

Abstract
Ataxia–telangiectasia confers a significant increase in the development of several cancer types, most commonly leukemia and lymphoma. However, as the natural history for these patients is evolving and their lifespan is increasing, there is the potential for the development of additional uncommon tumors in an already rare patient population. We report the first case, to our knowledge, of an incidental retroperitoneal tumor in a 26-year-old woman undergoing evaluation for hepatic dysfunction. The mass was suspicious for retroperitoneal sarcoma, but proved to be an extramedullary hematopoietic pseudotumor after extensive pathologic evaluation. The changing landscape of neoplasms associated with ataxia–telangiectasia is discussed with emphasis on previously underreported benign and malignant tumors.
Keywords: Extramedullary hematopoietic pseudotumor, retroperitoneal sarcoma, ataxia–telangiectasia
Introduction
First described in 1941,1 ataxia–telangiectasia (A-T) is a rare, autosomal recessive disorder with myriad characteristics, most notably cerebellar degeneration, telangiectasias, immunodeficiency, and cancer susceptibility.2 The age of onset and rate of disease progression can vary widely, with typical onset of ataxia and telangiectasias at 1–4 years of age and 6 years of age, respectively.3 Average life expectancy for A-T patients is 26 years,4 and the most common causes of death are chronic lung disease and cancer.2
Following extensive molecular and genetic evaluation, the genetic defect responsible for A-T was localized to chromosome 11 in 1988,5 and ATM was subsequently identified as the culprit gene in 1995.6 Although the molecular mechanisms of A-T pathophysiology are not fully elucidated, ATM is known to play a critical role in regulation of the cell cycle, apoptosis, and response to DNA damage.7 Given the critical importance of the ATM gene in cellular function and DNA repair, it is clear that A-T patients are at high risk for various cancers, and the risk of malignancy is estimated at 61–184 times greater than the general population.8
Although several reports have demonstrated a clear link between A-T and hematological malignancies, an association with solid tumors is less established. Here, we describe the first reported case of a retroperitoneal tumor arising in a patient with A-T. The mass was suspicious for a retroperitoneal sarcoma, but proved to be an extramedullary hematopoietic pseudotumor after thorough radiographic and pathologic evaluation. To our knowledge, this is the first reported case of extramedullary hematopoietic pseudotumor in an A-T patient and highlights the potential changing spectrum of neoplasms in A-T patients who are enjoying a longer lifespan because of improvements in multimodality care.
Case report
The patient is a 26-year-old woman with a childhood diagnosis of A-T, who suffers from the typical manifestations of the disease, including spastic paresis and frequent pulmonary infections as well as insulin-resistant diabetes mellitus, polycystic ovarian syndrome, osteopenia, and non-alcoholic steatohepatitis. She provided written informed consent for her case information to be published in the medical literature. It was during a hepatic ultrasound for evaluation of her liver disease that an incidental left retroperitoneal mass was discovered. Sonographic images of the abdomen demonstrated a 5.5 cm hypoechoic mass with internal vascularity in the left perirenal space which was potentially suspicious for malignancy (Figure 1(a)).
Figure 1.

Results of initial imaging evaluation. (a) Longitudinal ultrasound image demonstrating an oval hypoechoic mass superficial to the left kidney with mild mass effect on the left kidney. (b) Axial T2 fat saturation MRI shows the lesion as a fluid signal oval mass exerting mass effect upon the left kidney. Extension of intermediate signal along the left kidney in the perirenal space is also seen. (c) Axial T1 fat saturation MRI demonstrating heterogeneous, predominantly peripheral, and progressive enhancement of the left perirenal mass. The mass extends laterally beyond the posterior renal fascia and invades the intercostal musculature (white arrow). (d) Axial image from a subsequent CT of the abdomen with IV contrast shows loss of the fat place between the intercostal musculature and the low-density left perirenal mass.
Further evaluation with a gadolinium-enhanced magnetic resonance imaging (MRI) revealed a 4.9 ×2.9 × 5.1 cm enhancing retroperitoneal mass with T2 hyperintensity which exerted mass effect on the left kidney (Figure 1(b)). In addition, MRI demonstrated heterogeneous enhancement, restricted diffusion, and invasion into Gerota’s fascia, all of which were concerning for malignancy (Figure 1(c)).
An initial percutaneous biopsy was non-diagnostic, so the patient was referred for surgical evaluation. Computed tomography (CT) of the chest, abdomen, and pelvis, obtained for formal staging evaluation and surgical planning, revealed no evidence of metastases and confirmed the MRI findings of an extra-renal mass abutting the kidney with no clear evidence of invasion of the renal parenchyma (Figure 1(d)). A repeat image-guided needle biopsy was performed, but was again non-diagnostic despite several expert consultations. The biopsy material consisted of abundant myxoid stroma with a paucicellular proliferation of spindle to stellate cells, rare larger atypical cells with hyperchromatic nuclei, and background chronic inflammation. Immunohistochemical stains for MDM2, CDK4, keratin, desmin, SOX10, CD34, and MUC4 were negative, and FISH testing was negative for MDM2 gene amplification. The lack of staining for MDM2 and CDK4, as well as the negative FISH result, argued against a diagnosis of well-differentiated liposarcoma. Overall, the findings were non-specific, and the lesion was unable to be classified beyond a morphologically low-grade spindle cell neoplasm.
Given the patient’s significant comorbid conditions including restrictive lung disease, frailty, and dependent functional status (with corresponding high risk for post-surgical complications), the decision was made initially to observe the mass with active surveillance; 3 months after the second biopsy, repeat MRI showed interval tumor enlargement with irregular enhancement, thickening of Gerota’s fascia, and new mass effect on the abdominal wall (Figure 2).
Figure 2.
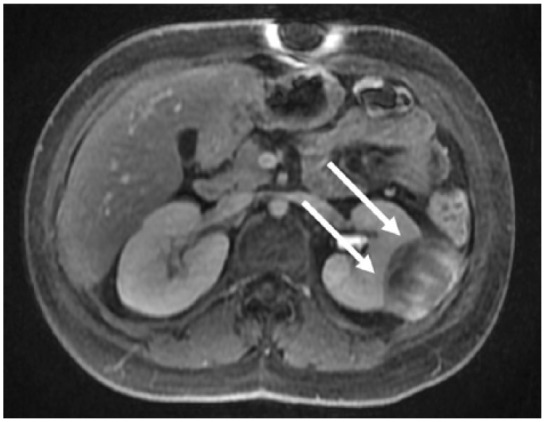
Results of repeat imaging studies. Axial T1 fat saturation post contrast image from a follow-up MRI performed 3 months after initial presentation showing progression of disease in the index left perirenal mass with greater enhancement and increased mass effect upon the left kidney (white arrows).
Based on the follow-up MRI results, the decision was made to proceed with surgical excision using a left subcostal approach. Intraoperatively, the mass was found to be contiguous with and adherent to the left kidney. Based on these intra-operative findings, the mass was not amenable to separation from the kidney without excision of kidney parenchyma. Therefore, a left partial nephrectomy was performed, and no local/regional disease extension was noted. As shown in Figure 3(a)–(c), the mass was immediately adjacent to the renal capsule and had a similar histologic appearance to the prior core biopsy. Although the diagnosis of soft tissue myxoma was previously considered, immunohistochemical staining for CD61 was positive, highlighting rare large atypical cells which were consistent with megakaryocytes (Figure 3(d) and (e)). Given the constellation of clinical and histopathologic findings, a final diagnosis of extramedullary hematopoietic pseudotumor was ultimately rendered. Although the patient’s postoperative course was notable for persistent fevers and fatigue, no clear infectious etiology was identified. Despite a relatively prolonged postoperative course, the patient made a full recovery and was discharged home after approximately 2 weeks with return to baseline functional status. Following the diagnosis of extramedullary hematopoietic pseudotumor, the patient underwent a bone marrow biopsy to rule out bone marrow pathology. Despite the association of extramedullary hematopoietic pseudotumor with myelofibrosis, bone marrow studies showed normocellular marrow with adequate trilineal hematopoiesis, no significant reticulin fibrosis, and no increase in blasts.
Figure 3.

Resection gross specimen and histopathology. (a) Gross resection specimen showing relationship between mass (double arrow) and renal parenchyma (single arrow). (b) Abundant myxoid material with scattered foci of inflammatory cells (H&E, 40×). (c) Relationship between the mass (left) and renal cortex (right) (H&E, 40×). (d) Focus of inflammatory cells with erythroid precursors (arrowhead) (H&E, 200×). (e) Larger atypical cell consistent with a megakaryocyte (arrow) (H&E, 400×).
Discussion
Although rare, the phenomenon of non-hepatosplenic extramedullary hematopoiesis (NHS-EMH) has been well described in the non-fetal population, but has never been previously reported in the A-T population. Much of the data pertaining to NHS-EMH come from a Mayo Clinic series of 27 patients between 1975 and 2002 published in 2003.9 In the study by Koch et al.,9 NHS-EMH was most frequently associated with myelofibrosis with myeloid metaplasia and thalassemia, though several other associations were also documented. The underlying connection appears to be chronic anemia, with an average hemoglobin concentration measured in their series at 9.5 g/dL. The patient described in this report had a preoperative hemoglobin concentration consistently around 12 g/dL, suggesting that she should be at lower risk for EMH if the association of chronic anemia with NHS-EMH pseudotumor is mechanistically related. However, given the risk of hematologic malignancy with A-T, the development of NHS-EMH may represent an additional manifestation of myeloid pathology secondary to as yet unidentified causes.
As previously noted, the connection between cancer and A-T is well described. Specifically, observational data suggest that the cumulative incidence of cancer increases with age in A-T, measured at 10.4% at 10 years, 22.6% at 20 years, 29.9% at 30 years, and 38.2% at 40 years.10 The most common cancers are leukemia and lymphoma, but there is no reported association between A-T and NHS-EMH, which importantly is a benign tumor. A large, retrospective analysis published by Morrel and colleagues8 in 1986 reviewed 263 patients with A-T and identified 52 individual cancers that arose. Of those 52 cancers identified, the breakdown was as follows: 31 lymphoma (60%), 14 leukemia (27%), 2 hepatocellular carcinoma (4%), 2 carcinomas of the parotid gland (4%), 1 ovarian carcinoma (2%), 1 cerebellar astrocytoma (2%), and 1 uterine leiomyosarcoma (2%). A subsequent retrospective study was recently completed by Suarez et al. from a French registry of A-T patients with similar results.10 In their cohort of 279 patients, they identified 69 patients with cancer (25%). In descending order of prevalence, they noted 38 cases of non-Hodgkin’s lymphoma, 12 Hodgkin’s lymphoma, 11 leukemia, 3 breast carcinoma, 2 gastric carcinoma, 2 hepatocellular carcinomas, and 1 thyroid carcinoma. One patient with Hodgkin’s lymphoma was diagnosed with a synchronous glioma. These two observational studies support the observation that patients with A-T are at highest risk of leukemia and lymphoma, whereas solid tumors are rarer and tend to occur in older A-T patients, notably with an average age at diagnosis of 31.4 years.10
Evaluation of tumors in A-T can be difficult due to the underlying defect in the ATM gene and the resultant susceptibility to ionizing radiation. While there is reasonable concern that A-T patients should limit exposure to plain film X-rays and CT scans, no clear association between A-T and complications from exposure to diagnostic radiographs has been reported,11 and it is important to avoid a delay in diagnosis from under-utilization of diagnostic imaging. Therefore, in the evaluation of A-T patients, CT imaging can be employed if indicated for appropriate evaluation and management of suspicious masses. Furthermore, ionizing radiation can frequently be avoided by employing MR imaging to evaluate and characterize suspicious lesions and for surgical planning. Specific to this case, T1-weighted images are useful in determining proximity to adjacent retroperitoneal organs, while T2-weighted images can aid in defining local muscular invasion.12
Careful diagnostic workup is also critical for retroperitoneal masses, especially in A-T. Unlike the general population where radiation therapy (RT) has a role in treating retroperitoneal tumors, RT is contraindicated in A-T because of the risk of severe and even lethal toxicities.13 Therefore, accurate tissue diagnosis is required to guide the proper surgical resection and thereby oncologic outcome with strategies to limit post-surgical morbidity and mortality.
In this case, prior to the final diagnosis of an extramedullary hematopoietic pseudotumor, the leading diagnostic consideration based on the percutaneous biopsy results was soft tissue myxoma, another benign, mesenchymal neoplasm also characterized by bland spindle to stellate cells in myxoid stroma.14 These benign tumors are generally non-recurrent, although certain variants have demonstrated a small risk of local, non-destructive recurrence.15 Overall, there seems to be no correlation between A-T and retroperitoneal masses, although recent research has identified an interaction between the ATM protein and FUS/TLS (currently designated as DDIT3), a translocated gene identified in certain human liposarcomas.16 Despite this molecular connection, primary sarcomas arising in A-T patients are limited to two case reports—notably uterine leiomyosarcoma17 and Epstein–Barr virus (EBV)-associated laryngeal leiomyosarcoma.18 Though the propensity for cancer is a hallmark of A-T, the development of either primary sarcoma or extramedullary hematopoietic pseudotumor is equally rare, emphasizing the need for careful multidisciplinary management of A-T patients who present with suspicious solid tumors.
Conclusion
Patients with A-T are at significantly increased risk of various cancer types, most notably leukemia and lymphoma. As more A-T patients live longer, other less common tumors are being identified. Given the association of A-T with several diseases that can lead to anemia, extramedullary hematopoietic pseudotumor should be on the differential diagnosis for atypical tumors arising in A-T, particularly in the retroperitoneum. Evaluation with comprehensive imaging modalities is essential, as is tissue diagnosis to aid definitive diagnosis and surgical planning. Given the complexities of caring for patients with A-T, evaluation, management, and treatment should be carried out in specialty centers where appropriate resources are available.
Acknowledgments
S.J.J. was involved in conception and design, literature review, and writing the manuscript. T.A.P., C.P.B., M.A.D., and C.P.E. participated in analysis and interpretation of data and critical revision of the article. R.J.C. was involved in conception and design, analysis and interpretation of data, writing the manuscript, and critical revision of the manuscript.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the Dr Mark Starr Family Fund.
References
- 1. Louis-Bar D. Sur un syndrome progress if comprenant des télangiectasies capillaires cutanées et conjonctivales symétriques a disposition naevoïde et des troubles cérébelleux. Confin Neurol 1941; 4(21): 32–42. [Google Scholar]
- 2. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, et al. Ataxia telangiectasia: a review. Orphanet J Rare Dis 2016; 11(1): 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatti R, Perlman S. Ataxia telangiectasia. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds) Genereviews((r)). Seattle, WA: University of Washington, 1993. [Google Scholar]
- 4. Crawford TO, Skolasky RL, Fernandez R, et al. Survival probability in ataxia telangiectasia. Arch Dis Child 2006; 91(7): 610–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gatti RA, Berkel I, Boder E, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature 1988; 336(6199): 577–580. [DOI] [PubMed] [Google Scholar]
- 6. Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995; 268(5218): 1749–1753. [DOI] [PubMed] [Google Scholar]
- 7. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14(4): 197–210. [PubMed] [Google Scholar]
- 8. Morrell D, Cromartie E, Swift M. Mortality and cancer incidence in 263 patients with ataxia telangiectasia. J Natl Cancer Inst 1986; 77(1): 89–92. [PubMed] [Google Scholar]
- 9. Koch CA, Li CY, Mesa RA, et al. Nonhepatosplenic extramedullary hematopoiesis: associated diseases, pathology, clinical course, and treatment. Mayo Clin Proc 2003; 78(10): 1223–1233. [DOI] [PubMed] [Google Scholar]
- 10. Suarez F, Mahlaoui N, Canioni D, et al. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: a report from the French national registry of primary immune deficiencies. J Clin Oncol 2015; 33(2): 202–208. [DOI] [PubMed] [Google Scholar]
- 11. McGrath-Morrow SA, Gower WA, Rothblum-Oviatt C, et al. Evaluation and management of pulmonary disease in ataxia-telangiectasia. Pediatr Pulmonol 2010; 45(9): 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Storm FK, Mahvi DM. Diagnosis and management of retroperitoneal soft-tissue sarcoma. Ann Surg 1991; 214(1): 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tamminga RY, Dolsma WV, Leeuw JA, et al. Chemo- and radiosensitivity testing in a patient with ataxia telangiectasia and Hodgkin disease. Pediatr Hematol Oncol 2002; 19(3): 163–171. [DOI] [PubMed] [Google Scholar]
- 14. Stout AP. Myxoma, the tumor of primitive mesenchyme. Ann Surg 1948; 127(4): 706–719. [PubMed] [Google Scholar]
- 15. Fletcher CDM, World Health Organization and International Agency for Research on Cancer. The WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press, 2013, p. 468. [Google Scholar]
- 16. Gardiner M, Toth R, Vandermoere F, et al. Identification and characterization of FUS/TLS as a new target of ATM. Biochem J 2008; 415(2): 297–307. [DOI] [PubMed] [Google Scholar]
- 17. Gatti RA, Nieberg R, Boder E. Uterine tumors in ataxia-telangiectasia. Gynecol Oncol 1989; 32(2): 257–260. [DOI] [PubMed] [Google Scholar]
- 18. Reyes C, Abuzaitoun O, De Jong A, et al. Epstein-Barr virus-associated smooth muscle tumors in ataxia-telangiectasia: a case report and review. Hum Pathol 2002; 33(1): 133–136. [DOI] [PubMed] [Google Scholar]