

Abstract
A heterohexameric complex composed of minichromosome maintenance protein 2–7 (MCM2–7), which acts as a key replicative enzyme in eukaryotes, is crucial for initiating DNA synthesis only once per cell cycle. The MCM complex remains inactive through the G1 phase, until the S phase, when it is activated to initiate replication. During the transition from the G1 to S phase, the MCM undergoes multisite phosphorylation, an important change that promotes subsequent assembly of other replisome members. Phosphorylation is crucial for the regulation of MCM activity and function. MCMs can be phosphorylated by multiple kinases and these phosphorylation events are involved not only in DNA replication but also cell cycle progression and checkpoint response. Dysfunctional phosphorylation of MCMs appears to correlate with the occurrence and development of cancers. In this review, we summarize the currently available data regarding the regulatory mechanisms and functional consequences of MCM phosphorylation and seek the probability that protein kinase inhibitor can be used therapeutically to target MCM phosphorylation in cancer.
Keywords: MCM, Phosphorylation, DNA replication, Checkpoint response, Cell cycle
Background
DNA is replicated via a multi-protein machinery comprising DNA polymerase, helicase, primase, circular sliding clamps, a pentameric clamp loader, single-strand binding protein (SSB) and other components [1–5]. This machinery is often referred to as a “replisome”. Initiation of DNA replication in each cell cycle is fundamental to maintain genomic integrity and stability. Key to initiation is the formation of pre-replicative complexes (pre-RCs) in late M/early G1 phase through the recruitment of MCM2–7 in an origin recognition complex (ORC)-, Cdc6-, and Cdt1-dependent manner [6–9]. After this key step, Dbf4-dependent kinase (DDK) and cyclin-dependent kinases (Cdks) phosphorylate MCM2–7, leading to the recruitment of Cdc45 and GINS (Go, Ichi, Ni, and San) to form the CMG (Cdc45–MCMs–GINS) replicative helicase complex. The CMG replicative helicase complex has a robust helicase activity [10–13]. In addition, emerging studies suggest that MCM2–7 plays a critical role not only in replication, but also in transcription [14, 15], replication checkpoint [16–18], and RNA splicing [19]. As MCMs also belong to the ATPases associated with diverse cellular activities (AAA+) family, they display ATPase activity [20]. Moreover, owing to the crucial function of MCMs, the regulatory mechanisms that modulate and control its activity are diverse and complex, particularly, the phosphorylation mechanism.
Multiple phosphorylation sites were distributed on the MCM2–7 subunits. The biological and functional consequence of MCM phosphorylation appears to be correlated with specific kinases and their phosphosites. Some MCM subunits undergo dynamic phosphorylation in a cell cycle-specific manner, which may be consistent with their cell-cycle-specific functions [21–25]. Aberrant phosphorylation of MCMs disrupts DNA replication and cell cycle progression, leading to diseases or cancers [26–31]. Several reviews have been published on MCMs. However, few specifically discuss the role of phosphorylation on MCM function. Here, we highlight the function and mechanism of MCM2–7 protein phosphorylation in human cancer cells.
Phosphorylation of MCMs by Cdc7
Cell division cycle 7 (Cdc7) is an evolutionary conserved serine-threonine kinase that promotes the initiation of DNA replication by targeting the functional substrate MCM2–7 protein [32–35]. Similar to Cdk, Cdc7 is activated by its regulatory subunits: Dbf4 and Drf1 in human [36, 37]. Cdc7 is found to be up-regulated in various cancers and has been characterized as an independent prognostic marker and a potential therapeutic target [38–41].
Cdc7 preferentially phosphorylates MCM2 as well as other MCM subunits (Table 1). Although there is agreement regarding specific phosphosites, each study has also identified additional sites. Differences in cell line, experimental design, or detection sensitivity may contribute to inconsistency of results among studies. In general, Cdc7 phosphorylation of MCMs is essential for the initiation of DNA replication. Tsuji et al. identified three Cdc7-dependent MCM2 phosphosites (Ser-27/41/139), both in vivo and in vitro [21]. A triple alanine substitution at these three sites in MCM2 did not support DNA replication in HeLa cells. This suggests that Cdc7 phosphorylation of MCM2 was essential for the initiation of DNA replication. In addition, this study revealed that MCM2 accumulated on chromatin early in the G1 phase before Cdc7 phosphorylation during the G1/S phase. Phosphorylation of MCM2 did not affect the chromatin loading of MCM complex. However, another study by Chuang et al. suggested that Cdc7 phosphorylated MCM2 at Ser-5 prior to chromatin loading. As a result, MCM2, along with other MCM subunits accumulates with the chromatin during cell cycle re-entry [42]. However, both of the research groups concurred that Cdc7 phosphorylation of MCM2 had no effect on MCM complex formation [21, 42]. The difference between studies may indicate that biological and functional consequences of MCM2 phosphorylation by Cdc7 is regulated in a phosphosite-dependent manner. This finding is consistent with a study by Montagnoli et al. In this study, the authors demonstrated that Cdc7 phosphorylation of MCM2 isoforms showed different a affinity for chromatin, although their variable properties were similar during the cell cycle [24]. In addition, this study identified seven phosphosites in the N-terminus of MCM2 by Cdc7 (Ser-40/53/108), Cdk1/Cdk2 (Ser-13/27/41), and CK2 (Ser-139) in vitro. In cells, the MCM2 protein was phosphorylated on all of these sites. However, only Ser-40/53/108 was Cdc7-dependent in vivo. In non-synchronized cells, pSer-53 MCM2 was detected both in the soluble and chromatin-enriched fractions, whereas phosphorylated MCM2 at Ser-40 and Ser-108 was only detected in the soluble fractions. However, in cells homogeneously arrested in S-phase by hydroxyurea (HU), pSer-108 and pSer-40 MCM2 were detected in chromatin-associated fractions. In addition, Ser-108 has also been reported as an ataxia-telangiectasia-mutated (ATM)/ATM- and Rad3-related (ATR) kinase phosphosite on MCM2 [43, 44]. This may reflect an overlapping regulatory function between Cdc7 and ATM/ATR in replication fork machinery under replication stress. Certain studies [45–47] also confirm this phenomenon. Furthermore, Montagnoli et al. demonstrated that MCM2 phosphorylation at Ser-41 (putative CDK-dependent site) and Ser-139 (putative CK2-dependent site) were not affected by reducing Cdc7 [24]. In the study by Tsuji et al., the two sites were Cdc7-dependent phosphosites [21]. This may imply that multiple kinases were involved in N-terminal phosphorylation of multiple sites in MCM2. However, results obtained from the two groups suggested that Ser-40 and Ser-108 in MCM2 were Cdc7-dependent phosphosites, both in vivo and in vitro [48]. However, functional consequences of the two phosphosites remained unclear in their study. In contrast to MCM2, phosphorylation of MCM4 remains less-studied. Masai et al. demonstrated that Cdc7-mediated N-terminal phosphorylation of MCM4 contributes to initiation of DNA replication and cell growth by promoting chromatin loading of Cdc45, a key replisome member [22]. Furthermore, N-terminal phosphorylation of MCM2, MCM4, and MCM6 might be redundant, but esssential in the initiation of DNA replication. Because the combination of MCM2, -4, and -6N-terminal mutations resulted in the loss of cell viability, these mutations alone did not affect DNA replication or growth [22]. MCM subunits have similar function in replication regulation, but they are not identical. A recent study revealed that Cdc7/Dbf4-dependent hyperphosphorylation of MCM4, but not MCM2, correlates with replication initiation [49]. Thus, a majority of MCMs phosphosites undergo dynamic phosphorylation mediated by Cdc7 in a cell-cycle-specific manner. This phosphorylation pattern is consistent with the role of MCM2–7 in the initiation of DNA replication, which confirms that genomic duplication occurs once per cell cycle.
Table 1.
Summary of MCM phosphorylation in human cancer cells
| Protein | Kinase | Phosphorylation site or domain | Biological and functional significance | References |
|---|---|---|---|---|
| MCM2 | Cdc7 | S27, S41, S139 | Suggested to be essential for initiation of DNA replication and ATPase activity of MCM complex | [16] |
| Cdc7 | S5 | Promotes pre-RC assembly during cell cycle re-entry | [37] | |
| Cdc7 | S40, S53, S108 | Regulates MCM2’s chromatin loading in a site-dependent manner | [19] | |
| Cdc7 | S40, S108 | Unclear | [43] | |
| MCM4 | Cdc7 | N-terminal | Promotes MCM4’s interaction with Cdc45 on chromatin. Suggested to be essential for initiation of DNA replication and cell growth | [17] |
| MCM3 | Cdk2/CycE | T722 | Promotes MCM3’s chromatin loading. Regulates S phase checkpoint activation | [12] |
| MCM4 | Cdk2 | S3, T7, S32, S54, T110 | Decreases chromatin loading of MCM complex to avoid re-replication during mitosis | [45] |
| Cdk1, Cdk2 | S3, T7, T19, S32, S54, S88, T110 | Inhibits chromatin loading and helicase activity of MCM complex. Blocks DNA replication through inactivation of MCM complex | [20, 46–49, 51] | |
| MCM7 | Cdk2/CycE, Cdk1/CycB | S121 | Promotes MCM complex formation. Regulates S checkpoint activation and mitotic exit | [13] |
| MCM2 | ATR | S108 | Responds to replication stress and stabilizes replication forks | [39] |
| ATR | S108 | Responds to replication stress | [38] | |
| MCM3 | ATM | S535 | Responds to replication stress | [38] |
| ATM, ATR | S728 | Responds to replication stress | [58] | |
| MCM6 | ATR | S13 | Responds to replication stress | [59] |
| MCM2 | SIK1 | Unclear | Activates helicase activity of MCM complex during DNA replication | [61] |
| MCM3 | DAPK | S160 | Unclear | [62] |
| MCM4 | EBV-PK | T19, T110 | Blocks DNA replication through inactivation of DNA unwinding by the MCM4/6/7 complex. Leads to cell growth arrest | [26] |
| MCM7 | p56Lyn | Tyr600 | Promotes MCM complex formation, chromatin loading, DNA synthesis and cancer cell proliferation. Pathologically correlates with poor survival of breast cancer patients | [23] |
| Akt | Unclear | Increases MCM7’s chromatin loading and MCM complex formation. Promotes DNA replication and cancer cell proliferation | [24] | |
| ILK | Unclear | Inhibits MCM7’s chromatin loading and cancer cell growth | [25] |
Phosphorylation of MCMs by Cdk
Cell cycle progression through each phase is tightly regulated by Cdks and their regulatory proteins, cyclins. Alterations in these proteins, lead to uncontrolled cell division, which is a characteristic of many cancers. Several lines of evidences have shown that MCMs are substrates of Cdks and some Cdk-dependent phosphosites on MCMs have been identified and characterized (Table 1). Moreover, a few phosphosites are stimulated by Cdk and Cdc7 simultaneously [22], indicating the functional crosstalk between the two classes of kinases. In general, Cdk-mediated phosphorylation of MCMs preferentially contributes to the cell cycle regulation. A study by Li et al. [17] revealed that Cdk2/CycE phosphorylation of MCM3 Thr-722 promoted its chromatin loading. Excessive MCM3 chromatin loading activated the checkpoint pathway, which as a result blocked the S phase entry, but did not affect mitotic exit. Similarly, phosphorylation of MCM7 also affects its function in cell cycle procession. In a study conducted by Wei et al. [18], it was found that both Cdk2/CycE and Cdk1/CycB phosphorylate MCM7 at Ser-121. Moreover, overexpression of the wild type (WT) MCM7, but not the MCM7-S121 mutant, resulted in an S phase block. This then activates the checkpoint kinase 1 (Chk1) checkpoint pathway through single-stranded DNA (ssDNA) accumulation in a p53-dependent manner. Phosphorylation of MCM7 at Ser-121 also contributes to the formation of MCM complex for a proper mitotic exit. Taken together, Cdk phosphorylation of MCM7 plays an important role in both activation of S phase checkpoint and regulation of proper M phase progression. Besides, the MCM7-S121 mutant, but not wild type MCM7, binds more efficiently to the chromatin [18]. This may indicate that the phosphorylation of MCM7 hinders its loading onto DNA. Similarly, Cdk1/2-dependent phosphorylation of MCM4 at multiple sites decreases the binding of MCM complex to DNA, avoiding re-replication during mitosis [50, 51]. Therefore, phosphorylation of MCM7 at Ser-121 may also contribute to proper mitotic exit by inhibiting the MCM complex association with chromatin. Additionally, MCM4 phosphorylation not only induced the MCM complex release from chromatin, but also inactivated the complex. Previous studies indicated that MCM4 phosphorylation at specific sites leads to loss of subassembly MCM4/6/7 DNA helicase activity [52, 53], which is necessary for initiating replication [54, 55]. Cdk-mediated phosphorylation of MCM4 may also be a critical checkpoint in the cell cycle because MCM4 phosphorylation levels at several Cdk sites, stimulated by HU or ultraviolet (UV) irradiation, correlated inversely with the level of DNA synthesis to some extent [56]. In addition to Cdk, ATR-Chk1 is also involved in this phosphorylation [56]. Another study by Komamura-Kohno demonstrated that some Cdk-dependent sites of MCM4 were differentially phosphorylated during the cell cycle [25], indicating MCM4 phosphorylation may have several distinct and site-specific roles. Interestingly, studies on the nuclear localization of chromatin-bound MCM4, phosphorylated at the Ser-3 and Ser-32 by Cdk2, showed that the nuclear localization was not generally colocalized with replicating DNA [25]. Similarly, Tsuji et al. have also reported that chromatin-bound Cdc7/Dbf4 phosphorylated MCM2 did not co-localize with replication foci during G1/S and S phase [21]. One explanation for this discrepancy is that MCM helicase work at a distance from replication forks as a rotary motor that pumps DNA along its helical axis by simple rotation [57, 58].
Phosphorylation of MCMs by ATM/ATR
Replication stress, commonly known as the slowing down or stalling of replication forks, is a major source of mutations that contribute to genomic instability and tumorigenesis [59, 60]. This threat triggers a DNA damage response (DDR). ATM and ATR, the master regulators of DDR, phosphorylate substrates to stabilize the DNA replication fork and activate cell cycle checkpoints. The checkpoint signaling pathways slow cell cycle progression, thus allowing the cell to recover and prevent inappropriate entry into mitosis [61, 62]. Several studies have suggested that ATM/ATR-dependent checkpoint pathways are directly linked to the subunits of the MCM complex (Table 1). It was suggested by Shi et al. [63] that ATM phosphorylated human MCM3 C-terminal at Ser-728, in response to DNA damage. The MCM3 phosphorylated form was preferentially localized to the nucleoplasmic fraction, and the phosphorylation did not alter its association with chromatin and other MCM subunits. One interpretation of this finding is that the chromatin-bound MCM3 C-terminal is inaccessible to ATM because of special structural changes, which may allow chromatin-bound MCM3 to escape checkpoint-mediated inhibition or may prevent chromatin loading of nucleoplasmic MCM3 during DNA damage [63]. ATR also contributed to MCM3 C-terminal phosphorylation in response to DNA replication stress. However, ATM still accounts for a major part of phosphorylation [63]. Therefore, ATM and ATR may contribute differently to MCM subunits phosphorylation under replication stress. Also, it has been reported that ATM phosphorylated MCM3 at Ser-535 in response to ionizing radiation (IR) [43]. Whereas ATR phosphorylated MCM2 at Ser-108 in response to multiple forms of DNA damage and stalling of replication forks including: IR, UV light, HU, and polyamides [43, 44]. In contrast to most ATM/ATR substrates, pSer-108 MCM2 was detected in the absence of exogenous DNA damage [43]. This corresponds with other reports on MCM2 phosphorylated at Ser-108 by Cdc7 [24, 48, 49]. In a recent study, Ser-13 at MCM6 was also reported to be a novel putative ATR target site in response to replication stress [64]. The identification of these ATM/ATR-dependent phosphosites suggests that MCM phosphorylation may be required for cells to repair DNA damage, restart replication, and recover from arrest coordinating with the checkpoint under replication stress. In concordance with this hypothesis, Izawa et al. reported that HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2), an E3 ligase critical for DNA damage repair pathways, regulates DNA replication progression and origin firing by facilitating MCM2 phosphorylation under replication stress [65].
Phosphorylation of MCMs by other kinases
Other than the three classes of kinases Cdc7, Cdk, and ATM/ATR, several additional kinases were also shown to be involved in MCM phosphorylation (Table 1). Among these, p56Lyn-, Akt-, and integrin-linked kinase (ILK)-mediated MCM7 phosphorylation primarily correlates with the development of human cancer. MCM7 phosphorylation at Tyr-600 mediated by epidermal growth factor receptor (EGFR)-p56Lyn-axis promotes MCM complex assembly and chromatin loading, consequently enhancing DNA synthesis and cancer cell proliferation [28]. Furthermore, the Tyr-600 phosphorylation of MCM7 correlates with poor survival of breast cancer patients [28]. Similarly, Akt-dependent phosphorylation of MCM7, mediated by receptor for activated C kinase 1 (RACK1), also facilitates association of MCM7 with chromatin and MCM complex formation. As a result, this promotes DNA replication and cell proliferation in non-small cell lung cancer [29]. In contrast to these oncogenic roles, MCM7 phosphorylation mediated by the integrin ɑ7 (ITGA7)-ILK axis reduces MCM7 chromatin association thus inhibiting cell growth. This suggests that phosphorylation may be a critical event in the ITGA7 tumor suppression signaling pathway [30]. However, specific Akt-dependent and ILK-dependent phosphosites for MCM7 remain unclear. Phosphosites of MCM7 and the signaling pathways involved may affect the role of MCM7 phosphorylation in cancer development. In addition to MCM7, MCM2 was shown to be a substrate of salt-induciblekinase1 (SIK1) [66], MCM3 was reported to be phosphorylated by death-associated protein kinase (DAPK) at Ser-160 [67], and MCM4 was phosphorylated by Epstein–Barr virus-encoded protein kinase (EBV-PK) at Thr-19 and Thr-110 [31]. Among these, SIK1-dependent MCM2 phosphorylation, mediated by Sld5, is required for MCM helicase activity, but it does not affect the chromatin association of MCM2 [66]. The authors identified five SIK1-dependent phosphosites on MCM2 in vitro; however, specific phosphosites in cells need further identification. In HeLa cells, EBV-PK phosphorylates MCM4 and shares at least two of the same sites (Thr-19 and Thr-110) with Cdk2. This results in the loss of MCM4/6/7 subassembly’s enzyme activity, which leads to cell growth arrest [31]. In addition, most of the pThr-110 MCM4 detaches from chromatin; however, about half of pThr-19 MCM4 is bound to chromatin [31]. However, the overall chromatin-bound MCM did not change, indicating that other mechanisms may be involved. Besides, EBV-PK might also phosphorylate MCM6 and additional sites of MCM4 to block DNA replication in Epstein–Barr virus (EBV)-infected cells [31]. In contrast to the MCM2 and MCM4 phosphorylation, the consequences of DAPK-mediated MCM3 phosphorylation remains to be elucidated [67].
Conclusions
MCM proteins are regulated by multiple kinases and the consequences of these phosphorylation events reflect the critical integration of DNA replication with cell cycle and the checkpoint response (Fig. 1). However, the temporal sequence of different phosphorylation events and the precise function of phosphorylation at different sites need further investigation. Many phosphosites on MCMs identified in vitro also need further identification in cells (Table 2). Current results indicate that the MCM phosphorylation may contribute to its function by affecting MCM complex formation, chromatin binding, and (or) the helicase activity. Phosphorylation of specific sites is likely to trigger detachment of the MCM complex from chromatin. The majority of chromatin loading and phosphorylation events are regulated in a cell cycle-dependent manner. Recently, it has be found that several MCM dephosphorylation events, mediated by Rap1-interacting factor 1 (RIF1)-protein phosphatase 1 (PP1) [49, 68] and phosphatase and tensin homolog deleted on chromosome ten (PTEN) [69], contribute to the strict regulation of DNA replication and replisome stability. Collectively, the phosphorylation and dephosphorylation of MCM proteins regulate cell cycle progression and protect genomic stability, whereas dysfunctional phosphorylation events appear to correlate with the occurrence and development of cancers (Fig. 2). Recently, protein kinase inhibitors targeting essential cell cycle regulators or major signaling pathways have become a major field of investigation for new therapeutic strategies. Drugs targeting MCM phosphorylation may be a potential method for cancer therapy. Evidence has shown that Cdc7-selective inhibitors may decrease MCM2 phosphorylation, inhibit DNA synthesis, and cancer cell viability [70–78]. Further investigation of the functional significance of MCMs phosphorylation in cancer cells is required as it may contribute to the development of novel cancer targets.
Fig. 1.
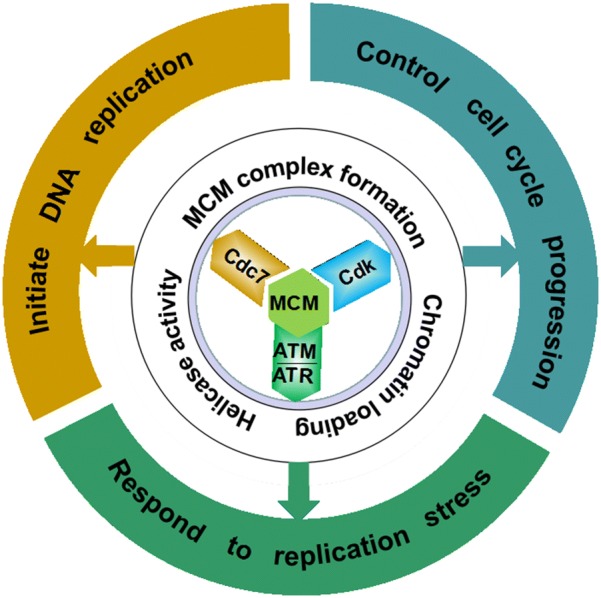
Schematic diagram summarizing the roles of MCM phosphorylation mediated by three classes of kinases Cdc7, Cdk and ATM/ATR. Although many functional crosstalks exist between MCM kinases-mediated phosphorylation events, evidence shows that Cdc7-dependent MCM phosphorylation primarily promotes the initiation of DNA replication, Cdk-dependent MCM phosphorylation mainly contributes to cell cycle progression regulation, and ATM/ATR-dependent MCM phosphorylation primarily responds to replication stress. MCM phosphorylation contributes to these various functions primarily by affecting MCM complex formation, chromatin binding, and (or) helicase activity
Table 2.
Summary of potential phosphosites on MCMs
Fig. 2.
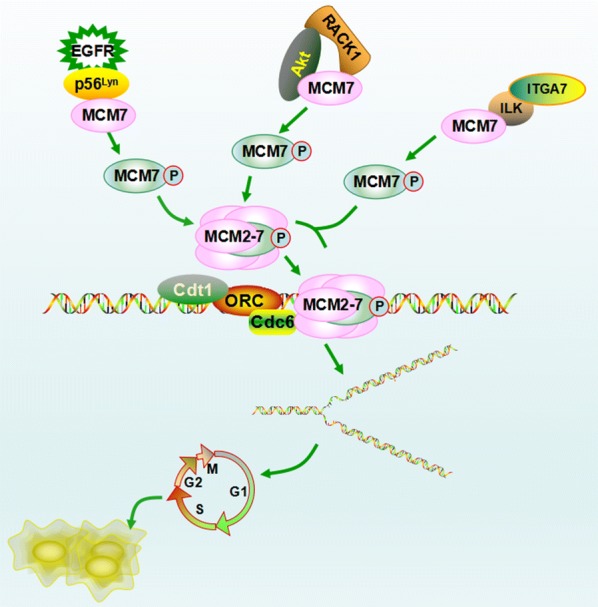
Roles of MCM phosphorylation mediated by p56Lyn, Akt and ILK in cancer development. Phosphorylation of MCM7 mediated by EGFR-p56Lyn and RACK1-Akt promotes MCM complex assembly and chromatin loading, therefore enhancing DNA synthesis and cancer cell proliferation. In contrast, MCM7 phosphorylation mediated by ITGA7-ILK axis reduces MCM7 chromatin association, inhibiting cell growth
Authors’ contributions
All authors have contributed to manuscript writing. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 81372497 to H.-T. Xu) and Program for Liaoning Excellent Talents in University (Grant No. LR2015067 to H.-T. Xu). We thank Dr. Yuchen Han (China Medical University) for her kind advice and support for the manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ATM
ataxia-telangiectasia-mutated kinase
- ATR
ATM- and Rad3-related kinase
- Cdc6
cell division cycle 6
- Cdc7
cell division cycle 7
- Cdc45
cell division cycle 45
- Cdk
cyclin-dependent kinase
- Cdt1
Cdc10 dependent transcript 1
- Chk1
checkpoint kinase 1
- CMG
Cdc45–MCMs–GINS
- DAPK
death-associated protein kinase
- DDK
Dbf4-dependent kinase
- DDR
DNA damage response
- EBV
Epstein–Barr virus
- EBV-PK
EBV-encoded protein kinase
- EGFR
epidermal growth factor receptor
- GINS
Go, Ichi, Ni, and San
- HERC2
HECT and RLD domain containing E3 ubiquitin protein ligase 2
- HU
hydroxyurea
- ILK
integrin-linked kinase
- IMEs
immortalized human diploid mammary epithelials
- IMFs
immortalized human fibroblasts
- ITGA7
integrin α7
- IR
ionizing radiation
- MEFs
mouse embryo fibroblasts
- NHDF
human dermal fibroblast
- ORC
origin recognition complex
- PP1
protein phosphatase 1
- pre-RCs
pre-replicative complexes
- PTEN
phosphatase and tensin homolog deleted on chromosome ten
- RACK1
receptor for activated C kinase 1
- RIF1
Rap1-interacting factor 1
- SIK1
salt-induciblekinase1
Contributor Information
Liangru Fei, Email: liangrufei@hotmail.com.
Hongtao Xu, Phone: 86-18900911667, Email: xuht@cmu.edu.cn.
References
- 1.Leman AR, Noguchi E. The replication fork: understanding the eukaryotic replication machinery and the challenges to genome duplication. Genes-Basel. 2013;4:1–32. doi: 10.3390/genes4010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waga S, Stillman B. The DNA replication fork in eukaryotic cells. Annu Rev Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 3.Barry ER, Bell SD. DNA replication in the archaea. Microbiol Mol Biol Rev. 2006;70:876–887. doi: 10.1128/MMBR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg P, Burgers PMJ. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol. 2005;40:115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- 5.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 6.Duzdevich D, Warner MD, Ticau S, Ivica NA, Bell SP, Greene EC. The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol Cell. 2015;58:483–494. doi: 10.1016/j.molcel.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell SP, Kaguni JM. Helicase loading at chromosomal origins of replication. Csh Perspect Biol. 2013;5:a010124. doi: 10.1101/cshperspect.a010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evrin C, Fernandez-Cid A, Riera A, Zech J, Clarke P, Herrera MC, et al. The ORC/Cdc6/MCM2–7 complex facilitates MCM2–7 dimerization during prereplicative complex formation. Nucleic Acids Res. 2014;42:2257–2269. doi: 10.1093/nar/gkt1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Cid A, Riera A, Tognetti S, Herrera MC, Samel S, Evrin C, et al. An ORC/Cdc6/MCM2–7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol Cell. 2013;50:577–588. doi: 10.1016/j.molcel.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 10.Bruck I, Dhingra N, Kaplan DL. A positive amplification mechanism involving a kinase and replication initiation factor helps assemble the replication fork helicase. J Biol Chem. 2017;292:3062–3073. doi: 10.1074/jbc.M116.772368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izumi M, Mizuno T, Yanagi K, Sugimura K, Okumura K, Imamoto N, et al. The Mcm2–7-interacting domain of human mini-chromosome maintenance 10 (Mcm10) protein is important for stable chromatin association and origin firing. J Biol Chem. 2017;292:13008–13021. doi: 10.1074/jbc.M117.779371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeeles JTP, Deegan TD, Janska A, Early A, Diffley JFX. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015;519:431–435. doi: 10.1038/nature14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruck I, Kaplan DL. Conserved mechanism for coordinating replication fork helicase assembly with phosphorylation of the helicase. Proc Natl Acad Sci USA. 2015;112:11223–11228. doi: 10.1073/pnas.1509608112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheu YJ, Stillman B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature. 2010;463:113–127. doi: 10.1038/nature08647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder M, Huang XY, Zhang JJ. The minichromosome maintenance proteins 2–7 (MCM2–7) are necessary for RNA polymerase II (Pol II)-mediated transcription. J Biol Chem. 2009;284:13466–13472. doi: 10.1074/jbc.M809471200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stead BE, Brandl CJ, Sandre MK, Davey MJ. Mcm2 phosphorylation and the response to replicative stress. Bmc Genet. 2012;13:36. doi: 10.1186/1471-2156-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li JH, Deng M, Wei Q, Liu T, Tong XM, Ye X. Phosphorylation of MCM3 protein by cyclin E/Cyclin-dependent kinase 2 (Cdk2) regulates its function in cell cycle. J Biol Chem. 2011;286:39776–39785. doi: 10.1074/jbc.M111.226464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Q, Li JH, Liu T, Tong XM, Ye X. Phosphorylation of minichromosome maintenance protein 7 (MCM7) by cyclin/cyclin-dependent kinase affects its function in cell cycle regulation. J Biol Chem. 2013;288:19715–19725. doi: 10.1074/jbc.M112.449652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen ZH, Yu YP, Michalopoulos G, Nelson J, Luo JH. The DNA replication licensing factor miniature chromosome maintenance 7 is essential for rna splicing of epidermal growth factor receptor, c-Met, and platelet-derived growth factor receptor. J Biol Chem. 2015;290:1404–1411. doi: 10.1074/jbc.M114.622761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bochman ML, Schwacha A. The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiol Mol Biol R. 2009;73:652–683. doi: 10.1128/MMBR.00019-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuji T, Ficarro SB, Jiang W. Essential role of phosphorylation of MCM2 by Cdc7/Dbf4 in the initiation of DNA replication in mammalian cells. Mol Biol Cell. 2006;17:4459–4472. doi: 10.1091/mbc.e06-03-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masai H, Taniyama C, Ogino K, Matsui E, Kakusho N, Matsumoto S, et al. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J Biol Chem. 2006;281:39249–39261. doi: 10.1074/jbc.M608935200. [DOI] [PubMed] [Google Scholar]
- 23.Tan BCM, Liu HA, Lin CL, Lee SC. Functional cooperation between FACT and MCM is coordinated with cell cycle and differential complex formation. J Biomed Sci. 2010;17:11. doi: 10.1186/1423-0127-17-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, et al. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J Biol Chem. 2006;281:10281–10290. doi: 10.1074/jbc.M512921200. [DOI] [PubMed] [Google Scholar]
- 25.Komamura-Kohno Y, Karasawa-Shimizu K, Saitoh T, Sato M, Hanaoka F, Tanaka S, et al. Site-specific phosphorylation of MCM4 during the cell cycle in mammalian cells. FEBS J. 2006;273:1224–1239. doi: 10.1111/j.1742-4658.2006.05146.x. [DOI] [PubMed] [Google Scholar]
- 26.Ren XH, Li J, Xia B, Liu W, Yang XF, Hong WX, et al. Phosphoproteomic analyses of L-02 liver cells exposed to trichloroethylene. Toxicol Mech Method. 2015;25:459–466. doi: 10.3109/15376516.2015.1045655. [DOI] [PubMed] [Google Scholar]
- 27.Bonda DJ, Evans TA, Santocanale C, Llosa JC, Vina J, Bajic VP, et al. Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging-Us. 2009;1:382–388. doi: 10.18632/aging.100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang TH, Huo LF, Wang YN, Xia WY, Wei YK, Chang SS, et al. Epidermal growth factor receptor potentiates MCM7-mediated DNA replication through tyrosine phosphorylation of Lyn kinase in human cancers. Cancer Cell. 2013;23:796–810. doi: 10.1016/j.ccr.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fei LR, Ma YN, Zhang MY, Liu XF, Luo Y, Wang CC, et al. RACK1 promotes lung cancer cell growth via an MCM7/RACK1/Akt signaling complex. Oncotarget. 2017;8:40501–40513. doi: 10.18632/oncotarget.17120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han YC, Yu YP, Nelson J, Wu CY, Wang H, Michalopoulos GK, et al. Interaction of integrin-linked kinase and miniature chromosome maintenance 7-mediating integrin alpha 7 induced cell growth suppression. Cancer Res. 2010;70:4375–4384. doi: 10.1158/0008-5472.CAN-09-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kudoh A, Daikoku T, Ishimi Y, Kawaguchi Y, Shirata N, Iwahori S, et al. Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4–MCM6–MCM7 complex during Epstein–Barr virus productive replication. J Virol. 2006;80:10064–10072. doi: 10.1128/JVI.00678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deegan TD, Yeeles JTP, Diffley JFX. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016;35:961–973. doi: 10.15252/embj.201593552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramer MD, Suman ES, Richter H, Stanger K, Spranger M, Bieberstein N, et al. Dbf4 and Cdc7 proteins promote DNA replication through interactions with distinct Mcm2–7 protein subunits. J Biol Chem. 2013;288:14926–14935. doi: 10.1074/jbc.M112.392910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francis LI, Randell JCW, Takara TJ, Uchima L, Bell SP. Incorporation into the prereplicative complex activates the Mcm2–7 helicase for Cdc7-Dbf4 phosphorylation. Gene Dev. 2009;23:643–654. doi: 10.1101/gad.1759609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheu YJ, Kinney JB, Lengronne A, Pasero P, Stillman B. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc Natl Acad Sci USA. 2014;111:E1899–E1908. doi: 10.1073/pnas.1404063111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamura R, Fukatsu R, Kakusho N, Cho YS, Taniyama C, Yamazaki S, et al. Molecular mechanism of activation of human Cdc7 kinase: bipartite interaction with Dbf4/activator of S phase kinase (ASK) activation subunit stimulates ATP binding and substrate recognition. J Biol Chem. 2011;286:23031–23043. doi: 10.1074/jbc.M111.243311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshizawa-Sugata N, Ishii A, Taniyama C, Matsui E, Arai K, Masai H. A second human Dbf4/ASK-related protein, Drf1/ASKL1, is required for efficient progression of S and M phases. J Biol Chem. 2005;280:13062–13070. doi: 10.1074/jbc.M411653200. [DOI] [PubMed] [Google Scholar]
- 38.Li Q, Xie WF, Wang N, Li CK, Wang MD. CDC7-dependent transcriptional regulation of RAD54L is essential for tumorigenicity and radio-resistance of glioblastoma. Transl Oncol. 2018;11:300–306. doi: 10.1016/j.tranon.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melling N, Muth J, Simon R, Bokemeyer C, Terracciano L, Sauter G, et al. Cdc7 overexpression is an independent prognostic marker and a potential therapeutic target in colorectal cancer. Diagn Pathol. 2015;10:125. doi: 10.1186/s13000-015-0360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaafari-Ashkavandi Z, Ashraf MJ, Abbaspoorfard AA. Overexpression of CDC7 in malignant salivary gland tumors correlates with tumor differentiation. Braz J Otorhinolaryngol. 2017 doi: 10.1016/j.bjorl.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huggett MT, Tudzarova S, Proctor I, Loddo M, Keane MG, Stoeber K, et al. Cdc7 is a potent anti-cancer target in pancreatic cancer due to abrogation of the DNA origin activation checkpoint. Oncotarget. 2016;7:18495–18507. doi: 10.18632/oncotarget.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chuang LC, Teixeira LK, Wohlschlegel JA, Henze M, Yates JR, Mendez J, et al. Phosphorylation of Mcm2 by Cdc7 promotes pre-replication complex assembly during cell-cycle re-entry. Mol Cell. 2009;35:206–216. doi: 10.1016/j.molcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci USA. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez TF, Phillips JW, Karanja KK, Polaczek P, Wang CM, Li BC, et al. Replication stress by Py-Im polyamides induces a non-canonical ATR-dependent checkpoint response. Nucleic Acids Res. 2014;42:11546–11559. doi: 10.1093/nar/gku866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tudzarova S, Mulholland P, Dey A, Stoeber K, Okorokov AL, Williams GH. p53 controls CDC7 levels to reinforce G1 cell cycle arrest upon genotoxic stress. Cell Cycle. 2016;15:2958–2972. doi: 10.1080/15384101.2016.1231281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamada M, Watanabe K, Mistrik M, Vesela E, Protivankova I, Mailand N, et al. ATR-Chk1-APC/C-Cdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Gene Dev. 2013;27:2459–2472. doi: 10.1101/gad.224568.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng AN, Fan CC, Lo YK, Kuo CL, Wang HC, Lien IH, et al. Cdc7-Dbf4-mediated phosphorylation of HSP90-S164 stabilizes HSP90-HCLK2-MRN complex to enhance ATR/ATM signaling that overcomes replication stress in cancer. Sci Rep-Uk. 2017;7:17024. doi: 10.1038/s41598-017-17126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charych DH, Coyne M, Yabannavar A, Narberes J, Chow S, Wallroth M, et al. Inhibition of Cdc7/Dbf4 kinase activity affects specific phosphorylation sites on MCM2 in cancer cells. J Cell Biochem. 2008;104:1075–1086. doi: 10.1002/jcb.21698. [DOI] [PubMed] [Google Scholar]
- 49.Alver RC, Chadha GS, Gillespie PJ, Blow JJ. Reversal of DDK-mediated MCM phosphorylation by Rif1-PP1 regulates replication initiation and replisome stability independently of ATR/Chk1. Cell Rep. 2017;18:2508–2520. doi: 10.1016/j.celrep.2017.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu YH, Ishimi Y, Tanudji M, Lees E. Human CDK2 inhibition modifies the dynamics of chromatin-bound minichromosome maintenance complex and replication protein A. Cell Cycle. 2005;4:1254–1263. doi: 10.4161/cc.4.9.1975. [DOI] [PubMed] [Google Scholar]
- 51.Moritani M, Ishimi Y. Inhibition of DNA binding of MCM2–7 complex by phosphorylation with cyclin-dependent kinases. J Biochem. 2013;154:363–372. doi: 10.1093/jb/mvt062. [DOI] [PubMed] [Google Scholar]
- 52.Ishimi Y, Komamura-Kohno Y, You ZY, Omori A, Kitagawa M. Inhibition of Mcm 4,6,7 helicase activity by phosphorylation with cyclin A/Cdk2. J Biol Chem. 2000;275:16235–16241. doi: 10.1074/jbc.M909040199. [DOI] [PubMed] [Google Scholar]
- 53.Ishimi Y, Komamura-Kohno Y. Phosphorylation of Mcm4 at specific sites by cyclin-dependent kinase leads to loss of Mcm 4,6,7 helicase activity. J Biol Chem. 2001;276:34428–34433. doi: 10.1074/jbc.M104480200. [DOI] [PubMed] [Google Scholar]
- 54.Ishimi Y. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J Biol Chem. 1997;272:24508–24513. doi: 10.1074/jbc.272.39.24508. [DOI] [PubMed] [Google Scholar]
- 55.Lee JK, Hurwitz J. Processive DNA helicase activity of the minichromosome maintenance proteins 4, 6, and 7 complex requires forked DMA structures. Proc Natl Acad Sci USA. 2001;98:54–59. doi: 10.1073/pnas.98.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ishimi Y, Komamura-Kohno Y, Karasawa-Shimizu K, Yamada K. Levels of MCM4 phosphorylation and DNA synthesis in DNA replication block checkpoint control. J Struct Biol. 2004;146:234–241. doi: 10.1016/j.jsb.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 57.Sakakibara N, Kelman LM, Kelman Z. Unwinding the structure and function of the archaeal MCM helicase. Mol Microbiol. 2009;72:286–296. doi: 10.1111/j.1365-2958.2009.06663.x. [DOI] [PubMed] [Google Scholar]
- 58.Laskey RA, Madine MA. A rotary pumping model for helicase function of MCM proteins at a distance from replication forks. EMBO Rep. 2003;4:26–30. doi: 10.1038/sj.embor.embor706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitao H, Iimori M, Kataoka Y, Wakasa T, Tokunaga E, Saeki H, et al. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018;109:264–271. doi: 10.1111/cas.13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iyer DR, Rhind N. The intra-S checkpoint responses to DNA damage. Genes-Basel. 2017;8:74. doi: 10.3390/genes8020074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaudhury I, Koepp DM. Recovery from the DNA replication checkpoint. Genes (Basel) 2016;7:94. doi: 10.3390/genes7110094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi YL, Dodson GE, Mukhopadhyay PS, Shanware NP, Trinh AT, Tibbetts RS. Identification of carboxyl-terminal MCM3 phosphorylation sites using polyreactive phosphospecific antibodies. J Biol Chem. 2007;282:9236–9243. doi: 10.1074/jbc.M609256200. [DOI] [PubMed] [Google Scholar]
- 64.Wagner SA, Oehler H, Voigt A, Dalic D, Freiwald A, Serve H, et al. ATR inhibition rewires cellular signaling networks induced by replication stress. Proteomics. 2016;16:402–416. doi: 10.1002/pmic.201500172. [DOI] [PubMed] [Google Scholar]
- 65.Izawa N, Wu WW, Sato K, Nishikawa H, Kato A, Boku N, et al. HERC2 interacts with claspin and regulates DNA origin firing and replication fork progression. Cancer Res. 2011;71:5621–5625. doi: 10.1158/0008-5472.CAN-11-0385. [DOI] [PubMed] [Google Scholar]
- 66.Joshi K, Shah VJ, Maddika S. GINS complex protein Sld5 recruits SIK1 to activate MCM helicase during DNA replication. Cell Signal. 2016;28:1852–1862. doi: 10.1016/j.cellsig.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 67.Bialik S, Berissi H, Kimchi A. A high throughput proteomics screen identifies novel substrates of death-associated protein kinase. Mol Cell Proteomics. 2008;7:1089–1098. doi: 10.1074/mcp.M700579-MCP200. [DOI] [PubMed] [Google Scholar]
- 68.Hiraga S, Ly T, Garzon J, Horejsi Z, Ohkubo Y, Endo A, et al. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 2017;18:403–419. doi: 10.15252/embr.201641983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feng JW, Liang J, Li JJ, Li YQ, Liang H, Zhao XY, et al. PTEN controls the DNA replication process through MCM2 in response to replicative stress. Cell Rep. 2015;13:1295–1303. doi: 10.1016/j.celrep.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 70.Erbayraktar Z, Alural B, Erbayraktar RS, Erkan EP. Cell division cycle 7-kinase inhibitor PHA-767491 hydrochloride suppresses glioblastoma growth and invasiveness. Cancer Cell Int. 2016;16:88. doi: 10.1186/s12935-016-0364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li W, Zhao XL, Shang SQ, Shen HQ, Chen X. Dual inhibition of Cdc7 and Cdk9 by PHA-767491 suppresses hepatocarcinoma synergistically with 5-fluorouracil. Curr Cancer Drug Tar. 2015;15:196–204. doi: 10.2174/1568009615666150212112753. [DOI] [PubMed] [Google Scholar]
- 72.Kurasawa O, Homma M, Oguro Y, Miyazaki T, Mori K, Uchiyama N, et al. 2-Aminomethylthieno[3,2-d]pyrimidin-4(3H)-ones bearing 3-methylpyrazole hinge binding moiety: highly potent, selective, and time-dependent inhibitors of Cdc7 kinase. Bioorgan Med Chem. 2017;25:3658–3670. doi: 10.1016/j.bmc.2017.04.044. [DOI] [PubMed] [Google Scholar]
- 73.FitzGerald J, Murillo LS, O’Brien G, O’Connell E, O’Connor A, Wu K, et al. A high through-put screen for small molecules modulating MCM2 phosphorylation identifies ryuvidine as an inducer of the DNA damage response. PLoS ONE. 2014;9:e98891. doi: 10.1371/journal.pone.0098891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koltun ES, Tsuhako AL, Brown DS, Aay N, Arcalas A, Chan V, et al. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg Med Chem Lett. 2012;22:3727–3731. doi: 10.1016/j.bmcl.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 75.Reichelt A, Bailis JM, Bartberger MD, Yao GM, Shu H, Kaller MR, et al. Synthesis and structure-activity relationship of trisubstituted thiazoles as Cdc7 kinase inhibitors. Eur J Med Chem. 2014;80:364–382. doi: 10.1016/j.ejmech.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 76.Irie T, Asami T, Sawa A, Uno Y, Hanada M, Taniyama C, et al. Discovery of novel furanone derivatives as potent Cdc7 kinase inhibitors. Eur J Med Chem. 2017;130:406–418. doi: 10.1016/j.ejmech.2017.02.030. [DOI] [PubMed] [Google Scholar]
- 77.Natoni A, Coyne MR, Jacobsen A, Rainey MD, O’Brien G, Healy S, et al. Characterization of a dual CDC7/CDK9 inhibitor in multiple myeloma cellular models. Cancers (Basel). 2013;5:901–918. doi: 10.3390/cancers5030901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sasi NK, Tiwari K, Soon FF, Bonte D, Wang T, Melcher K, et al. The potent Cdc7-Dbf4 (DDK) kinase inhibitor XL413 has limited activity in many cancer cell lines and discovery of potential new DDK inhibitor scaffolds. PLoS ONE. 2014;9:e113300. doi: 10.1371/journal.pone.0113300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.