

Abstract
The GPI (Glycosylphosphatidylinositol) biosynthetic pathway is a multistep conserved pathway in eukaryotes that culminates in the generation of GPI glycolipid which in turn anchors many proteins (GPI-APs) to the cell surface. In spite of the overall conservation of the pathway, there still exist subtle differences in the GPI pathway of mammals and other eukaryotes which holds a great promise so far as the development of drugs/inhibitors against specific targets in the GPI pathway of pathogens is concerned. Many of the GPI structures and their anchored proteins in pathogenic protozoans and fungi act as pathogenicity factors. Notable examples include GPI-anchored variant surface glycoprotein (VSG) in Trypanosoma brucei, GPI-anchored merozoite surface protein 1 (MSP1) and MSP2 in Plasmodium falciparum, protein-free GPI related molecules like lipophosphoglycans (LPGs) and glycoinositolphospholipids (GIPLs) in Leishmania spp., GPI-anchored Gal/GalNAc lectin and proteophosphoglycans in Entamoeba histolytica or the GPI-anchored mannoproteins in pathogenic fungi like Candida albicans. Research in this active area has already yielded encouraging results in Trypanosoma brucei by the development of parasite-specific inhibitors of GlcNCONH2-β-PI, GlcNCONH2-(2-O-octyl)-PI and salicylic hydroxamic acid (SHAM) targeting trypanosomal GlcNAc-PI de-N-acetylase as well as the development of antifungal inhibitors like BIQ/E1210/gepinacin/G365/G884 and YW3548/M743/M720 targeting the GPI specific fungal inositol acyltransferase (Gwt1) and the phosphoethanolamine transferase-I (Mcd4), respectively. These confirm the fact that the GPI pathway continues to be the focus of researchers, given its implications for the betterment of human life.
Keywords: Glycosylphosphatidylinositol (GPI), GPI inhibitor, Candida albicans, Gwt1, Mcd4
Introduction
GPI anchor discovered in 1976 [1] is a complex glycolipid synthesized in the endoplasmic reticulum by a conserved pathway involving at least 19 gene products with a common core structure of EtNP-6Manα1-2Manα1-6Manα1-4GlcNα1-6myo-inositol-phospholipid [Figure 1] [2,3]. After its synthesis, GPI anchor is post-translationally transferred to the proteins carrying specific C-terminal GPI signal sequences (GPI-APs) in the endoplasmic reticulum. This is followed by their targeting to the cell membrane/cell wall through the secretory pathway. GPI-APs make up approximately 0.5% of cellular proteins in organisms as diverse as Saccharomyces cerevisiae, Caenorhabditis elegans and Plasmodium falciparum [4]. These are widely distributed in eukaryotes ranging from protozoans to mammals [5–7] and a few are reported even in some species of archaebacteria [4,8]. The GPI anchor as such and the GPI-APs play many vital and diverse roles in eukaryotes, owing to which GPI biosynthesis is either essential or important in eukaryotes [2,9,10]. Perhaps more noteworthy is the fact that GPI anchors and the GPI-APs have been used as virulence tools by pathogens especially protozoans and fungi to evade human immune response [11–14], and as such are the determining factors for various human diseases and disorders. Therein lies the great scope for specific targeting of the GPI pathway as there are species-specific differences in the GPI pathway among eukaryotes [Figure 2] which can be exploited for the development of antiprotozoan/antifungal drugs.
Figure 1.

Overview of GPI biosynthesis in yeast.
Notes: GPI biosynthesis is initiated on the cytoplasmic side of the ER by the transfer of N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to membrane bound phosphatidylinositol (PI), forming N-acetylglucosaminylphosphatidylinositol (GlcNAc-PI). This reaction is catalyzed by GPI-GlcNAc transferase complex (GPI-GnT) comprising of six core subunits – Gpi3, Gpi2, Gpi15, Gpi19, Gpi1 and Eri1, of which Gpi3 acts as the catalytic subunit. GlcNAc-PI is then de-N-acetylated to glucosaminylphosphatidylinositol (GlcN-PI) by GlcNAc-PI de-N-acetylase (Gpi12), followed by flipping of GlcN-PI to the lumenal side of the ER by yet to be identified flippase. All subsequent reactions of GPI biosynthesis take place on the lumenal side of the ER. GlcN-PI is acylated, usually by palmitoyl group, on the 2-position of inositol by acyl-CoA dependent inositol acyltransferase (Gwt1) generating GlcN-(acyl)PI. First and second mannoses are then added sequentially to the GlcN-(acyl)PI by the GPI-α1,4mannosyltransferase-I (Gpi14/Pbn1) and GPI-α1,6mannosyltransferase-II (Gpi18/Pga1) respectively, leading to the formation of Man-Man-GlcN-(acyl)PI intermediate. This is followed by the transfer of phosphoethanolamine group (EtNP) to the 2-position of first mannose by the GPI-phosphoethanolaminetransferase-I (Mcd4) generating Man-(EtNP)Man-GlcN-(acyl)PI. Third and fourth mannose additions are subsequently carried out by the GPI-α1,2mannosyltransferase-III (Gpi10) and GPI-α1,2mannosyltransferase-IV (Smp3) respectively, resulting in the formation of Man-Man-Man-(EtNP)Man-GlcN-(acyl)PI. Phosphoethanolamine moieties are then added sequentially to the 6-position of third and second mannose respectively by the GPI-phosphoethanolaminetransferase-III (Gpi13/Gpi11) and GPI-phosphoethanolaminetransferase-II (Gpi7/Gpi11), resulting in the formation of complete GPI precursor, Man-(EtNP)Man-(EtNP)Man-(EtNP)Man-GlcN-(acyl)PI. Dolichol-phosphate-mannose (Dol-P-Man) and phosphatidylethanolamine (PE) serve as the donors for the mannoses and the phosphoethanolamine groups respectively in the mannosylation and phosphoethanolamine transfer reactions. Finally, complete GPI precursor is transferred to the C-termini of the proteins bearing GPI signal sequences with the help of GPI transamidase (GPI-T). This multisubunit enzyme complex comprises of five subunits, namely, Gpi8, Gaa1, Gpi17, Gpi16 and Gab1, of which Gpi8 acts as the catalytic subunit. GPI transamidase covalently links the preformed GPI precursor to the protein through the amino group of the phosphoethanolamine moiety attached to the third mannose (Ref. 48). Gwt1 and Mcd4 inhibitors, which are the central focus of this review, are also shown in the schematic with details in the text.
Figure 2.

Species-specific variations in the mature GPI anchors of eukaryotes.
Note: [As per Ref. 49 & 50; GalNAc – N-acetylgalactosamine, Gal – galactose].
Antiprotozoan inhibitors
Perhaps in no other class of organisms GPI anchoring is as essential as in pathogenic protozoans like Trypanosoma, Plasmodium, Leishmania, or Entamoeba. Here, GPI structures and their anchored proteins play critical virulence roles. Notable examples include GPI-anchored variant surface glycoprotein (VSG) in Trypanosoma brucei [15], GPI-anchored merozoite surface protein 1 (MSP1) and MSP2 in Plasmodium falciparum [11], protein-free GPI related molecules like lipophosphoglycans (LPGs) and glycoinositolphospholipids (GIPLs) in Leishmania spp [14]. and the GPI-anchored Gal/GalNAc lectin and proteophosphoglycans in Entamoeba histolytica [12].
Studies for selective targeting of the protozoan GPI pathway were mostly carried out on GlcNAc-PI de-N-acetylase, which catalyzes the second conserved step of GPI biosynthesis. It shows subtle differences in terms of substrate specificity from its mammalian counterpart [16], and has been validated as a drug target in Trypanosoma brucei chemically as well as genetically [14,17]. Ferguson and his group studied trypanosomal de-N-acetylase using different substrate analogs and came up with two potential inhibitors, GlcNCONH2-β-PI (2-deoxy-2-ureido-D-Glcβ1-6D-myo-inositol-1-HPO4-sn-1,2-dipalmitoylglycerol) and GlcNCONH2-(2-O-octyl)-PI (2-deoxy-2-ureido-D-Glcα1-6D-(2-O-octyl)-myo-inositol-1-HPO4-sn-1,2-dipalmitoylglycerol) [18]. Both were found to be relatively ineffective in the HeLa system but equally potent in the trypanosomal system with IC50 of 8 nM. GlcNCONH2-β-PI along with GalNCONH2-PI (2-deoxy-2-ureido-D-Galα1-6D-myo-inositol-1-HPO4-sn-1,2-dipalmitoylglycerol) have also been shown to effectively inhibit the Plasmodium GlcNAc-PI de-N-acetylase (IC50 ~ 0.2 μM), but not the human enzyme [19]. Presumably, these inhibitors bind and initiate the reaction in the same way as is true for the normal substrate. However, inhibitor halts the reaction progress through the formation of stable enzyme-inhibitor complex, and thereby inactivates the enzyme [18,19]. The fact that GlcNAc-PI de-N-acetylase (PIG-L/Gpi12) is a zinc metalloenzyme [20], forms the basis of screening compounds with a zinc-binding hydroxamic acid or carboxylic acid moieties as probable inhibitors. Although such substrate-based inhibitors have been found to be effective against the trypanosomal enzyme, their potency is not enough, moreover, they do not possess drug-like physiochemical properties [21]. Further, screening of the alternative zinc-binding fragments that could potentially act as potent inhibitors of the trypanosomal GlcNAc-PI de-N-acetylase led to the identification of salicylic hydroxamic acid (SHAM) [Table 1] as a non-substrate analog inhibitor of the trypanosomal de-N-acetylase [22]. It exhibits high ligand efficiency of 0.57 kcal per mol per non-hydrogen atom and in spite of the modest potency with IC50 of 63 ± 4 μM, it can still serve as a potential lead for the development of more potent parasite-specific inhibitors [22].
Table 1.
Target and chemical structure of inhibitors.
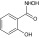
Antifungal inhibitors
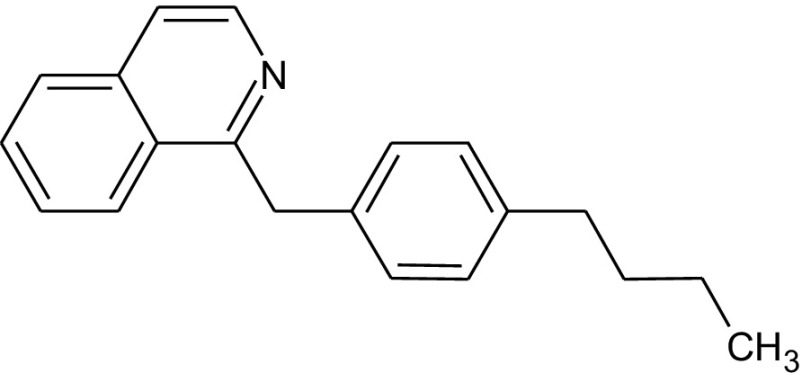
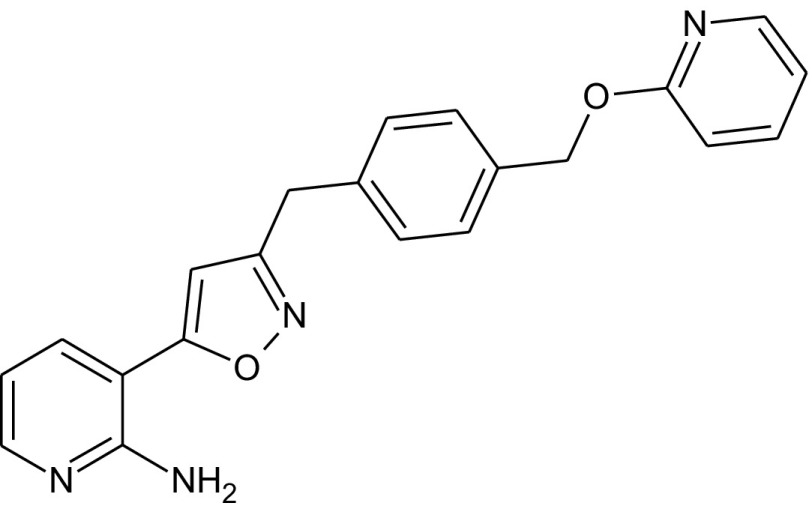
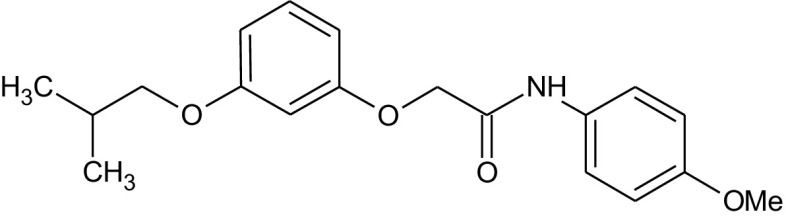
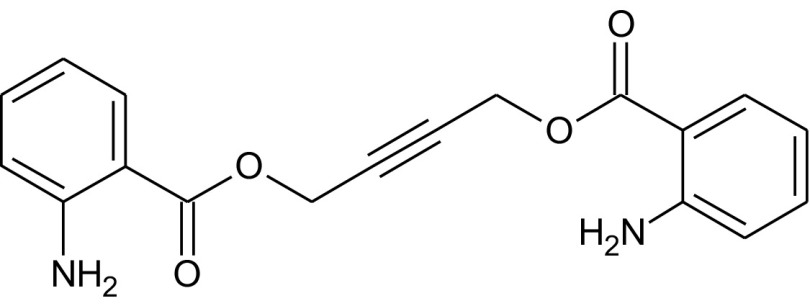
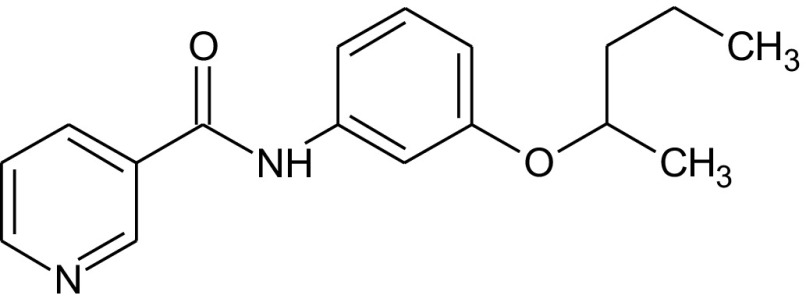
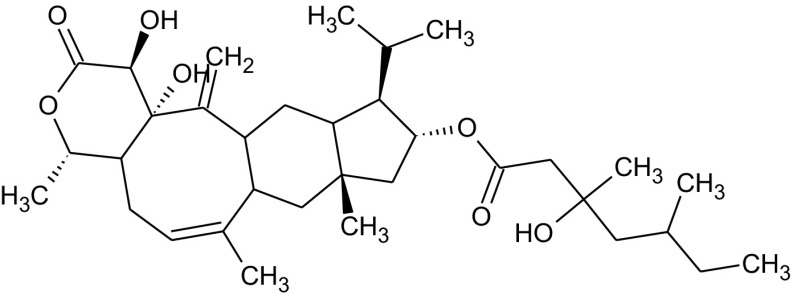
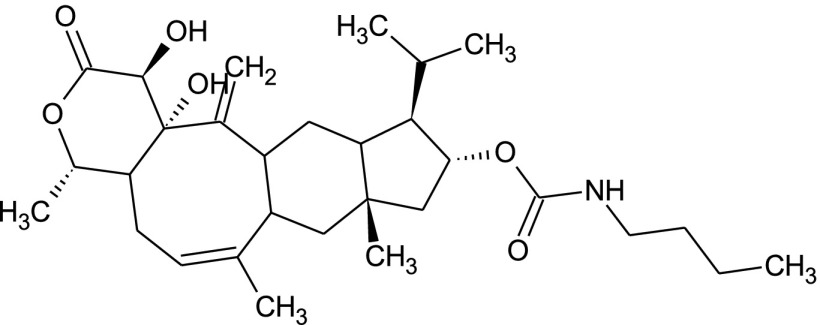
Fungi are the second class of organisms, where significant progress has been achieved in terms of the development of selective inhibitors targeting the GPI pathway. GPI biosynthesis is indispensable not only for the integrity of fungal cell wall in general [10,23], but also for the pathogenesis and virulence of pathogenic fungi such as Candida albicans [13,24–30]. Also, one has no other option than to look out for new antifungal drugs with novel modes of action to counter the problems of narrow activity range, side effects and increased drug resistance associated with the presently available antifungal drugs like azoles and polyenes [31]. To date, two novel targets in the GPI biosynthetic pathway, Gwt1 and Mcd4 have been exploited for the development of antifungal inhibitors. Gwt1 catalyzes inositol acylation whereas Mcd4 catalyzes phosphoethanolamine addition to first mannose. Gwt1 inhibitors include BIQ (1-[4-butylbenzyl] isoquinoline), E1210 (3-(3-{4-[(pyridin-2-yloxy) methyl] benzyl} isoxazol-5-yl) pyridin-2-amine), gepinacin (N-[4-methoxyphenyl]-2-{3-[2-methylpropoxy]phenoxy}acetamide), G365 (but-2-yne-1,4-diylbis[2-aminobenzoate]) and G884 (N-{3-[(pentan-2-yl)oxy]phenyl}pyridine-3-carboxamide), whereas Mcd4 inhibitors include YW3548/M743 ((1S,4S,4aS,6Z,7aS,8aS,10R,11R,11aS,12aS,13aS)-1,13a-dihydroxy-11-isopropyl-4,7,8a-trimethyl-13-methylene-2-oxo-1,2,4,4a,5,7a,8,8a,9,10,11,11a,12,12a,13,13a-hexadecahydroindeno[5′,6′:4,5]cycloocta[1,2-c]pyran-10-yl-3-hydroxy-3,5-dimethylheptanoate) and M720 ((1S,4S,4aS,6Z,7aS,8aS,10R,11R,11aS,12aS,13aS)-1,13a-dihydroxy-11-isopropyl-4,7,8a-trimethyl-13-methylene-2-oxo-1,2,4,4a,5,7a,8,8a,9,10,11,11a,12,12a,13,13a-hexadecahydroindeno[5′,6′:4,5]cycloocta[1,2-c]pyran-10-ylbutylcarbamate) [Table 1].
Tsukahara et al. [32]. carried out one of the earlier studies for the identification of GPI specific inhibitory drug and its target through the medicinal genetics approach which involves three steps: (a) Identification of a phenotypic marker involved in a pathological condition and developing assay for the same, which in this case was to look out for cell wall localization of GPI-anchored mannoprotein. It is worth mentioning here that the GPI-anchored cell wall mannoproteins act as important virulence markers for the pathogenic yeasts like Candida albicans as these are required for adhesion to the host cells. (b) Screening of small molecules to find out the ones that affect the expected phenotype and (c) Identification of the target. With this objective in the mind, they developed a reporter construct by fusing a bacterial cephalosporinase with Cwp2 (a GPI-anchored cell wall mannoprotein in Saccharomyces cerevisiae). The fusion protein, cephalosporinase-Cwp2 localizes to the yeast cell wall in a GPI dependent manner. As such, assaying the activity of cephalosporinase in the cell wall fraction provided information about the content and localization of GPI-anchored mannoprotein in the yeast cell wall. Screening of the chemical library for the potential GPI inhibitors was then done by assaying for cephalosporinase activity in the cell wall fraction of the strain carrying the reporter construct [32]. Based on this set procedure, BIQ [Table 1] was identified as the GPI specific inhibitory drug with effects ranging from growth defect to reduced target cell adherence in both Saccharomyces cerevisiae and Candida albicans. Fungal GPI specific inositol acyltransferase, Gwt1, was identified as the target protein of BIQ based on the following observations: (a) increase in the copy number of GWT1 or so to say its enhanced expression overcame BIQ induced phenotypes, (b) deletion of GWT1 and BIQ treatment of wild-type yeast cells produced similar phenotypes and (c) point mutations in GWT1 (V397I and G132R) conferred resistance to BIQ [32].
BIQ was found to be metabolically unstable, moderately active against Candida albicans (MIC – 1.56 μg/ml) but inactive against Aspergillus fumigatus [33]. As such, various other compounds were synthesised and screened to look out for the ones with better efficacy and broad spectrum of activity. This led to the development of E1210 [Table 1] [34]. In comparison to clinically available antifungals like azoles, echinocandins and amphotericin B, E1210 showed profound in vitro broad spectrum antifungal activity against a variety of pathogenic yeasts, filamentous fungi, and dermatophytes, notably, Candida spp. (MIC90 of ≤0.008–0.06 μg/ml), Aspergillus fumigatus (MIC90 of 0.13 μg/ml) as well as azole/echinocandin/amphotericin B resistant Candida, Aspergillus, Fusarium and Scedosporium species [34–37]. Further, it was found to be highly active against Pseudallescheria boydii (MIC of 0.03–0.13 μg/ml), Paecilomyces lilacinus (MIC of 0.06 μg/ml) and Scedosporium prolificans (MIC of 0.03 μg/ml) [34]. Not only that, E1210 has shown promise by being active in various in vivo murine models of oropharyngeal candidiasis, disseminated candidiasis, pulmonary aspergillosis and disseminated fusariosis [38,39]. Any newly discovered drug, no matter how much efficacious it would be, is useless if it is not target specific. The first evidence of E1210 target specificity has been provided when it was demonstrated that E1210 inhibits inositol acylation of GlcN-PI by yeast membranes carrying overexpressed Candida albicans or Aspergillus fumigatus Gwt1, which was not so in case of similarly overexpressed human counterpart, PIG-W [40]. Since Gwt1 catalyzes one of the critical steps in the generation of the active GPI precursor, its inhibition significantly impacts the assembly, translocation or maturation of the GPI-anchored proteins. Many of these GPI-anchored proteins play critical virulence roles in the pathogenesis of Candida albicans, such as maintenance of cell wall integrity, adherence to host cell, and yeast to hyphal transition for invasiveness [40]. As expected, E1210 mediated inhibition of fungal Gwt1 leads to reduced cell surface expression of GPI-anchored adhesin, Als1, in Candida albicans [40]. Also, other virulence factors like germ tube formation and biofilm formation have been found to be significantly affected in E1210 treated Candida albicans cells [40]. As E1210 has shown promise both in in vitro and in vivo systems followed by successful toxicological and metabolic studies in experimental animals, it has now entered phase 1 clinical trials in the name of APX001 [41].
Gepinacin (for GPI acylation inhibitor) [Table 1] was identified initially as a false positive compound while screening for heat shock protein inhibitors. Like E1210, it specifically targets fungal Gwt1. The target of gepinacin was identified on the basis of genetic and biochemical evidence as was true for E1210 [42]. Since E1210 and gepinacin share a common target, it is not surprising that both share similarity in effects as well, like reduced target cell adherence, growth defect, reduced switching to the more invasive hyphal form, reduced secretion of lytic enzymes and changes in the architecture of the cell wall, which play critical roles in the disease cycle of pathogenic fungi like Candida albicans. Gepinacin has been found to be potent against a wide range of fungal species including Candida albicans isolates resistant to fluconazole/caspofungin/amphotericin B [42]. What is different, though not surprising, is the finding that gepinacin induces ER stress. ER stress is induced due to a block in the trafficking of GPI-anchored proteins, as has been observed in the case of Gas1 – a GPI-anchored protein in Saccharomyces cerevisiae [42]. Normally, Golgi-processed mature form of Gas1 is predominantly present in the yeast cells relative to the precursor, immature form found in the ER. Gepinacin treatment alters this distribution to the extent that now ER localized immature form constitutes the predominant species [42]. This observation is further supported by the evidence that the trafficking and the maturation of carboxypeptidase Y (CPY) – a non-GPI-anchored protein remained unaltered in the gepinacin treated cells [42]. Further proof that gepinacin specifically alters trafficking of the GPI-anchored proteins is provided by the evidence that ER integral membrane protein, Emp24, which interacts specifically with the GPI-anchored proteins for their ER to Golgi exit, has shown altered localization in gepinacin treated Saccharomyces cerevisiae cells [42]. Consequent to ER stress, there is activation of unfolded protein response pathways in gepinacin treated cells, as monitored in a reporter assay by the enhanced expression of GFP under the control of unfolded protein response element of KAR2 [42]. Another notable finding which has therapeutic ramifications is that gepinacin treatment exposes β-glucans on the cell surface due to thinning out of outer mannoprotein coat mostly contributed by GPI-anchored proteins. This, in turn, enhances immunogenicity due to greater recognition of pathogens, as has been observed by the enhanced secretion of cytokine, TNFα, when gepinacin treated Candida albicans cells were incubated with the mouse macrophage cell line (RAW264.7) [42].
Mann et al. [43] identified G365 and G884 [Table 1] as Gwt1 inhibitors after screening a library of synthetic compounds by Candida albicans fitness test (CaFT). CaFT is based on the idea that the effect of a bioactive compound on its target can be seen prominently in a so called haploid – a condition of haploinsufficiency well known in Saccharomyces cerevisiae. In Candida albicans, a CaFT library of 5400 heterozygous strains was created (i.e. 5400 loci were made heterozygous separately with locus-specific bar codes). After co-culturing of a pooled CaFT library with a given bioactive compound, the relative abundance of each strain, assessed by the locus-specific PCR amplification followed by microarray hybridization, provided information about the target specificity of the compound in question. The authors validated their result genetically through mutational analysis and biochemically by performing acylation assays in the cell-free systems [43]. The mutational analysis revealed that single amino acid substitutions of G132W & F238C in Gwt1 that confer Saccharomyces cerevisiae resistance to G884, have been found to provide cross-resistance to gepinacin as well, with G132R also mapped in BIQ resistant GWT1 mutants. It emphasizes that probably Gwt1 inhibitors share a target binding site with a common mode of action, in spite of the structural differences that exist among them [43]. As is true for gepinacin, effects of both G365 and G884 can be seen in three ways: (a) growth suppression and alteration in the virulence characteristics of the pathogenic fungi as seen by the effectiveness of these inhibitors against Candida albicans and Aspergillus fumigatus (MIC – 4 μg/ml), (b) enhancement of the host immune response by exposure of otherwise masked immunoreactive β-glucans and (c) minimal cytotoxicity observed even at the highest drug concentrations tested in multiple human cell lines of HeLa, HepG2 and THLE-2 [43].
The structural diversity seen in Gwt1 inhibitors ends in Mcd4 inhibitors as all of them identified till date like YW3548, BE49385A and M743 are structurally identical [Table 1] except that they are identified independently by different groups [43–45]. It all started with the discovery of YW3548, a naturally occurring terpenoid lactone, possessing GPI specific inhibitory activity. It causes retention of the GPI-anchored protein, Gas1 in yeast ER without affecting the trafficking of non-GPI-anchored secretory proteins like carboxypeptidase Y (CPY) and invertase as monitored by pulse-chase experiments using [35S] cysteine/methionine as pulse [44]. Since Man2-GlcN-acylPI accumulates majorly in YW3548 treated yeast cells, the authors proposed that YW3548 inhibits addition of third mannose to Man2-GlcN-acylPI GPI precursor. This proposal was, however, proved to be incorrect by Kinoshita and his group, who showed that it is actually the modification of first mannose with phosphoethanolamine that is inhibited by YW3548 [46]. This step is catalysed by Mcd4 and PIG-N respectively in fungi and humans. YW3548 mediated inhibition is not observed in protozoans as the modification of the first mannose with phosphoethanolamine does not occur in them [44,46,47]. However, mammals are least affected by YW3548 mediated inhibition as they can still process Man2-GlcN-acylPI intermediate in the absence of phosphoethanolamine group on first mannose, which is not the case for yeast and related fungi, and as such explains antimycotic activity of YW3548. It was not until the work of Mann et al. [43] that further studies on YW3548 in the name of M743 and M720 were carried out. They identified M743 as Mcd4 inhibitor after screening natural product extracts through CaFT approach as was done in the case of G365 and G884, with M720 as semisynthetic plasma stable analog of M743. Genetic evidence for the target validation was provided when whole genome sequencing of drug-resistant yeast mutants revealed that all the amino acid substitutions of Q679P, G792C, F800L and P810L/Q conferring drug resistance map to MCD4 locus [43]. As is true for Gwt1 inhibitors, Mcd4 inhibitors affect growth, morphogenesis, physiology and virulence of fungi which in addition to other molecular events include induction of ER stress and unmasking of inner β-glucans for better immune recognition. However, it is noteworthy that as compared to G365 and G884, M743 is more potent against a wide range of pathogenic fungi like Candida albicans (MIC – 0.5 μg/mL), Candida parapsilosis (MIC – 0.5 μg/mL), Candida glabrata (MIC – 0.5 μg/mL), Candida krusei (MIC – 1.0 μg/mL), Candida lusitaniae (MIC – 0.25 μg/mL) and Aspergillus fumigatus (MIC – 0.25 μg/mL) as confirmed by spot assays. Besides this, the plasma stable and an equally potent analog of M743, M720 has been found to be effective in the murine model of candidiasis as well [43]. Considerable drug synergy was also observed in both Saccharomyces cerevisiae and Candida albicans by the simultaneous use of Gwt1 and Mcd4 inhibitors like G884 and M720 or G365 and M720. Nevertheless, these desirable effects come with a cost as both M743 and M720 showed considerable cytotoxicity in multiple human cell lines of HeLa, HepG2, and THLE-2 [43].
All in all, considering the therapeutic significance of GPI-specific antimycotic drugs, it is only a matter of time before one sees them replacing conventional antifungals like azoles, polyenes and echinocandins that are currently in clinical use.
Conclusion
Although the core structure of GPI is conserved, there are differences in the timing of acylation/deacylation, lipid chains anchoring GPI into the lipid bilayer, modification of hexoses etc. Besides this, differences have been observed in the enzymatic mechanism and substrate requirement. Moreover, GPI anchoring in different organisms has a different level of significance, per se. It is essential for viability in yeasts and protozoa, but not in mammals. That means one can specifically target the GPI pathway of one organism without affecting others as has been proved till now. Also, targeting a conserved pathway whose end products presumably perform many vital and diverse roles in the living cell may perhaps yield better results. The species-specific differences in the GPI pathway of pathogens and their mammalian hosts at the enzyme level have been exploited for the development of antifungal GPI inhibitors as described in the review. For any compound to qualify as a drug, it must be potent, target specific with negligible human toxicity. These antimycotic GPI inhibitors are promising so far as the target specificity is concerned. Further, experimental evidence from multiple human cell lines show that these inhibitors, especially those targeting fungal Gwt1, are least cytotoxic. Toxicokinetic studies of E1210/APX001 in experimental animals have been successful and E1210/APX001 has entered phase 1 clinical trials. The main challenge in front of the scientific community is to improve their potency. It is true that GPI inhibitors are novel and relatively effective, even in situations where conventional antifungals have failed. But, it is also a reality that most, if not all, of these inhibitors possess just fungistatic activity, which perhaps limits their scope. The problem is further compounded by the lack of three dimensional structures of the target enzymes, which would have helped a great deal in the rational drug design to optimise the potency and specificity of the inhibitors. In spite of these challenges that are likely to be resolved in due course of time, the area of specific targeting of fungal pathogens by the development of GPI inhibitors is promising and seems to have a great future.
Disclosure of interest
The authors report no conflicts of interest.
Acknowledgements
We greatly acknowledge Prof. Sneha Sudha Komath as she has been our mentor and source of inspiration. We also thank ACD labs whose free tool has been used for drawing of chemical structures.
References
- [1].Ikezawa H, Yamanegi M, Taguchi R, et al. . Studies on phosphatidylinositol phosphodiesterase (phospholipase C type) of Bacillus cereus. Purification, properties and phosphatase-releasing activity. Biochim Biophys Acta. 1976;450:154–164. [PubMed] [Google Scholar]
- [2].Ferguson MA. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci. 1999;112:2799–2809. [DOI] [PubMed] [Google Scholar]
- [3].Ikezawa H. Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol Pharm Bull. 2002;25:409–417. 10.1248/bpb.25.409 [DOI] [PubMed] [Google Scholar]
- [4].Eisenhaber B, Bork P, Eisenhaber F. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: analysis of protein sequence data from complete genomes. Protein Eng. 2001;14:17–25. 10.1093/protein/14.1.17 [DOI] [PubMed] [Google Scholar]
- [5].Ferguson MAJ, Low MG, Cross GAM. Glycosyl-sn- 1,2-dimyristylphosphatidylinositol is covalently linked to Trypanosoma brucei variant surface glycoprotein. J Biol Chem. 1985;260:14547–14555. [PubMed] [Google Scholar]
- [6].Ferguson MAJ, Williams AF. Cell-surface anchoring of proteins via glycosyl-phosphatidylinositol structures. Annu Rev Biochem. 1988;57:285–320. 10.1146/annurev.bi.57.070188.001441 [DOI] [PubMed] [Google Scholar]
- [7].Morita N, Nakazato H, Okuyama H, et al. . Evidence for a glycosylinositol phospholipid anchored alkaline phosphatase in the aquatic plant Spirodela oligorrhiza. Biochim Biophys Acta. 1996;1290:53–62. 10.1016/0304-4165(95)00185-9 [DOI] [PubMed] [Google Scholar]
- [8].Kobayashi T, Nishizaki R, Ikezawa H. The presence of GPI-linked protein(s) in an archaeobacterium, Sulfolobus acidocaldarius, closely related to eukaryotes. Biochim Biophys Acta. 1997;1334:1–4. [DOI] [PubMed] [Google Scholar]
- [9].Kawagoe K, Kitamura D, Okabe M, et al. . Glycosylphosphatidylinositol-anchor deficient mice: implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:3600–3606. [PubMed] [Google Scholar]
- [10].Leidich SD, Drapp DA, Orlean PA. Conditionally lethal yeast mutant blocked at the first step in glycosyl phosphatidylinositol anchor synthesis. J Biol Chem. 1994;269:10193–10196. [PubMed] [Google Scholar]
- [11].Itzstein MV, Plebanski M, Cooke BM, et al. . Hot, sweet and sticky: the glycobiology of Plasmodium falciparum. Trends Parasitol. 2008;24:210–218. 10.1016/j.pt.2008.02.007 [DOI] [PubMed] [Google Scholar]
- [12].Vats D, Vishwakarma RA, Bhattacharya S, et al. . Reduction of cell surface glycosylphosphatidylinositol conjugates in Entamoeba histolytica by antisense blocking of E. histolytica GlcNAc-phosphatidylinositol deacetylase expression: Effect on cell proliferation, endocytosis, and adhesion to target cells. Infect Immun. 2005;73:8381–8392. 10.1128/IAI.73.12.8381-8392.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Martinez-Lopez R, Monteoliva L, Diez-Orejas R, et al. . The GPI-anchored protein CaEcm33p is required for cell wall integrity, morphogenesis and virulence in Candida albicans. Microbiology. 2004;150:3341–3354. 10.1099/mic.0.27320-0 [DOI] [PubMed] [Google Scholar]
- [14].Chang T, Milne KG, Guther MLS, et al. . Cloning of Trypanosoma brucei and Leishmania major genes encoding the GlcNAc-Phosphatidylinositol de- N -acetylase of glycosylphosphatidylinositol biosynthesis that is essential to the African sleeping sickness parasite. J Biol Chem. 2002;277:50176–50182. 10.1074/jbc.M208374200 [DOI] [PubMed] [Google Scholar]
- [15].Hong Y, Kinoshita T. Trypanosome glycosylphosphatidylinositol biosynthesis. Korean J Parasitol. 2009;47:197–204. 10.3347/kjp.2009.47.3.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sharma DK, Smith TK, Weller CT, et al. . Differences between the trypanosomal and human GlcNAc-PI de-N- acetylases of glycosylphosphatidylinositol membrane anchor biosynthesis. Glycobiology. 1999;9:415–422. 10.1093/glycob/9.4.415 [DOI] [PubMed] [Google Scholar]
- [17].Smith TK, Crossman A, Brimacombe JS, et al. . Chemical validation of GPI biosynthesis as a drug target against African sleeping sickness. EMBO J. 2004;23:4701–4708. 10.1038/sj.emboj.7600456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Smith TK, Crossman A, Borissow CN, et al. . Specificity of GlcNAc-PI de-N-acetylase of GPI biosynthesis and synthesis of parasite-specific suicide substrate inhibitors. EMBO J. 2001;20:3322–3332. 10.1093/emboj/20.13.3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Smith TK, Gerold P, Crossman A, et al. . Substrate specificity of the Plasmodium falciparum glycosylphosphatidylinositol biosynthetic pathway and inhibition by species specific suicide substrates. Biochemistry. 2002;41:12395–12406. [DOI] [PubMed] [Google Scholar]
- [20].Urbaniak MD, Crossman A, Chang T, et al. . The N-acetyl-D-glucosaminylphosphatidylinositol de-N-acetylase of glycosylphosphatidylinositol biosynthesis is a zinc metalloenzyme. J Biol Chem. 2005;280:22831–22838. 10.1074/jbc.M502402200 [DOI] [PubMed] [Google Scholar]
- [21].Abdelwahab NZ, Crossman AT, Sullivan L, et al. . Inhibitors incorporating zinc-binding groups target the GlcNAc-PI de-N-acetylase in Trypanosoma brucei, the causative agent of African sleeping sickness. Chem Biol Drug Des. 2012;79:270–278. 10.1111/j.1747-0285.2011.01300.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Urbaniak MD, Capes AS, Crossman A, et al. . Fragment screening reveals salicylic hydroxamic acid as an inhibitor of Trypanosoma brucei GPI GlcNAc-PI de-N-acetylase. Carbohydr Res. 2014;387:54–58. 10.1016/j.carres.2013.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Brul S, King A, Vander Vaart JM, et al. . The incorporation of mannoproteins in the cell wall of S. cerevisiae and filamentous Ascomycetes. Antonie van Leeuwenhoek. 1997;72:229–237. 10.1023/A:1000429208049 [DOI] [PubMed] [Google Scholar]
- [24].Grimme SJ, Colussi PA, Taron CH, et al. . Deficiencies in the essential Smp3 mannosyltransferase block glycosylphosphatidylinositol assembly and lead to defects in growth and cell wall biogenesis in Candida albicans. Microbiology. 2004;150:3115–3128. 10.1099/mic.0.27254-0 [DOI] [PubMed] [Google Scholar]
- [25].Victoria GS, Kumar P, Komath SS. The Candida albicans homologue of PIG-P, CaGpi19p: gene dosage and role in growth and filamentation. Microbiology. 2010;156:3041–3051. 10.1099/mic.0.039628-0 [DOI] [PubMed] [Google Scholar]
- [26].Victoria GS, Yadav B, Hauhnar L, et al. . Mutual co-regulation between GPI-N-acetylglucosaminyltransferase and ergosterol biosynthesis in Candida albicans. Biochem J. 2012;443:619–625. 10.1042/BJ20120143 [DOI] [PubMed] [Google Scholar]
- [27].Hoyer LL, Scherer S, Shatzman AR, et al. . Candida albicans ALS1: domains related to a Saccharomyces cerevisiae sexual agglutinin separated by a repeating motif. Mol Microbiol. 1995;15:39–54. 10.1111/mmi.1995.15.issue-1 [DOI] [PubMed] [Google Scholar]
- [28].Gaur NK, Klotz SA. Expression, cloning, and characterization of a Candida albicans gene, ALA1, that confers adherence properties upon Saccharomyces cerevisiae for extracellular matrix proteins. Infect Immun. 1997;65:5289–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cormack BP, Ghori N, Falkow S. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science. 1999;285:578–582. 10.1126/science.285.5427.578 [DOI] [PubMed] [Google Scholar]
- [30].Staab JF, Bradway SD, Fidel PL, et al. . Adhesive and mammalian transglutaminase substrate properties of Candida albicans Hwp1. Science. 1999;283:1535–1538. 10.1126/science.283.5407.1535 [DOI] [PubMed] [Google Scholar]
- [31].Liu N, Wang C, Su H, et al. . Strategies in the discovery of novel antifungal scaffolds. Future Med Chem. 2016;8:1435–1454. 10.4155/fmc-2016-0020 [DOI] [PubMed] [Google Scholar]
- [32].Tsukahara K, Hata K, Nakamoto K, et al. . Medicinal genetics approach towards identifying the molecular target of a novel inhibitor of fungal cell wall assembly. Mol Microbiol. 2003;48:1029–1042. 10.1046/j.1365-2958.2003.03481.x [DOI] [PubMed] [Google Scholar]
- [33].Nakamoto K, Tsukada I, Tanaka K, et al. . Synthesis and evaluation of novel antifungal agents-quinoline and pyridine amide derivatives. Bioorg Med Chem Lett. 2010;20:4624–4626. 10.1016/j.bmcl.2010.06.005 [DOI] [PubMed] [Google Scholar]
- [34].Miyazaki M, Horii T, Hata K, et al. . In vitro activity of E1210, a novel antifungal, against clinically important yeasts and molds. Antimicrob Agents Chemother. 2011;55:4652–4658. 10.1128/AAC.00291-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pfaller MA, Hata K, Jones RN, et al. . In vitro activity of a novel broad-spectrum antifungal, E1210, tested against Candida spp. as determined by CLSI broth microdilution method. Diagn Microbiol Infect Dis. 2011;71:167–170. 10.1016/j.diagmicrobio.2011.05.001 [DOI] [PubMed] [Google Scholar]
- [36].Pfaller MA, Duncanson F, Messer SA, et al. . In vitro activity of a novel broad-spectrum antifungal, E1210, tested against Aspergillus spp. determined by CLSI and EUCAST broth microdilution methods. Antimicrob Agents Chemother. 2011;55:5155–5158. 10.1128/AAC.00570-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Castanheira M, Duncanson FP, Diekema DJ, et al. . Activities of E1210 and comparator agents tested by CLSI and EUCAST broth microdilution methods against Fusarium and Scedosporium species identified using molecular methods. Antimicrob Agents Chemother. 2012;56:352–357. 10.1128/AAC.05414-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hata K, Horii T, Miyazaki M, et al. . Efficacy of oral E1210, a new broad-spectrum antifungal with a novel mechanism of action, in murine models of candidiasis, aspergillosis, and fusariosis. Antimicrob Agents Chemother. 2011;55:4543–4551. 10.1128/AAC.00366-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wiederhold NP, Najvar LK, Fothergill AW, et al. . The investigational agent E1210 is effective in treatment of experimental invasive candidiasis caused by resistant Candida albicans. Antimicrob Agents Chemother. 2015;59:690–692. 10.1128/AAC.03944-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Watanabe NA, Miyazaki M, Horii T, et al. . E1210, a new broad-spectrum antifungal, suppresses Candida albicans hyphal growth through inhibition of glycosylphosphatidylinositol biosynthesis. Antimicrob Agents Chemother. 2012;56:960–971. 10.1128/AAC.00731-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Osherov N, Kontoyiannis DP. The anti-Aspergillus drug pipeline: is the glass half full or empty? Med Mycol. 2017;55:118–124. 10.1093/mmy/myw060 [DOI] [PubMed] [Google Scholar]
- [42].Mclellan CA, Whitesell L, King OD, et al. . Inhibiting GPI anchor biosynthesis in fungi stresses the endoplasmic reticulum and enhances immunogenicity. ACS Chem Biol. 2012;7:1520–1528. [DOI] [PubMed] [Google Scholar]
- [43].Mann PA, McLellan CA, Koseoglu S, et al. . Chemical genomics-based antifungal drug discovery: targeting glycosylphosphatidylinositol (GPI) precursor biosynthesis. ACS Infect Dis. 2015;1:59–72. 10.1021/id5000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sütterlin C, Horvath A, Gerold P, et al. . Identification of a species specific inhibitor of glycosylphosphatidylinositol synthesis. EMBO J. 1997;16:6374–6383. 10.1093/emboj/16.21.6374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kushida H, Nakajima S, Uchiyama S, et al. . Antifungal substances BE-49385 and process for their production. U.S. Patent 5, 928, 910 1999
- [46].Hong Y, Maeda Y, Watanabe R, et al. . Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. J Biol Chem. 1999;274:35099–35106. 10.1074/jbc.274.49.35099 [DOI] [PubMed] [Google Scholar]
- [47].McConville MJ, Ferguson MAJ. The structure, biosynthesis and function of glycosylated phosphatidylinositols in the parasitic protozoa and higher eukaryotes. Biochem J. 1993;294:305–324. 10.1042/bj2940305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Orlean P, Menon AK. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res. 2007;48:993–1011. 10.1194/jlr.R700002-JLR200 [DOI] [PubMed] [Google Scholar]
- [49].Fujita M, Kinoshita T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett. 2010;584:1670–1677. 10.1016/j.febslet.2009.10.079 [DOI] [PubMed] [Google Scholar]
- [50].Paulick MG, Bertozzi CR. The glycosylphosphatidylinositol anchor: A complex membrane-anchoring structure for proteins. Biochemistry. 2008;47:6991–7000. 10.1021/bi8006324 [DOI] [PMC free article] [PubMed] [Google Scholar]