

Pathogenic bacteria that establish chronic infections in immunocompromised patients frequently undergo adaptation or selection for traits that are advantageous for their growth and survival. Clinical isolates of Pseudomonas aeruginosa, a Gram-negative, opportunistic bacterial pathogen, exhibit a temporal transition from a motile to a nonmotile phenotype through loss of flagellar motility during the course of chronic infection.
KEYWORDS: Pseudomonas aeruginosa, phagocytosis, PIP3, flagellar motility
ABSTRACT
Pathogenic bacteria that establish chronic infections in immunocompromised patients frequently undergo adaptation or selection for traits that are advantageous for their growth and survival. Clinical isolates of Pseudomonas aeruginosa, a Gram-negative, opportunistic bacterial pathogen, exhibit a temporal transition from a motile to a nonmotile phenotype through loss of flagellar motility during the course of chronic infection. This progressive loss of motility is associated with increased resistance to both antibiotic and immune clearance. We have previously shown that loss of bacterial motility enables P. aeruginosa to evade phagocytic clearance both in vitro and in vivo and fails to activate the phosphatidylinositol 3-kinase (PI3K)/Akt-dependent phagocytic pathway. Therefore, we tested the hypothesis that clearance of phagocytosis-resistant bacteria could be induced by exogenously pretreating innate immune cells with the Akt-activating molecule phosphatidylinositol-(3,4,5)-trisphosphate (PIP3). Here, we demonstrate that PIP3 induces the uptake of nonmotile P. aeruginosa by primary human neutrophils >25-fold, and this effect is phenocopied with the use of murine phagocytes. However, surprisingly, mechanistic studies revealed that the induction of phagocytosis by PIP3 occurs because polyphosphoinositides promote bacterial binding by the phagocytes rather than bypassing the requirement for PI3K. Moreover, this induction was selective since the uptake of other nonmotile Gram-negative, but not Gram-positive, bacteria can also be induced by PIP3. Since there is currently no treatment that effectively eradicates chronic P. aeruginosa infections, these findings provide novel insights into a potential methodology by which to induce clearance of nonmotile pathogenic bacteria and into the endogenous determinants of phagocytic recognition of P. aeruginosa.
INTRODUCTION
Over the course of chronic infections, bacterial pathogens undergo phenotypic and genetic changes that facilitate their persistence (1). One of the established temporal adaptations of bacteria in chronic infections is the transition from a motile to a nonmotile lifestyle (1–3). This is perhaps best described from longitudinal studies of cystic fibrosis (CF) patients with chronic respiratory infections, which revealed that isolates of the motile Gram-negative bacterium Pseudomonas aeruginosa consistently demonstrate progressive loss of flagellar motility due to downregulation or mutation (4, 5). This transition from a motile to nonmotile lifestyle is correlated with increased bacterial burdens and poor prognoses for CF patients (1) and is also observed at other sites of infection, including ocular infections and burn wounds (6–8).
The emergence of nonmotile P. aeruginosa during chronic infection has a profound impact on the ability to eradicate the bacteria. Functional innate immunity mediated by professional phagocytic cells (neutrophils and macrophages) is essential for effective clearance of P. aeruginosa. This is supported by clinical observations that neutropenic patients are very susceptible to P. aeruginosa infections, while immunocompetent individuals, in the absence of immunocompromised tissue, are largely resistant, and this finding has been recapitulated in animal models (9–12). Thus, the previous findings that loss of flagellar swimming motility by P. aeruginosa, regardless of flagellar expression, confers in vitro and in vivo resistance (∼100-fold) to phagocytic clearance provided key insights into how nonmotile bacteria evade intrinsic innate immune defenses (13, 14). Additionally, nonmotile P. aeruginosa achieves antibiotic tolerance that exceeds standard-of-care clinical treatments, particularly upon formation of microcolonies or biofilms (15, 16). Since chronic P. aeruginosa infections cannot currently be eradicated due to their phagocytic and antibiotic resistance, there is an obvious need to identify novel mechanisms and methodologies by which to promote their clearance.
The current studies were initially guided by a previous insight that nonmotile P. aeruginosa fails to activate the mammalian phosphatidylinositol-3-kinase (PI3K)/Akt kinase signaling pathway, which is necessary for phagocytosis of P. aeruginosa (17, 18). The intracellular lipid kinase PI3K is activated following bacterial engagement at the phagocyte cell surface and phosphorylates PI-(4,5)-bisphosphate (PIP2) to PI-(3,4,5)-trisphosphate (PIP3) at the inner leaflet of the plasma membrane. PIP3 recruits and activates the master regulator kinase Akt, which facilitates the downstream signaling that is responsible for actin polymerization and cytoskeletal arrangement, thereby controlling host phagocytic activities (19). Therefore, since the PI3K/Akt pathway is preferentially activated by motile P. aeruginosa and efficiently mediates the clearance of this bacterium, we hypothesized that exogenous activation of this pathway could be a mechanism by which host cells could be induced to phagocytose nonmotile bacteria.
In this study, based on the aforementioned premise, we have identified a strategy to induce uptake of nonmotile P. aeruginosa bacteria by human and murine phagocytic cells using PIP3. Quantitative phagocytosis assays employed to assess the effect of pretreatment of phagocytes with PIP3 revealed that this methodology massively promotes the phagocytosis of nonmotile P. aeruginosa by human and murine phagocytes to a greater extent than other interventions reported. Importantly, and unexpectedly, mechanistic studies identified that while the induction of phagocytosis by PIP3 is dependent upon PI3K/Akt activity, it promotes phagocytosis by increasing bacterial binding by the phagocytes. We provide evidence that PIP3 induces the phagocytic uptake of nonmotile mutants of a variety of Gram-negative bacterial genera; however, there is specificity to this effect since PIP3 is ineffective at promoting the phagocytosis of motile P. aeruginosa and Gram-positive bacteria. These findings provide novel insights into molecular mechanisms that facilitate motility-dependent bacterial interactions with phagocytic cells and identify a methodology by which to induce the clearance of nonmotile bacteria that are present in chronic bacterial infections.
RESULTS
PIP3 induces phagocytosis of nonswimming P. aeruginosa.
We have previously shown that phagocytosis of P. aeruginosa requires PI3K and Akt activities and that motile bacteria preferentially activate the PI3K/Akt pathway to induce phagocytosis (17). Phagocytic uptake of every isolate of P. aeruginosa tested is dependent on PI3K activity, as quantitatively assessed by phagocytic inhibition following pretreatment of cells with LY-294002, a pharmacological inhibitor of PI3K (see Fig. S1A in the supplemental material). Thus, PI3K activity is necessary for uptake of P. aeruginosa, which led us to hypothesize that exogenous activation of this pathway with the use of the PI3K enzymatic product and Akt activator PIP3 could overcome the nonmotile P. aeruginosa evasion of phagocytosis. Therefore, we preincubated murine bone marrow-derived dendritic cells (BMDCs) in the absence or presence of PIP3 and then assessed their relative abilities, in parallel, to phagocytose two nonmotile mutants of PA14 P. aeruginosa, PA14 motAB motCD and flgK strains, of which the former has flagella but lacks motility and the latter lacks both flagella and swimming motility (13). Treatment of phagocytes with PIP3 prominently increased the uptake of both the motAB motCD and flgK bacteria (>10-fold compared to levels in untreated phagocytes), suggesting that the effect may be broadly applicable to nonmotile P. aeruginosa and, specifically, that the effect is not dependent upon flagellar expression (Fig. 1A).
FIG 1.

PIP3 induces uptake of nonmotile P. aeruginosa by BMDCs. Murine WT bone marrow-derived dendritic cells (BMDCs) were assayed for relative phagocytosis of the indicated P. aeruginosa PA14 strains (MOI of 10) without or after treatment of cells with 12 μM PIP3. The data are normalized to phagocytosis of bacterial strains without PIP3 treatment and represented as fold increase in phagocytosis. All data were analyzed using an unpaired t test with Welch's correction and are representative of at least two independent biological experiments (n ≥ 8). ***, P ≤ 0.0005; *, P ≤ 0.05; ns, not significant, for results compared to results without PIP3 treatment.
We then tested the specificity of PIP3 treatment for the phagocytic induction of nonswimming P. aeruginosa with regard to the contribution of two additional bacterial extracellular structures that promote alternative forms of motility and adherence of bacteria to epithelial cells (20–22). Thus, we investigated whether the O-antigen of lipopolysaccharide (LPS) and type IV pili are required for the PIP3 effect on nonmotile P. aeruginosa. To do so, we first employed the PA14 waaL motAB motCD mutant, which is nonmotile, lacks O antigen, and is still resistant to phagocytosis (21). Results of a gentamicin protection assay to quantitatively assess phagocytosis indicated that PIP3 phagocytic induction of nonswimming P. aeruginosa is independent of LPS O antigen since PIP3 significantly increased the uptake of PA14 waaL motAB motCD (Fig. 1B). Second, we assessed induction of phagocytic uptake of type IV pilus-deficient mutants that lack twitching motility but have intact swimming motility and have phagocytic susceptibility equivalent to that of wild-type (WT) bacteria (20, 23). Phagocytosis levels of all mutants tested that lack surface pili (pilA, pilB, and pilG) were comparable in PIP3-treated cells and the untreated groups, which contrasted with high phagocytic induction of nonswimming motAB motCD bacteria (Fig. 1C). Hence, treatment with PIP3 selectively induces the phagocytosis of nonswimming P. aeruginosa. Taken together, these data demonstrate that PIP3 treatment of phagocytes induces a significant uptake of nonmotile P. aeruginosa by BMDCs, independent of O antigen and pilin.
PIP3 induction of nonmotile P. aeruginosa can be competed with motile WT bacteria.
Our data (Fig. 1) support the observation that PIP3 significantly increased the uptake of nonmotile P. aeruginosa; however, the studies employing pilus-deficient bacteria (Fig. 1C) led us to assess the extent to which PIP3 affects the phagocytosis of motile WT PA14 bacteria. Pretreatment with PIP3 was notably effective, reconstituting the phagocytosis of nonmotile P. aeruginosa to ∼40% of the level for WT bacteria (Fig. 2A). As a control, histone, the PIP3 carrier protein, was insufficient to enhance the uptake of nonmotile motAB motCD bacteria and required associated PIP3 for the effect (Fig. 2B, inset). Surprisingly, there was no additional increase in phagocytosis of WT bacteria upon PIP3 treatment in comparison to the >10-fold increase in phagocytosis of nonmotile P. aeruginosa (Fig. 2B). Therefore, PIP3 preferentially induces uptake of nonmotile compared to motile P. aeruginosa.
FIG 2.

PIP3 preferentially induces uptake of nonmotile compared to that of motile P. aeruginosa. (A and B) Murine WT BMDCs were assayed for relative phagocytosis of the PA14 WT or motAB motCD strain (MOI of 10) without or after treatment of cells with 12 μM PIP3. The PIP3 was complexed with a histone carrier which, in itself, was ineffective at inducing phagocytosis (B, inset). (C) Murine BMDCs were pretreated with PIP3 and assayed for relative in vitro phagocytosis of motAB motCD bacteria (carbenicillin resistant) upon addition of increasing ratios of WT PA14 bacteria. For panel A, phagocytic uptake levels were normalized as percentages of mean WT PA14 phagocytosis in untreated cells. For panels B and C, the data were normalized to phagocytosis of PA14 motAB motCD without PIP3 treatment and are represented as fold increase in phagocytosis. All data were analyzed using one-way ANOVA with Tukey's post hoc analyses (A and C) or an unpaired t test with Welch's correction (B) and are representative of at least two independent biological experiments (n ≥8). ***, P ≤ 0.0005; **, P ≤ 0.005; ns, not significant, for results compared to results without PIP3 treatment.
Since phagocytosis of WT bacteria was not enhanced by PIP3 treatment, we tested whether the PIP3 treatment enables association of nonmotile bacteria to different binding sites (compared to sites for the WT) on the phagocytic cells or whether it enables access to the binding sites normally accessed by the WT bacteria. To address this, we performed a competition experiment with constant numbers of nonmotile motAB motCD bacteria (carbenicillin resistant [Carbr]) and increasing ratios of motile WT PA14 bacteria (carbenicillin sensitive), followed by quantitative evaluation of the internalized nonmotile (Carbr) bacteria. Importantly, PIP3-mediated uptake of nonmotile bacteria was dose-dependently competed by the presence of increasing numbers of WT bacteria (Fig. 2C). Therefore, since PIP3 does not induce the uptake of motile P. aeruginosa but does enable nonmotile bacteria to access binding sites that are competed by the presence of motile bacteria, the competition experiment supports the notion that PIP3 pretreatment makes the binding sites responsible for the uptake of motile bacteria accessible to nonmotile bacteria.
PIP3 integration into the phagocyte cell membrane is essential for phagocytic induction of nonmotile P. aeruginosa.
Next, we wanted to determine whether PIP3-mediated induction of phagocytosis was due to effects on the bacteria or the phagocytes. To address this, we first assessed whether PIP3 treatment of the bacteria could recapitulate the phagocytic induction of nonmotile bacteria observed with PIP3-pretreated phagocytes. Therefore, we pretreated either BMDCs or PA14 motAB motCD bacteria with PIP3 before initiating the phagocytosis assay. In contrast with PIP3 treatment of phagocytes, incubation of PIP3 exclusively with the bacteria was not sufficient to mediate phagocytic induction of nonmotile P. aeruginosa (Fig. 3A). This suggests that PIP3 treatment is specific to phagocytes and does not affect the bacteria directly. To extend this line of inquiry, we tested the hypothesis that the PIP3 phagocytic effect requires integration of the phospholipid into the plasma membrane of phagocytes. We coincubated PA14 motAB motCD with the soluble inositol phosphate, inositol 1,3,4,5-tetrakisphosphate (IP4), which has a structure similar to that of PIP3 but lacks acyl tails and does not stably integrate into membranes. In contrast to PIP3, IP4 did not induce the phagocytosis of PA14 motAB motCD and was even a modest competitor of phagocytosis (Fig. 3B). Together, these data demonstrate that PIP3 induction of phagocytosis requires phagocytic cell, rather than bacterial, treatment and integration of the phospholipid into the plasma membrane.
FIG 3.

PIP3 treatment of cells and integration in the plasma membrane are essential for phagocytic induction of nonmotile P. aeruginosa. (A) Murine WT BMDCs were assayed for relative phagocytosis of PA14 motAB motCD (MOI of 10) without or after treatment of bacteria (gray bar) or cells (white bar) with 12 μM PIP3. The data were normalized to phagocytosis of PA14 motAB motCD without PIP3 treatment and are represented as fold increase in phagocytosis. (B) Phagocytic uptake of P. aeruginosa PA14 motAB motCD (MOI of 10) by murine WT BMDCs in the absence or presence of 12 μM inositol 1,3,4,5-tetrakisphosphate (IP4). Phagocytic uptake was normalized as a percentage of the mean phagocytosis of motAB motCD bacteria in untreated cells. (C) The killing rate of PA14 WT and motAB motCD bacteria by BMDCs treated with PIP3 was assayed by a gentamicin protection assay. Following a 45-min incubation of bacteria with BMDCs (MOI of 10), gentamicin was added, and at the indicated time points following gentamicin addition (20, 40, and 60 min), cells were lysed to assess remaining CFU counts. The number of CFU recovered was plotted relative to recovery at 20 min after gentamicin addition. All data were analyzed using one-way ANOVA with Tukey's post hoc analyses (A) or using an unpaired t test with Welch's correction (B) and are representative of at least two independent biological experiments (n ≥8). ***, P ≤ 0.0005; *, P ≤ 0.05; ns, not significant, for results compared to those without PIP3 treatment.
Based on the results shown in Fig. 2 that PIP3 induces the uptake of nonmotile but not motile P. aeruginosa, an important consideration was investigating the relative intracellular killing rates for these bacteria in PIP3-treated BMDCs. Both genotypes of bacteria exhibited similar rates of intracellular killing following ingestion (Fig. 3C); this finding is consistent with previously derived intracellular bacterial killing kinetics (13). These results suggest that PA14 and PA14 motAB motCD are equally susceptible to killing by phagocytes pretreated with PIP3 and that the observed induction of phagocytosis is not an artifact of differential killing kinetics between the bacterial strains.
Polyphosphoinositides (PIP3 and PIP2) specifically elicit efficient phagocytic induction of nonmotile P. aeruginosa.
Based on the induction of phagocytosis by PIP3, we tested the specificity of PIP3 with regard to whether this effect could be recapitulated with other phospholipids. Equivalent molar concentrations of phospholipids with different head groups and identical tails were used in parallel to prime BMDCs, followed by quantitative phagocytosis assays of PA14 motAB motCD. Interestingly, the precursor to PIP3, PIP2, was as effective as PIP3 and elicited similar phagocytic induction of motAB motCD bacteria by BMDCs (Fig. 4A). In contrast, pretreating BMDCs with phosphatidylserine (PS), phosphatidylethanolamine (PE), or phosphatidylcholine (PC) was much less effective than treatment with PIP3 and PIP2 (Fig. 4A and B). These results suggest that polyphosphoinositides are far more potent at inducing the phagocytosis of nonmotile bacteria than the other phospholipids tested (PS, PE, and PC). This led us to test the breadth of the effect utilizing human cells and other genera of bacteria and, in light of the efficacy of PIP2, to define the governing mechanism of polyphosphoinositide-induced phagocytosis, particularly with regard to the role of the PI3K/Akt pathway.
FIG 4.

Phospholipid specificity of phagocytic induction of nonmotile P. aeruginosa. Murine WT BMDCs were assayed for relative phagocytosis of PA14 motAB motCD (MOI of 10) without or after treatment of cells with equivalent molar concentrations (12 μM) of PIP3, PIP2 or PS (A) and PIP3, PC, or PE (B). The data were normalized to phagocytosis of bacterial strains without treatment and are represented as fold increase in phagocytosis. All data were analyzed using one-way ANOVA with Tukey's post hoc analyses and are representative of at least two independent biological experiments (n ≥8). ***, P ≤ 0.0005; ns, not significant, for results compared to results with no treatment.
PIP3 induces uptake of nonmotile P. aeruginosa by primary human phagocytes.
To determine whether the described findings are exclusive to murine BMDCs or represent a widespread mechanism applicable to the human neutrophils and monocytes responsible for clearance of P. aeruginosa infections (9), we pretreated primary human phagocytes with PIP3 and tested their ability to phagocytose nonmotile P. aeruginosa. Human neutrophils and monocytes recapitulated the studies that employed murine cells, such that nonmotile PA14 motAB motCD was phagocytosed to a much greater extent (>10-fold) in PIP3-treated cells (Fig. 5A to C). Importantly, since P. aeruginosa is responsible for high incidences of chronic infections in CF patients, we decided to test whether PIP3 treatment of neutrophils directly derived from these patients would be effective for induction of phagocytosis of nonmotile P. aeruginosa. Indeed, PIP3 pretreatment of blood-derived neutrophils obtained from CF patients similarly promoted the uptake of motAB motCD bacteria (Fig. 5B). In addition to loss of bacterial motility, another conserved phenotypic adaptation observed in chronic CF infections includes the loss of O antigen, frequently described as conversion from smooth to rough LPS. Therefore, we tested a P. aeruginosa nonmotile mutant with rough LPS (PA14 waaL motAB motCD), which resulted in similar and effective phagocytic induction following pretreatment of healthy or CF human neutrophils with PIP3 (Fig. 5D and E). Thus, induction of nonmotile P. aeruginosa by PIP3 is effective regardless of a smooth or rough LPS phenotype. Importantly, these findings demonstrate that PIP3 induces the uptake of nonmotile P. aeruginosa by primary human phagocytes and therefore has broad applicability to phagocytic cells across multiple cell types and species.
FIG 5.

Primary human neutrophils and monocytes are induced by PIP3 to phagocytose nonmotile P. aeruginosa. Primary human neutrophils from healthy donors (A and D), primary human neutrophils from CF patients (B and E), and primary human monocytes from healthy donors (C) were assayed for relative phagocytosis of PA14 motAB motCD and PA14 waaL motAB motCD, as indicated, without or after, treatment of cells with 12 μM PIP3. The data were normalized to phagocytosis of bacterial strains without PIP3 treatment and are represented as fold increase in phagocytosis. An MOI of 10 was used for all experiments. All data were analyzed using an unpaired t test with Welch's correction and are representative of at least two independent biological experiments (for A, C, D, E, n ≥8; for B, n ≥4). ***, P ≤ 0.0005; **, P ≤ 0.005, for results compared to those in the absence of PIP3 treatment.
PIP3 induction of phagocytosis is effective against multiple genera of nonmotile Gram-negative bacteria.
Additional flagellated Gram-negative pathogens besides P. aeruginosa, such as Escherichia coli and Vibrio cholerae, also resist phagocytosis through the loss of swimming motility (14). Therefore, we sought to evaluate whether PIP3 treatment of murine BMDCs would overcome this evasion of phagocytosis by testing multiple nonmotile genera of Gram-negative bacteria. We performed similar assays using nonswimming flagellated V. cholerae (motX) and nonflagellated E. coli K-12 (fliC), genetic mutants that contain orthologous mutations to the P. aeruginosa mutants described previously (14, 24). In assays using V. cholerae and E. coli, priming BMDCs with PIP3 significantly increased the uptake of the bacteria compared to that in the untreated groups (Fig. 6). Moreover, we tested the effect of PIP3 on cells incubated with the opportunistic, nonflagellated Klebsiella pneumoniae (KPPR1) bacteria and observed robust induction of phagocytosis in PIP3-treated cells (Fig. 6). These results support a general mechanism by which PIP3 intervention enables enhanced recognition and uptake of nonmotile Gram-negative bacteria by phagocytic cells.
FIG 6.
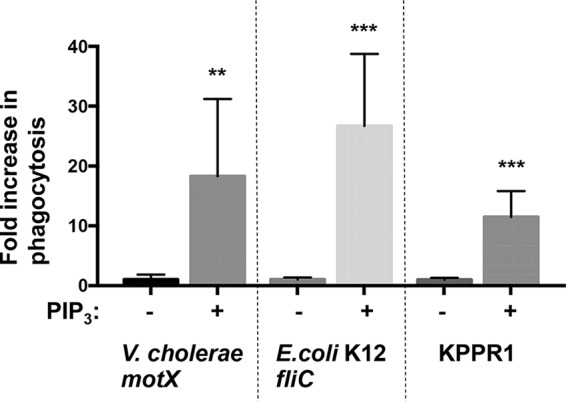
PIP3 broadly induces the phagocytosis of nonmotile Gram-negative pathogens. Murine WT BMDCs were assayed for relative in vitro phagocytosis of V. cholerae motX, K. pneumoniae KPPR1, or E. coli K-12 fliC (MOI of 10) by standard gentamicin protection assay after preincubation in the absence or presence of 12 μM PIP3. The data were normalized to phagocytosis of bacterial strains without PIP3 treatment and are represented as fold increase in phagocytosis. All data were analyzed using an unpaired t test with Welch's correction and are representative of at least two independent biological experiments (n ≥8). ***, P ≤ 0.0005; **, P ≤ 0.005, for results compared to those without PIP3 treatment.
Selectivity of PIP3-induced phagocytosis.
In order to address the selectivity of the PIP3 phagocytic induction, the analyses were expanded to assess Gram-positive, as well as Gram-negative, bacterial pathogens. Phagocytic assays employing two different nonmotile strains of Staphylococcus aureus (USA300 and rn4220) revealed no measurable phagocytic induction of these bacteria upon PIP3 treatment of cells (Fig. 7A). In fact, the treatment groups exhibited slightly lower uptake of both S. aureus bacterial strains than the untreated groups. As a control, we confirmed the dependence of S. aureus on the PI3K/Akt signaling pathway for phagocytosis by inhibiting PI3K with LY-294002 (Fig. S1B). To test whether the resistance to PIP3-induced phagocytosis was specific to S. aureus, we extended these studies to incorporate Bacillus subtilis strain JH642. PIP3-treated cells exhibited lower uptake of B. subtilis than untreated cells (Fig. 7B). Taken together, these data support the notion that PIP3 induction of phagocytosis is selective to Gram-negative bacteria.
FIG 7.

PIP3 selectively induces phagocytosis of nonmotile P. aeruginosa but not S. aureus or B. subtilis. (A and B) Murine WT BMDCs were assayed for relative in vitro phagocytosis of S. aureus USA300 and rn4220 (MOI of 1) or B. subtilis JH642 (MOI of 10), as indicated, by standard gentamicin protection assay after preincubation in the absence or presence of 12 μM PIP3. (C) Phagocytic uptake of P. aeruginosa PA14 motAB motCD (MOI of 10) with murine TLR4−/− BMDCs pretreated with 12 μM PIP3. (D) Murine BMDCs were pretreated with PIP3 and assayed for relative in vitro phagocytosis of PA14 flgK bacteria and USA300 upon mixing the bacteria in a 1:1 ratio. The data were normalized to phagocytosis of bacterial strains (either PA14 flgK or USA300) incubated alone without PIP3 treatment and are represented as fold increase in phagocytosis. All data were analyzed using an unpaired t test with Welch's correction (A to C) or one-way ANOVA with Tukey's post hoc analyses (D) and are representative of at least two independent biological experiments (n ≥8). ***, P ≤ 0.0005, for results compared to those without PIP3 treatment.
Since the phagocytosis of Gram-negative bacteria, as opposed to that of Gram-positive bacteria, is preferentially induced by PIP3, we hypothesized that the LPS recognition receptor Toll-like receptor 4 (TLR4) could potentially mediate this specificity. Therefore, we evaluated the contribution of TLR4 to our system by performing parallel phagocytosis assays in BMDCs that genetically lack TLR4. However, we observed significant induction of uptake of motAB motCD bacteria by PIP3-treated TLR4−/− BMDCs, similar to that with WT BMDCs, suggesting that the PIP3 effect is independent of TLR4 expression (Fig. 7C).
In order to further test the selectivity of the PIP3 treatment for uptake of P. aeruginosa rather than S. aureus, we mixed cultures of nonmotile P. aeruginosa (PA14 flgK) with S. aureus (USA300) in equal numbers and monitored the uptake of both bacteria by BMDCs in the presence or absence of PIP3 pretreatment. Intriguingly, the number of recovered USA300 CFU after coincubation with PA14 flgK and BMDCs was not significantly different than that when USA300 was incubated alone with both untreated and PIP3-treated cells. In contrast, uptake of flgK bacteria by BMDCs primed with PIP3 was still induced upon coincubation with USA300 S. aureus bacteria (Fig. 7D). This indicates that PA14 flgK is not able to confer additional susceptibility to S. aureus uptake in trans and that the PIP3-treated BMDCs are able to preferentially phagocytose nonmotile P. aeruginosa from a mixed bacterial culture (in the presence of Gram-positive bacteria). Overall, we inferred that the PIP3 effect is selective for uptake of nonmotile P. aeruginosa and other Gram-negative bacteria, independent of TLR4 expression.
PIP3-induced phagocytosis is dependent upon PI3K and Akt activity.
Based on our observations that PIP3 treatment induces substantial internalization of nonmotile P. aeruginosa and since PI3K and Akt are necessary for phagocytosis of P. aeruginosa (Fig. S1A) (17), we hypothesized that addition of PIP3 may promote Akt activation. To assess this, relative Akt activation was quantitatively measured by intracellular staining for phosphorylated Akt (phospho-Akt) followed by fluorescence-activated cell sorting (FACS) analyses. Indeed, PIP3-treated cells exhibited a significant increase in Akt activation following coincubation with bacteria compared to the level in control cells lacking PIP3 treatment (Fig. 8A). However, this result does not distinguish between two possibilities for how PIP3 treatment acts to increase uptake of nonmotile P. aeruginosa. One possible explanation is that exogenous PIP3 treatment of cells bypasses the requirement for PI3K kinase to activate Akt and promote phagocytosis. An alternative, though not mutually exclusive, potential mechanism is that PIP3 potentiates an earlier step in the phagocytic pathway such that PI3K activity is still required. To test the former hypothesis, that exogenous addition of PIP3 would bypass the requirement of PI3K and directly activate Akt to induce uptake of nonmotile P. aeruginosa, PI3K or Akt was inhibited with LY-294002 or Akt inhibitor VIII, respectively, and the uptake of PA14 motAB motCD by PIP3-treated BMDCs was quantitatively assessed. Unexpectedly, the PIP3 effect on phagocytic induction of nonmotile P. aeruginosa was abrogated upon inhibition of PI3K activity (Fig. 8B). Inhibition of Akt, which is downstream of PIP3, also abolished the phagocytic induction of PA14 motAB motCD in PIP3-treated cells, supporting the idea that phagocytosis in PIP3-induced cells still occurs through Akt (Fig. 8C). As a positive control for blockade of phagocytosis, cytochalasin D (CytoD), an inhibitor of actin polymerization, was used (Fig. 8B). These data strongly support the notion that the induction of phagocytosis by PIP3 still requires PI3K and Akt activities. Since the addition of PIP3 does not bypass the requirement for PI3K for phagocytosis, we sought the potential contribution of an alternative mechanism at an earlier step in the phagocytic pathway that could account for the PIP3 effect on induction of phagocytosis of nonmotile bacteria.
FIG 8.

PI3K and Akt activities are required for PIP3-induced phagocytosis of nonmotile P. aeruginosa. (A) Murine BMDCs were coincubated with PA14 motAB motCD bacteria for 45 min after pretreatment of cells in the absence or presence of 12 μM PIP3. Following intracellular staining for phospho-Akt, Akt activation was quantified by FACS analyses. Mean fluorescence intensity (MFI) was normalized to untreated cells (set as 1, y axis). (B and C) Phagocytic uptake of PA14 motAB motCD (MOI of 10) by gentamicin protection assay with murine WT BMDCs pretreated with 12 μM PIP3 with or without 100 μM LY-294002, 5 μM Akt inhibitor VIII, or 10 μM cytochalasin D, as indicated. The data were normalized to phagocytosis of PA14 motAB motCD without PIP3 treatment and are represented as fold increase in phagocytosis. Data were analyzed using one-way ANOVA with Tukey's post hoc analyses and are representative of at least two independent biological experiments (A, n = 4; B and C, n = 8). ***, P ≤ 0.0005; ns, not significant.
PIP3 markedly increases phagocyte cell surface interactions with nonmotile Gram-negative bacteria.
In an effort to determine the mechanistic basis for the PIP3 effect on phagocytic induction of nonmotile Gram-negative bacteria, we assessed the total association and binding of bacteria by phagocytic cells. First, to visualize the interactions that occur between P. aeruginosa and BMDCs by fluorescence microscopy, murine BMDCs were primed with PIP3, followed by incubation in the presence or absence of Akt inhibitor VIII, and subsequently incubated with green fluorescent protein (GFP)-expressing PA14 motAB motCD. Representative images demonstrate that cells pretreated with PIP3 associate with these bacteria to a much greater extent than untreated cells (Fig. 9A). Importantly, blocking phagocytosis in PIP3-treated cells by inhibition of Akt (Fig. 9A) did not diminish the total binding of nonmotile P. aeruginosa by the BMDCs, thereby supporting the notion that PIP3 can potentiate cell surface interactions with these bacteria.
FIG 9.

PIP3 significantly increases binding of nonmotile bacteria to phagocytes. (A) Representative images of WT BMDCs pretreated with 12 μM PIP3 with or without 5 μM Akt inhibitor VIII and incubated with GFP-expressing PA14 motAB motCD (green) at an MOI of 10 (magnification, ×40). The nuclei of BMDCs are stained with 4′,6′-diamidino-2-phenylindole (blue), and cells are viewed by differential interference contrast overlay. (B) Relative association of GFP-expressing PA14 WT or motAB motCD bacteria with PIP3-pretreated murine WT BMDCs, quantitatively measured by FACS analyses. PIP3-dependent increases in bacterial association with BMDCs are shown as the fold increase compared to the level of association in the absence of PIP3 pretreatment. The relative association of GFP-expressing PA14 WT or motAB motCD bacteria to BMDCs in the absence of PIP3 pretreatment is also shown (B, inset). (C) WT BMDCs either untreated or pretreated with PIP3 were assayed for relative cellular cytotoxicity following infection with the PA14 WT or motAB motCD strain (MOI of 10) for 45 min. BMDC cellular cytotoxicity was analyzed by propidium iodide (PI) staining followed by flow cytometric analysis. (D) Flow cytometry to assay KPPR1-YFP bacterial association with murine WT BMDCs pretreated with PIP3. The data were normalized in the same manner as described for panel B. All data were analyzed using an unpaired t test with Welch's correction (B and D) or one-way ANOVA with Tukey's post hoc analyses (C) and are representative of at least two independent biological experiments (n ≥ 8). ***, P ≤ 0.0005; **, P ≤ 0.005; *, P ≤ 0.05; ns, not significant, for results compared to those without PIP3 treatment.
As quantitative metrics to measure cell surface association of the bacteria with the phagocytes, we employed two complementary assays: FACS analyses, which provided an alternative and quantitative technique by which to validate the microscopy data, and analysis of bacterial type III secretion system (T3SS)-induced cytotoxicity, which is dependent upon interaction of the bacteria with the cell surface but not upon phagocytic uptake (25). Of note, both the nonmotile and motile PA14 bacteria are equally capable of T3SS activity, but the nonmotile bacteria elicit less cytotoxicity by virtue of less cellular association (25). Consistent with previous data (13, 14), GFP-expressing PA14 WT bacteria exhibited significantly greater association with untreated cells than motAB motCD bacteria (9B, inset). However, consistent with the phagocytosis data (Fig. 2), the fold increases in bacterial binding (Fig. 9B) and induced cytotoxicity (Fig. 9C) following PIP3 pretreatment of the cells were both far greater for the nonmotile bacteria than for the motile bacteria. The PIP3 treatment induced similar fold increases in binding and phagocytosis of the nonmotile P. aeruginosa, quantitatively supporting the increased binding as a predominant contributing factor to the increased phagocytosis. Intriguingly, PIP3 pretreatment of the phagocytes also elicited small but significant increases in binding and cytotoxicity by the motile bacteria (Fig. 9B and C) that were not reflected in measurable increases in phagocytosis (Fig. 1C and 2B); it is not clear if this is reflective of differing sensitivities of the assays or of an additional rate-limiting step within the phagocytic process. Last, K. pneumoniae, which inherently lacks flagellar motility, recapitulated the observations derived from nonmotile P. aeruginosa, such that the total association of fluorescent bacteria with PIP3-treated cells as measured by FACS analysis was significantly greater than that in untreated cells (Fig. 9D). These data support our gentamicin protection assays and reveal (i) that PIP3 promotes phagocytosis by increasing overall bacterial binding by the phagocytes and (ii) that the PIP3-induced phagocytosis is dependent on both PI3K and Akt activities and, specifically, does not bypass the requirement for PI3K. Thus, PIP3 preferentially supports cell surface association, and associated elicited cytotoxicity, by nonmotile bacteria in comparison to its effect on motile bacteria.
DISCUSSION
Flagellar motility has long been recognized as a major colonization and virulence factor in many bacterial pathogens, including P. aeruginosa. While flagellar motility is necessary for initial establishment of infection (23), it is well accepted that clinical isolates recovered from patients suffering from chronic P. aeruginosa infections have downregulated flagellar gene expression or no flagella entirely, resulting in loss of motility (4, 5). This nonmotility phenotype observed in clinical strains isolated from established infections is likely reflective of the selective pressure exhibited by the immune system to clear motile bacteria by phagocytosis. In this regard, phagocytic clearance of bacteria by neutrophils and macrophages is the major mechanism of host control of P. aeruginosa (9), and while flagellar motility is a potent activator of phagocytosis by P. aeruginosa, the nonswimming bacteria evade uptake by phagocytes (13, 14). Indeed, effective phagocyte function in immunocompetent individuals is typically sufficient to protect them from establishment of chronic P. aeruginosa infections (11). We reasoned that if we could boost the ability of phagocytes to bind and engulf nonmotile bacteria closer to the levels that they achieve with motile bacteria, then bacterial clearance would drastically improve, and similar levels of protection might be achieved. In this work, we have described a methodology and identified a potential mechanism by which uptake of these nonmotile bacteria is enhanced by pretreating immune cells with phospholipids (PIP3 and PIP2).
PI3K activity is broadly required for internalization of P. aeruginosa, as well as that of other bacterial genera, by phagocytic cells. These data were recapitulated by the PI3K dependence for phagocytosis of all motile and nonmotile strains tested in this report. Moreover, these data also complement and extend previous reports exploring PI3K dependence using epithelial and phagocytic cells with P. aeruginosa (17, 26, 27), as well as several other bacteria, including Escherichia coli K1, Listeria monocytogenes, and Helicobacter pylori (28–30). Previously, we determined that the PI3K/Akt signal transduction pathway was responsible for motility-induced uptake of P. aeruginosa such that flagellar motility results in Akt activation and ultimately phagocytosis (17). Moreover, murine alveolar macrophages that lack PTEN phosphatase, which converts PIP3 to PIP2, exhibit enhanced phagocytosis of P. aeruginosa (31). Therefore, taking these observations into account, our initial hypothesis was that exogenous activation of Akt by pretreating immune cells with PIP3 would stimulate the uptake of the phagocytosis-resistant nonmotile P. aeruginosa.
Indeed, using multiple independent nonmotile mutants of P. aeruginosa, we report substantial (10- to 30-fold) induction of phagocytosis upon priming of human and murine cells with PIP3. Notably, the magnitude of the PIP3 induction of phagocytosis is much greater than that observed with other interventions meant to increase uptake of bacteria. For instance, Leid et al. reported that uptake of P. aeruginosa biofilms (PAO1 and FRD1 strains) lacking alginate (algD deletion bacteria) by human leukocytes was increased about 4-fold after exogenous treatment with gamma interferon (IFN-γ), while uptake of wild-type bacteria was unaffected by the treatment (32). Similarly, another study demonstrated that treatment of human blood neutrophils or monocyte-derived macrophages with resolvin D1 increased phagocytosis of the P. aeruginosa clinical strain RP73 ∼3-fold (33).
Although our original premise was that phagocytic induction by PIP3 was through direct activation of Akt, a series of informative experiments altered our hypothesis. Notably, since our data revealed that addition of PIP3 does not bypass the requirement for PI3K and that PIP2 also elicited a level of induction of phagocytosis similar to that of PIP3, it is unlikely that PIP2 works by getting converted to PIP3. Additionally, we observe that the polyphosphoinositides induce cell surface binding of the bacteria. Since the effect elicited by lipids was specific to the phosphoinositides tested (PIP3 and PIP2) in comparison with other lipids tested, we speculate that the phosphatidyl inositol head groups and the negative charge mediate the interaction. Thus, we believe that the most likely mechanism is that the PIP3 integrated in the host membrane after exogenous treatment binds directly to bacteria, facilitating the internalization into cells. However, an alternative scenario is that the addition of PIP3 might affect phagocytic cell membrane permeability or fluidity or even the distribution and clustering of phagocytic receptors on the membrane, thereby promoting productive binding and uptake of bacteria. Since the PIP3 effect is completely abrogated upon inhibition of either Akt or PI3K activities in PIP3-treated cells, this supports the notion that the requirement for PI3K is not bypassed, despite addition of the direct Akt activator PIP3, and thus that the stimulus is likely not working directly upon Akt. Instead, the main driver of our phagocytic phenotype upon PIP3 pretreatment seems to be at the level of bacterial association to the outside of cells, which occurs in an Akt-independent manner. Thus, the increase in bacterial binding to cells results in increased uptake of those bacteria though the addition of PIP3 may also accelerate the kinetics of internalization.
Interestingly, our results support the idea that PIP3 pretreatment of cells is effective only for uptake of Gram-negative bacteria and not Gram-positive bacteria. In fact, the uptake of S. aureus and B. subtilis modestly decreased upon PIP3 treatment of cells. Moreover, the mixed-culture experiment where nonmotile P. aeruginosa was preferentially phagocytosed in the presence of S. aureus suggests that there are no soluble factors being secreted into the extracellular environment that would alter the phagocytic activity of the BMDCs. This selectivity in the response might be accounted for by the inherent differences between these bacteria, such as the chemical composition of the structural (cell) envelopes, that would facilitate binding/interaction with eukaryotic cells. The outer shell of Gram-positive bacteria consists of a thick cell wall (peptidoglycan) containing teichoic acids and lipoteichoic acids (LTA) while, in contrast, exclusive to Gram-negative bacteria is an additional outer membrane with lipopolysaccharides, lipoproteins, and pore-forming proteins (porins) outside their thin cell walls (34). Therefore, it is conceivable that these unique structures and potential interacting bacterial ligands (adhesins) associated with Gram-negative bacteria mediate a productive interaction with the PIP3-treated cells which enables their uptake, contrasting with the interaction of Gram-positive bacteria. With the use of nonmotile P. aeruginosa mutants lacking O antigen ligase, we provide the key insight that the PIP3 induction of phagocytosis is independent of the presence of O antigen on the LPS. We also demonstrate that TLR4 on phagocytic cells is not responsible for PIP3 selectivity toward Gram-negative bacteria since TLR4-deficient BMDCs were still induced by PIP3 to phagocytose nonmotile P. aeruginosa. Moreover, it is worth noting that both groups of bacteria have an overall negative charge since surface charge is an important factor for mediating bacterial attachment to surfaces (including cells). However, the negative charge on Gram-positive bacteria is conferred due to teichoic acids, whereas the Gram-negative bacteria are negatively charged due to the presence of lipopolysaccharides in the outer membrane (34).
In addition to gaining insights about the mechanisms by which PIP3 enhances uptake of nonmotile bacteria, this study is also informative regarding the mechanistic requirements for phagocytosis of P. aeruginosa. Previous work in the lab illustrated that bacterial swimming motility as a function of flagellar rotation is utilized by phagocytes to recognize bacteria for uptake. Specifically, the motility promotes engagement between the bacteria and the cell surface of the phagocyte, which results in a stimulus for internalization. From the results of the competition experiment with motile wild-type and nonflagellated (flgK) P. aeruginosa, we inferred that both genotypes of bacteria are able to access the same binding sites on the cell surface in PIP3-treated cells. Since the bacteria devoid of flagella (and flagellar motility) are taken up by PIP3-treated cells, this supports the concept that flagellar motility enables engagement with the cell but that the flagellum is not the ligand recognized by phagocytic cells for uptake; the exact bacterial ligand(s) supporting phagocytic uptake is currently unknown. From these studies, we speculate that P. aeruginosa and other Gram-negative bacteria favor binding to negatively charged molecules on the cell surface. Consistent with this notion, Kierbel et al. demonstrated that PIP3-rich membrane sites can facilitate motile P. aeruginosa (PAK) invasion into epithelial cells (27). However, given the subcellular distribution of PIP3 within phagocytes and, specifically, the lack of polyphosphoinositides on the cell surface, it appears unlikely that they are endogenous ligands that mediate bacterial uptake. Therefore, this study paves the way for further identification of cellular determinants for phagocytosis of P. aeruginosa, especially with regard to charge-mediated effects in conjunction with as yet poorly described phagocytic receptor(s).
In conclusion, there is a great need to develop and optimize methodologies to effectively eradicate nonmotile bacteria in chronic infections. With the use of a variety of nonmotile bacterial genera and bacterial mutants, including P. aeruginosa, we demonstrate that PIP3 pretreatment of phagocytic cells helps to overcome the phagocytic evasion exhibited by nonmotile bacteria. Importantly, our data support a model whereby increased bacterial association to phagocytic cells upon PIP3 treatment is mainly responsible for the enhanced internalization of nonswimming bacteria. Overall, our findings provide key insights into poorly understood determinants of phagocytic recognition of bacteria and reveal the basis for a potential therapeutic intervention whereby plasma membrane-anchored polyanions, potentially phospholipids, could be leveraged to mediate bacteria clearance.
MATERIALS AND METHODS
Bacteria.
All bacteria used in this study are listed in Table S1 in the supplemental material and have been previously published. B. subtilis JH642 and many of the P. aeruginosa strains on the PAO1 and PA14 backgrounds, except PA14 waaL motAB motCD (21), were provided by G. O'Toole and D. Hogan (Dartmouth Medical School, Hanover, NH) (13, 14, 25, 35, 36). P. aeruginosa PAK and PA103 strains were provided by B. Kazmierczak (Yale University, New Haven, CT) (37). The remaining bacteria used are as follows (source): E. coli K-12 fliC (E. coli genetic stock center), S. aureus USA300 (ATCC) and rn4220 (A. Cheung, Dartmouth Medical School, Hanover, NH), V. cholerae O395 motX (24), and K. pneumoniae KPPR1 (M. Wargo, University of Vermont, Burlington, VT) (38, 39). Bacteria were cultured overnight at 37°C and subsequently subcultured to log-phase growth for 2 h in Luria Bertani (LB) broth, and the concentrations were determined by optical density at 600 nm.
Cells.
Bone marrow-derived dendritic cells (BMDCs) were cultured from C57BL/6 WT mice (Charles River Laboratories) or C3H/HeJ TLR4-knockout (TLR4−/−) mice (Jackson Laboratories) as previously described (40). Six- and 7-day-old BMDCs were used for these studies. Blood was collected from healthy volunteers and patients with CF by venipuncture into heparinized tubes. Donor blood samples were procured from three CF patients with the following cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations: R1162X/W1282G, E60X/A455E, and ΔF508/ΔF508. Neutrophils were enriched with a Ficoll-Paque Plus (GE Healthcare Biosciences) discontinuous gradient followed by dextran sedimentation as previously described (21). Human peripheral blood monocytes were kindly provided by P. Guyre (Dartmouth Medical School, Hanover, NH).
Ethics statement.
The Dartmouth Committee for the Protection of Human Subjects (CPHS) approved procurement and use of human cells in this study (22781 and 22634). All samples were obtained with informed consent and subsequently deidentified. Animal use was conducted in strict compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council (41) and was approved by the Dartmouth Institutional Animal Care and Use Committee (IACUC).
Gentamicin protection assay.
Phagocytosis of live bacteria was performed as previously described (21). Nonadherent phagocytic cells (2.5 × 105) were preincubated with exogenous PIP3 (P-3916; Echelon Biosciences) with the use of an established protocol (42). Briefly, 12 μM PIP3 was loaded onto 6 μM histone carrier (P-9C2; Echelon Biosciences) (5 min at room temperature [RT]) and then incubated with BMDCs in serum-free Hanks balanced salt solution (HBSS) for 10 min at RT. Excess lipids were then removed by centrifugation prior to incubation of cells with the bacteria in HBSS. Similarly, phagocytic cells were preincubated with equivalent molar concentrations (12 μM) of phosphatidyl-(4,5)-bisphosphate (PIP2), phosphatidylserine (PS), phosphatidylcholine (PC), and phosphatidylethanolamine (PE) (Avanti Polar Lipids); histone carrier was also used for all of these treatments. In the experiment involving direct incubation of bacteria with PIP3 (Fig. 3A), free PIP3 was washed out by layering bacteria on a sucrose pad (0.3 M sucrose in 1× phosphate-buffered saline [PBS]) after the 10-min incubation and pelleting the bacteria through the sucrose by centrifugation.
For inhibitor studies, 2.5 × 105 cells were preincubated in 100 μM LY-294002 (LY9908; Sigma), 5 μM Akt inhibitor VIII (124018; Calbiochem), 10 μM cytochalasin D (C8273; Sigma), or serum-free HBSS for 15 min at RT before being coincubated with bacteria. PIP3 was added to the cells as described above 5 min after the initial start of the incubation with the inhibitors. The BMDCs were then cocultured in the presence of the inhibitors with the bacterial strains indicated in the figures (at multiplicities of infection [MOI] of 10 or 1 as specified in the figure legends) for 45 min at 37°C. For studies using inositol 1,3,4,5-tetrakisphosphate (IP4) (Q-1345; Echelon Biosciences), 12 μM phosphoinositol was added to the cells at the same time as the bacteria.
For all conditions, after the 45-min coincubation with bacteria, 100 μg/ml gentamicin was added to the cells for 20 min at 37°C. The cells were then washed twice with HBSS and lysed with 500 μl of 0.1% Triton X-100 solution or 0.1% Tween 20 solution (for B. subtilis only) in 1× PBS. Lysates were plated on LB plates and incubated overnight at 37°C. For phagocytosis assays, the number of recovered CFU on LB plates is represented as the percentage of the mean of WT bacteria phagocytosed or fold increase in phagocytosis (normalized to phagocytosis of bacterial strains without PIP3 pretreatment), as indicated in the figures, to quantitatively compare relative phagocytosis levels.
Microscopy.
WT BMDCs were incubated with GFP-expressing PA14 motAB motCD at an MOI of 10 for 45 min as previously described (13). Where indicated, WT BMDCs were pretreated with 12 μM PIP3 with or without 5 μM Akt inhibitor VIII before incubation with PA14 motAB motCD expressing GFP (as described above). Microscopy was performed on an inverted Zeiss Axiovert 200 microscope with a 40× objective.
FACS-based bacterial association assay for fluorescent bacteria.
WT BMDCs were incubated with GFP-positive (GFP+) PA14 WT, GFP+ PA14 motAB motCD, or yellow fluorescent protein (YFP)-positive (YFP+) Klebsiella pneumoniae KPPR1 (M. Wargo, University of Vermont, Burlington, VT) bacterial strains for 45 min at 37°C following published protocols (13, 14). WT BMDCs were pretreated with 12 μM PIP3 before incubation with fluorescent bacteria in HBSS. Fluorescence-activated cell sorting (FACS) analyses were subsequently used to quantitatively measure the association of fluorescent bacteria with the BMDCs, with nonassociated bacteria excluded by gating on scatter. In order to combine data derived from multiple independent experiments, the fluorescence values from each experiment were normalized to the mean fluorescence intensity (MFI) of uninfected BMDCs, with this background fluorescence subsequently subtracted from the data in which fluorescent bacteria were employed.
To assess cellular cytotoxicity using FACS analysis, WT BMDCs were untreated or pretreated with 12 μM PIP3 before incubation with PA14 WT or motAB motCD bacteria (MOI of 10) for 45 min at 37°C. At 45 min postinfection, cells were washed twice with HBSS, followed by staining with propidium iodide ([PI] 1:200, diluted in HBSS) for 5 min on ice, followed by flow cytometric analysis.
Akt activation assay.
Akt activation assays were performed with previously described reagents and methodology (21). Akt activation (phospho-Akt levels) was quantified by FACS analyses.
Statistical analyses.
Means ± standard deviations (SD) derived from multiple independent experiments with technical replicates are shown for each graph. Sample sizes for each experiment are noted in the figure legends. As indicated in the legends, unpaired Student's t test with Welch's correction or one-way analysis of variance (ANOVA) with Tukey's post hoc analyses was performed using Prism, version 7.02, to determine statistical significance of the data. Statistical significance is represented in the figures by asterisks.
Supplementary Material
ACKNOWLEDGMENTS
We thank Henry Higgs, Matthew Wargo, George O'Toole, Deborah Hogan, Alix Ashare, William Rigby, Iviana Torres, Petra Sergent, Sladjana Skopelja-Gardner, Haley Hazlett, and Amanda Nymon (Geisel School of Medicine at Dartmouth) for reagents and discussion. This work was facilitated by the Dartmouth Lung Biology Translational Research Core and the DartLab Immune Monitoring Lab.
This work was supported by grants from the National Institutes of Health (NIH) (P30 RR032136-01, R03AI135358, and R21 AI121820) and the Cystic Fibrosis Foundation Research Development Program (STANTO19R0 and STANTO11R0) (to B.B.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We have no conflicts of interest to report.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00215-18.
REFERENCES
- 1.Hauser AR, Jain M, Bar-Meir M, McColley SA. 2011. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev 24:29–70. doi: 10.1128/CMR.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, Molin S. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 3.Winstanley C, O'Brien S, Brockhurst MA. 2016. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol 24:327–337. doi: 10.1016/j.tim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luzar MA, Thomassen MJ, Montie TC. 1985. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect Immun 50:577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahenthiralingam E, Campbell ME, Speert DP. 1994. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun 62:596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner KH, Everett J, Trivedi U, Rumbaugh KP, Whiteley M. 2014. Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet 10:e1004518. doi: 10.1371/journal.pgen.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran VB, Fleiszig SM, Evans DJ, Radke CJ. 2011. Dynamics of flagellum- and pilus-mediated association of Pseudomonas aeruginosa with contact lens surfaces. Appl Environ Microbiol 77:3644–3652. doi: 10.1128/AEM.02656-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans DJ, Fleiszig SM. 2013. Why does the healthy cornea resist Pseudomonas aeruginosa infection? Am J Ophthalmol 155:961–970 e962. doi: 10.1016/j.ajo.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. 2009. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun 77:5300–5310. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurahashi K, Sawa T, Ota M, Kajikawa O, Hong K, Martin TR, Wiener-Kronish JP. 2009. Depletion of phagocytes in the reticuloendothelial system causes increased inflammation and mortality in rabbits with Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 296:L198–L209. doi: 10.1152/ajplung.90472.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrews T, Sullivan KE. 2003. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev 16:597–621. doi: 10.1128/CMR.16.4.597-621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanamaru A, Tatsumi Y. 2004. Microbiological data for patients with febrile neutropenia. Clin Infect Dis 39(Suppl 1):S7–S10. doi: 10.1086/383042. [DOI] [PubMed] [Google Scholar]
- 13.Amiel E, Lovewell RR, O'Toole GA, Hogan DA, Berwin B. 2010. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect Immun 78:2937–2945. doi: 10.1128/IAI.00144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovewell RR, Collins RM, Acker JL, O'Toole GA, Wargo MJ, Berwin B. 2011. Step-wise loss of bacterial flagellar torsion confers progressive phagocytic evasion. PLoS Pathog 7:e1002253. doi: 10.1371/journal.ppat.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan W, Bernier SP, Kuchma SL, Hammond JH, Hasan F, O'Toole GA. 2010. Aminoglycoside resistance of Pseudomonas aeruginosa biofilms modulated by extracellular polysaccharide. Int Microbiol 13:207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciofu O, Tolker-Nielsen T, Jensen PO, Wang H, Hoiby N. 2015. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliv Rev 85:7–23. doi: 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 17.Lovewell RR, Hayes SM, O'Toole GA, Berwin B. 2014. Pseudomonas aeruginosa flagellar motility activates the phagocyte PI3K/Akt pathway to induce phagocytic engulfment. Am J Physiol Lung Cell Mol Physiol 306:L698–L707. doi: 10.1152/ajplung.00319.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovewell RR, Patankar YR, Berwin B. 2014. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 306:L591–L603. doi: 10.1152/ajplung.00335.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weichhart T, Saemann MD. 2008. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis 67(Suppl 3):iii70–iii74. doi: 10.1136/ard.2008.098459. [DOI] [PubMed] [Google Scholar]
- 20.Bertrand JJ, West JT, Engel JN. 2010. Genetic analysis of the regulation of type IV pilus function by the Chp chemosensory system of Pseudomonas aeruginosa. J Bacteriol 192:994–1010. doi: 10.1128/JB.01390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demirdjian S, Schutz K, Wargo MJ, Lam JS, Berwin B. 2017. The effect of loss of O-antigen ligase on phagocytic susceptibility of motile and non-motile Pseudomonas aeruginosa. Mol Immunol 92:106–115. doi: 10.1016/j.molimm.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn HP. 1997. The type-4 pilus is the major virulence-associated adhesin of Pseudomonas aeruginosa—a review. Gene 192:99–108. doi: 10.1016/S0378-1119(97)00116-9. [DOI] [PubMed] [Google Scholar]
- 23.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 24.Martinez RM, Dharmasena MN, Kirn TJ, Taylor RK. 2009. Characterization of two outer membrane proteins, FlgO and FlgP, that influence vibrio cholerae motility. J Bacteriol 191:5669–5679. doi: 10.1128/JB.00632-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, Berwin B. 2013. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun 81:2043–2052. doi: 10.1128/IAI.00054-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kierbel A, Gassama-Diagne A, Mostov K, Engel JN. 2005. The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol Biol Cell 16:2577–2585. doi: 10.1091/mbc.e04-08-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kierbel A, Gassama-Diagne A, Rocha C, Radoshevich L, Olson J, Mostov K, Engel J. 2007. Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J Cell Biol 177:21–27. doi: 10.1083/jcb.200605142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ireton K, Payrastre B, Chap H, Ogawa W, Sakaue H, Kasuga M, Cossart P. 1996. A role for phosphoinositide 3-kinase in bacterial invasion. Science 274:780–782. doi: 10.1126/science.274.5288.780. [DOI] [PubMed] [Google Scholar]
- 29.Kwok T, Backert S, Schwarz H, Berger J, Meyer TF. 2002. Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect Immun 70:2108–2120. doi: 10.1128/IAI.70.4.2108-2120.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy MA, Prasadarao NV, Wass CA, Kim KS. 2000. Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem 275:36769–36774. doi: 10.1074/jbc.M007382200. [DOI] [PubMed] [Google Scholar]
- 31.Hubbard LL, Wilke CA, White ES, Moore BB. 2011. PTEN limits alveolar macrophage function against Pseudomonas aeruginosa after bone marrow transplantation. Am J Respir Cell Mol Biol 45:1050–1058. doi: 10.1165/rcmb.2011-0079OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. 2005. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol 175:7512–7518. doi: 10.4049/jimmunol.175.11.7512. [DOI] [PubMed] [Google Scholar]
- 33.Codagnone M, Cianci E, Lamolinara A, Mari VC, Nespoli A, Isopi E, Mattoscio D, Arita M, Bragonzi A, Iezzi M, Romano M, Recchiuti A. 2018. Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol 11:35–39. doi: 10.1038/mi.2017.36. [DOI] [PubMed] [Google Scholar]
- 34.Mai-Prochnow A, Clauson M, Hong J, Murphy AB. 2016. Gram positive and Gram negative bacteria differ in their sensitivity to cold plasma. Sci Rep 6:38610. doi: 10.1038/srep38610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toutain CM, Zegans ME, O'Toole GA. 2005. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J Bacteriol 187:771–777. doi: 10.1128/JB.187.2.771-777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuchma SL, Ballok AE, Merritt JH, Hammond JH, Lu W, Rabinowitz JD, O'Toole GA. 2010. Cyclic-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J Bacteriol 192:2950–2964. doi: 10.1128/JB.01642-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrity-Ryan L, Kazmierczak B, Kowal R, Comolli J, Hauser A, Engel JN. 2000. The arginine finger domain of ExoT contributes to actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect Immun 68:7100–7113. doi: 10.1128/IAI.68.12.7100-7113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ubags ND, Vernooy JH, Burg E, Hayes C, Bement J, Dilli E, Zabeau L, Abraham E, Poch KR, Nick JA, Dienz O, Zuniga J, Wargo MJ, Mizgerd JP, Tavernier J, Rincon M, Poynter ME, Wouters EF, Suratt BT. 2014. The role of leptin in the development of pulmonary neutrophilia in infection and acute lung injury. Crit Care Med 42:e143–e151. doi: 10.1097/CCM.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ubags ND, Burg E, Antkowiak M, Wallace AM, Dilli E, Bement J, Wargo MJ, Poynter ME, Wouters EF, Suratt BT. 2016. A comparative study of lung host defense in murine obesity models. Insights into neutrophil function. Am J Respir Cell Mol Biol 55:188–200. doi: 10.1165/rcmb.2016-0042OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amiel E, Alonso A, Uematsu S, Akira S, Poynter ME, Berwin B. 2009. Pivotal advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J Leukoc Biol 85:595–605. doi: 10.1189/jlb.1008631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academy Press, Washington, DC. [Google Scholar]
- 42.Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. 2002. A PtdInsP3- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol 4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.