

Staphylococcus aureus contains a certain subclass of lipoproteins, the so-called lipoprotein-like lipoproteins (Lpl's), that not only represent Toll-like receptor 2 (TLR2) ligands but are also involved in host cell invasion. Here we addressed the question of which factors contribute to Lpl-mediated invasion of epithelial cells and keratinocytes.
KEYWORDS: Staphylococcus aureus, invasion, Lpl, lipoprotein, TLR2, HEK293, HaCaT, host cell invasion
ABSTRACT
Staphylococcus aureus contains a certain subclass of lipoproteins, the so-called lipoprotein-like lipoproteins (Lpl's), that not only represent Toll-like receptor 2 (TLR2) ligands but are also involved in host cell invasion. Here we addressed the question of which factors contribute to Lpl-mediated invasion of epithelial cells and keratinocytes. For this purpose, we compared the invasiveness of USA300 and its Δlpl mutant under different conditions. In the presence of the matrix proteins IgG, fibrinogen (Fg), and fibronectin (Fn), and of fetal bovine serum (FBS), the invasion ratio was increased in both strains, and always more in USA300 than in its Δlpl mutant. Interestingly, when we compared the invasion of HEK-0 and HEK-TLR2 cells, the cells expressing TLR2 showed a 9-times-higher invasion frequency. When HEK-TLR2 cells were additionally stimulated with a synthetic lipopeptide, Pam3CSK4 (P3C), the invasion frequency was further increased. A potential reason for the positive effect of TLR2 on invasion could be that TLR2 activation by P3C also activates F-actin formation. Here we show that S. aureus invasion depends on a number of factors, on the host side as well as on the bacterial side.
INTRODUCTION
Staphylococcus aureus is an opportunistic Gram-positive human-pathogenic bacterial species that causes serious community-acquired and nosocomial infections (1). S. aureus possesses an arsenal of virulence factors (i.e., adhesins, invasins, enzymes, toxins) that contribute to the pathogenesis of infection, promoting colonization, dissemination, and transmission (2–5). Previous studies have shown that S. aureus has the ability to invade and persist within nonprofessional phagocytic cells (NPPCs), such as epithelial cells (6, 7), endothelial cells (8, 9), osteoblasts (10), and fibroblasts (11, 12). Major invasion factors of S. aureus include the fibronectin binding proteins (FnBPs), which trigger invasion by bridging S. aureus with the host cell receptor integrin α5β1 (6, 13). FnBPs also bind to human Hsp60, thereby contributing to efficient S. aureus internalization by epithelial cells (14). Another invasion factor is the staphylococcal autolysin (Atl) (15), which binds to heat shock cognate protein 70 (Hsc70) and triggers invasion (3). The interaction of extracellular adherence protein (Eap) with an unidentified cellular receptor also prompts S. aureus internalization (5). It is assumed that the basic mechanism for S. aureus internalization by NPPCs is based on the adhesion of the pathogen to the host cell, resulting in signal transduction, tyrosine kinase activity, cytoskeletal rearrangement (16), and, finally, internalization of the bacteria into the host cells.
Recently, the lpl gene cluster has been shown to trigger the invasion of NPPCs, such as keratinocytes and cancer cells, by S. aureus (17, 18). Lpl's (lipoprotein-like lipoproteins) are lipoproteins (Lpp) encoded on a pathogenicity island named νSaα (19). This island is present in most S. aureus strains. However, highly epidemic strains carry a larger number of tandem lpl genes (as many as 10) than other strains (17, 20). The Lpl's are homologous, sharing about 60% similarity. Since the Lpl's are lipoproteins, they also trigger Toll-like receptor 2 (TLR2) signaling (17). The lipidation and maturation of the Lpp is important for TLR2 activation, as evidenced by the fact that the Δlgt mutant (with the gene encoding the diacylglyceryl transferase enzyme deleted), lacking lipidation of pre-Lpp, does not activate TLR2 (21, 22). Among the TLRs, TLR2 has been shown to play a crucial role in host signaling to S. aureus (21, 23). Previous reports have shown that TLR2 activation contributed to bacterial uptake by phagocytic cells through the activation of scavenger receptors (24, 25). However, it remains unclear whether TLR2 affects the invasion of NPPCs by S. aureus and whether Lpl's are involved in the invasion mechanism.
Here we show that the Lpl's play a crucial role in host cell invasion and that activation of the TLR2 receptor enhances the invasion of NPPCs by S. aureus about 10-fold.
RESULTS
S. aureus invades HaCaT cells more frequently at the stationary-growth phase than at the log phase.
S. aureus USA300, its Δlpl mutant, and the complemented mutant USA300Δlpl(pTX30::lpl) were collected at early-log phase (4 h) or stationary phase (16 h) for invasion assays in HaCaT cells. All the strains showed similar generation times (Fig. 1A). Before infection, the bacterial cells were always adjusted to a multiplicity of infection (MOI) of 30. Interestingly, we observed that the frequency of invasion by stationary-phase cells (16 h) was generally higher than that by log-phase cells (4 h) (Fig. 1B). In both cases, the invasion frequency of the Δlpl mutant was lower than that of the parent (3 times lower for the 4-h culture and 2.4 times lower for the 16-h culture). Because of the higher invasion frequency of stationary-phase cells, we used 16-h cultures of S. aureus in all subsequent experiments. In general, it can be said that the lpl cluster increased the invasion frequency in HaCaT cells about 3-fold. Although reports that TLR2 is expressed in HaCaT cells have been published (26, 27), we do not think that TLR2 is functional in this cell line, since we observed no response when these cells were stimulated with Pam3CSK4 (P3C), a synthetic tripalmitoylated lipopeptide that mimics the acylated amino terminus of bacterial lipoproteins, or with whole S. aureus USA300 cells at an MOI of 30 (see Fig. S2 in the supplemental material).
FIG 1.

Effects of bacterial growth phases on invasion. (A) Growth curves of wild-type S. aureus USA300, the Δlpl mutant, and the complemented mutant USA300 Δlpl(pTX30::lpl). The bacteria were precultivated aerobically in TSB overnight at 37°C, inoculated into fresh TSB at an OD578 of 0.1, and cultivated at 37°C for 24 h under constant shaking at 160 rpm. The OD578 of the cultures was monitored over time and was measured every hour during the first 8 h and after 24 h. (B) Effect of the S. aureus growth phase on the invasion of HaCaT cells. A total of 106 HaCaT cells were infected with USA300, its Δlpl mutant, or the complemented mutant USA300Δlpl(pTX30::lpl) at an MOI of 30. The host cells were infected for 1.5 h, followed by lysostaphin treatment for another 1.5 h to kill adherent S. aureus cells. All experiments were performed at least in triplicate in three independent replications. Error bars indicate standard deviations. Statistical significance was calculated by using Student's t test (ns, no statistical difference; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Matrix proteins enhance S. aureus invasion.
We hypothesized that the Lpl interacts directly or indirectly with a receptor of the host cell and that this receptor triggers the invasion. Relative to that with the control, the frequency of invasion was increased ≈3-fold in the presence of immunoglobulin G (IgG), ≈50-fold with fibrinogen (Fg), and ≈100-fold with fibronectin (Fn), but not with laminin (LMN) (Fig. 2A). Among these proteins, the invasion frequency was highest with Fn, which was not unexpected, since Fn is one of the major bridging proteins involved in invasion (6). Slightly higher invasion was achieved with fetal bovine serum (FBS), which caused 203-fold-higher invasion with the parent strain and 124-fold-higher invasion with the Δlpl mutant. In all invasion assays, the parent strain, USA300, was 2- to 10-fold more invasive than its Δlpl mutant, indicating that Lpl's contribute significantly to invasion (Fig. 2A). The results also show that Fg, Fn, and FBS, in particular, generally increased invasion, independently of the presence or absence of Lpl's.
FIG 2.

Enhancement of bacterial invasion by IgG, fibrinogen, fibronectin, and fetal bovine serum. (A) The effects of matrix proteins laminin (LMN), IgG, fibrinogen (Fg), and fibronectin (Fn), and of fetal bovine serum (FBS), on the invasion of HaCaT cells by S. aureus were investigated. A total of 106 HaCaT cells were stimulated either with 10% FBS or with 10 μg/ml of IgG, Fg, or Fn and were then infected with USA300 or its Δlpl mutant at an MOI of 30. The controls (C) were infected with bacteria only. The host cells were infected for 1.5 h, followed by lysostaphin treatment for another 1.5 h to kill adherent S. aureus cells. All experiments were performed at least in triplicate in three independent replications. Error bars indicate standard deviations. Statistical significance was calculated by using 2-way ANOVA (ns, no statistical difference; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (B) Far-Western blotting was performed with different matrix proteins to determine the possible binding partners. A polyacrylamide gel was loaded with 10 μg of BSA, Fg, Fn, IgG, LMN, PLG, or Vn. Proteins were blotted onto a nitrocellulose blotting membrane and were subsequently probed with 100 μg Lpl1. For immunoblotting, an anti-Lpl1 antibody was used as the first antibody, and goat-anti-rabbit IgG was used as the secondary antibody. Detection was performed with a BCIP/NBT solution.
Next, we investigated whether Lpl's can bind to one of the typical matrix proteins. For this purpose, we performed binding studies, using far-Western blotting, with isolated Lpl1 as a representative Lpl. It turned out that Lpl1 bound to Fg, IgG, and LMN, but not to bovine serum albumin (BSA), Fn, or vitronectin (Vn) (Fig. 2B). Interestingly, Lpl1 did not bind to Fn, although Fn generally increased the frequency of invasion. We also carried out far-Western blotting with matrix proteins and the SitC lipoprotein as a control. Although SitC is one of the most abundant lipoproteins in S. aureus (28), it showed no interaction (see Fig. S3 in the supplemental material). The results suggest that Lpl1 might need a bridging molecule present in FBS in order to interact with one as-yet-unknown host receptor.
TLR2 enhances bacterial invasion.
To study the potential role of TLR2 in Lpl-dependent invasion, we used the human embryonic kidney cell line HEK293. HEK293 cells do not express TLR2 and are referred to here as HEK-0 cells. In order to investigate the impact of TLR2 on invasion, we transfected this cell line with a plasmid expressing TLR2; the transfected cells are referred to as HEK-TLR2 cells. The infection frequency with HEK-0 cells was roughly 550 bacteria/105 host cells for USA300 and roughly 100 bacteria/105 host cells for its Δlpl mutant (Fig. 3A). With HEK-TLR2 cells, the situation was completely changed. The frequency of invasion with USA300 was almost 5,000 bacteria/105 host cells, about 9-fold higher than that with HEK-0 cells. Furthermore, there was no difference between the parent and its Δlpl mutant (Fig. 3A). To rule out the possibility that the transfection in HEK-TLR2 cells affects invasion, we also tested HEK cells with a Flag tag vector as a control (HEK-FLAG cells). There was no significant difference in S. aureus invasion frequency between HEK-0 and HEK-FLAG cells (Fig. 3A).
FIG 3.
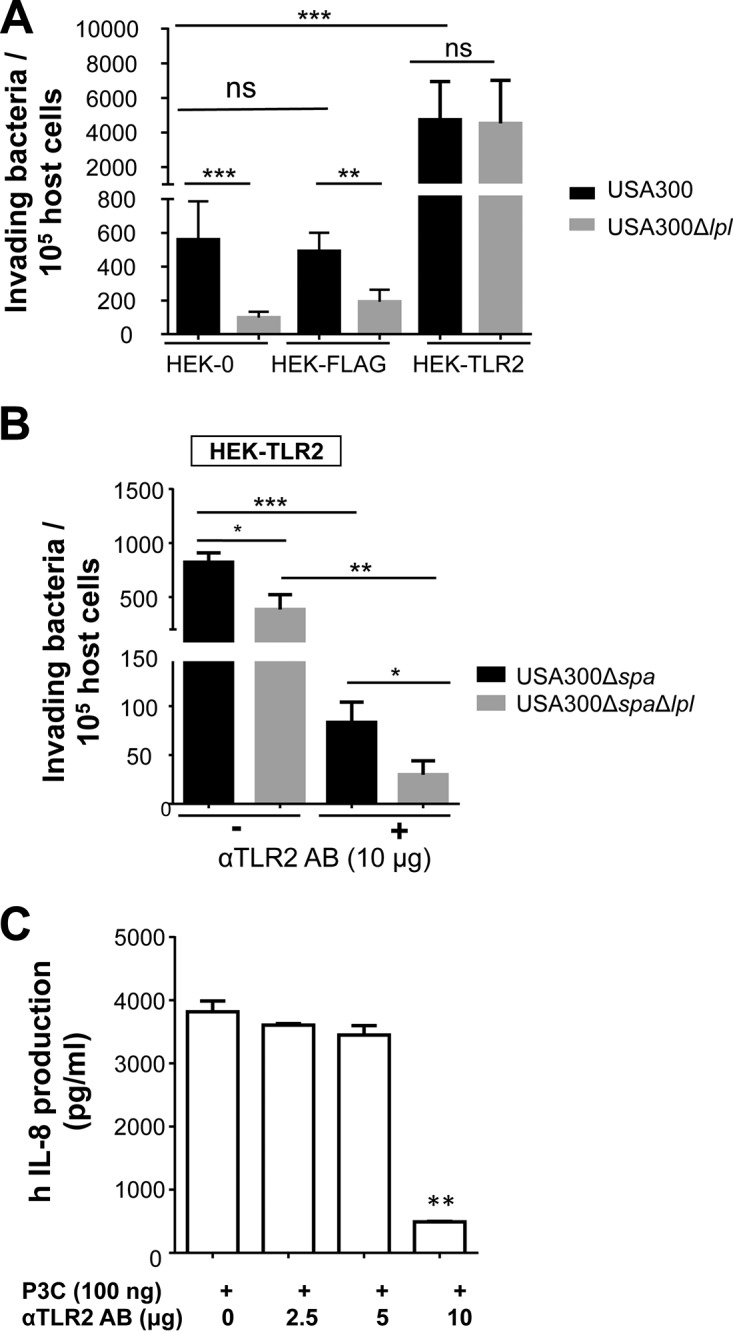
TLR2 enhances S. aureus invasion independently of Lpl's. (A) A total of 106 HEK293 cells either untransfected (HEK-0) or transfected with the human TLR2 gene (HEK-TLR2) or the FLAG tag gene (HEK-FLAG) were infected with USA300 or its Δlpl mutant at an MOI of 30. (B) Additionally, 105 cells were blocked with 10 μg/ml anti-TLR2 antibody (αTLR2 AB). The host cells were infected for 1.5 h, followed by lysostaphin treatment for another 1.5 h to kill adherent S. aureus cells. (C) Detection of IL-8 levels in HEK-TLR2 cells after incubation with anti-TLR2 antibodies. Before the addition of 100 ng P3C, 105 HEK-TLR2 cells were incubated for 1 h at 37°C with function-blocking antibodies (2.5 μg, 5.0 μg, and 10.0 μg) directed against TLR2. IL-8 levels were detected after 18 h of stimulation. All experiments were performed at least in triplicate in three independent replications. Error bars indicate standard deviations. Statistical significance was calculated by using Student's t test (ns, no statistical difference; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
This means that in the absence of TLR2 (HEK-0 cells), Lpl's contribute significantly to host cell invasion, while in HEK-TLR2 cells, invasion is generally considerably increased and the effect of Lpl's is abrogated. Since there was no difference in invasion between USA300 and its Δlpl mutant in HEK-TLR2 cells, it is unlikely that TLR2 is the receptor for Lpl's.
To further investigate the role of TLR2 in cellular invasion, we blocked TLR2 function by preincubating the cells with 10 μg anti-TLR2 antibody. Indeed, invasiveness was roughly 9-fold decreased in the presence of the antibody (Fig. 3B). To avoid any effect of the interaction of the antibody with protein A, we used a mutant in which the gene encoding staphylococcal protein A (SpA) was deleted (USA300Δspa) and the corresponding double mutant (USA300ΔspaΔlpl) for this experiment. The anti-TLR2 antibody was used at a concentration of 10 μg, because at this concentration, the P3C-triggered production of human interleukin 8 (hIL-8) was significantly inhibited (Fig. 3C); P3C, a TLR2 ligand, is a synthetic lipopeptide that mimics the acylated amino terminus of bacterial lipoproteins. In the spa knockout background, both USA300 and USA300Δlpl showed 9-fold-reduced invasion frequencies in HEK-TLR2 cells, suggesting that SpA also plays a role in S. aureus invasion (Fig. 3B and C).
Activation of TLR2 with P3C enhanced bacterial invasion.
Next, we investigated whether 10 ng IL-8, a proinflammatory cytokine and product of TLR2 stimulation, can influence bacterial invasion. As shown in Fig. 4A, IL-8 had no impact on the bacterial invasion of HEK-0 cells. However, when we stimulated HEK-TLR2 cells with the TLR2 agonist P3C, the frequency of invasion by USA300 was about 2-fold increased, while invasion by USA300Δlpl was not triggered by P3C (Fig. 4B). This result indicates that Lpl-mediated invasion is triggered by activation of TLR2.
FIG 4.
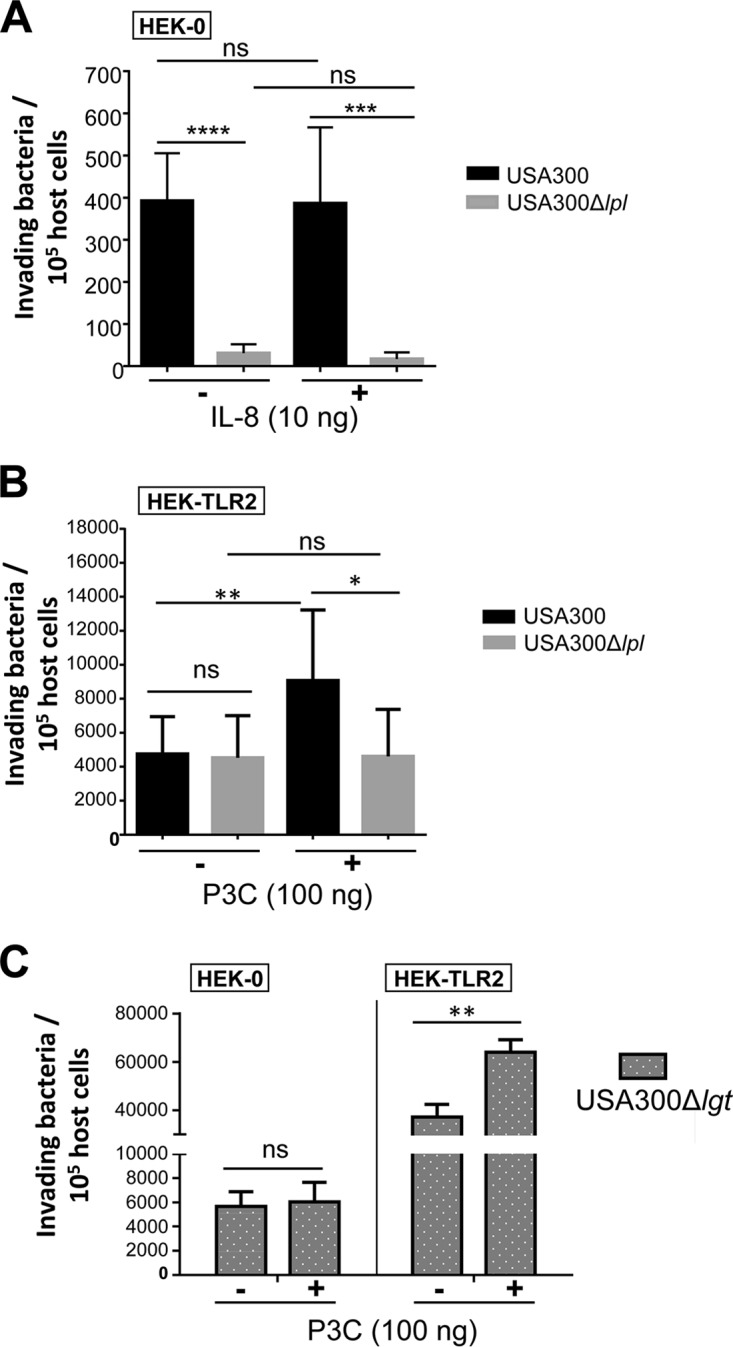
TLR2 activation by triacylated synthetic peptides (P3C) enhanced S. aureus invasion. (A) Rate of invasion of HEK-0 cells by S. aureus with IL-8 stimulation. A total of 106 HEK-0 cells were stimulated with 10 ng IL-8 for 45 min before infection with USA300 or its Δlpl mutant at an MOI of 30 and the performance of the bacterial invasion assay. (B) Rate of invasion of HEK-TLR2 cells by S. aureus with P3C stimulation. Before infection with USA300 or its Δlpl mutant, 106 HEK-TLR2 cells were stimulated with 100 ng P3C. (C) Rate of invasion of HEK-0 and HEK-TLR2 cells by the S. aureus Δlgt mutant. A total of 106 HEK-0 or HEK-TLR2 cells were infected with USA300Δlgt at an MOI of 30 with or without supplementation with 100 ng of P3C. The host cells were infected for 1.5 h, followed by lysostaphin treatment for another 1.5 h to kill adherent S. aureus cells. All experiments were performed at least in triplicate in three independent replications. Error bars indicate standard deviations. Statistical significance was calculated by using Student's t test (ns, no statistical difference; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Since USA300 contains 67 lipoproteins containing the lipid moiety, which is the TLR2 ligand, we next carried out invasion assays with USA300Δlgt in HEK-TLR2 cells to investigate the effect of P3C. As demonstrated previously, the Δlgt mutant is unable to lipidate prolipoproteins and therefore is not responsive to TLR2 (21, 29). As shown in Fig. 4C, P3C enhanced the invasion of HEK-TLR2 cells, but not HEK-0 cells, by USA300Δlgt. However, USA300Δlgt showed an invasion frequency approximately 10-fold higher than that of wild-type USA300 (Fig. 4B and C).
Activation of TLR2 by P3C enhanced F-actin formation.
The question is how P3C-triggered TLR2 activation could enhance S. aureus invasion. One possibility is that actin polymerization was involved. Therefore, we investigated the effect of TLR2 activation on actin polymerization or filaments (F-actin). HEK-0 and HEK-TLR2 cells were stimulated with P3C or IL-8 for 2 h prior to determination of the amount of F-actin formation. Indeed, P3C significantly induced F-actin formation in HEK-TLR2 cells, but not in HEK-0 cells (Fig. 5A and B), suggesting that TLR2 activation by P3C also activates F-actin formation. IL-8 showed no effect. According to this result, F-actin activation may benefit USA300 with respect to invasion.
FIG 5.
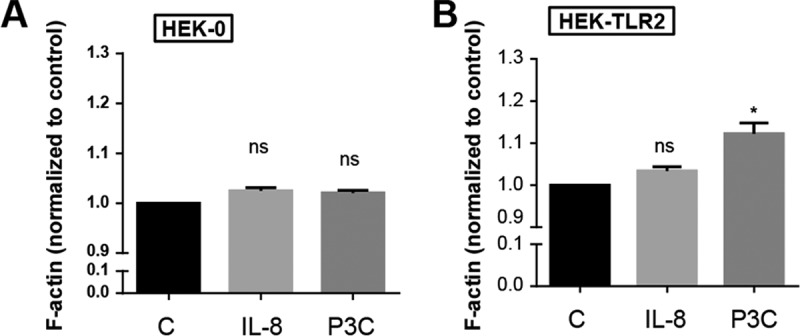
TLR2 activation by triacylated synthetic peptides (P3C) caused F-actin formation. A 384-well black microtiter plate was seeded with HEK-0 cells (A) or HEK-TLR2 cells (B) for 24 h prior to incubation with 10 ng of IL-8 or P3C for 2 h. F-actin was stained with ActinGreen 488 ReadyProbes reagent (Thermo Fisher). The amount of F-actin formation in treated cells was determined by measurement at 495 nm for excitation and 518 nm for emission, with measurements normalized to those for the untreated control (C). The experiments we carried out in triplicate in two independent replications. Statistical significance was calculated by using Student's t test (*, P < 0.05; ns, no significance).
DISCUSSION
S. aureus contains around 55 to 70 Lpp, depending on the strain (20). Approximately 40% of the Lpp are involved in the transport of ions and other nutrients as part of ABC transporters. However, there is a small class of Lpp, the so-called lipoprotein-like lipoproteins (Lpl's), that are encoded in tandem on the νSAα pathogenicity island (19). Previous results have shown that Lpl's contribute to the invasion of keratinocytes and cancer cells by S. aureus (17, 18). Lpl's also act as cell cycle modulins by delaying the G2/M phase transition in HeLa cells (18). It appears that the G2 phase delay is used by S. aureus for invasion and propagation within the host (30).
Host cell invasion is considered one of the most critical factors in the development of chronic infections and long-term persistence, because intracellular bacteria can escape antibiotic treatment as well as host immune clearance (31). Therefore, it is important to analyze the factors that play important roles in bacterial invasion. To understand which factors contribute to Lpl-mediated invasion of epithelial cells and keratinocytes, our model strains were the community-acquired methicillin-resistant S. aureus (CA-MRSA) strain USA300, its Δlpl mutant, in which the entire lpl operon was deleted, and the complemented mutant USAΔlpl(pTX30-lpl) (17). In HaCaT cells, we always observed a significantly lower invasion frequency for the Δlpl mutant than for the parent strain, USA300. Our results also show that different growth phases of S. aureus influence the invasion ratio. These findings can be explained by the diverse expression levels of surface-associated or secreted proteins at different stages, such as log phase or stationary phase (32). We observed a 100-fold-higher S. aureus invasion frequency at stationary phase than at the early-exponential phase, suggesting that more adhesion and invasion factors were expressed in the later growth phase. Indeed, some excreted cytoplasmic proteins (ECP), such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and adolase (FbaA), which enhance bacterial adhesion to epithelial cells, are released at higher levels in the stationary phase (33, 34). More recently, it was found that staphylococcal lipases enhanced S. aureus invasion of keratinocytes and that the lipases were expressed at higher levels in the late-stationary phase (35). The invasion rate of 4-h-cultured S. aureus cells was extremely low (around 10 bacteria per 105 HaCaT cells). We think it is interesting that the bacterial growth phase affects the invasion rate. There are reports of studies in which S. aureus was grown to early-exponential phase (optical density at 600 nm [OD600], 0.3); however, in those publications, a much higher MOI of 100 was used (36). We thought that the use of an MOI of only 30 reflected in vivo conditions more accurately.
In order to investigate the contribution of TLR2 to host cell invasion, we compared the invasion of HEK-0 cells with that of HEK-TLR2 cells (Fig. 3A). Surprisingly, in HEK-TLR2 cells, the rate of invasion was roughly 10-fold higher, indicating that TLR2 contributes to invasion. To confirm this result, we blocked TLR2 with specific anti-TLR2 antibodies. To rule out any interaction of staphylococcal protein A (SpA), an IgG binding protein, with the antibody, we used a spa deletion mutant (USA300Δspa) as the parental strain and the double mutant USA300ΔspaΔlpl. Indeed, in the presence of the blocking antibody, the invasion rate was significantly decreased, suggesting that TLR2 really contributes to invasion (Fig. 3B). However, we also observed that the invasion rate of the spa mutant, USA300Δspa, was roughly 4 times lower than that of USA300 (Fig. 3A and B). This result suggests that SpA also contributes to invasion, in accordance with the earlier observation that SpA mediates invasion across airway epithelial cells through the activation of RhoA GTPase signaling and proteolytic activity (37).
Another question that we addressed was whether the stimulation of TLR2 by P3C increased the invasion rate in HEK-TLR2 cells. Indeed, we saw an increased invasion rate for USA300 but not for USA300Δlpl (Fig. 4B). Then we carried out the same experiment with USA300Δlgt. The lgt mutant is unable to lipidate prolipoproteins and therefore cannot activate TLR2 (21). In HEK-0 cells, the presence or absence of P3C made no difference in invasion. However, in HEK-TLR2 cells, we saw two effects: (i) the invasion rate was generally roughly 8 times higher than that in HEK-0 cells, and (ii) in the presence of P3C, the invasion rate was boosted another 3 times (Fig. 4B). Another observation was that USA300Δlgt was almost 10 times more invasive in HEK-0 cells than was the parent, USA300 (Fig. 4A and C). We had already observed this effect earlier (21). An explanation is that in the lgt mutant, the lipoproteins are not tightly anchored in the membrane and are easily released into the supernatant, probably together with other microbe-associated molecular patterns (MAMPs), such as DNA and RNA. Although the lgt mutant cannot stimulate TLR2, it may stimulate DNA and RNA receptors. Nevertheless, the increased invasion rate for HEK-TLR2 cells indicates that TLR2 plays a crucial role in invasion, which is further boosted by TLR2 activation. It has been shown previously that diacylated lipopeptides, such as FSL-1 or P2C, enhance phagocytosis through TLR2 activation in macrophages (25, 38). While it was shown that TLR2 contributes to the invasion of macrophages, nothing is known about the nonprofessional phagocytes. Here we demonstrate that TLR2 also enhances the invasion of epithelial cell lines and that the triacylated synthetic lipopeptide P3C boosts uptake.
There are some reports that TLR2 is an important factor for the progress of phagocytosis in macrophages, microglia, and neutrophils (39–41). TLR2 was expressed at higher levels in mast cells infected with S. aureus than in uninfected cells (42). Furthermore, activation of murine keratinocytes by the lipoprotein SitC triggered massive intracellular accumulation of TLR2 (43). The question is how TLR2 contributes to host cell invasion. It could be that TLR2 activates the Rac pathway, which subsequently stimulates actin polymerization (44, 45). It is known that F-actin polymerization plays an active role in bacterial entry into host cells. If host cells are treated with cytochalasins, inhibitors of F-actin formation, invasion by Salmonella is inhibited (46). The invasion-associated Salmonella protein SipA forms a complex with T-plastin that requires the presence of F-actin to induce membrane ruffling and bacterial internalization (47). Fibronectin also activates F-actin polymerization to form fibronectin fibrils, suggesting that Fn-mediated invasion depends on cytoskeleton rearrangement (48, 49). Therefore, we investigated whether the stimulation of TLR2 by P3C affects F-actin formation (Fig. 5). Indeed, we found that P3C slightly stimulated F-actin formation in HEK-TLR2 cells, suggesting that the positive effect of TLR2 on invasion is, at least in part, based on F-actin formation.
We observed that TLR2 boosts invasion by S. aureus USA300 but that at the same time, IL-8 is produced by HEK-TLR2 cells. IL-8 is the major cytokine, besides IL-6 and tumor necrosis factor alpha (TNF-α), that is produced in the HEK-TLR2 cell line; IL-2, IL-4, IL-10, IL-12(p70), gamma interferon, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor are not secreted above baseline levels (50–52). Therefore, we tested whether IL-8 alone has an impact on invasion. We found that IL-8 alone has no effect, supporting the finding that it is the activation of TLR2 and downstream effects that contribute to invasion. It has been shown that IL-8 can induce neutrophil adhesion (53); however, in murine macrophages, phagocytosis of Listeria monocytogenes was induced through activation of phosphatidylinositol 3-kinase (PI3K) and Rac1, not by cytokine release (54). It was also observed that the invasiveness of S. aureus strongly depends on the type of host cells, such as epithelial and endothelial cells, keratinocytes, fibroblasts, or osteoblasts (55), since Fn and TLR2 are expressed differently in different host cell types.
Conclusion.
In this study, we show that bacterial invasion depends on both the host cells and the bacterial strains used. Fibronectin is still a major factor that can boost S. aureus invasion almost 100-fold. However, the other factor is TLR2 activation, which also boosts invasion by S. aureus, most likely by activation of F-actin polymerization. Lpl's also play a crucial role in invasion by S. aureus, besides well-known factors such as Fn, Alt, Spa, and Eap. With this study, we shed some light on the complex system of bacterial invasion of host cells.
MATERIALS AND METHODS
Bacterial strains and human cell lines.
The bacterial strains used in this study are listed in Table S1 in the supplemental material. Bacteria were aerobically grown at 37°C in tryptic soy broth (TSB). When appropriate, the media were supplemented with tetracycline (25 μg/ml; Carl Roth, Karlsruhe, Germany). The deletion of the tandem lipoprotein cluster (comprising 10 lpl genes) by allelic replacement in S. aureus USA300 and the construction of the complementing plasmid pTX30::lpl, expressing all 10 lpl genes under the control of the xylose-inducible promoter, have been described previously (17). HaCaT cells (a human keratinocyte line) were obtained from the Department of Dermatology at the University of Tübingen and were cultured in Dulbecco's modified Eagle medium (DMEM) (Thermo Fisher, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Biochrom AG, Berlin, Germany) and 1% penicillin-streptomycin (Thermo Fisher, Waltham, MA, USA). HEK293, a human embryonic kidney cell line, is referred to here as HEK-0 when cultured in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin. HEK293 cells with the transfected human TLR2 gene are referred to here as HEK-TLR2 cells and were cultured in DMEM supplemented with 10% FBS, 10 μg/ml Normocin (InvivoGen, San Diego, CA, USA), and 10 μg/ml blasticidin (InvivoGen, San Diego, CA, USA). As a control, we used HEK293 cells with the transfected FLAG tag (HEK-FLAG cells); they were cultured in DMEM supplemented with 10% FBS (Biochrom AG, Berlin, Germany), 1% penicillin-streptomycin (Thermo Fisher, Waltham, MA, USA), and 0.8% G418-BC (Biochrom AG, Berlin, Germany). The cells were cultured at 37°C with 5% CO2 supplementation.
Growth curves.
S. aureus USA300, its Δlpl mutant, and the complemented mutant USA300Δlpl(pTX30::lpl) were first precultivated aerobically in TSB overnight at 37°C; this culture was used to inoculate fresh TSB (optical density at 578 nm [OD578], 0.1), which was cultivated at 37°C for 24 h under constant shaking at 160 rpm. The OD578 of the cultures was monitored over time and was measured every hour during the first 8 h and after 24 h.
Invasion assay.
For bacterial infection, 2.5 × 105 HaCaT cells were seeded into 24-well plates and were incubated at 37°C under 5% CO2 for 48 h. For HEK, HEK-FLAG, and HEK-TLR2 cells, 5 × 105 cells were incubated for 24 h. The human cells were washed twice with DMEM without any supplements prior to incubation with USA300, its Δlpl mutant, or the complemented mutant USA300Δlpl(pTX30::lpl) at an MOI of 30. For bacterial preparation, the bacterial cells were cultured aerobically for 4 h (exponential phase) or 16 h (stationary phase) in TSB and were washed twice with phosphate-buffered saline (PBS). For stimulation, bacterial pellets were resuspended in DMEM–F-12 medium and were incubated with host cells for 1.5 h. In some specific cases, invasion was accompanied by other substrates as indicated in Results and in the figure legends. For the detection of invading bacteria, cells were treated with 2.5 μg/ml lysostaphin (Sigma-Aldrich, Taufkirchen, Germany) for an additional 1.5 h to remove extracellular bacteria. The host cells were lysed with 0.1% Triton X-100, 0.5% trypsin, and 0.3 mg/ml DNase in PBS. Portions (10 μl) of several dilutions were dropped onto agar plates and were incubated overnight at 37°C. The numbers of internalized bacteria were determined based on the calculation of bacterial colonies grown on the agar plates.
Purification of Lpl1(−sp)-His.
Lpl1(−sp)-His, a lipoprotein without the lipid moiety, was isolated from the cytoplasmic fractions of SA113Δlgt(pTX30::lpl1-his) as described previously (17, 18). Briefly, the clone was first cultivated aerobically at 37°C in the absence of xylose and glucose (BO-medium) until an OD578 of ∼0.5 was reached; then 0.5% xylose was added to induce protein expression, and cultivation was continued for 4 h. The bacterial cells were harvested by centrifugation at 4,000 × g and 4°C. The cell pellets were washed twice with Tris buffer (20 mM Tris, 100 mM HCl [pH 8.0]) and were resuspended in Tris buffer containing a protease inhibitor tablet (Merck, Darmstadt, Germany) and lysostaphin (30 μg/ml; Sigma-Aldrich, Taufkirchen, Germany), followed by 2 h of incubation at 37°C to disrupt the cell wall. After ultracentrifugation (at 235,000 × g for 45 min at 4°C), the cytoplasmic fractions were harvested. For purification, Lpl1-His was incubated with Ni-nitrilotriacetic acid (NTA) Superflow beads (Qiagen, Germany) overnight at 6°C under mild rotation at 20 rpm. One volume of Ni-NTA beads was washed twice with 5 volumes of extraction buffer (Tris buffer containing 0.25% Triton X-100 and 20 mM imidazole); subsequently, the beads were washed four times with 5 volumes of the same buffer containing 40 mM imidazole, and finally, the protein with the His tag was eluted with the same buffer containing 250 mM imidazole. The purified proteins were concentrated by a centrifugal ultrafilter unit with a molecular mass cutoff of 10 kDa (Sartorius AG, Göttingen, Germany). Finally, Lpl1(−sp)-His purification was checked by SDS-PAGE (see Fig. S1 in the supplemental material). SitC-His was purified as described previously (28).
Far-Western blotting studies of the binding of Lpl1 with matrix proteins.
The binding of Lpl1 with different matrix proteins was studied by far-Western blotting as described previously (33). Briefly, a polyacrylamide gel was loaded with 10 μg of each of the following proteins: bovine serum albumin (BSA), fibrinogen (Fg), fibronectin (Fn), immunoglobulin G (IgG), laminin (LMN), plasminogen (PLG), and vitronectin (Vn) (all from Sigma, Taufkirchen, Germany). Proteins were transferred to a nitrocellulose blotting membrane (Bio-Rad, USA) and were incubated with 100 μg purified Lpl1-His. For immunoblotting, an anti-Lpl1 antibody (29) was used as the first antibody, and goat-anti-rabbit IgG (Sigma, Taufkirchen, Germany) was used as the secondary antibody. The reaction was detected with a 5-bromo-4-chloro-3-indolylphosphate (BCIP)/nitroblue tetrazolium (NBT) solution (Sigma, Taufkirchen, Germany) according to the manufacturer′s instructions.
Blocking of HEK-TLR2 cells by a TLR2 antibody.
HEK-TLR2 cells were preincubated with 2.5 μg, 5.0 μg, or 10.0 μg anti-TLR2 antibody (clone TL2.1; eBioscience, Frankfurt, Germany) for 1 h, followed by stimulation with 100 ng/ml P3C (EMC, Tübingen, Germany). After 18 h of incubation, the supernatant was harvested, and human IL-8 was measured using a BD OptEIA enzyme-linked immunosorbent assay (ELISA) kit (BD Biosciences, CA, USA) according to the manufacturer's instructions.
F-actin measurement.
A total of 5 × 104 HEK-0 or HEK-TLR2 cells in 50 μl were seeded into a 384-well black cell culture microplate (Greiner, Germany) for 24 h prior to incubation with 10 ng of the cytokine IL-8 or the synthetic P3C peptide for 2 h. F-actin levels were measured using the ActinGreen 488 ReadyProbes reagent (Thermo Fisher). The treated cells were washed with Dulbecco's PBS (DPBS), permeabilized with 0.25% (vol/vol) Triton X-100, stained with dye for 30 min, and washed twice again with DPBS, and fluorescence was measured at 495 nm for excitation and at 518 nm for emission using a Tecan reader.
Statistical analysis.
Student t tests or analysis of variance (ANOVA) was employed when appropriate to compare the differences between means. All the statistical analysis was performed with GraphPad Prism. The significance level was set as follows: a P value of >0.05 was considered not significant. In figures, significant differences are indicated as described in the legends.
Supplementary Material
ACKNOWLEDGMENTS
We thank Patrick Ebner and Nimerta Kumari for kindly providing USA300Δspa and USA300ΔspaΔlpl. We thank Dorothee Kretschmer and Cordula Gekeler for kindly providing the HEK-FLAG cells.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (grants TR-SFB34 and SFB766).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00343-18.
[This article was published on 23 July 2018 with a byline in which Francesca Barletta's name incorrectly appeared as Francesca Barletta Solari. The byline was updated in the current version, posted on 15 August 2018.]
REFERENCES
- 1.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Alva-Murillo N, López-Meza JE., Ochoa-Zarzosa A. 2014. Nonprofessional phagocytic cell receptors involved in Staphylococcus aureus internalization. Biomed Res Int 2014:538546. doi: 10.1155/2014/538546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, Heilmann C. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol 12:1746–1764. doi: 10.1111/j.1462-5822.2010.01506.x. [DOI] [PubMed] [Google Scholar]
- 4.Zecconi A, Scali F. 2013. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunol Lett 150:12–22. doi: 10.1016/j.imlet.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Haggar A, Hussain M, Lonnies H, Herrmann M, Norrby-Teglund A, Flock JI. 2003. Extracellular adherence protein from Staphylococcus aureus enhances internalization into eukaryotic cells. Infect Immun 71:2310–2317. doi: 10.1128/IAI.71.5.2310-2317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinha B, Francois PP, Nusse O, Foti M, Hartford OM, Vaudaux P, Foster TJ, Lew DP, Herrmann M, Krause KH. 1999. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin α5β1. Cell Microbiol 1:101–117. doi: 10.1046/j.1462-5822.1999.00011.x. [DOI] [PubMed] [Google Scholar]
- 7.Bayles KW, Wesson CA, Liou LE, Fox LK, Bohach GA, Trumble WR. 1998. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect Immun 66:336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menzies BE, Kourteva I. 1998. Internalization of Staphylococcus aureus by endothelial cells induces apoptosis. Infect Immun 66:5994–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogawa SK, Yurberg ER, Hatcher VB, Levitt MA, Lowy FD. 1985. Bacterial adherence to human endothelial cells in vitro. Infect Immun 50:218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Usui A, Murai M, Seki K, Sakurada J, Masuda S. 1992. Conspicuous ingestion of Staphylococcus aureus organisms by murine fibroblasts in vitro. Microbiol Immunol 36:545–550. doi: 10.1111/j.1348-0421.1992.tb02054.x. [DOI] [PubMed] [Google Scholar]
- 11.Ellington JK, Reilly SS, Ramp WK, Smeltzer MS, Kellam JF, Hudson MC. 1999. Mechanisms of Staphylococcus aureus invasion of cultured osteoblasts. Microb Pathog 26:317–323. doi: 10.1006/mpat.1999.0272. [DOI] [PubMed] [Google Scholar]
- 12.Jevon M, Guo C, Ma B, Mordan N, Nair SP, Harris M, Henderson B, Bentley G, Meghji S. 1999. Mechanisms of internalization of Staphylococcus aureus by cultured human osteoblasts. Infect Immun 67:2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha B, Fraunholz M. 2010. Staphylococcus aureus host cell invasion and post-invasion events. Int J Med Microbiol 300:170–175. doi: 10.1016/j.ijmm.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 14.Dziewanowska K, Carson AR, Patti JM, Deobald CF, Bayles KW, Bohach GA. 2000. Staphylococcal fibronectin binding protein interacts with heat shock protein 60 and integrins: role in internalization by epithelial cells. Infect Immun 68:6321–6328. doi: 10.1128/IAI.68.11.6321-6328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Götz F, Heilmann C, Stehle T. 2014. Functional and structural analysis of the major amidase (Atl) in Staphylococcus. Int J Med Microbiol 304:156–163. doi: 10.1016/j.ijmm.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Dziewanowska K, Patti JM, Deobald CF, Bayles KW, Trumble WR, Bohach GA. 1999. Fibronectin binding protein and host cell tyrosine kinase are required for internalization of Staphylococcus aureus by epithelial cells. Infect Immun 67:4673–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen MT, Kraft B, Yu W, Demircioglu DD, Hertlein T, Burian M, Schmaler M, Boller K, Bekeredjian-Ding I, Ohlsen K, Schittek B, Götz F. 2015. The νSaα specific lipoprotein like cluster (lpl) of S. aureus USA300 contributes to immune stimulation and invasion in human cells. PLoS Pathog 11:e1004984. doi: 10.1371/journal.ppat.1004984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen MT, Deplanche M, Nega M, Le Loir Y, Peisl L, Götz F, Berkova N. 2016. Staphylococcus aureus Lpl lipoproteins delay G2/M phase transition in HeLa cells. Front Cell Infect Microbiol 6:201. doi: 10.3389/fcimb.2016.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 20.Shahmirzadi SV, Nguyen MT, Götz F. 2016. Evaluation of Staphylococcus aureus lipoproteins: role in nutritional acquisition and pathogenicity. Front Microbiol 7:1404. doi: 10.3389/fmicb.2016.01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoll H, Dengjel J, Nerz C, Götz F. 2005. Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73:2411–2423. doi: 10.1128/IAI.73.4.2411-2423.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen MT, Götz F. 2016. Lipoproteins of Gram-positive bacteria: key players in the immune response and virulence. Microbiol Mol Biol Rev 80:891–903. doi: 10.1128/MMBR.00028-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451. doi: 10.1016/S1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 24.Doyle SE, O'Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, Lane TF, Cheng G. 2004. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med 199:81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mae M, Iyori M, Yasuda M, Shamsul HM, Kataoka H, Kiura K, Hasebe A, Totsuka Y, Shibata K. 2007. The diacylated lipopeptide FSL-1 enhances phagocytosis of bacteria by macrophages through a Toll-like receptor 2-mediated signalling pathway. FEMS Immunol Med Microbiol 49:398–409. doi: 10.1111/j.1574-695X.2007.00218.x. [DOI] [PubMed] [Google Scholar]
- 26.Köllisch G, Kalali BN, Voelcker V, Wallich R, Behrendt H, Ring J, Bauer S, Jakob T, Mempel M, Ollert M. 2005. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 114:531–541. doi: 10.1111/j.1365-2567.2005.02122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pivarcsi A, Koreck A, Bodai L, Szell M, Szeg C, Belso N, Kenderessy-Szabo A, Bata-Csorgo Z, Dobozy A, Kemeny L. 2004. Differentiation-regulated expression of Toll-like receptors 2 and 4 in HaCaT keratinocytes. Arch Dermatol Res 296:120–124. doi: 10.1007/s00403-004-0475-2. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen MT, Uebele J, Kumari N, Nakayama H, Peter L, Ticha O, Woischnig AK, Schmaler M, Khanna N, Dohmae N, Lee BL, Bekeredjian-Ding I, Götz F. 2017. Lipid moieties on lipoproteins of commensal and non-commensal staphylococci induce differential immune responses. Nat Commun 8:2246. doi: 10.1038/s41467-017-02234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen MT, Hanzelmann D, Härtner T, Peschel A, Götz F. 2016. Skin-specific unsaturated fatty acids boost the Staphylococcus aureus innate immune response. Infect Immun 84:205–215. doi: 10.1128/IAI.00822-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alekseeva L, Rault L, Almeida S, Legembre P, Edmond V, Azevedo V, Miyoshi A, Even S, Taieb F, Arlot-Bonnemains Y, Le Loir Y, Berkova N. 2013. Staphylococcus aureus-induced G2/M phase transition delay in host epithelial cells increases bacterial infective efficiency. PLoS One 8:e63279. doi: 10.1371/journal.pone.0063279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wright JA, Nair SP. 2010. Interaction of staphylococci with bone. Int J Med Microbiol 300:193–204. doi: 10.1016/j.ijmm.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohler C, Wolff S, Albrecht D, Fuchs S, Becher D, Buttner K, Engelmann S, Hecker M. 2005. Proteome analyses of Staphylococcus aureus in growing and non-growing cells: a physiological approach. Int J Med Microbiol 295:547–565. doi: 10.1016/j.ijmm.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Ebner P, Rinker J, Götz F. 2016. Excretion of cytoplasmic proteins in Staphylococcus is most likely not due to cell lysis. Curr Genet 62:19–23. doi: 10.1007/s00294-015-0504-z. [DOI] [PubMed] [Google Scholar]
- 34.Ebner P, Prax M, Nega M, Koch I, Dube L, Yu W, Rinker J, Popella P, Flötenmeyer M, Götz F. 2015. Excretion of cytoplasmic proteins (ECP) in Staphylococcus aureus. Mol Microbiol 97:775–789. doi: 10.1111/mmi.13065. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen MT, Luqman A, Bitschar K, Hertlein T, Dick J, Ohlsen K, Bröker B, Schittek B, Götz F. 2017. Staphylococcal (phospho)lipases promote biofilm formation and host cell invasion. Int J Med Microbiol doi: 10.1016/j.ijmm.2017.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Ridley RA, Douglas I, Whawell SA. 2012. Differential adhesion and invasion by Staphylococcus aureus of epithelial cells derived from different anatomical sites. J Med Microbiol 61:1654–1661. doi: 10.1099/jmm.0.049650-0. [DOI] [PubMed] [Google Scholar]
- 37.Soong G, Martin FJ, Chun J, Cohen TS, Ahn DS, Prince A. 2011. Staphylococcus aureus protein A mediates invasion across airway epithelial cells through activation of RhoA GTPase signaling and proteolytic activity. J Biol Chem 286:35891–35898. doi: 10.1074/jbc.M111.295386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah P, Fatehchand K, Patel H, Fang H, Justiniano SE, Mo X, Jarjoura D, Tridandapani S, Butchar JP. 2013. Toll-like receptor 2 ligands regulate monocyte Fcγ receptor expression and function. J Biol Chem 288:12345–12352. doi: 10.1074/jbc.M113.449983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Assinger A, Laky M, Schabbauer G, Hirschl AM, Buchberger E, Binder BR, Volf I. 2011. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J Thromb Haemost 9:799–809. doi: 10.1111/j.1538-7836.2011.04193.x. [DOI] [PubMed] [Google Scholar]
- 40.Fang L, Wu HM, Ding PS, Liu RY. 2014. TLR2 mediates phagocytosis and autophagy through JNK signaling pathway in Staphylococcus aureus-stimulated RAW264.7 cells. Cell Signal 26:806–814. doi: 10.1016/j.cellsig.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 41.Ribes S, Ebert S, Regen T, Agarwal A, Tauber SC, Czesnik D, Spreer A, Bunkowski S, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R. 2010. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect Immun 78:865–871. doi: 10.1128/IAI.01110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rocha-de-Souza CM, Berent-Maoz B, Mankuta D, Moses AE, Levi-Schaffer F. 2008. Human mast cell activation by Staphylococcus aureus: interleukin-8 and tumor necrosis factor alpha release and the role of Toll-like receptor 2 and CD48 molecules. Infect Immun 76:4489–4497. doi: 10.1128/IAI.00270-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Müller P, Müller-Anstett M, Wagener J, Gao Q, Kaesler S, Schaller M, Biedermann T, Götz F. 2010. The Staphylococcus aureus lipoprotein SitC colocalizes with Toll-like receptor 2 (TLR2) in murine keratinocytes and elicits intracellular TLR2 accumulation. Infect Immun 78:4243–4250. doi: 10.1128/IAI.00538-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Machesky LM, Hall A. 1997. Role of actin polymerization and adhesion to extracellular matrix in Rac- and Rho-induced cytoskeletal reorganization. J Cell Biol 138:913–926. doi: 10.1083/jcb.138.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. 2000. Toll-like receptor 2-mediated NF-κB activation requires a Rac1-dependent pathway. Nat Immunol 1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 46.Finlay BB, Falkow S. 1988. Comparison of the invasion strategies used by Salmonella cholerae-suis, Shigella flexneri and Yersinia enterocolitica to enter cultured animal cells: endosome acidification is not required for bacterial invasion or intracellular replication. Biochimie 70:1089–1099. doi: 10.1016/0300-9084(88)90271-4. [DOI] [PubMed] [Google Scholar]
- 47.Zhou D, Mooseker MS, Galan JE. 1999. An invasion-associated Salmonella protein modulates the actin-bundling activity of plastin. Proc Natl Acad Sci U S A 96:10176–10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pankov R, Cukierman E, Katz BZ, Matsumoto K, Lin DC, Lin S, Hahn C, Yamada KM. 2000. Integrin dynamics and matrix assembly: tensin-dependent translocation of α5β1 integrins promotes early fibronectin fibrillogenesis. J Cell Biol 148:1075–1090. doi: 10.1083/jcb.148.5.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohashi T, Kiehart DP, Erickson HP. 2002. Dual labeling of the fibronectin matrix and actin cytoskeleton with green fluorescent protein variants. J Cell Sci 115:1221–1229. [DOI] [PubMed] [Google Scholar]
- 50.Razonable RR, Henault M, Lee LN, Laethem C, Johnston PA, Watson HL, Paya CV. 2005. Secretion of proinflammatory cytokines and chemokines during amphotericin B exposure is mediated by coactivation of toll-like receptors 1 and 2. Antimicrob Agents Chemother 49:1617–1621. doi: 10.1128/AAC.49.4.1617-1621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quevedo-Diaz MA, Song C, Xiong Y, Chen H, Wahl LM, Radulovic S, Medvedev AE. 2010. Involvement of TLR2 and TLR4 in cell responses to Rickettsia akari. J Leukoc Biol 88:675–685. doi: 10.1189/jlb.1009674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohta K, Ishida Y, Fukui A, Mizuta K, Nishi H, Takechi M, Kamata N. 2014. Toll-like receptor (TLR) expression and TLR mediated interleukin-8 production by human submandibular gland epithelial cells. Mol Med Rep 10:2377–2382. doi: 10.3892/mmr.2014.2507. [DOI] [PubMed] [Google Scholar]
- 53.Lomakina EB, Waugh RE. 2006. Dynamics of increased neutrophil adhesion to ICAM-1 after contacting immobilized IL-8. Ann Biomed Eng 34:1553–1563. doi: 10.1007/s10439-006-9172-y. [DOI] [PubMed] [Google Scholar]
- 54.Shen Y, Kawamura I, Nomura T, Tsuchiya K, Hara H, Dewamitta SR, Sakai S, Qu H, Daim S, Yamamoto T, Mitsuyama M. 2010. Toll-like receptor 2- and MyD88-dependent phosphatidylinositol 3-kinase and Rac1 activation facilitates the phagocytosis of Listeria monocytogenes by murine macrophages. Infect Immun 78:2857–2867. doi: 10.1128/IAI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strobel M, Pfortner H, Tuchscherr L, Volker U, Schmidt F, Kramko N, Schnittler HJ, Fraunholz MJ, Löffler B, Peters G, Niemann S. 2016. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin Microbiol Infect 22:799–809. doi: 10.1016/j.cmi.2016.06.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.