) (Fig. 2.A.i).
Fig. 2:
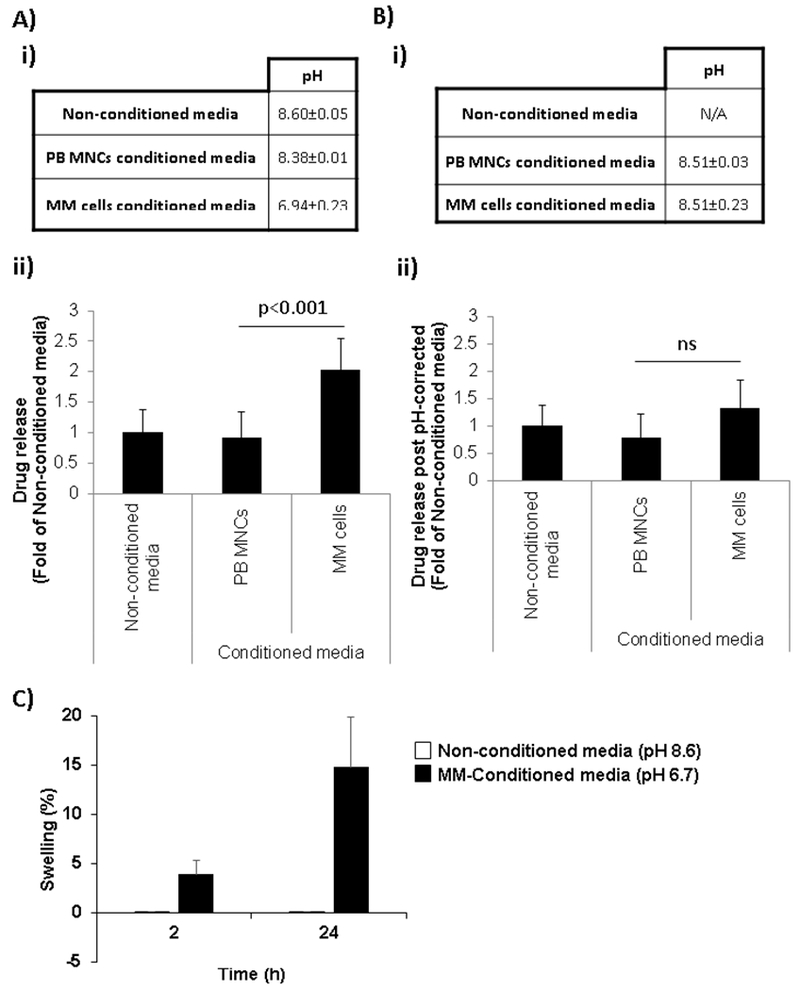
In vitro drug release from anti-CD38 chitosan NPs to different environments. A) i) pH of non-conditioned media and conditioned media from PB MNCs and MM cells after 72h in culture (n=3). ii) Effect of conditioned media pH on drug release from anti-CD38 chitosan NPs. B) i) Corrected pH of conditioned media from PB MNCs and MM cells after 72h in culture (n=3). ii) Effect of conditioned media with corrected pH on drug release from anti-CD38 chitosan NPs. C) Swelling of anti-CD38 chitosan NPs in non-conditioned media and MM-conditioned media over 24 h.
The in vitro release of a model fluorescent drug (doxorubicin) from anti-CD38 NPs was analyzed in the conditioned media of MM cells and normal PB MNCs. The acidic tumor microenvironment induced a 2-fold increase release of drug from the anti-CD38 NPs compared to non-conditioned media and normal PB MNCS-conditioned media (Fig. 2.A.ii).
To corroborate that the increase drug release was due to the change in pH, all the conditioned media were corrected to the pH of non-conditioned media and drug release was evaluated again. Note that the pH-corrected was around 8.51 for both MM-conditioned media and normal PB MNCS-conditioned media (Fig. 2.B.i). The release in the pH-corrected conditioned media (MM and PB-MNCs) were non-significantly different than in non-conditioned media (Fig. 2.B.ii), reflecting that the acidic tumor environment induced faster release of the drug compared to neutral-basic pH (8.5). In addition, it’s important to note that while the conditioned media from PB MNCs was very consistent (8.38), the conditioned media from MM cells showed more variability in different cell lines (Fig. S2.A.i), with indirect correlation where the lower the pH the more drug is released from the anti-CD38 NPs (Fig. S2.A.ii). An effect that can be reversed by correcting the pH of the media (Fig. S2.B.i), with proportional reduction of drug release to a level of non-significant difference compared to the conditioned media of normal PB MNCs (Fig. S2.B.i).
Swelling of anti-CD38 chitosan NPs in different environments
To further confirm our hypothesis that the acidic tumor environment induces swelling of the chitosan NPs and therefore induces a faster drug release, anti-CD38 chitosan NPs were incubated in non-conditioned media (pH 8.6) and MM conditioned-media (pH 6.7) for 24h. Swelling was measured and we found that while anti-CD38 chitosan NPs have an increase in swelling in MM-conditioned media (Q=3.88 ± 1.43 at 2h and Q=14.74 ± 5.12 at 24h), anti-CD38 chitosan NPs did not change in non-conditioned media (Q=0.04 ± 0.02 at 2h and Q=0.06 ± 0.03 at 24h) (Fig. 2.C).
Kinetics of binding/uptake of anti-CD38 chitosan NPs to MM cells
First, we tested the expression of CD38 in three MM cell lines and found that although all the cell lines showed a high expression compared to the corresponding isotype, the different cell lines showed different CD38 expression (Fig. 3.A). Then, we tested the binding/uptake of anti-CD38 chitosan NPs and non-targeted NPs at 2 h and compared with the CD38 expression in these cell lines. We found that while the binding/uptake of the particles was directly correlated with the level of expression of CD38 for anti-CD38 chitosan NPs, non-targeted chitosan NPs did not show any correlation with CD38 expression and binding/uptake in all cell lines was very low (Fig. 3.B).
Fig. 3:
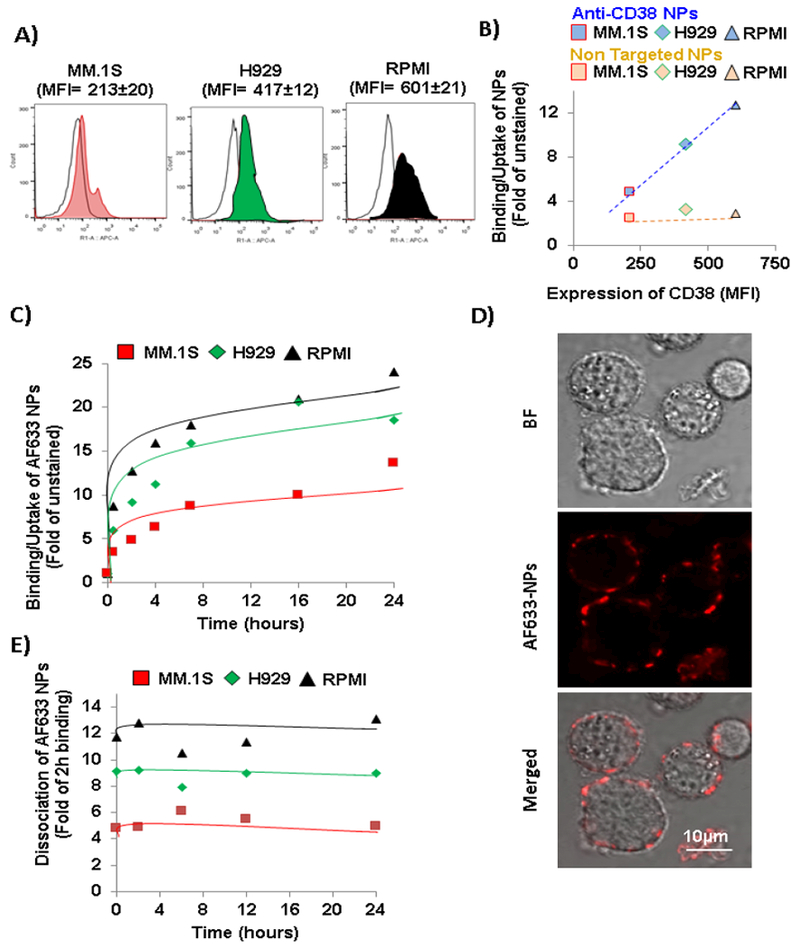
Kinetics of binding/uptake of anti-CD38 chitosan NPs to MM cells. A) Flow cytometry representative histograms of CD38 expression measured as fold of MFI of anti-CD38 to isotype controls in MM cells. B) Correlation of anti-CD38 NPs binding/uptake at 2 h with CD38 expression in MM cells compared to the non-correlation of non-targeted NPs with CD38 expression. C) Kinetics of binding/uptake of anti-CD38 chitosan NPs over a period of time of 24 h to MM cells. D) Fluorescent imaging of AF633 anti-CD38 chitosan NPs after 2 h of binding/uptake to MM1s cells (Bright field (BF); AF633, Red). Scale bar= 10 μm. E) Kinetics of dissociation of anti-CD38 chitosan NPs over a period of time of 24 h to MM cells after 2 h of binding/uptake.
Next, we determined the kinetics of binding/uptake for the anti-CD38 chitosan NPs in the three MM cell lines over a period of time of 24 h. We found that the binding/uptake is very fast and in a short period of a round 2 - 4 h the binding/uptake reach a plateau (Fig. 3.C). Fig. 3.D demonstrated the binding/uptake of the anti-CD38 chitosan NPs (red) to the surface MM.1S cells at 2 h post-treatment.
In addition, we study the potential dissociation of the anti-CD38 chitosan NPs from the MM cells after binding/uptake, and found that the particles did not dissociate from the MM cells over 24 h of incubation (Fig. 3.E).
Specificity of binding/uptake of anti-CD38 chitosan NPs to MM cells in vitro
The binding/uptake of anti-CD38 chitosan NPs to five MM cell lines and primary MM cell isolated from five MM patients was significant (3-fold) higher than the binding/uptake in non-targeted NPs (Fig. 4.A). The binding/uptake of the anti-CD38 chitosan NPs was highly consistent in the cell lines and the primary samples, while the binding/uptake of the non-targeted NPs to the different cell lines was more variable (Fig. S3.A and B). For normal controls, we used mononuclear cells isolated from the peripheral blood of five normal subjects (PB MNCs,) and from the BM of five normal subjects (BM MNCs). We found that the binding/uptake of the anti-CD38 chitosan NPs to the normal PB MNCs and BM MNCs was significantly (4-folds and 10-folds, respectively) lower than the binding/uptake to MM cells (Fig. 4.A). The binding/uptake of both the anti-CD38 chitosan NPs and the non-targeted NPs was constantly low with little variability between samples of PB MNCs (Fig. S3.C) and BM MNCs (Fig. S3.D).
Fig. 4:
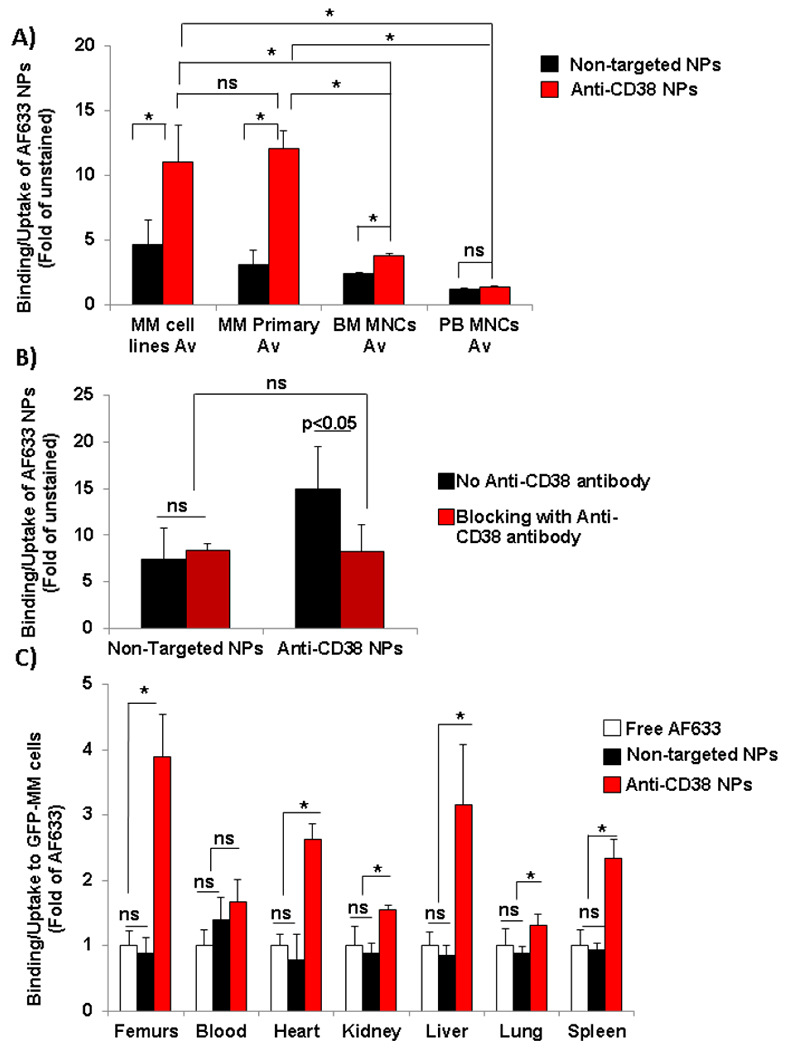
Specificity of binding/uptake of anti-CD38 chitosan NPs to MM cells. A) Comparison of specificity of binging of anti-CD38 chitosan NPs versus non-targeted chitosan NPs to MM cell lines (Av, n=5), CD138+ cells isolated from MM patients (Av, n=5), normal mononuclear cells isolated from the peripheral blood of normal subjects (PB MNCs, Av, n=5) and normal plasma cells isolated from the BM of normal subjects (BM MNCs, Av, n=5) analyzed by flow cytometry, *p<0.05. B) Mechanism of binding/uptake of the anti-CD38 targeted and non-targeted NPs by blocking or not with free anti-CD38 antibody, *p<0.05. C) Comparison of the biodistribution of anti-CD38 chitosan NPs, non-targeted chitosan NPs and free deactivated AF633 to MM cells (GFP+) in different organs: femurs, blood, heart, kidney, liver, lung and spleen, analyzed by fluorescent signal (fold of MFI of AF633) by flow cytometry, *p<0.05. (n=5).
We further confirm the effect of increasing concentrations of anti-CD38 antibody on MM cells and found that anti-CD38 antibody did not affect the survival of MM cells (Fig. S3.E).
Specificity of binding/uptake of anti-CD38 chitosan NPs to MM cells in vivo
We further study the specificity of anti-CD38 NPs binding/uptake to MM cells in vivo. MM-bearing mice were injected with free AF633 dye, non-targeted AF633-NPs and anti-CD38 AF633-NPs, and its binding/uptake to MM cells was tested in different organs. We found that free AF633 and non-targeted particles have the same random distribution (no significance differences) in the MM cells (GFP+) of all the organs. However, the binding/uptake of the anti-CD38 chitosan NPs was significantly higher in MM cells in the BM (femurs) and other extramedullary organs (heart, kidney, liver, lung and spleen), with around 4.5-fold increase of targeted compared to non-targeted NPs in the femurs and 1.5 to 3-fold increase in the other organs (Fig. 4.C). In the blood, we found that there was no significance difference between the binding/uptake of the anti-CD38 chitosan NPs compared to non-targeted and free AF633, probably due to same accessibility of NPs (targeted and non-targeted) to the cells in the circulation.
Effect of bortezomib-loaded anti-CD38 chitosan NPs on proliferation, cell cycle, and apoptosis in MM cells
The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ amounts to 5 nM for 48 h on proliferation of MM1s, H929, RPMI cells and PB MNCs was analyzed by MTT. We found that BTZ 5 nM as a free drug have a modest effect (20% killing) on MM cells with no effect on normal mononuclear cells from peripheral blood (PB MNCs). While empty chitosan particles showed no effect on survival of MM cells or PB MNCs, BTZ encapsulated in non-targeted and anti-CD38 chitosan NPs (5nM) had a robust effect (70% killing) in MM cells with no effect on PBMNCs (Fig. 5.A). The effects on individual cell lines was consistent with low variability (Fig. S4.A).
Fig. 5:
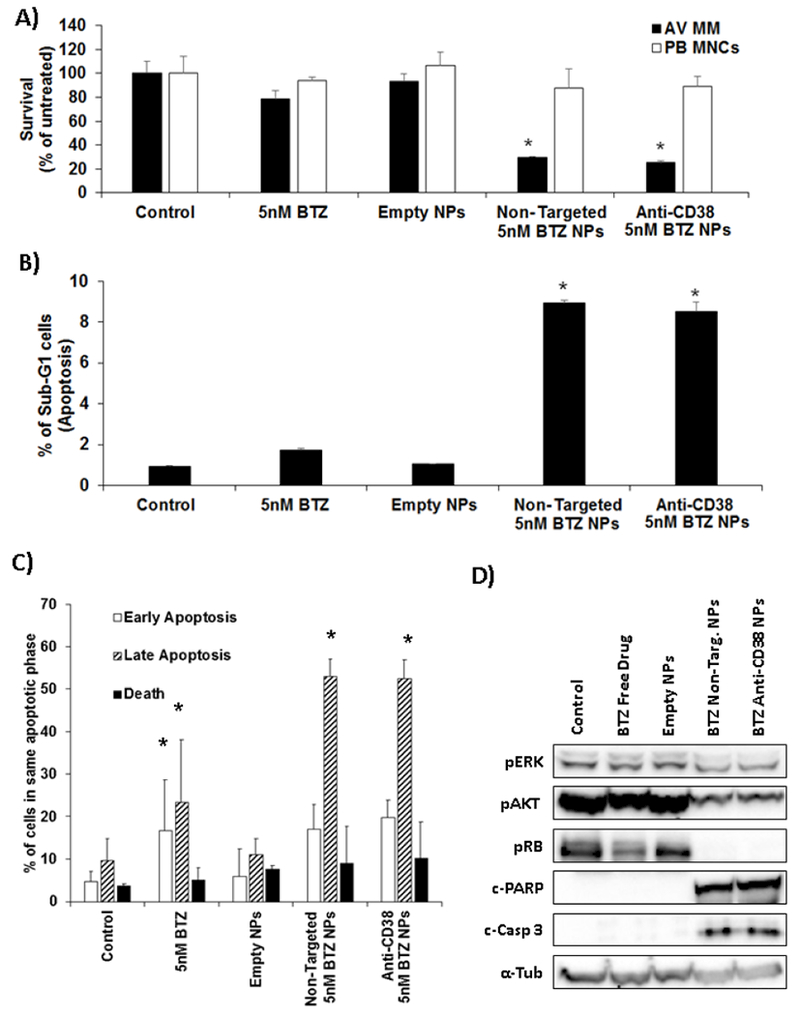
Effect of bortezomib-loaded anti-CD38 chitosan NPs on proliferation, cell cycle, and apoptosis. The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h on: A) proliferation of MM cells and PB MNCs analyzed by MTT, *p<0.05; B) % Sub-G1 population of H929 cells measured by Propidium Iodide stained of DNA, *p<0.05; C) Apoptosis of MM cells by FITC-Annexin-V and Propidium Iodide, *p<0.05. D) Survival–associated molecules (pERK and pAKT) after 6h of treatment, cell cycle–associated molecule (pRB), as well as, apoptotic–associated molecules (cleaved PARP and cleaved caspase 3) after for 12h of treatment, in H929 cells were measured by immunoblotting.
We further investigated the same therapy regimens on cell cycle and apoptosis. We found that BTZ as a free drug induced G0-G1 arrest, which was demonstrated also by BTZ encapsulated in non-targeted and targeted NPs, while empty chitosan NPs showed no effect (Fig. S4.B). However, BTZ encapsulated in non-targeted and targeted NPs revealed an important increase in subG1 cells (apoptosis) compared to empty NPs and free BTZ (Fig. 5.B). BTZ as a free drug induced increase early and late apoptosis, non-targeted and targeted NPs showed a significant increase in the fraction of late apoptosis compared to free drug (Fig. 5.C). The apoptosis results were studied in three MM cell lines (H929, Fig. S4.Ci, U266, Fig. S4.Cii and RPMI, Fig. S4.Ciii), which they show similar trends where late apoptosis was increased in the BTZ-loaded NPs compared to free drug.
Alternatively, we also performed toxicity at 48 h after a 2h pulse (where the non-targeted and anti-CD38 NPs bind differently) to test if there is a difference due to targeting. We found that after a 2 h pulse, while BTZ 5 nM as a free drug and empty NPs have no effect on MM cells, anti-CD38 chitosan NPs (5nM) had a significantly higher effect (55.19 ± 14.81) than non-targeted NPs (79.56 ± 22.25) in MM cells (Fig. S5).
Finally, these effects were confirmed on survival, cell cycle, and apoptosis signaling molecules and we found that BTZ encapsulated in non-targeted and targeted NPs suppressed more potently the phosphorylation levels pAKT and pERK, induced down-regulation of the expression of protein involved in cell cycle transition, such as pRb, and increased cleaved caspase 3 and PARP compared to free drug, while vehicle control and empty NPs did not have an effect (Fig. 5.D).
Enhanced bortezomib uptake lead to improved proteasome activity inhibition
To explain the enhanced effect of the encapsulated BTZ, we tested the effect of the BTZ encapsulation in the NPs on its ability to inhibit the proteasome activity. The effect of BTZ loaded anti-CD38 NPs and non-targeted NPs on proteasome activity inhibition was compared to free BTZ. We found that BTZ as a free drug have a moderate effect (50% inhibition) on MM cells; while BTZ encapsulated in non-targeted and targeted NPs had a more robust effect (68% and 75% inhibition, respectively) (Fig. 6.A). These results are in agreement with our previous results showing a more robust effect of the NPs on survival and apoptosis of MM cells compared to free drug (Fig. 5).
Fig. 6:
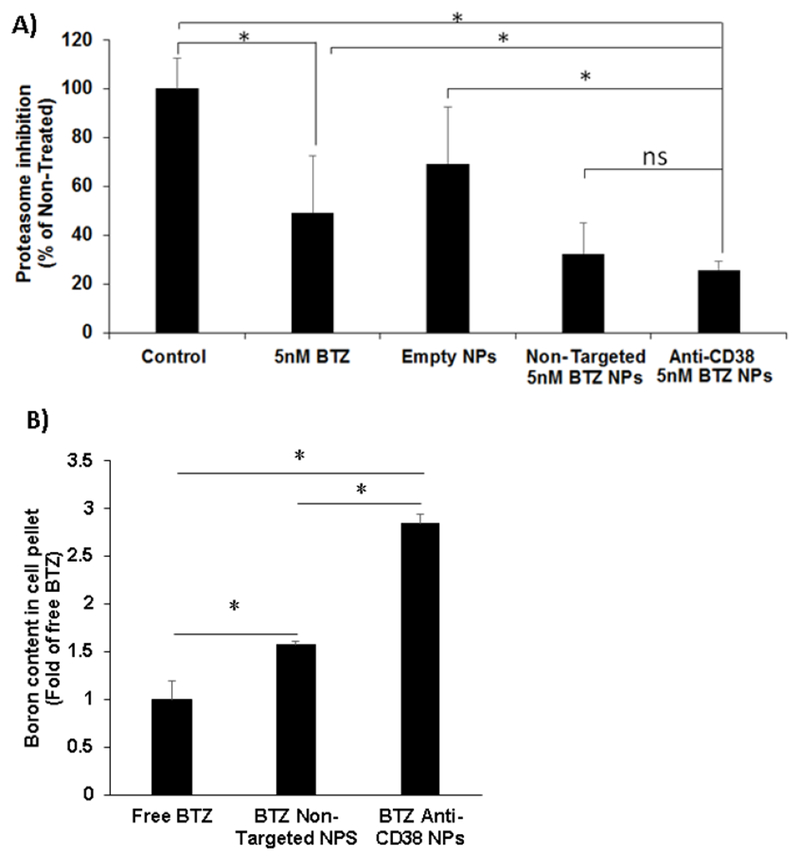
Enhanced bortezomib uptake and proteasome inhibition by anti-CD38 chitosan NPs. A) The effect of no treatment (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 2 h on proteasome activity, *p<0.05. B) Cell pellet boron concentration analyzed by ICP-OES after no treatment (control), BTZ as a free drug (100 nM), non-targeted chitosan NPs loaded with BTZ equivalent amounts to 100 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 100 nM for 1.5 h, *p<0.05.
To explain the effect on the proteasome activity, we tested the effect of BTZ encapsulation in NPs on its accumulation in the cells, as reflected by the boron content of the cells using elemental analysis of the cells by ICP-OES. Elemental analysis showed that the boron content of cells treated with BTZ encapsulated in anti-CD38 NPs showed a significant 3-fold higher boron content than cells treated with free BTZ and 2-fold higher than cells treated with BTZ-loaded non-targeted NPs (Fig. 6.B).
Enhanced proteasome activity inhibition by anti-CD38 chitosan NPs endocytic internalization
To understand the mechanism of the uptake of anti-CD38 chitosan NPs we tested the sub-cellular co-localization of the NPs with the endosomal markers (rab5+). We found that anti-CD38 chitosan NPs (red) were taken up by the MM cells via endocytosis and transferred into early endosomes (rab5+) and co-localization was detected at 6 and 24 h (Fig. 7.A).
Fig. 7:
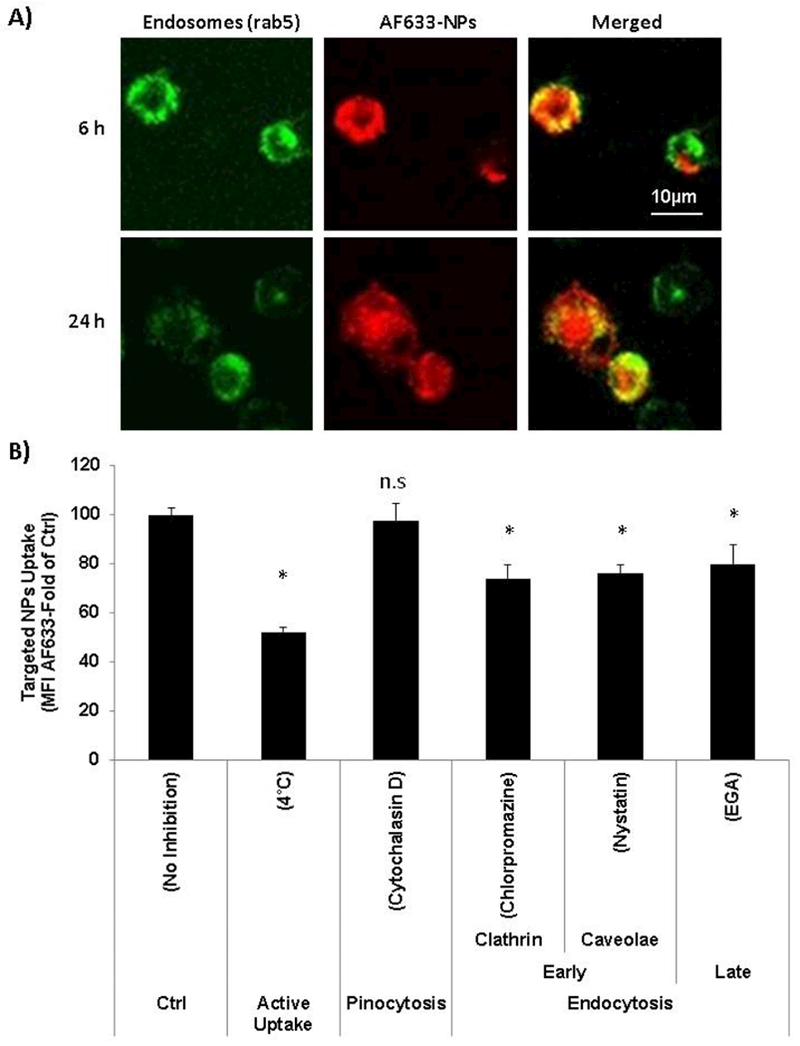
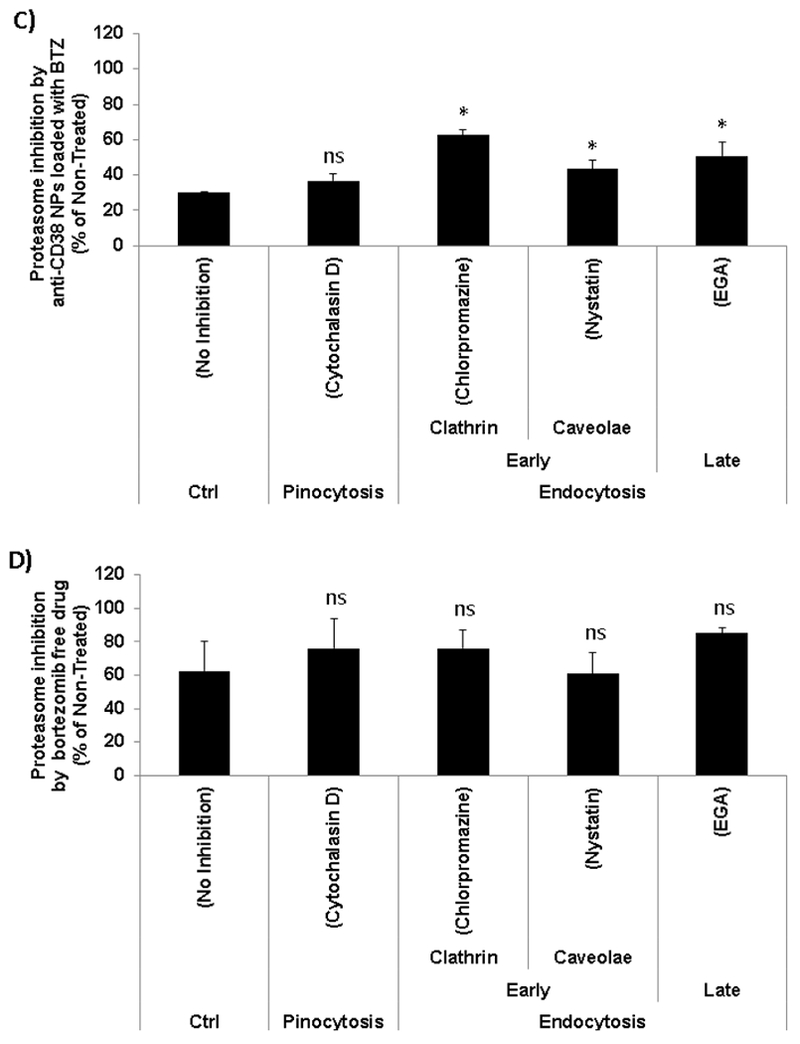
Enhanced proteasome activity inhibition by anti-CD38 chitosan NPs endocytic internalization. A) Confocal micrographs of the MM cells showing intracellular location of AF633 anti-CD38 in red, early endosomes expressing GFP (rab5+), and merged images showing co-localization of the red NPs in the green early endosomes at 2 and 24 hours; Scale bar= 10 μm. B) Anti-CD38 chitosan NPs uptake in the presence of macropinocitosys and endocytosis inhibitors. H929 cells were pre-incubated with the following inhibitors cytochalasin D 1μM, chlorpromazine 2μM, nystatin 0.5μg/ml, and EGA 2.5μM for 30 min. Two control samples were used with no inhibitors (No inhibition) at either 37°C or 4°C, *p<0.05. C) The effect of anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM uptake in the presence of macropinocitosys and endocytosis inhibitors (cytochalasin D 1μM, chlorpromazine 2μM, nystatin 0.5μg/ml, and EGA 2.5μM for 30 min) on proteasome activity, *p<0.05. D) The effect of BTZ as a free drug (5 nM) in the presence of macropinocitosys and endocytosis inhibitors (cytochalasin D 1μM, chlorpromazine 2μM, nystatin 0.5μg/ml, and EGA 2.5μM for 30 min) on proteasome activity, *p<0.05.
Incubation of MM cells at 4°C before treatment with anti-CD38 NPs significantly (50%) decreased their uptake, indicating it is an active uptake process (Fig. 7.B). To further investigate the mechanism of active uptake of the anti-CD38 NPs, we tested the contribution of macropinocytosis, early endocytosis pathways (mediated by clathrin or caveolae), and late endocytosis internalization, by specific inhibitors of the different uptake routs [30, 31]. We preliminary screened all the inhibitors on survival of MM cells to determine a concentration that does not induce cell death. Pre-incubation at 37°C with cytochalasin D 1μM (Fig. S6.A), chlorpromazine 2μM (Fig. S6.B), nystatin 0.5μg/ml (Fig. S6.C), and EGA 2.5μM (Fig. S6.D) for 30 min was found to have no effect on cell survival. Then, we found that while inhibition of macropinocytosis (by Cytochalasin D) did not affect the uptake of anti-CD38 NPs, inhibition of early endocytosis clathrin-mediated (by chlorpromazine) or caveolae-mediated (by nystatin), and inhibition of late endocytosis (by EGA) significantly (about 25% each) decreased the uptake of the anti-CD38 NPs (Fig.7.B).
We further corroborated the effect of inhibition of each of the uptake routes on the proteasome inhibition induced by the BTZ-loaded anti-CD38 NPs. It was shown that MM cells treated with BTZ-loaded anti-CD38 NPs (without inhibition of the uptake routs) induced about 70% inhibition in the proteasome activity. While inhibition of macropinocytosis did not affect the proteasome inhibition induced by the BTZ-loaded anti-CD38 NPs, inhibition of the early (clathrin- and caveolae-mediated) and late endocytosis interfered with proteasome inhibition activity of the BTZ-loaded anti-CD38 NPs (Fig. 7.C). We also confirmed that MM cells treated with free BTZ (without inhibition of the uptake routs) induced about 40% inhibition in the proteasome activity and demonstrated that the inhibition of macropinocytosis or endocytosis pathways led to any significant effect on proteasome activity (Fig. 7.D).
Effect of bortezomib-loaded anti-CD38 chitosan NPs on drug resistance induced by the tumor microenvironment
Stromal cells play a critical role in cell adhesion-mediated drug resistance (CAM-DR) in MM [17]. Co-culture of MM cells with stromal cells derived from MM patients induced resistance in the MM cells to treatment with BTZ (5nM) as a free drug, compared to mono-culture of MM cells alone. In contrast, encapsulated BTZ in anti-CD38 NPs induced a significantly more profound killing effect in the MM mono-culture, and overcame the stroma-induced resistance in MM cells (Fig. 8.A).
Fig. 8:
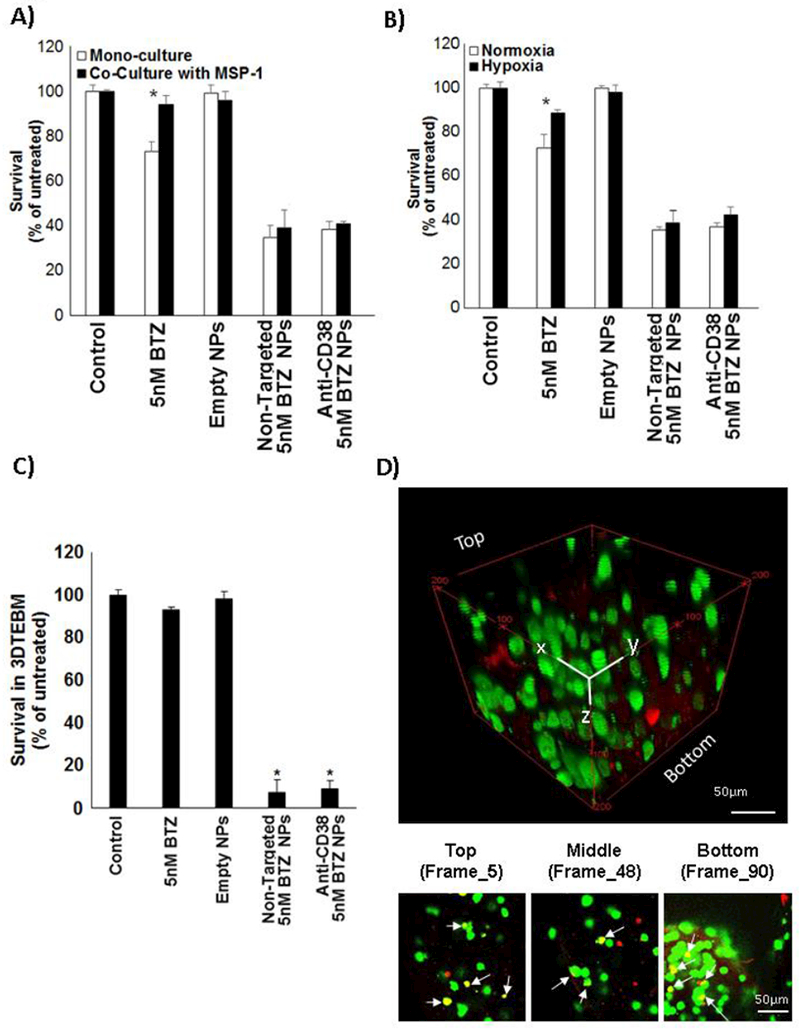
Effect of bortezomib-loaded anti-CD38 chitosan NPs on drug resistance. A) The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h on: A) cell adhesion-mediated drug resistance in co-culture with MSP-1 (myeloma-derived stroma); and B) hypoxia-mediated drug resistance, *p<0.05. C) The effect of vehicle (control), BTZ as a free drug (10 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 10 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 10 nM for 48 h on survival of MM cells cultured in 3DTEBM, *p<0.05. D) Confocal microscopy images of MM cells cultured (green) in 3DTEBM after 24h treatment with AF633 anti-CD38 chitosan NPs (red), shown by a Z-Stack rotated view, and images at different depths of the z-stack to show the co-localization (white arrows) of NPs and MM cells in yellow; Scale bar= 50 μm.
Similarly, hypoxia in the microenvironment was shown to play an important role in cell drug resistance in MM[25]. Incubation of MM cells in hypoxic conditions (1% O2) induced resistance in the MM cells to treatment with BTZ (5nM) as a free drug, compared to MM cells cultured in normoxic conditions (21% O2). In contrast, encapsulated BTZ in anti-CD38 NPs induced a significantly more profound killing effect in the normoxic MM cells, and overcame the hypoxia-induced resistance in MM cells (Fig. 8.B).
To better mimic the tumor microenvironment, we have previously shown that a more realistic 3D tissue engineered bone marrow (3DTEBM) culture model, derived from the BM of MM patients, can recreate better the pathophysiology and drug resistance of MM which resembles tissue depth with oxygen and drug gradients, as well as, recreated the tumor microenvironment [20, 21]. BTZ (10nM) as free drug had a very modest effect on survival of MM cells (8% killing). In contrast, encapsulated BTZ induced a significant and robust effect with a 93% killing in the 3DTEBM (Fig. 8.C). We further detected the co-localization of NPs (red) and the MM cells (green) at different depths in the 3D cultures, indicating that the NP can penetrate the 3D culture and deliver BTZ (Fig. 8.D).
Effect of the anti-CD38 chitosan NPs on the efficacy in inhibition of tumor progression and reduction of the side effects of bortezomib in vivo
Tumor bearing mice were treated with vehicle, BTZ as a free drug (1 mg/kg once a week), non-targeted NPs loaded with BTZ (1 mg/kg once a week), and anti-CD38 NPs loaded with BTZ (1 mg/kg once a week). Tumor progression was followed in the 4 groups for 5 weeks by BLI.
BTZ treatment was efficacious in reducing tumor size in all the treatment-groups (free drug, BTZ-loaded non-targeted NPs and BTZ-loaded anti-CD38 NPs) compared to vehicle treatment. However, treatment with BTZ-loaded anti-CD38 NPs reduced the tumor size to significantly lower tumor size compared to non-targeted NPs and free drug-groups. And no significant difference was observed between BTZ as free-drug and BTZ-loaded in non-targeted NPs (Fig. 9.A).
Fig. 9:
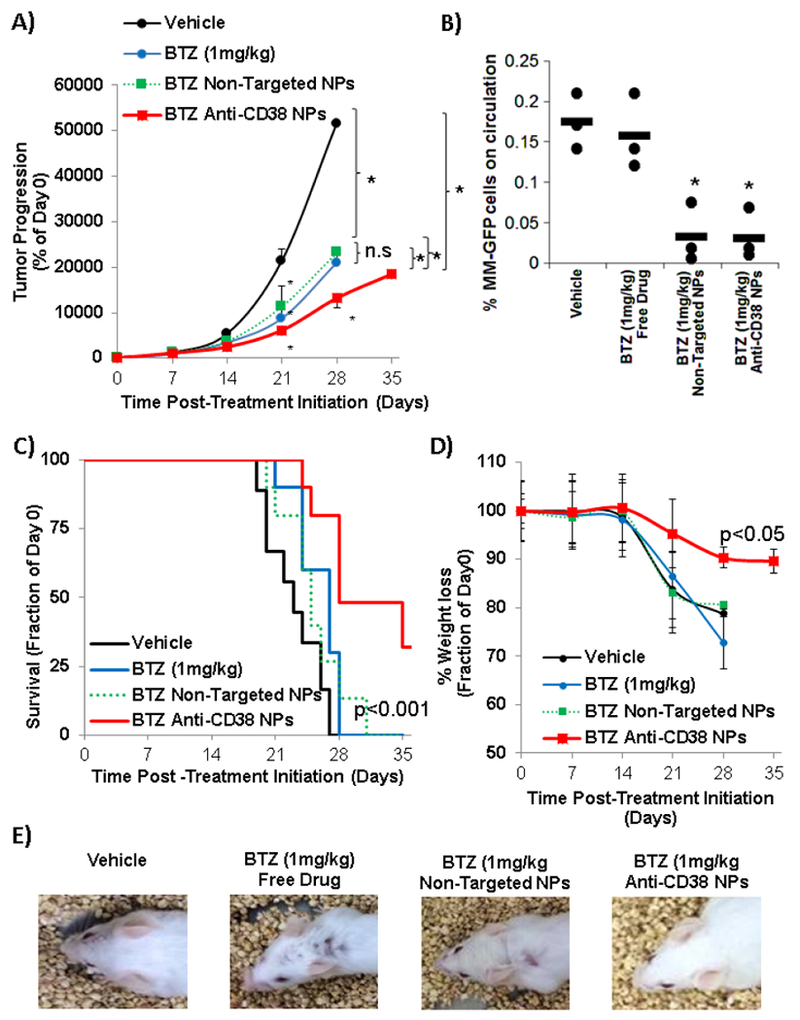
Inhibition of tumor progression and reduction of the side effects of bortezomib-loaded anti-CD38 chitosan NPs in vivo. The effect of vehicle, BTZ as a free drug (1 mg/kg once a week), non-targeted chitosan NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week), and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week) on: A) tumor progression shown by quantification of BLI*p<0.05; B) number of MM cells on circulation, *p<0.05; C) survival shown by Kaplan-Meier survival curves (p value targeted compared to other treatments); D) % weight loss, and E) hair loss (representative images of one mice from each group).
In addition, the number of circulating MM cells in a blood sample taken from each group at day 25 after the beginning of the treatment revealed a significant reduction of MM cells in the BTZ-loaded NPs groups compared to free drug (Fig. 9.B).
Importantly, treatment with BTZ-loaded anti-CD38 NPs improved overall survival of the MM-bearing mice (32% surviving at day 35), compared to vehicle group (all the group died by day 28), the free BTZ (all the group died by day 29), and BTZ-loaded non-targeted NPs (all the group died by day 31) (Fig. 9.C).
Moreover, we tested the effect of different formulations on the weight loss in the animals. Treatment anti-CD38 NPs resulted in 10% loss in body mass during the 5 weeks treatment period, whereas the free BTZ and non-targeted NPs-groups demonstrated >20% weight loss, and they were consequently sacrificed (Fig. 9.D). Moreover, in both the free BTZ and non-targeted NPs-groups a significant hair loss was observed compared to the vehicle-treated (control) group, while such hair loss was not observed in the anti-CD38 NPs group (Fig. 9.E).
In addition, 3 mice were taken from each group at day 25 post treatment, and specimens of the BM, spleen, liver, brain, spinal-cord and intestine were fixed and pathologically evaluated for histological tissue damage. No apparent histopathologic changes in the tissues, including femurs, spinal cord, liver, spleen, kidney and intestine was observed in any of the groups (Fig. S7).
DISCUSSION
The establishment of more effective treatments that can circumvent chemoresistance in MM is a priority. BTZ, the first proteasome inhibitor approved by the FDA for the treatment of MM, remains one of the most potent proteasome inhibitors available [32]. MM cells exhibit a greater sensitivity to proteasome activity inhibition compared to healthy cells, resulting in an accumulation of pro-apoptotic proteins, cyclins, and cyclin-dependent kinase inhibitors, while decreasing NF-κB activity within tumor cells, which ultimately results in cell cycle arrest and apoptosis [33, 34]. However, despite its success in the clinic, BTZ still possesses limitations related to dose limiting side effects [35, 36]. Therefore, novel approaches that reduce the systemic toxicity of BTZ while enhancing or improving its anti-tumor efficacy is of utmost importance.
In this study, we developed NPs-mediated targeted delivery of BTZ to MM cells specifically to improve the therapeutic index relative to free drug; though increased efficacy and specificity to MM cells and reducing the side effects in normal tissue; by targeting the NPs to CD38 which is overexpressed on MM cells.
NPs drug delivery systems allow the delivery of larger doses of chemotherapies and increase the drug bioavailability into targeted areas, thus sparing healthy tissues [13]. Chitosan has been widely used in drug delivery systems. NPs synthesized from chitosan have gained prominence due to their large drug loading capacity, superior adsorption capabilities, and long shelf life. Chitosan also possesses an abundance of hydroxyl and amino functional groups, allowing for NPs to be synthesized by physical and/or chemical crosslinking [37]. Instead of using harsh conditions in the formulation of chitosan NPs, our method is determined by ionotropic gelation, which simply involves the interaction of an ionic polymer with oppositely charge ion to initiate crosslinking [38]. TPP is a polyanion, which interacts with the cationic chitosan by electrostatic forces [39]. These chitosan NPs were further rationally targeted to MM cells by streptavidin-biotin linkage to anti-CD38 antibody (Fig. 1).
We have fine-tuned the production of anti-CD38 chitosan NPs with a small size (50nm) to allow better penetration to tumors [14, 15, 40], and we showed that these NPs were stable at different storage conditions and periods (Fig. 1). All the studies performed considered only NPs crosslinked with TPP 0.25mg/ml with size 50 nm and high ζ-potential, other NPs sizes or stabilities might deeply influence their kinetics, binding and uptake and further in vivo results.
The chitosan NPs showed BTZ release in the first 8 h, and preferential drug release in MM tumor microenvironment compared to normal tissue microenvironment, and this release was pH dependent (Fig. 1 & 2). We have previously shown that chitosan swelling (as a first step for drug release) is pH dependent, in which chitosan swelling is increased in acidic pH and enhance release for its encapsulated materials [41], due to hydration of the protonated amine groups under acidic conditions [42]. Tumor microenvironment is reported to be more acidic than normal healthy tissue [27-29], and our model demonstrated similar pH in the MM cultures to these reported in the literature. We further confirm that anti-CD38 NPs swelling was pH dependent, and that acidic tumor microenvironment induced increase swelling compared to normal microenvironment. This explains our findings that the chitosan NPs in this study had preferential release in the acidic tumor microenvironment in MM compared with normal microenvironment. This is an interesting phenomenon which demonstrates another aspect of specificity of the NPs to tumor environment.
Several strategies have been developed to improve the delivery of chemotherapies to MM by targeting different moieties expressed on MM cells, including: (1) Very Late Antigen-4 (VLA-4), an integrin receptor expressed on cancer of hematopoietic origin such as MM, has led to very promising results with several different drug combinations [43-45]. However, the limitation with targeting VLA4-4 stems from its heterogeneous expression on myeloma cells, with even negligible expression in some MM cells, as well as its high expression in many other normal cells [43, 45]. (2) ATP-binding cassette (ABC) drug transporters, such as ABCG2 (breast cancer resistance protein) was used to target MM cancer stem cells and deliver placitaxel. These NPs inhibited tumor growth, increased survival by inducing apoptotic pathways, and showed less toxic side effects in comparison with the placitaxel treatment [46, 47]. However, the expression of the ABC in cancer cells is variable [48]. (3) Targeting of the bone marrow microenvironment by using alendronate PLGA-PEG NPs, which did not target the MM cell but the general bone microenvironment. This approach failed to showed specificity to MM, in which non-targeted and targeted NPs showed the same efficacy in vivo [49].
CD38 is a cell surface marker with low expression on various hematopoietic cells, but it was shown to be highly expressed on malignant MM cells [50]. Due to its high expression on MM cells, it is used as a marker for identification of MM cells [51-53], and there are several indications supporting the notion that CD38 plays significant roles in the progression of MM [50, 54]. Moreover, CD38 has been used as a therapeutic target in MM; anti-CD38 monoclonal antibodies are showing promising results of selective and efficient treatment of MM in preclinical studies and in early clinical trials [55-59]. Moreover, we have recently shown that while the expression of several plasma cell markers such as CD138, CD56, and CD20 changed in different stages of the disease, CD38 is constantly expressed on all forms of MM cells including differentiated MM cells in progressive MM models, as well as on stem cell-like MM cells in minimal residual disease models [60]. We are the first to propose the use of CD38 as a targeting moiety for a nanoparticle drug delivery system.
The in vitro binding/uptake studies demonstrated that CD38 expression was high throughout different MM cells lines and primary MM cells, and that the anti-CD38 chitosan NPs were effectively uptaken by all MM cells, with correlation to the expression of CD38 (while non-targeted NPs were not correlated to CD38 expression and binding was low), with minimal dissociation after binding/uptake (Fig. 3). The uptake of the anti-CD38 chitosan NPs to MM cells (cell lines and primary) was significantly higher than their uptake in normal cells (4-fold of BM MNCs, and 10-fold of PB MNC), and higher than the binding/uptake of the non-targeted NPs (3-fold in vitro, 4.5-fold in bone marrow, and 1.5 to 3-fold in other organs), demonstrating that the anti-CD38 chitosan NPs are selective and specific to MM (Fig. 4). It should be noted that anti-CD38 NPs are slightly up-taken by a CD38+ population in mononuclear cells; however, the higher expression of CD38 in MM cells makes that anti-CD38 NPs were higher up-taken with 4 and 10-fold compared to MNCs [53].
The in vitro cellular studies with BTZ-loaded anti-CD38 chitosan NPs demonstrated enhanced cytotoxicity and apoptosis on MM cells compared to similar concentration of BTZ as free drug; demonstrated by induction of more profound cell cycle arrest, and apoptosis leading to cell death through inactivation of proliferative cell signaling (MAPK, PI3K pathways, cell cycle) and activation of pro-apoptotic pathways (PARP and Caspase-3) (Fig. 5).
To further investigate the mechanism of the increased efficacy of the BTZ-loaded anti-CD38 chitosan NPs, we evaluated the effect of BTZ-loaded NPs compared to free drug in proteasome activity inhibition. We found that BTZ-loaded NPs were significantly more potent in the proteasome activity inhibition than free BTZ. To study this phenomenon, we evaluated first the accumulation of BTZ in MM cells by testing their elemental boron content (since BTZ includes a boron atom) and found that boron content in MM cells treated with BTZ-loaded anti-CD38 NPs was 3-fold higher than MM cells treated with BTZ as free drug and 2-fold higher than BTZ-loaded non-targeted NPs (Fig. 6). This explains the results of higher proteasome inhibition, and consequently the higher cell death induced by the BTZ-loaded NPs.
In order to explain the higher bortezomib content of the cells when treated with BTZ-loaded NPs, we investigated the mechanism of uptake of the anti-CD38 NPs. We hypothesized that endocytosis is the preferential internalization route for the anti-CD38 NPs. Endocytosis is a general term for the internalization of different components by the invagination of the plasma membrane and the formation of vesicles and vacuoles through membrane fission [61]. To confirm that the process involved in the internalization of the anti-CD38 chitosan NPs was endocytosis, we have shown that the NPs distribution in the cells co-localized with the distribution of endosomes (detected by rab5). Moreover, endocytosis is an active energy-dependent process [62, 63], and stopping the energy production in the cells by pre-incubation at 4 °C induced significant 50% reduction of the anti-CD38 NPs uptake, again as a confirmation that it is endocytosis-dependent (Fig. 7). These results are in agreement with previously reported in the literature, demonstrating the active uptake of other types of NPs through endocytosis [64-66].
To further investigate the specific routes of uptake by endocytosis, we tested the contribution of macropinocytosis, early endocytosis pathways (clathrin or caveolae-mediated), and late endocytosis internalization. Late endocytosis refers to the endocytosis process in which late endosomes are involved. It’s important to differentiate between early and late endocytosis due to several differences biological features, including Rab5 is exchanged for Rab7 in late endosomes and different inhibitors are specific for each phase [30, 31]. We found that macropinocytosis did not play a role in the uptake of anti-CD38 NPs. Rather, we found that the anti-CD38 NPs were uptaken through clathrin-mediated and caveolae-mediated early endocytosis which progressed to late endocytosis and delivery of the NPs content into the cells (Fig. 7.B). It has been previously characterized that after CD38 ligation to mAbs internalization through endocytosis pathways takes place, where specifically CD38 intracellular movements go from early endosomes to late endosomes [67]. Since the anti-CD38 chitosan NPs tested in our work have a specific surface functionalization, it is conceivable that they entered cells by a receptor-mediated endocytic pathway.
Finally, the internalization through endocytic pathways of anti-CD38 NPs was correlated to the enhanced proteasome activity inhibition of these NPs in MM cells. Treatment of MM cells with clathrin and caveolae-mediated early endocytosis, or late endocytosis inhibitors (but not macropinocytosis inhibitors) significantly reduced the proteasome inhibition of BTZ-loaded anti-CD38 chitosan NPs (Fig. 7.C). When BTZ is administered as a free drug, it penetrated through passive diffusion into MM cells as well as in normal cells, and inhibition of the different endocytosis routes did not induce any change in its ability to inhibit the proteasome (Fig. 7.D).
The interaction between the proteasome and the endocytic pathway has been described before in the context of internalization and delivery of content of viruses such as Kaposi’s Sarcoma-associated herpesvirus, influenza virus, and adeno-associated virus [68-70]. Furthermore, two recent papers have investigated the cytotoxic effect of NPs and their direct effect on proteasome activity. Phukan et al have shown that high doses (1.0 μg/μl) of silica-coated magnetic nanoparticles were reported to be cytotoxic in neurons by induction of ROS generation and proteasome activity reduction [71]. Armand et al have also shown that long-term exposure (2 months) of titanium dioxide nanoparticles to A549 epithelial alveolar cells modifies the cellular content of proteins involved in intra- and extracellular trafficking and proteasome activity [72].
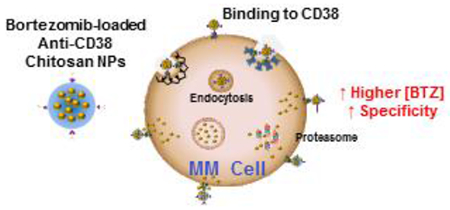
It is noteworthy that, BTZ-loaded anti-CD38 chitosan NPs bind to CD38+ MM cells, and BTZ can be released directly from the NPs attached to the surface and anti-CD38 NPs entered CD38+ MM cells via the endocytic pathway. Through both mechanisms BTZ is presented at a higher drug concentration to the proteasome in MM cells compared to the free-penetration by passive diffusion of the free drug to both MM and normal cells (a summary of that suggested mechanism is illustrated in the cartoon in Fig. 10).
Fig. 10:
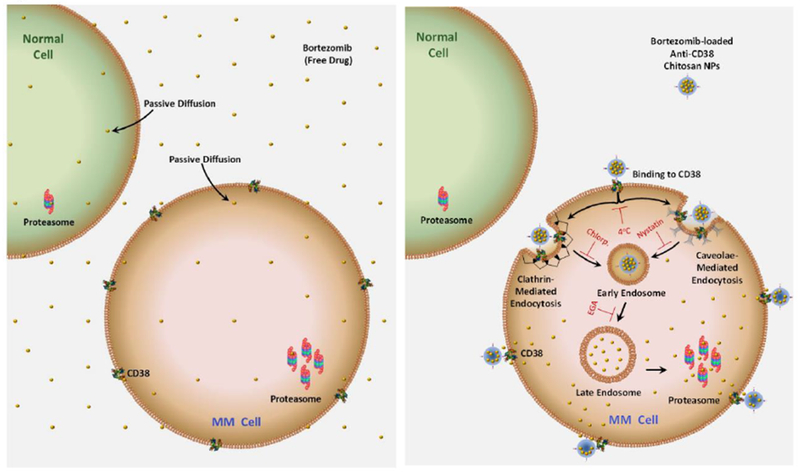
Schematic representation of BTZ cellular uptake by normal and MM cells. (Left) Free BTZ penetrates into all cells by passive diffusion and preferentially inhibits the overexpressed proteasome in MM cells, even at a low drug concentration. (Right) BTZ-loaded anti-CD38 chitosan NPs bind to CD38+ MM cells, and BTZ can be released directly from the NPs attached to the surface or anti-CD38 NPs entered CD38+ MM cells via the endocytic pathway (inhibited by 4°C incubation). Clathrin- (inhibited by chlorpromazine, chlorp.), caveolae-mediated endocytosis (inhibited by nystatin) incorporated anti-CD38 chitosan NPs into early endosomes, which will be transformed into late endosomes (inhibited by EGA). Through both mechanisms BTZ is presented at a higher drug concentration to the proteasome based on higher availability of BTZ inside the cells through either diffusion from the anti-CD38 NPs that bind to the surface or the endocytosed anti-CD38 NPs.
Then, we tested the role of NPs drug delivery on drug resistance induced by the tumor microenvironment. We have previously reported that the tumor microenvironment (including CAM-DR and hypoxia) plays a crucial role in the resistance of MM cells to BTZ [53, 73, 74]. In this study, we found that BTZ-loaded anti-CD38 NPs proved to be more efficacious than free BTZ and overcame CAM-DR and hypoxia-mediated drug resistance (Fig. 8.A & B). In addition, 3D culture systems are gaining strength as in vitro systems to assess and predict drug sensitivity in myeloma [20, 75, 76]. Therefore, we used a relevant 3D model made from bone marrow supernatants of MM patients, which recreates the tumor microenvironment, to evaluate NPs as drug delivery systems and predict better the in vivo performance of NPs. The 3DTEBM model corroborated the increase cytotoxicity of anti-CD38 chitosan NPs on MM cells relative to free drug and allowed to corroborated co-localization of the NPs in the MM cells (Fig. 8.C & D). These results establish the significance of targeting MM cells as well as their interactions with the microenvironment in the design of more effective novel therapeutic approaches. However, it is important to mention that both BTZ-loaded chitosan NPs (anti-CD38 chitosan NPs and non-targeted chitosan NPs) were equally effective in vitro increasing cytotoxicity, apoptosis, and overcoming drug resistance, unless a 2 h pulse (where the binding/uptake of both NPs was different) was used in a 48 h toxicity assay, where we indeed detect differences and BTZ anti-CD38 NPs were more potent than non-targeted NPs (Fig. S5). This is most likely due to the long incubation periods needed for the cytotoxic effects in vitro, which eliminates the kinetic advantage of the binding/uptake of the anti-CD38 NPs over the non-targeted NPs in the in vitro stationary system. This is in the contrast to the in vivo system where the fast binding/uptake kinetics is crucial, due to elimination of the unbound particles.
Indeed, anti-CD38 NPs were further supported in the in vivo studies which demonstrated that although both NPs were equally effective in vitro, the anti-CD38 targeted NPs had a markedly improved tumor growth inhibition, improved overall survival, and reduction of side effects compared to the non-targeted NPs (Fig. 9). For our in vivo study, we used the MM.1S injected model to develop MM-bearing mice with tumors mainly in the bone marrow recreating the pathophysiology of the disease including the BM microenvironment; with the treatment performed once a week at a dose 1mg/kg BTZ intravenously similar to human clinical protocols. In vivo, the BTZ-loaded anti-CD38 NPs were more efficacious in delaying tumor progression in the BM and circulating tumor cells, improved overall survival, and reduction of systemic side effects measured by body weight loss compared to non-targeted chitosan NPs and free drug, and lower toxicity such as hair loss (Fig. 9), with no histological toxicity in any the organs.
The major limitation of our studies was than although anti-CD38 chitosan NPs were able to show significant inhibition of tumor growth and improved overall survival compared to free drug, the results could be improved. It should be taken into consideration that due to the low toxicity of the BTZ-loaded anti-CD38 NPs, higher doses could be used in future experiments to achieve a better tumor growth inhibition and overall survival, as well as, initiate treatment at earlier times points to improve therapeutic outcome.
While this study focuses solely on the encapsulation of BTZ into anti-CD38 chitosan NPs, the methodology presented could also be applied to other therapeutic or diagnostic molecules, especially other proteasome inhibitors for improved efficacy and safety profile.
In conclusion, we have developed and characterized anti-CD38 chitosan NPs for the delivery of BTZ in MM showing preferential BTZ release in tumor-microenvironment, specific binding to MM cells, and an improved drug cellular uptake through BTZ diffusion from the surface and endocytosed NPs, which translated in enhanced proteasome inhibition and robust cytotoxic effect on MM cells. Furthermore, the anti-CD38 chitosan NPs specifically delivered therapeutic agents to MM cells improving therapeutic efficacy and reducing side effects in vivo. We also report the enhancement of the efficacy and specificity of proteasome inhibitors due to higher accumulation of BTZ through BTZ diffusion from the surface and endocytosis-driven delivery of NPs. The findings in this manuscript are the basis for a provisional patent application, an IND application, and future clinical trials to test the BTZ-loaded anti-CD38 NPs as a novel therapeutic approach in MM.
Supplementary Material
Fig. S1: Characterization of bortezomib-loaded anti-CD38 chitosan NPs. A) BTZ HPLC detection peak with retention time 2 minutes, λ = 270 nm. B) BTZ calibration curve formed by plotting the AUC of BTZ HPLC peak for the concentration range of bortezomib (0 to 1.5 mM). C) Effect of intermediate (50 μM) and high-dose (1 mM) BTZ on i) size and ii) stability of anti-CD38 chitosan NPs. D) Effect of time preservation on i) size and ii) stability of empty or BTZ loaded anti-CD38 chitosan NPs.
Fig. S2: In vitro drug release from anti-CD38 chitosan NPs to different environments. A) i) pH of non-conditioned media and conditioned media from 3 independent PB MNCs patient samples and 3 MM cell lines after 72h in culture. ii) Effect of conditioned media pH on drug release from anti-CD38 chitosan NPs. B) i) Corrected pH of conditioned media from 3 independent PB MNCs and 3 MM cell lines after 72h in culture. ii) Effect of conditioned media with corrected pH on drug release from anti-CD38 chitosan NPs.
Fig. S3: Specificity and variability of binding/uptake of anti-CD38 chitosan NPs. Binging of anti-CD38 chitosan NPs, non-targeted chitosan NPs and unstained control NPs to A) MM cell lines (MM1S, H929, OPM1, RPMI, U266), B) CD138+ cells isolated from MM patients (n=5), C) mononuclear cells isolated from the peripheral blood of normal subjects (PB MNCs, n=5), D) and normal plasma cells isolated from the BM of normal subjects (BM MNCs, n=5) analyzed by flow cytometry. E) Effect of anti-CD38 antibody (0.5 – 5 μg/ml) on proliferation of MM cells for 48 h analyzed by MTT.
Fig. S4: Effect of bortezomib-loaded anti-CD38 chitosan NPs on proliferation, cell cycle, and apoptosis. The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h on: A) Proliferation of MM1s, H929, RPMI cells and PB MNCs analyzed by MTT, *p<0.05; B) Cell cycle of H929 cells measured by Propidium Iodide stained of DNA, *p<0.05; and C) Apoptosis of H929 (i), U266 (ii) and RPMI (iii) cells by FITC-Annexin-V and Propidium Iodide.
Fig. S5: The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h after a 2 h pulse on proliferation of MM1s cells analyzed by MTT, *p<0.05.
Fig. S6: Effect of macropinocytosis and endocytosis inhibitors on survival of MM cells. A) Incubation with macropinocytosis inhibitor Cytochalasin D (0 – 1.5 μM) for 30 min. B) Incubation with early endocytosis chlathrin-mediated inhibitor Chlorpromazine (0 – 3 μM) for 30 min. C) Incubation with early endocytosis caveolae-mediated inhibitor Nystatin (0 – 1 μg/ml) for 30 min. D) Incubation with late endocytosis inhibitor EGA (0 – 5 μM) for 30 min.
Fig. S7: Histological tissue damage of bortezomib-loaded anti-CD38 chitosan NPs in vivo. The effect of vehicle, BTZ as a free drug (1 mg/kg once a week), non-targeted chitosan NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week), and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week) on histological tissue damage on femur, spinal cord, liver, spleen, kidney, intestine after 25 days of treatment.
ACKNOWLEDGEMENTS
We want to thank Remya Nair from the Nano Research Facility (NRF), School of Engineering and Applied Science at Washington University in St. Louis, for her help with TEM analysis. This research was partially supported by the 2016 Multiple Myeloma Research Foundation (MMRF) Research Fellow Award and the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) and the National Cancer Institute (NCI) of the NIH under Award Number U54CA199092.
Dr. Azab receives research support from Verastem, Selexys, Karyopharm, Cell Works, Cleave Bioscience, Glycomimetics, Abbvie and Vasculox; and is the founder and owner of Targeted Therapeutics LLC and Cellatrix LLC, however there has no contribution of the aforementioned entities to the current study. Dr. de la Puente is a co-founder of Cellatrix LLC, however, there has been no contribution to the current study. Dr. Azab and Dr. de la Puente have a provisional patent application on the technology described in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHORSHIP
Contribution: P.P. designed the study, performed experiments, analyzed, interpreted the data and wrote the manuscript; A.J, M.L.J, R.G, K. A., S.S., and C.E, performed research and analyzed data; B.M, J.S. C.F, J.K, D.K, R.V, N.N.S., and S.A. analyzed and interpreted data; A.K.A., supervised the study, interpreted the data and edited the manuscript. All authors reviewed and approved the manuscript.
CONFLICT OF INTERESTS
Other authors state no conflicts of interest.
REFERENCES
- [1]. Siegel RL Miller KD Jemal A Cancer statistics, 2016 CA: a cancer journal for clinicians 2016. 66 7–30 [DOI] [PubMed] [Google Scholar]
- [2]. Kyle RA Rajkumar SV Multiple myeloma The New England journal of medicine 2004. 351 1860–1873 [DOI] [PubMed] [Google Scholar]
- [3]. Gonsalves WI Milani P Derudas D Buadi FK The next generation of novel therapies for the management of relapsed multiple myeloma Future oncology (London, England) 2017. 13 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Gonsalves WI Rajkumar SV Gertz MA Dispenzieri A Lacy MQ Buadi FK Dingli D Go RS Leung N Kapoor P Hayman SR Lust JA Russell SJ Zeldenrust SR Hwa YL Kourelis TV Kyle RA Kumar SK Clinical course and outcomes of patients with multiple myeloma who relapse after autologous stem cell therapy Bone marrow transplantation 2016. 51 1156–1158 [DOI] [PubMed] [Google Scholar]
- [5].de la Puente P, Muz B, Azab F, Luderer M, Azab AK. Molecularly Targeted Therapies in Multiple Myeloma. Leukemia Research and Treatment. 2014;2014:976567. doi: 10.1155/2014/976567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. de la Puente P Azab AK Contemporary drug therapies for multiple myeloma Drugs of today (Barcelona, Spain: 1998) 2013. 49 563–573 [DOI] [PubMed] [Google Scholar]
- [7]. Adams J The proteasome: a suitable antineoplastic target Nature reviews. Cancer 2004. 4 349–360 [DOI] [PubMed] [Google Scholar]
- [8].LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC. Proteasome Inhibitor PS-341 Inhibits Human Myeloma Cell Growth <strong><em>in Vivo</em></strong> and Prolongs Survival in a Murine Model. Cancer Research. 2002;62:4996. [PubMed] [Google Scholar]
- [9]. Field-Smith A Morgan GJ Davies FE Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma Therapeutics and clinical risk management 2006. 2 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10]. Cao B Li J Mao X Dissecting bortezomib: development, application, adverse effects and future direction Current pharmaceutical design 2013. 19 3190–3200 [DOI] [PubMed] [Google Scholar]
- [11]. Miguel JFS Richardson P Sonneveld P Schuster M Irwin D Stadtmauer E Facon T Harousseau J Ben-Yehuda D Lonial S Goldschmidt H Reece D Blade J Boccadoro M Cavenagh J Dalton W Boral A Schenkein D Anderson K Frequency, Characteristics, and Reversibility of Peripheral Neuropathy (PN) in the APEX Trial Blood 2005. 106 366–366 [Google Scholar]
- [12]. Ashley JD Stefanick JF Schroeder VA Suckow MA Kiziltepe T Bilgicer B Liposomal bortezomib nanoparticles via boronic ester prodrug formulation for improved therapeutic efficacy in vivo Journal of medicinal chemistry 2014. 57 5282–5292 [DOI] [PubMed] [Google Scholar]
- [13].de la Puente P, Azab AK. Nanoparticle delivery systems, general approaches and their implementation in Multiple Myeloma. European journal of haematology. 2017 doi: 10.1111/ejh.12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Maeda H The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting Advances in enzyme regulation 2001. 41 189–207 [DOI] [PubMed] [Google Scholar]
- [15]. Jain RK Stylianopoulos T Delivering nanomedicine to solid tumors Nature reviews. Clinical oncology 2010. 7 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Fang J Nakamura H Maeda H The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect Advanced drug delivery reviews 2011. 63 136–151 [DOI] [PubMed] [Google Scholar]
- [17]. de la Puente P Quan N Hoo RS Muz B Gilson RC Luderer M King J Achilefu S Salama NN Vij R Azab AK Newly established myeloma-derived stromal cell line MSP-1 supports multiple myeloma proliferation, migration, and adhesion and induces drug resistance more than normal-derived stroma Haematologica 2016. 101 e307–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Azab AK Quang P Azab F Pitsillides C Thompson B Chonghaile T Patton JT Maiso P Monrose V Sacco A Ngo HT Flores LM Lin CP Magnani JL Kung AL Letai A Carrasco R Roccaro AM Ghobrial IM P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment Blood 2012. 119 1468–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].de la Puente P, Muz B, Jin A, Azab F, Luderer M, Salama NN, Azab AK. MEK inhibitor, TAK-733 reduces proliferation, affects cell cycle and apoptosis, and synergizes with other targeted therapies in multiple myeloma. Blood cancer journal. 2016;6:e399. doi: 10.1038/bcj.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. de la Puente P Muz B Gilson RC Azab F Luderer M King J Achilefu S Vij R Azab AK 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma Biomaterials 2015. 73 70–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. de la Puente P Azab AK 3D tissue-engineered bone marrow: what does this mean for the treatment of multiple myeloma? Future oncology (London, England) 2016. 12 1545–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. de la Puente P Ludena D Cell culture in autologous fibrin scaffolds for applications in tissue engineering Experimental cell research 2014. 322 1–11 [DOI] [PubMed] [Google Scholar]
- [23]. de la Puente P Ludena D Fernandez A Aranda JL Varela G Iglesias J Autologous fibrin scaffolds cultured dermal fibroblasts and enriched with encapsulated bFGF for tissue engineering Journal of biomedical materials research. Part A 2011. 99 648–654 [DOI] [PubMed] [Google Scholar]
- [24]. de la Puente P Ludena D Lopez M Ramos J Iglesias J Differentiation within autologous fibrin scaffolds of porcine dermal cells with the mesenchymal stem cell phenotype Experimental cell research 2013. 319 144–152 [DOI] [PubMed] [Google Scholar]
- [25]. de la Puente P Azab F Muz B Luderer M Arbiser J Azab AK Tris DBA palladium overcomes hypoxia-mediated drug resistance in multiple myeloma Leukemia & lymphoma 2016. 57 1677–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].de la Puente P, Weisberg E, Muz B, Nonami A, Luderer M, Stone RM, Melo JV, Griffin JD, Azab AK. Identification of ILK as a novel therapeutic target for acute and chronic myeloid leukemia. Leukemia research. 2015 doi: 10.1016/j.leukres.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Wojtkowiak JW Verduzco D Schramm KJ Gillies RJ Drug resistance and cellular adaptation to tumor acidic pH microenvironment Molecular pharmaceutics 2011. 8 2032–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Raghunand N Gillies RJ pH and drug resistance in tumors Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy 2000. 3 39–47 [DOI] [PubMed] [Google Scholar]
- [29]. Abdullah SY Ali MK Sabha MM Type-B lactic acidosis associated with progressive multiple myeloma Saudi medical journal 2015. 36 239–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Gillespie EJ Ho CL Balaji K Clemens DL Deng G Wang YE Elsaesser HJ Tamilselvam B Gargi A Dixon SD France B Chamberlain BT Blanke SR Cheng G de la Torre JC Brooks DG Jung ME Colicelli J Damoiseaux R Bradley KA Selective inhibitor of endosomal trafficking pathways exploited by multiple toxins and viruses Proceedings of the National Academy of Sciences of the United States of America 2013. 110 E4904–4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Dutta D Donaldson JG Search for inhibitors of endocytosis: Intended specificity and unintended consequences Cellular Logistics 2012. 2 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Dick LR Fleming PE Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy Drug discovery today 2010. 15 243–249 [DOI] [PubMed] [Google Scholar]
- [33]. Mitsiades N Mitsiades CS Poulaki V Chauhan D Fanourakis G Gu X Bailey C Joseph M Libermann TA Treon SP Munshi NC Richardson PG Hideshima T Anderson KC Molecular sequelae of proteasome inhibition in human multiple myeloma cells Proceedings of the National Academy of Sciences of the United States of America 2002. 99 14374–14379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Hideshima T Mitsiades C Akiyama M Hayashi T Chauhan D Richardson P Schlossman R Podar K Munshi NC Mitsiades N Anderson KC Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341 Blood 2003. 101 1530–1534 [DOI] [PubMed] [Google Scholar]
- [35]. Curran MP McKeage K Bortezomib: a review of its use in patients with multiple myeloma Drugs 2009. 69 859–888 [DOI] [PubMed] [Google Scholar]
- [36]. Arastu-Kapur S Anderl JL Kraus M Parlati F Shenk KD Lee SJ Muchamuel T Bennett MK Driessen C Ball AJ Kirk CJ Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events Clinical cancer research : an official journal of the American Association for Cancer Research 2011. 17 2734–2743 [DOI] [PubMed] [Google Scholar]
- [37]. Lopez-Leon T Carvalho EL Seijo B Ortega-Vinuesa JL Bastos-Gonzalez D Physicochemical characterization of chitosan nanoparticles: electrokinetic and stability behavior Journal of colloid and interface science 2005. 283 344–351 [DOI] [PubMed] [Google Scholar]
- [38]. Alsarra IA Evaluation of crosslinked chitosan hydrogel beads as a carrier for prolonged delivery of diclofenac sodium: in vitro and in vivo studies Bollettino chimico farmaceutico 2004. 143 359–364 [PubMed] [Google Scholar]
- [39]. Kawashima Y Handa T Kasai A Takenaka H Lin SY Ando Y Novel method for the preparation of controlled-release theophylline granules coated with a polyelectrolyte complex of sodium polyphosphate-chitosan Journal of pharmaceutical sciences 1985. 74 264–268 [DOI] [PubMed] [Google Scholar]
- [40]. Torchilin V Tumor delivery of macromolecular drugs based on the EPR effect Advanced drug delivery reviews 2011. 63 131–135 [DOI] [PubMed] [Google Scholar]
- [41]. Azab AK Orkin B Doviner V Nissan A Klein M Srebnik M Rubinstein A Crosslinked chitosan implants as potential degradable devices for brachytherapy: in vitro and in vivo analysis Journal of controlled release : official journal of the Controlled Release Society 2006. 111 281–289 [DOI] [PubMed] [Google Scholar]
- [42].Bhumkar DR, Pokharkar VB. Studies on effect of pH on cross-linking of chitosan with sodium tripolyphosphate: a technical note. AAPS PharmSciTech. 2006;7:E50. doi: 10.1208/pt070250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kiziltepe T, Ashley JD, Stefanick JF, Qi YM, Alves NJ, Handlogten MW, Suckow MA, Navari RM, Bilgicer B. Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacy in vivo. Blood cancer journal. 2012;2:e64. doi: 10.1038/bcj.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Ashley JD Stefanick JF Schroeder VA Suckow MA Alves NJ Suzuki R Kikuchi S Hideshima T Anderson KC Kiziltepe T Bilgicer B Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo J Control Release 2014. 196 113–121 [DOI] [PubMed] [Google Scholar]
- [45]. Soodgupta D Pan D Cui G Senpan A Yang X Lu L Weilbaecher KN Prochownik EV Lanza GM Tomasson MH Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma Mol Cancer Ther 2015. 14 1286–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46]. Yang C Xiong F Wang J Dou J Chen J Chen D Zhang Y Luo S Gu N Anti-ABCG2 monoclonal antibody in combination with paclitaxel nanoparticles against cancer stem-like cell activity in multiple myeloma Nanomedicine (Lond) 2014. 9 45–60 [DOI] [PubMed] [Google Scholar]
- [47]. Yang C He X Song L Zhan X Zhang Y Dou J Gu N Gamma-Fe2O3 nanoparticles increase therapeutic efficacy of combination with paclitaxel and anti-ABCG2 monoclonal antibody on multiple myeloma cancer stem cells in mouse model Journal of biomedical nanotechnology 2014. 10 336–344 [DOI] [PubMed] [Google Scholar]
- [48]. Szakacs G Annereau JP Lababidi S Shankavaram U Arciello A Bussey KJ Reinhold W Guo Y Kruh GD Reimers M Weinstein JN Gottesman MM Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells Cancer cell 2004. 6 129–137 [DOI] [PubMed] [Google Scholar]
- [49]. Swami A Reagan MR Basto P Mishima Y Kamaly N Glavey S Zhang S Moschetta M Seevaratnam D Zhang Y Liu J Memarzadeh M Wu J Manier S Shi J Bertrand N Lu ZN Nagano K Baron R Sacco A Roccaro AM Farokhzad OC Ghobrial IM Engineered nanomedicine for myeloma and bone microenvironment targeting Proceedings of the National Academy of Sciences of the United States of America 2014. 111 10287–10292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]. Chillemi A Zaccarello G Quarona V Lazzaretti M Martella E Giuliani N Ferracini R Pistoia V Horenstein AL Malavasi F CD38 and bone marrow microenvironment Front Biosci (Landmark Ed) 2014. 19 152–162 [DOI] [PubMed] [Google Scholar]
- [51]. Rawstron AC Orfao A Beksac M Bezdickova L Brooimans RA Bumbea H Dalva K Fuhler G Gratama J Hose D Kovarova L Lioznov M Mateo G Morilla R Mylin AK Omede P Pellat-Deceunynck C Perez Andres M Petrucci M Ruggeri M Rymkiewicz G Schmitz A Schreder M Seynaeve C Spacek M de Tute RM Van Valckenborgh E Weston-Bell N Owen RG San Miguel JF Sonneveld P Johnsen HE N. European Myeloma Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders Haematologica 2008. 93 431–438 [DOI] [PubMed] [Google Scholar]
- [52]. San Miguel JF Gutierrez NC Mateo G Orfao A Conventional diagnostics in multiple myeloma European journal of cancer (Oxford, England : 1990) 2006. 42 1510–1519 [DOI] [PubMed] [Google Scholar]
- [53]. Muz B de la Puente P Azab F Luderer MJ King J Vij R Azab AK A CD138-independent strategy to detect minimal residual disease and circulating tumour cells in multiple myeloma British journal of haematology 2016. 173 70–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54]. Malavasi F Deaglio S Funaro A Ferrero E Horenstein AL Ortolan E Vaisitti T Aydin S Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology Physiological reviews 2008. 88 841–886 [DOI] [PubMed] [Google Scholar]
- [55].Flemming A. Deal watch: J&J and Genmab deal to push forward CD38 as a blood cancer target. Nature reviews. Drug discovery. 2012;11:822. doi: 10.1038/nrd3890. [DOI] [PubMed] [Google Scholar]
- [56]. de Weers M Tai YT van der Veer MS Bakker JM Vink T Jacobs DC Oomen LA Peipp M Valerius T Slootstra JW Mutis T Bleeker WK Anderson KC Lokhorst HM van de Winkel JG Parren PW Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors Journal of immunology (Baltimore, Md.: 1950) 2011. 186 1840–1848 [DOI] [PubMed] [Google Scholar]
- [57]. van der Veer MS de Weers M van Kessel B Bakker JM Wittebol S Parren PW Lokhorst HM Mutis T Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab Haematologica 2011. 96 284–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58]. Green DJ Orgun NN Jones JC Hylarides MD Pagel JM Hamlin DK Wilbur DS Lin Y Fisher DR Kenoyer AL Frayo SL Gopal AK Orozco JJ Gooley TA Wood BL Bensinger WI Press OW A preclinical model of CD38-pretargeted radioimmunotherapy for plasma cell malignancies Cancer research 2014. 74 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher V, Vallee F, Zhou-Liu Q, Ferrari P, Pouzieux S, Lahoute C, Dumontet C, Plesa A, Chiron M, Lejeune P, Chittenden T, Park PU, Blanc V. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent anti-tumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-14-0695. [DOI] [PubMed] [Google Scholar]
- [60].Muz B, de la Puente P, Azab F, Luderer M, Azab A. Hypoxia promotes stem cell -like phenotype in multiple myeloma cells. Blood cancer journal. 2014 doi: 10.1038/bcj.2014.82. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61]. Huotari J Helenius A Endosome maturation The EMBO journal 2011. 30 3481–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. Journal of extracellular vesicles. 2014;3 doi: 10.3402/jev.v3.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63]. Dutta D Donaldson JG Search for inhibitors of endocytosis: Intended specificity and unintended consequences Cell Logist 2012. 2 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64]. Panyam J Zhou WZ Prabha S Sahoo SK Labhasetwar V Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2002. 16 1217–1226 [DOI] [PubMed] [Google Scholar]
- [65]. Gilleron J Querbes W Zeigerer A Borodovsky A Marsico G Schubert U Manygoats K Seifert S Andree C Stoter M Epstein-Barash H Zhang L Koteliansky V Fitzgerald K Fava E Bickle M Kalaidzidis Y Akinc A Maier M Zerial M Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape Nature biotechnology 2013. 31 638–646 [DOI] [PubMed] [Google Scholar]
- [66]. Wang Q Liu P Gong T Sun X Duan Y Zhang Z Uptake mechanism and endosomal fate of drug-phospholipid lipid nanoparticles in subcutaneous and in situ hepatoma Journal of biomedical nanotechnology 2014. 10 993–1003 [DOI] [PubMed] [Google Scholar]
- [67]. Funaro A Reinis M Trubiani O Santi S Di Primio R Malavasi F CD38 functions are regulated through an internalization step Journal of immunology (Baltimore, Md.: 1950) 1998. 160 2238–2247 [PubMed] [Google Scholar]
- [68]. Widjaja I de Vries E Tscherne DM Garcia-Sastre A Rottier PJ de Haan CA Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step Journal of virology 2010. 84 9625–9631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Greene W, Zhang W, He M, Witt C, Ye F, Gao SJ. The ubiquitin/proteasome system mediates entry and endosomal trafficking of Kaposi’s sarcoma-associated herpesvirus in endothelial cells. PLoS pathogens. 2012;8:e1002703. doi: 10.1371/journal.ppat.1002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70]. Douar AM Poulard K Stockholm D Danos O Intracellular trafficking of adeno-associated virus vectors: routing to the late endosomal compartment and proteasome degradation Journal of virology 2001. 75 1824–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Phukan G, Shin TH, Shim JS, Paik MJ, Lee J-K, Choi S, Kim YM, Kang SH, Kim HS, Kang Y, Lee SH, Mouradian MM, Lee G. Silica-coated magnetic nanoparticles impair proteasome activity and increase the formation of cytoplasmic inclusion bodies in vitro. Scientific Reports. 2016;6:29095. doi: 10.1038/srep29095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72]. Armand L Biola-Clier M Bobyk L Collin-Faure V Diemer H Strub J-M Cianferani S Van Dorsselaer A Herlin-Boime N Rabilloud T Carriere M Molecular responses of alveolar epithelial A549 cells to chronic exposure to titanium dioxide nanoparticles: A proteomic view Journal of Proteomics 2016. 134 163–173 [DOI] [PubMed] [Google Scholar]
- [73]. Azab AK Runnels JM Pitsillides C Moreau AS Azab F Leleu X Jia X Wright R Ospina B Carlson AL Alt C Burwick N Roccaro AM Ngo HT Farag M Melhem MR Sacco A Munshi NC Hideshima T Rollins BJ Anderson KC Kung AL Lin CP Ghobrial IM CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy Blood 2009. 113 4341–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74]. Muz B de la Puente P Azab F Luderer M Azab AK The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies Molecular cancer research : MCR 2014. 12 1347–1354 [DOI] [PubMed] [Google Scholar]
- [75]. Kirshner J Thulien KJ Martin LD Debes Marun C Reiman T Belch AR Pilarski LM A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma Blood 2008. 112 2935–2945 [DOI] [PubMed] [Google Scholar]
- [76]. Calimeri T Battista E Conforti F Neri P Di Martino MT Rossi M Foresta U Piro E Ferrara F Amorosi A Bahlis N Anderson KC Munshi N Tagliaferri P Causa F Tassone P A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells Leukemia 2011. 25 707–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Characterization of bortezomib-loaded anti-CD38 chitosan NPs. A) BTZ HPLC detection peak with retention time 2 minutes, λ = 270 nm. B) BTZ calibration curve formed by plotting the AUC of BTZ HPLC peak for the concentration range of bortezomib (0 to 1.5 mM). C) Effect of intermediate (50 μM) and high-dose (1 mM) BTZ on i) size and ii) stability of anti-CD38 chitosan NPs. D) Effect of time preservation on i) size and ii) stability of empty or BTZ loaded anti-CD38 chitosan NPs.
Fig. S2: In vitro drug release from anti-CD38 chitosan NPs to different environments. A) i) pH of non-conditioned media and conditioned media from 3 independent PB MNCs patient samples and 3 MM cell lines after 72h in culture. ii) Effect of conditioned media pH on drug release from anti-CD38 chitosan NPs. B) i) Corrected pH of conditioned media from 3 independent PB MNCs and 3 MM cell lines after 72h in culture. ii) Effect of conditioned media with corrected pH on drug release from anti-CD38 chitosan NPs.
Fig. S3: Specificity and variability of binding/uptake of anti-CD38 chitosan NPs. Binging of anti-CD38 chitosan NPs, non-targeted chitosan NPs and unstained control NPs to A) MM cell lines (MM1S, H929, OPM1, RPMI, U266), B) CD138+ cells isolated from MM patients (n=5), C) mononuclear cells isolated from the peripheral blood of normal subjects (PB MNCs, n=5), D) and normal plasma cells isolated from the BM of normal subjects (BM MNCs, n=5) analyzed by flow cytometry. E) Effect of anti-CD38 antibody (0.5 – 5 μg/ml) on proliferation of MM cells for 48 h analyzed by MTT.
Fig. S4: Effect of bortezomib-loaded anti-CD38 chitosan NPs on proliferation, cell cycle, and apoptosis. The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h on: A) Proliferation of MM1s, H929, RPMI cells and PB MNCs analyzed by MTT, *p<0.05; B) Cell cycle of H929 cells measured by Propidium Iodide stained of DNA, *p<0.05; and C) Apoptosis of H929 (i), U266 (ii) and RPMI (iii) cells by FITC-Annexin-V and Propidium Iodide.
Fig. S5: The effect of vehicle (control), BTZ as a free drug (5 nM), empty chitosan NPs, non-targeted chitosan NPs loaded with BTZ equivalent amounts to 5 nM, and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 5 nM for 48 h after a 2 h pulse on proliferation of MM1s cells analyzed by MTT, *p<0.05.
Fig. S6: Effect of macropinocytosis and endocytosis inhibitors on survival of MM cells. A) Incubation with macropinocytosis inhibitor Cytochalasin D (0 – 1.5 μM) for 30 min. B) Incubation with early endocytosis chlathrin-mediated inhibitor Chlorpromazine (0 – 3 μM) for 30 min. C) Incubation with early endocytosis caveolae-mediated inhibitor Nystatin (0 – 1 μg/ml) for 30 min. D) Incubation with late endocytosis inhibitor EGA (0 – 5 μM) for 30 min.
Fig. S7: Histological tissue damage of bortezomib-loaded anti-CD38 chitosan NPs in vivo. The effect of vehicle, BTZ as a free drug (1 mg/kg once a week), non-targeted chitosan NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week), and anti-CD38 chitosan NPs loaded with BTZ equivalent amounts to 1 mg/kg (once a week) on histological tissue damage on femur, spinal cord, liver, spleen, kidney, intestine after 25 days of treatment.