

Abstract
Staphylococcal species are a leading cause of bacterial drug-resistant infections and associated mortality. One strategy to combat bacterial drug resistance is to revisit compromised targets, and to circumvent resistance mechanisms using structure-assisted drug discovery. The folate pathway is an ideal candidate for this approach. Antifolates target an essential metabolic pathway, and the necessary detailed structural information is now available for most enzymes in this pathway. Dihydropteroate synthase (DHPS) is the target of the sulfonamide class of drugs, and its well characterized mechanism facilitates detailed analyses of how drug resistance has evolved. Here, we surveyed clinical genetic sequencing data in S. aureus to distinguish natural amino acid variations in DHPS from those that are associated with sulfonamide resistance. Five mutations were identified, F17L, S18L, T51M, E208K, and KE257_dup. Their contribution to resistance and their cost to the catalytic properties of DHPS were evaluated using a combination of biochemical, biophysical and microbiological susceptibility studies. These studies show that F17L, S18L, and T51M directly lead to sulfonamide resistance while unexpectedly increasing susceptibility to trimethoprim, which targets the downstream enzyme dihydrofolate reductase. The secondary mutations E208K and KE257_dup restore trimethoprim susceptibility closer to wild-type levels while further increasing sulfonamide resistance. Structural studies reveal that these mutations appear to selectively disfavor the binding of the sulfonamides by sterically blocking an outer ring moiety that is not present in the substrate. This emphasizes that new inhibitors must be designed that strictly stay within the substrate volume in the context of the transition state.
Keywords: infectious disease, antibiotics, drug discovery, fitness cost, kinetics, drug susceptibility, bacterial genetics
Introduction
Sulfonamides are the oldest class of synthetic antibiotics. They have been in clinical use for over 75 years, with proven efficacy against many microbial infections (Domagk, 1935; Bermingham and Derrick, 2002). Sulfonamides target the enzyme dihydropteroate synthase (DHPS) that catalyzes a key step in microbial folate biosynthesis, the production of 7,8-dihydropteroate from para-aminobenzoic acid (pABA) and dihydropterin pyrophosphate (DHPP). Sulfonamides exert their antimicrobial action in two ways, by directly competing with the substrate pABA and through the formation of pterin-sulfa dead-end metabolic products (Roland et al., 1979). Prokaryotes and lower eukaryotes rely on this pathway for the de novo synthesis of folate that is a critically important cell metabolite, and disruption of folate biosynthesis therefore severely curtails their growth. In contrast, higher eukaryotes obtain folate directly from their diet and have dispensed with the pathway. The universal presence of DHPS in lower organisms and its absence in higher organisms explains why sulfonamides have been successful as broad-spectrum antimicrobials (Bermingham and Derrick, 2002).
Today, sulfonamides are mainly used in a fix dose combination with trimethoprim (TMP), a dihydrofolate reductase (DHFR) inhibitor. Co-trimoxazole, a combination of sulfamethoxazole (SMX), and TMP, is the most commonly prescribed. This cheap and orally bioavailable combination is used as a second-line therapy to treat a wide variety of bacterial infections including urinary tract infections (UTIs), bronchitis, traveler's diarrhea, and methicillin-resistant Staphylococcus aureus (MRSA) infections. Application of co-trimoxazole prophylaxis to prevent Pneumocystis jirovecii infections in immunosuppressed patients, such as those undergoing intensive cancer chemotherapy or with advanced HIV infections, has also emerged as a particularly important clinical application (Bermingham and Derrick, 2002).
The emergence of multidrug and pan resistant bacterial pathogens is an alarming and increasing phenomenon that requires immediate action (Boucher et al., 2009). To tackle this problem, we are revisiting previously identified antimicrobial targets and applying new strategies to develop inhibitors that are less prone to resistance mechanisms. Key to this approach is gaining an improved understanding of the targets' biochemical mechanisms, active site structures and resistance mechanisms. In many ways, DHPS is the perfect candidate for such an approach. Structurally and mechanistically, DHPS has been well characterized. The crystal structures of DHPS have been determined from 15 microbial species within the last 20 years, and more recent structural and computational studies from our group have revealed the ordered SN1 catalytic mechanism and the detailed configuration of the near transition state (Yun et al., 2012). These new insights have already enabled us to generate pyridazine derivatives with improved DHPS inhibition, identify allosteric inhibitors that hinder product release, and develop inhibitory pterin-sulfa conjugates (Zhao et al., 2012, 2016; Hammoudeh et al., 2014).
In this study, we focus on the structural and mechanistic basis of sulfonamide resistance in S. aureus DHPS (SaDHPS), and analyze how recurring resistance mutations can selectively disfavor the binding of sulfonamides while retaining the necessary fitness of the enzyme. Crystal structures of DHPS containing pterin•PPi•Mg•pABA and pterin•PPi•Mg•sulfonamide have revealed that pABA and sulfonamides occupy the same locale in the near transition state (Yun et al., 2012). Our focus will be on this locale and how the resistance mutations modulate its structure and dynamics to selectively disfavor the binding of the drug. Our studies proceeded in three stages; (1) bioinformatics analysis to identify the key resistance mutations across organisms, (2) biochemical, resistance and fitness analyses of these mutations in DHPS from S. aureus, and (3) structural and computational analyses to understand the mechanistic basis of these mutations. Our goal is to use these results to support ongoing drug discovery efforts toward this enzyme and to develop lead compounds that are not cross-resistant to sulfonamides.
Results
Primary and secondary mutations confer sulfonamide resistance
The increasing prevalence of MRSA during the past two decades and the associated sequencing of clinical isolates has generated a large dataset of SaDHPS sequence variations in the DHPS-encoding folP gene, including those that are found in sulfonamide resistant strains. We rigorously analyzed the available data up to and including 2014 to identify variations that are clearly associated with sulfonamide resistance. We identified two classes of resistance-associated mutations; primary mutations that are directly associated with sulfonamide resistance and secondary mutations that are only found in the presence of the primary mutations. An important goal of this analysis was to differentiate these mutations from the natural variations in SaDHPS that are present in sulfonamide susceptible strains but do not directly contribute to resistance. The results of this survey are summarized in Table 1. F17L, S18L, and T51M emerge as primary mutations; F17L occurs at the highest frequency, and S18L and T51M were each observed in less than 1% of sequences surveyed as single variants. E208K and KE257_dup (a duplication/insertion of Lys-Glu at position 257) were categorized as secondary mutations. E208K is found with both F17L and T51M, and KE257_dup is only found with F17L. The primary mutation S18L is not found with either of the two secondary mutations. In an earlier study, Hampele and coauthors identified 15 mutations among nine sulfonamide-resistant MRSA clinical isolates that are not present in the sulfonamide susceptible S. aureus Rosenbach 25923 strain (Hampele et al., 1997). Although this study also identified F17L, T51M, E208K and KE257_dup, our analysis showed that the 11 remaining mutations are found in sulfonamide susceptible S. aureus strain NCTC 8325 and are apparently natural polymorphisms in SaDHPS that do not contribute to sulfonamide resistance (Supplementary Table 1).
Table 1.
Survey of DHPS variants and known resistance to sulfonamides.
|
Sequence Background |
25923 | 8325* | 8325 | 25923 | 8325 | 8325 | 25923 | 8325 | 25923 | 8325 |
|---|---|---|---|---|---|---|---|---|---|---|
| Resistance Mutations | None | None | F17L KE257_dup |
T51M E208K |
T51M E208K |
F17L E208K |
F17L | F17L | T51M | S18L |
| % Sequences (n = 136) |
28 | 49 | 3 | 8 | 1.5 | 3.7 | 3 | 2 | 0.7 | 0.7 |
| Hampele Strain |
Group 1 | Group 2 | Group 3 | Group 4 | ||||||
| Hampele MIC (μg/mL) | 256–>1024 | 256–>1024 | >1024 | >1024 | ||||||
| Sulfonamide resistant | No | No | Yes | Yes | Yes | Yes | ND | ND | ND | ND |
Hampele strain group and MIC values have previously been published (Hampele et al., 1997).
Indicates that among the wild type folP genes surveyed and categorized as having an 8325 background, 31% lacked the natural variations T59S and L64M seen in the S. aureus strain Rosenbach 25923 (Supplementary Table 1).
A survey of other organisms was conducted to determine which of these mutations is conserved across species (Table 2). Mutations equivalent to F17L were found in Neisseria meningitidis and Escherichia coli, and mutations equivalent to T51M were found in Plasmodium species, Pneumocystis carinii, Mycobacterium leprae, and Streptococcus pneumoniae (Dallas et al., 1992; Fermer et al., 1995; Lane et al., 1997; Maskell et al., 1997; Wang et al., 1997b; Elena et al., 1998; Kazanjian et al., 1998; Mei et al., 1998; Kai et al., 1999; Williams et al., 2000; Pornthanakasem et al., 2016). A mutation homologous to E208K was also found in Plasmodium species but not in conjunction with any of the primary mutations (Pornthanakasem et al., 2016). We did not identify mutations equivalent to S18L or KE257_dup in other species. Alignment of DHPS sequences from S. aureus strains NCTC 8325 and Rosenbach 25923, and nine other microbial pathogens reveals that the primary mutations occur at highly conserved regions of the sequence while the secondary mutations occur in less conserved regions (Figure 1).
Table 2.
DHPS mutations associated with sulfonamide resistance in S. aureus, and their homologs in E. coli (Dallas et al., 1992), N. meningitidis (Fermer et al., 1995), B. anthracis (Yun et al., 2012), S. pneumoniae (Maskell et al., 1997), P. falciparum (Wang et al., 1997a), P. vivax (Pornthanakasem et al., 2016), M. leprae (Kai et al., 1999), and P. carinii (Mei et al., 1998).
| Classification | Mutation | Location | Similar Mutations in Other Microbial Species |
|---|---|---|---|
| Primary | F17L | Loop 1 | E. coli, N. meningitidis, B. anthracis |
| S18L | Loop 1 | None | |
| T51M | Loop 2 | S. pneumoniae*, P. falciparum*, P. vivax*, M. leprae*, P. carinii | |
| Secondary | E208K | α-helix Loop 7 | P. falciparum†, P. vivax† |
| KE257_Dup | α-helix 8 | None |
Indicate that mutations align with T51M and E208K, respectively, but are not strictly conserved (Figure 1).
Figure 1.

DHPS amino acid sequence alignment for S. aureus wild type representatives used in this study and nine other clinically relevant microbial pathogens. The five mutations that directly contribute to sulfonamide resistance are boxed in red. Amino acids that are 100% conserved, highly conserved and weakly conserved are highlighted in dark blue, light blue, and gray, respectively.
Thermal stabilities of sulfonamide resistant SaDHPS mutants
To explore how the resistance mutations identified in this study affect the stability of SaDHPS, the “wild type” enzyme from sulfonamide-susceptible strain Rosenbach 25923 and eight defined mutant enzymes were generated, expressed in E. coli and purified. Five of the mutant enzymes contained the single mutations F17L, S18L, T51M, E208K, and KE257_dup, and three contained the clinically observed double mutations F17L/E208K, T51M/E208K, and F17L/KE257_dup. The DHPS from sulfonamide susceptible S. aureus strain NCTC 8325 that harbors the 11 background variations not associated with sulfonamide resistance was also expressed and purified as a further control.
Thermal shift assays were employed to measure the denaturation temperatures (TM) of the purified proteins and to assess whether the mutations affect their stabilities (Table 3). These experiments were performed using Sypro-Orange that fluoresces when exposed to the hydrophobic interior of unfolded proteins upon denaturation. The SaDHPS primary mutations did not significantly alter the TM, falling within 1°C of wild type 25923. In contrast, the secondary mutations E208K and KE257_dup both result in a 3–4°C drop in TM, revealing that these secondary mutations destabilize the protein structure. Addition of the F17L or T51M mutations to E208K maintains the 3–4°C drop in TM whereas addition of the F17L mutation to KE257_dup restores the stability of the protein. These results are consistent with the SaDHPS crystal structure (Hampele et al., 1997). F17, S18, and T51 are in the two flexible loops 1 and 2 that are disordered in the absence of substrates and unlikely to contribute to the stability of the protein fold. In contrast, E208 is part of a salt bridge array involving R176, R204, and K207 that appears to stabilize this region of the protein. The structural basis for the reduction in stability produced by the KE257_dup mutation is not so obvious and it may simply result from its location at the dimer interface. However, the compensation in stability provided by F17L suggests that it may involve the dynamic allosteric communication between the interface and the active site that we previously described (Hammoudeh et al., 2014). The DHPS from NCTC 8325 is ~4°C more stable than wild type DHPS from Rosenbach 25923, indicating that the 11 background mutations generate an inherently more stable enzyme.
Table 3.
Changes in thermal stabilization of DHPS imparted by the observed sulfonamide resistant variations.
| DHPS Variant | TM (°C) | Δ TM (°C) | |
|---|---|---|---|
| Rosenbach 25923 | 38.63 ± 0.09 | ||
| NCTC 8325 | 42.48 ± 0.01 | ||
| Primary mutation | F17L | 39.14 ± 0.0 | 0.51 |
| S18L | 39.52 ± 0.003 | 0.89 | |
| T51M | 38.7 ± 0.09 | 0.07 | |
| Secondary mutation | E208K | 34.89 ± 0.04 | −3.74 |
| KE257_dup | 34.26 ± 0.1 | −4.37 | |
| Double mutation | F17L E208K | 34.77 ± 0.05 | −3.86 |
| F17L KE257_dup | 40.34 ± 0.14 | 1.71 | |
| T51M E208K | 35.67 ± 0.05 | −2.96 |
Changes in melting temperature (ΔTM) are relative to the Rosenbach 25923 wild type DHPS enzyme.
Catalytic properties of sulfonamide resistant SaDHPS mutants
We then analyzed the kinetic properties of the purified proteins (Table 4). The KM values for DHPP, pABA and SMX were measured using a colorimetric assay that monitors the release of pyrophosphate. The Ki values of SMX were derived from a radiometric assay that monitors the incorporation of 14C-labeled pABA into the 7,8-dihydropteroate product. The Kcat values for pABA and SMX were also derived from the colorimetric assay. The primary mutations F17L, S18L and T51M impart a slight increase in the KM for DHPP, but significantly larger increases for pABA. In contrast, the effects are reversed for the secondary mutations where the increases in the DHPP KM values are more pronounced than those for pABA. When the primary and secondary mutations are combined, they consistently lower the pABA KM values toward that of the wild type protein and increase the DHPP KM values to those seen in the secondary mutations alone. As anticipated, the KM and Ki values for SMX showed that the drug efficiently binds and inhibits the wild type enzyme. F17L, both alone and in combination with the two secondary mutations, decreases the binding and inhibition of SMX, but this was not the case with T51M where the effects were less obvious. S18L also significantly increased the KM for SMX but it was not possible to measure the Ki value for technical reasons. The same was true for the secondary mutations alone.
Table 4.
Kinetic characterization of S. aureus DHPS variants.
| saDHPS Variant | Native Substrate | SMX Antibiotic | Substrate Turnover | ||||
|---|---|---|---|---|---|---|---|
| Km DHPP (μM) | KmpABA (μM) | Km SMX (μM) | Ki SMX (μM) | KcatpABA (s−1) | Kcat SMX (s−1) | ||
| Wild Type | 10.0 (±1.4) | 3.1 (±0.9) | 5.9 (±0.2) | 1.3 (±0.5) | 4.5 (±0.4) | 2.2 (±0.1) | |
| Primary mutation | F17L | 18.3 (±4.7) | 40.2 (±6.1) | 202 (±54) | 94.1 (±23.7) | 1.7 (±0.3) | 1.0 (±0.1) |
| S18L | 10.2 (±1.8) | 26.2 (±0.2) | 140 (±40) | ND | 3.4 (±0.2) | 1.6 (±0.1) | |
| T51M | 12.1 (±2.6) | 29.8 (±13.2) | 3.9 (±1.6) | 10.0 (±0.68) | 1.9 (±0.2) | 0.8 (±0.1) | |
| Secondary mutation | E208K | 34.5 (±2.2) | 15.7 (±4.5) | ND | ND | 3.1 (±0.0) | 1.5 (±0.1) |
| KE257_Dup | 21.1 (±1.6) | 5.3 (±3.0) | ND | ND | 1.8 (±0.0) | 0.9 (±0.2) | |
| Double mutants | F17L E208K | 21.3 (±6.8) | 13.1 (±2.6) | 167.5 (±43.6) | 362.1 (±33.3) | 0.5 (±0.0) | 0.3 (±0.1) |
| T51M E208K | 38.1 (±1.5) | 9.2 (±3.5) | 5.7 (±3.0) | 29.0 (±2.7) | 0.5 (±0.1) | 0.3 (±0.0) | |
| F17L KE257_Dup | 22.3 (±5.6) | 14.3 (±3.3) | 26.0 (±6.0) | 158.9 (±31.7) | 3.6 (±0.3) | 2.1 (±0.2) | |
DHPP, pABA and SMX KM and Kcat values were measured using a colorimetric assay that monitors pyrophosphate release. A radiometric assay that monitors 14C-labeled pABA incorporation into 7,8-dihydropteroate was used to determine the SMX Ki values.
The kinetic data confirmed that SMX is a bona fide substrate of DHPS, although the turnover rates with the natural substrate pABA, as reflected in the Kcat values, were consistently lower for all the variants. The individual mutations, both primary and secondary, decreased the turnover rates for both ligands, which confirms that the catalytic efficiency is compromised by each mutation. Somewhat surprisingly, apart from F17L/KE257_dup, there was an additive decrease in turnover rates for both pABA and SMX when the primary and secondary mutations are combined. Thus, the “rescue” of the pABA KM by the secondary mutations does not translate into a similar rescue of Kcat.
Resistant mutations and antibacterial MIC values
To determine the individual effects of the identified resistance mutations in S. aureus, we measured the MIC values of isogenic strains that only contained the mutant DHPS enzymes (Supplementary Table 1). It was first necessary to perform allelic replacements of the wild type folP gene with the mutant genes to generate the required strains in a USA300 AH1263 background, and this was successful for seven of the eight mutants that were biochemically analyzed. We failed to generate an isogenic strain containing the T51M/E208K double mutation after several attempts, and we therefore used the COL S. aureus strain that naturally harbors these mutations for select experiments. We and others have found that metabolic intermediates and nutrients in standard testing media can mask the action of antibiotics, including sulfonamides, and that minimal inhibitory concentration (MIC) determinations are more easily and accurately performed in minimal media (Zlitni et al., 2013; Zhao et al., 2016). We therefore measured the MICs of the nine S. aureus strains in minimal SSM9PR media lacking pABA and several products of folate biosynthesis (Reed et al., 2015). We used chloramphenicol (CAM) as the control antibiotic that does not act through the folate pathway, and it has an MIC of 3.1–4.2 μg/mL for all strains tested. The results are summarized in Table 5. The most notable increase in resistance by a primary mutation occurred in the T51M mutant with dapsone, which is a structurally distinct member of the sulfonamide class. It is therefore significant that a homologous loop 2 mutation is also observed in M. leprae and two malarial strains (Table 2), for which dapsone is a common treatment option (Matsuoka, 2010).
Table 5.
μg/ml MIC80 values for DHPS variants.
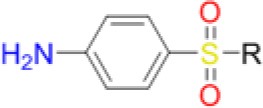 |
Primary mutation | Secondary mutation | Double mutation | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | WT | F17L | S18L | T51M | E208K | KE257_dup | F17L E208K | F17L KE257_dup | T51M E208K* | |
| Sulfacetamide |  |
25 | 41.7 | 20.8 | 12.5 | 50 | 41.7 | 200 | 133.3 | 200 – >200 |
| Sulfathiazole |  |
2.6 | 12.5 | 4.2 | 3.1 | 8.3 | 8.3 | 41.7 | 41.7 | 83.3 |
| Sulfamethoxazole |  |
4.2 | 16.7 | 10.4 | 6.3 | 8.3 | 8.3 | 50 | 41.7 | 66.7 |
| Sulfisoxazole | 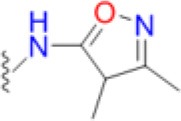 |
2.1 | 6.3 | 5.2 | 2.1 | 6.3 | 6.3 | 25 | 25 | 58 |
| Sulfapyridine | 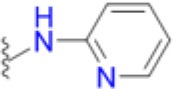 |
33.3 | 100 | 50 | 66.7 | 50 | 50 | 200 | 200 – >200 | >200 |
| Sulfadiazine | 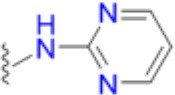 |
10.4 | 25 | 25 | 8.3 | 20.8 | 20.8 | 83.3 | 83.3 | 200 |
| Dapsone | 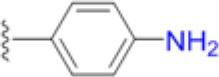 |
16.7 | 20.8 | 6.3 | 50 | 33.3 | 29.2 | 50 | 66.7 | 200 – >200 |
| Sulfamethoxypyridazine |  |
7.3 | 25.0 | 25.0 | 7.3 | 12.5 | 16.7 | 83.3 | 83.3 | 133.3 |
| Sulfadimethoxine |  |
2.6 | 12.5 | 4.2 | 3.6 | 8.3 | 8.3 | 33.3 | 25 | 66.7 |
| Sulfadoxine |  |
25 | 33.3 | 41.7 | 12.5 | 50 | 41.7 | 200 | 83.3 | 200 – >200 |
| Chloramphenicol | 4.2 | 4.2 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | |
| Trimethoprim | 1.6 | 0.065 | 0.024 | 0.16 | 1.6 | 1.0 | 0.16 | 0.20 | 2.1 | |
| § | ||||||||||
MIC80 values reported are an average of three experiments. Apart from T51M/E208K, all mutants were cloned via allelic replacement into an isogenic USA300 AH1263 panel.
T51M/E208K values were derived from the COL S. aureus strain (Supplementary Table 1).
The SMX/TMP combination MICs at the bottom of the Table represent co-trimoxazole at a ratio of 19:1.
Individually, the primary mutations increased the MIC for most of the 10 sulfonamides tested in this study, but not markedly. F17L had the greatest impact, but only 4- to 5-fold for three of the sulfonamides. S18L and T51M had minimal effect, and the same was true for the two secondary mutations, apart from T51M with dapsone. In contrast, the combination of primary and secondary mutations dramatically increased the MIC values for all 10 sulfonamides. The largest effect was observed with T51M/E208K (Table 5), but this may be related to strain variability because this double mutation was evaluated in the COL S. aureus strain rather than as an allelic replacement in USA300 AH1263.
TMP targets DHFR within the folate pathway downstream of DHPS, and changes in the TMP MIC may indicate fitness costs associated with DHPS mutations. The primary resistance mutations significantly decreased TMP MIC, but the secondary mutations alone had negligible effect. When combined, the secondary mutations decreased the effect of the F17L mutant. Addition of pABA to the testing media universally restored the TMP MICs closer to wild type for all the mutants. This could indicate that defects in catalysis caused by primary mutations in DHPS cause the downstream enzyme DHFR to become more crucial for overall pathway function, resulting in heightened susceptibility to DHFR inhibition as indicated by the lowered TMP MICs (Figure 2). The supplementation of pABA may boost DHPS catalytic output, thus returning the TMP MICs to wild type levels. Appreciable increases in the co-trimoxazole SMX/TMP combination MICs were only observed with the double mutants, likely because the heightened TMP susceptibility of primary mutants drives the combination MIC closer to wild type levels despite having resistance to SMX (Table 5).
Figure 2.
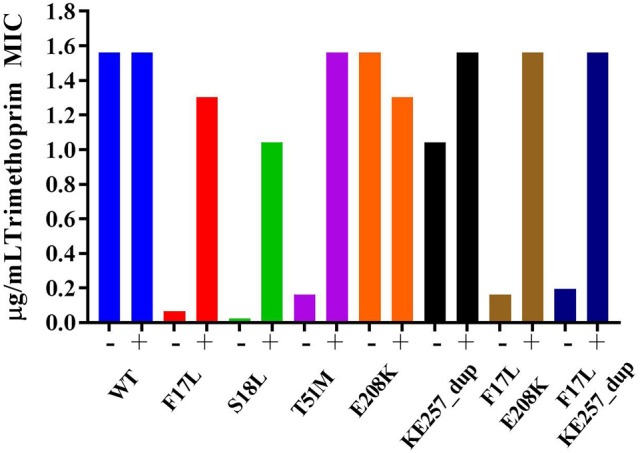
Trimethoprim susceptibility among mutants and influence of supplementation with 5 μg/mL pABA. The presence and absence of pABA in the testing media is indicated by + or – symbol, respectively. Data are representative of three independent experiments.
Fitness studies of the folP mutants
The kinetic data showed that the primary resistance mutations raise the KM of pABA and the secondary mutations lower the pABA KM back toward the value observed in the wild type enzyme. Furthermore, the TMP MIC testing revealed fitness consequences imposed by primary mutations at later stages of the folate biosynthesis pathway that could be restored by secondary mutation. Thus, the secondary mutations appear to restore the fitness of DHPS that is impaired by the primary mutations. To test whether these apparent changes in fitness of the isolated enzyme affect bacterial cell growth, the doubling times for the eight isogenic strains were measured (Figure 3). The strain harboring T51M had the slowest growth rate, but F17L did not have an altered growth rate. This was surprising because F17L had the highest consequence to pABA binding (Table 4). However, the neighboring mutation S18L also had a significantly increased doubling time relative to the wild type enzymes. The remaining four mutations had no significant effect on doubling time.
Figure 3.
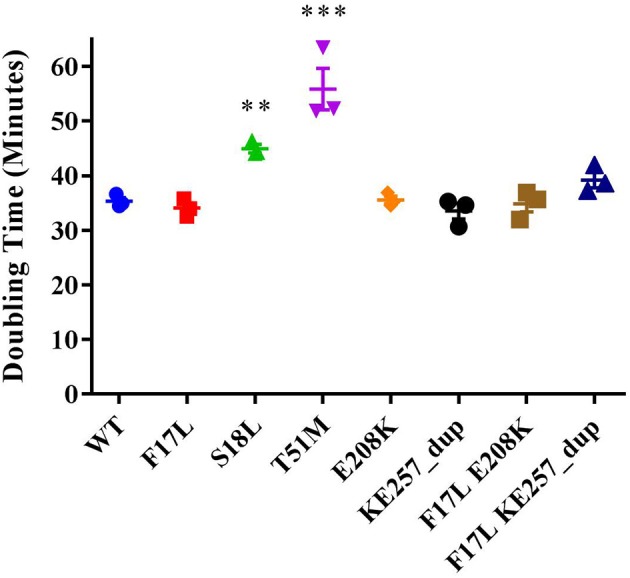
Doubling times for the isogenic USA300 folP variant strains. **indicates p < 0.05, *** indicates p < 0.001 via ANOVA (Dunnett's multiple comparisons).
To test whether the changes in fitness might become apparent in vivo, the isogenic S. aureus strains were subjected to a wax moth larvae infection study (Figure 4). In this model system, the larvae were infected with S. aureus strains from our isogenic panel of DHPS variants (Supplementary Table 1) and larval mortality rates were monitored. It was assumed that any significant decrease in the virulence of a particular strain would reflect the fitness cost associated with its introduced DHPS variant. Overall mortality rates at the end of the 72-h survival study indicated that the T51M mutant displayed the least virulence, consistent with the cell growth studies, but there were no significant changes in mortality-based virulence among any of the mutants according to Mantel Cox (Log-Rank) analysis. However, the wax moth larval model did respond as predicted in the presence of SMX. When administered 100 mg/kg SMX daily, the larvae could be rescued from the susceptible wild type strain, but every mutant strain was resistant to the antibiotic.
Figure 4.

Wax moth larvae rescue study with isogenic USA300 folP variants. All groups contain 32 larvae and each data point indicates an observed death. Percentage survival at the end of the study is listed at the top. *** indicates P < 0.001 via Mantel Cox (Log-Rank) survival analysis.
Structural and biophysical analyses of the pABA/SMX binding pocket
Having revealed the importance of Phe17, Ser18, and Thr51 in sulfa drug resistance, we re-examined their structural roles in the context of the near-transition state structure. We first modeled and energy minimized the structures of the SaDHPS near transition states in the presence of pABA and SMX using our previously determined crystal structures of YpDHPS (Yun et al., 2012). This proved to be very straightforward because the residues are highly conserved in the DHPS active site locale. Like all DHPS structures, loops 1 and 2 are disordered in the absence of substrates but become ordered in the near-transition state to create the pABA-binding pocket and to structurally and chemically optimize the substrates for catalysis (Yun et al., 2012). This SaDHPS active site locale is shown in Figure 5A, which highlights the roles of Phe17, Ser18, and Thr51. Phe17, together with Pro53, Phe172, and Lys203 create the pABA-binding pocket, with the side chain rings of Phe17, Pro53, and Phe172 forming edge-to-face interactions with the phenyl ring of pABA. The adjacent Ser18 does not interact with pABA but appears to stabilize loop1 in this region. Meanwhile, the hydroxyl group of Thr51 forms hydrogen bonds with the amino group of pABA and an oxygen of the pyrophosphate group that has been released from DHPP prior to the SN1 reaction that forms the product (Yun et al., 2012). Thr51 appears to help align the amino group for bond formation to the C11 carbon atom of the pterin substrate.
Figure 5.

DHPS active site locale. (A) The modeled S. aureus DHPS transition state based on the published Y. pestis structure (3TYZ) highlighting the pABA environment. The protein backbone is shown in pale green cartoon, the residues are in stick representation with green carbon, pABA and DHP are in stick representation with salmon and magenta carbons, respectively, and pyrophosphate is orange. The essential Mg2+ ion is shown as a fluorescent green ball. (B) The mutated salt bridge arrangement in the crystal structure of SaDHPS-F17L-E208K. The protein backbone is shown in purple cartoon, and the residues are in stick representation with purple carbons. (C) The mutated salt bridge arrangement in the crystal structure of the SaDHPS-F17L-E208K-1530 compound complex. The protein backbone is shown in yellow cartoon, the residues are in stick representation with yellow carbons, and compound 1530 is in stick representation with orange carbons. (D) The wild type SaDHPS salt bridge arrangement adjacent to the active site locale from the previously published crystal structure (1AD1). The coloring is the same as (A). In all figures, the dashed gray lines indicate salt-bridges and hydrogen bonds.
To gain more insights into the formation of the transition state ordered loop structure and the binding of pABA and sulfonamides, we used isothermal titration calorimetry (ITC). ITC revealed that, while pABA and pyrophosphate are both absolutely required to generate the pABA-binding pocket, the pterin moiety of DHPP is not necessary (Figure 6). This is consistent with the ordered loop structure that makes multiple conserved interactions with the enclosed pABA and pyrophosphate while the pterin moiety is independently accommodated in an adjacent preformed pocket (Figure 5A). The binding thermodynamics of SMX are almost identical to those of pABA (Figure 6), which is consistent with our published structures that show that both occupy the binding pocket created by loops 1 and 2 in almost identical fashion (Yun et al., 2012). In the presence of pyrophosphate, pABA and SMX have negative free energy (−6.9 and −6.8 kcal/mol respectively) reflecting a spontaneous binding interaction. The negative enthalpy measured in these studies is indicative of binding interactions driven by hydrogen bonding or π-stacking, again consistent with our structures. Finally, the significant entropic penalty associated with the binding of pABA and SMX is consistent with the observed ordering of loops 1 and 2.
Figure 6.

Isothermal titration calorimetric analysis of pABA or SMX binding to DHPS in the presence and absence of sodium pyrophosphate. Trace of the calorimetric titration of 19 × 2 μl aliquots of 500 μM pABA (A) or SMX (B) into 25 μM DHPS (top) and integrated binding isotherms (bottom). Red squares represent the heat of binding in the absence of sodium pyrophosphate. Black squares represent heat of binding in the presence of 10 mM sodium pyrophosphate. The solid black lines represent the best fit to a one site model. The derived thermodynamic parameters are shown as insets in the lower panel.
Structural analyses of SaDHPS F17L/E208K
We crystallized SaDHPS F17L/E208K and determined its structure in the absence of ligands (apo) and with our previously published (Zhao et al., 2016) inhibitory pterin-sulfisoxazole conjugate compound 1530 (Figures 5B,C, respectively). In the published SaDHPS wild type structures, Phe17 within loop1 is either distant from the active site locale or missing (Hampele et al., 1997). However, in our two F17L/E208K structures, F17L and loop1 adopt a conformation that is similar but not identical to the known conformation of loop1 in the near transition state (compare the position of Phe17 in Figure 5A with the positions of Leu17 in Figures 5B,C). We have previously shown that compound 1530 binds to the wild type DHPS active site locale in a similar fashion to the pterin substrate and SMX in the near transition state (Yun et al., 2012; Zhao et al., 2016). In the F17L/E208K mutant structure complexed with compound 1530, Leu17 is directly adjacent to, and may therefore sterically impact, the oxazole ring of sulfisoxazole (Figure 5C).
The structural consequences of the E208K mutation are apparent from our two structures. In the wild type SaDHPS structures, Glu208 forms a salt bridge with Arg176 and the adjacent Glu179 forms a salt bridge with Arg204 (Figure 5D). Arg176 and Arg204 also interact via a π-stacking interaction to stabilize this 4-residue cluster. When the E208K mutation is introduced, Arg176 relocates to form a salt bridge with Glu179 and Arg204 is displaced (Figures 5B,C). The structures suggest three ways in which the E208K mutation can contribute to resistance. First, the relocated Arg204 is adjacent to the oxazole ring in the 1530 complex (Figure 5C) and may sterically interfere with the transition state binding of sulfa drugs that have similar moieties (Table 5). The relocated Arg204 does not impact the phenyl ring of 1530 and should therefore have minimal impact on the binding of pABA that occupies the same location. Second, the relocated Arg204 may form a stabilizing salt bridge with the carboxyl group of pABA and thereby compensate for the negative impact on pABA binding of the F17L and T51M mutations. The equivalent of this interaction with the negatively charged sulfone of sulfisoxazole is visible in Figure 5C. Finally, disruption of the Arg176/Glu179/Arg204/Glu208 salt bridge substructure may introduce some local flexibility to facilitate the distortions of the pABA-binding site caused by the F17L, S18L, and T51M mutations. This is consistent with the thermal shift assay data for E208K (Table 3).
Modeling studies of F17L, S18L, and T51M in the transition state
We failed to obtain crystal structures of F17L, S18L, and T51M and we therefore turned to modeling and energy minimization to gain further insights into their roles in resistance. We introduced the three mutations independently into the two modeled SaDHPS transition state structures containing pABA or SMX, and performed energy minimization. The side chain of Leu17 is predicted to adopt the same rotamer in the pABA and SMX complexes that minimally impacts the transition state structure. However, while this rotamer maintains a favorable and close interaction with pABA, it sterically impacts the methylisoxazole ring of SMX. In the case of T51M, the mutation appears to have an indirect affect by impacting the location of Pro53 in loop2. As described above, Pro53 loosely forms part of the pABA binding pocket, but it forms a tight van der Waals interaction with the methylisoxazole ring of SMX. The modeling suggests that any movement of Pro53 toward the methylisoxazole ring caused by T51M would selectively disfavor the binding of SMX compared to pABA. Modeling with S18L did not yield a clear answer, but we suggest that it also acts indirectly to impact the position of the adjacent Phe17. The conclusions from these modeling studies are summarized in Figure 7.
Figure 7.

Close up view of the wild type and F17L mutant of SaDHPS in complex with pABA and sulfamethoxazole (SMX). (A) Wild type with pABA. (B) F17L with pABA. (C) Wild type with SMX. (D) F17L with SMX. Color scheme – wild type SaDHPS, green carbons and ribbon; F17L SaDHPS, violet carbons and ribbon; pABA, salmon carbons; SMX, cyan carbons. The complexes were modeled using the YpDHPS transition state complexes (PDB ID: 3TYZ and 3TZF).
Discussion
The output of novel antibiotic classes has been greatly reduced in recent years, and there is a crucial need for new orally bioavailable antimicrobials effective against pathogenic Staphylococci such as MRSA and vancomycin-resistant S. aureus (VRSA). The folate biosynthesis pathway remains a viable target for inhibitor development due to the essentiality of this pathway in microbes. Indeed, sulfonamides that target the DHPS enzyme in the folate pathway remain a useful treatment option for common infection types such as UTIs, skin and soft tissue infections, and osteomyelitis (Liu et al., 2011). Opportunities for new therapies that target the folate pathway are afforded by continuing advances in our understanding of the catalytic mechanisms of DHPS and other component enzymes such as 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (Shi et al., 2012; Dennis et al., 2016), and powerful new technologies to probe and exploit these mechanisms. In this study, we have analyzed the sequences of DHPS from sulfonamide resistant clinical isolates of S. aureus to identify the biochemical and structural basis of resistance. This information will be invaluable for developing new therapeutics that target DHPS and minimize the potential for resistance.
We have demonstrated using bioinformatics, biochemistry and microbiological analyses that five mutations in the enzyme DHPS directly contribute to sulfonamide resistance. We designated three of these mutations, Phe17, Ser18, and Thr51, as “primary” because one of these mutations is consistently present in resistant strains, and two mutations, E208K and KE257_dup, as “secondary” because they only appear in the presence of the primary mutations. Our crystallographic and modeling analyses reveal how these mutations achieve this resistance at the structural level. The three amino acids altered by primary mutations are highly conserved and have fundamental roles in creating the transition state structure in which two otherwise flexible loops become ordered by the pyrophosphate of DHPP and create a pocket that is bound and stabilized by pABA (Yun et al., 2012). ITC data reported here confirm this mechanism. Sulfonamides are exquisite mimics of pABA that can also engage this pocket, and we demonstrate by measuring the KM values that the primary mutations have a greater negative impact on sulfonamide binding compared to pABA. Previous studies have also reported this phenomenon (Fermer and Swedberg, 1997; Vedantam and Nichols, 1998; Gibreel and Sköold, 1999; Haasum et al., 2001; Levy et al., 2008). The secondary mutations partially restore pABA binding to the wild type levels but do not restore the catalytic efficiency that is significantly reduced by the primary mutations. This is consistent with a recent study on clinical mutations in Plasmodium species that showed similar additive increases in sulfonamide Ki and whole-cell resistance (Pornthanakasem et al., 2016).
Thus, the mechanism of resistance is based on increasing the KM of the sulfonamides compared to pABA, and the organism can apparently survive with a less efficient DHPS enzyme under drug selective pressure. Our structural studies reveal that the discrimination between the binding of sulfonamides compared to pABA is via the electron deficient outer ring, exemplified by methylisoxazole in SMX, that is required to generate the negative charge on the sulfone that mimics the carboxyl group of pABA. As shown in Figures 5, 7, all three primary mutations appear to impact this ring and therefore the binding of the entire drug. The same is also true for the E208K secondary mutation that leads to a reorganization of a salt bridge structure adjacent to the active site locale that also appears to disfavor the binding of sulfonamides via this outer ring moiety. The way in which KE257_dup functions at the dimer interface is less apparent. We suggest that it operates allosterically based on our previous studies that identified inhibitory compounds that bind at the dimer interface, rigidify the dimer and significantly slow down the release of product (Hammoudeh et al., 2014). Overall, the data are most consistent for the F17L/E208K double mutant. The kinetics, sulfonamide susceptibility, crystal structures and modeling all present a consistent picture of how these two mutations work in concert to efficiently produce high levels of resistance. This double mutation results in a high level of resistance and is frequently observed in sulfonamide resistant strains of S. aureus.
Avoiding resistance is a key component of any successful drug discovery regime, as has been shown with other notoriously mutable targets such as the essential NS3/4A protease component of the hepatitis C virus (HCV). Structural studies with this protease indicated that the most marked increases in resistance are seen among inhibitors that extend beyond the “substrate envelope,” or consensus volume assumed by native substrate binding (Romano et al., 2010). Our studies reveal the same basic mechanism of resistance in DHPS but also show the importance of secondary mutations that can partially offset the negative impact of primary resistance mutations on enzyme function. Developing inhibitors that only occupy the volume assumed by native substrates will continue to be a key strategy in our drug discovery efforts on DHPS and other key enzymes in the folate pathway (Yun et al., 2014). An important conclusion from our studies is that continued development of the sulfonamide scaffold focused on the ring extending beyond the native substrate binding pocket is fundamentally flawed. Historically, structure activity relationship efforts to improve the efficacy of sulfonamides have focused on the outer ring which also produces more favorable potency and ADME properties. Although sulfanilamide and sulfacetamide lack this ring moiety, they are significantly less potent, and all potent sulfonamides are therefore inherently vulnerable to this mechanism of resistance (Greenwood, 2010).
Our studies demonstrate that the biochemical data derived from the resistance mutations do not necessarily translate into cellular MIC or fitness measurements. The flow of precursors from other metabolic pathways and uptake of exogenous metabolites contributes to resistance and fitness within the folate pathway. Our results also reflect that there is direct interplay between the enzymes within the folate pathway. Thus, we observed a modulation of TMP growth inhibition by both primary DHPS mutations and exogenously provided pABA. This “pABA rescue” effect has been observed with novel inhibitors generated by our group (Zhao et al., 2016), and with MAC173979, a pABA biosynthesis inhibitor discovered by Zlitni et al. (2013). We also demonstrate that resistance to DHPS inhibitors increases sensitivity to TMP. This indirect consequence toward the susceptibility of the downstream enzyme DHFR is consistent with the mutual potentiation effects recently described between SMX and TMP (Minato et al., 2018). In these studies, TMP is shown to potentiate SMX by limiting production of the 7,8-dihydropteroate precursor DHPP, and SMX is shown to potentiate TMP by ultimately limiting 7,8-dihydrofolate production. The latter potentiation effect is similar to the results observed with the primary mutations in our study which hinder biochemical function.
Secondary mutations are not observed by themselves in our genetic survey of clinical isolates, perhaps due to its limited size. E208K when combined with F17L, clearly contributes to resistance and partially restores the binding of pABA, and one might expect these benefits to be present without F17L. Our data show that this is not the case and that E208K fails to provide an advantage under the selective pressure of sulfonamide treatment.
Class modification is a highly successful strategy to develop improved antimicrobial agents that continue to engage clinically validated targets, but are engineered to avoid limiting resistance mechanisms (Silver, 2011). Many groups, including ours, are applying this strategy toward the folate biosynthesis pathway, within which DHPS remains a promising target (Bermingham and Derrick, 2002; Lombardo et al., 2016). Numerous pathogenic species acquire sulfonamide resistance through equivalent mutations to those we have characterized in this study. The findings from our work describes the structural and biochemical basis of sulfonamide resistance in S. aureus, which can now be used for the future development of next generation DHPS inhibitors suitable for the treatment of many multidrug-resistant pathogens.
Materials and methods
Genetic survey of foLP variations contributing to sulfonamide resistance
Comparative analyses of the DHPS amino acid sequences were performed against a set of 56 S. aureus reference strains and 80 non-redundant S. aureus DHPS sequences from GenBank using BLAST (Benson et al., 2005; Pruitt et al., 2005; Boratyn et al., 2012). Sequence variances in DHPS were recorded and compared. Based on these analyses, sequences were separated into the two wild type backgrounds, and 8 subgroups containing at least one of the 5 variations that contribute to sulfonamide resistance or a combination of them. Sulfonamide susceptibility data for each isolate, where available, were associated with the sequencing data. Amino acid sequence alignments were performed using Clustal Omega (Goujon et al., 2010; Sievers et al., 2011).
SaDHPS expression and purification
The folP gene of S. aureus Rosenbach 25923 DHPS (EMBL accession number Z84573) and those of the F17L, S18L, T51M, E208K, KE257_dup, T51M/E208K, F17L/E208K, and F17L/KE257_dup mutants were synthesized into the pET16b vector by GenScript to produce the protein with a N-terminal 10xHis tag. These plasmids were used to transform competent E. coli BL21[DE3] cells. Resultant cell lines were used to inoculate 1L LB broth containing 50 μg/mL Ampicillin (added as a selective agent). These cultures were incubated at 37°C with shaking at 225 RPM until they reached an OD600 of 0.5. These cultures were then induced with 1 mM IPTG and incubated overnight at 18°C with shaking at 225 rpm. Cell pellets were collected with centrifugation at 3700 RCF and resuspended in a lysis buffer consisting of 50 mM Tris pH 8, 500 mM NaCl, 5 mM imidazole, lysozyme, and protease inhibitor cocktail (Roche 11-836-170-001). Cells were lysed by sonication and cell debris was cleared with centrifugation at 14,000 rpm. Crude lysate was further clarified by filtration through a 0.22 μm filter. DHPS was purified from crude lysate in two steps. Crude lysate was applied to a GE HisTrap HP 5 ml column at a rate of 0.2 ml/min. The column was then washed with 200 ml Buffer A consisting of 50 mM Tris, 500 mM NaCl, and 5 mM imidazole, pH 8. DHPS was eluted from the GE HisTrap HP 5 ml column using a gradient of 5–500 mM imidazole. Elution fractions were collected and those with an elevated UV spectrum at 280 were pooled. Pooled fractions were then applied to a HiPrep 16/60 Sephacryl S-200 HR column at a rate of 0.5 ml/min. The column was then washed with 2 column volumes elution buffer consisting of 50 mM HEPES, 150 mM NaCl, and 1 mM DTT at pH 7.6. Elution fractions were collected and examined via SDS-PAGE. Those fractions yielding a single band at approximately 32 kDa were pooled as a final product of purification. Samples were flash frozen using liquid nitrogen and stored for a maximum of 6 months at −80°C.
DHPS kinetic characterization
All kinetic characterization experiments were carried out in 50 mM HEPES with 10 mM MgCl2 at pH 7.6. Two kinetic analyses were employed. The first measures the pyrophosphate that is released by the DHPS reaction. The pyrophosphate is converted to orthophosphate using yeast inorganic pyrophosphatase, and the PiColor Lock Gold assay (Innova Biosciences) was used to detect orthophosphate. The KM values for the two substrates pABA and DHPP were individually measured by maintaining one of the substrates at a concentration that was at least 20-fold in excess of the established Kd.
The second kinetic analysis employed a radiometric assay that measures 14C-labeled pABA incorporation into the 7,8-dihydropteroate product. The reaction was carried out for 30 min at 37°C in a total volume of 100 and 10 μl aliquots were removed at intervals of 5 min. Ice-cold 50% acetic acid was added to stop the reaction. Reaction products were loaded onto PEI TLC cellulose plates (Analtech 205016) followed by development in 100 mM NaPO4, pH 7. Phos-Screen exposure was followed by Typhoon imaging. Inhibition constants were determined by maintaining substrate levels at their Kd. SMX was added at concentration ranges between 0 and 10 mM. The Ki values were determined using the one-site Fit Ki equation.
Thermal stability studies
The stability of wild-type and variant DHPS was assessed using a thermal stability assay. Sypro-Orange was added to a 10 μM sample of DHPS in the same buffer conditions used for kinetic studies, resulting in a final concentration of 5X. The fluorescence of the solution was then measured over a range of temperatures (23–99°C). Resultant data were fit to the Boltzmann equation resulting in the melting temperature of the protein (TM).
Isothermal titration studies
Binding of pABA or SMX to DHPS in the presence or absence of 10 mM sodium pyrophosphate was performed on an iTC200 (MicroCal) at 25°C. Wild type DHPS was dialyzed into 2 L ITC buffer (50 mM HEPES pH 7.6, 5 mM MgCl2) overnight at 4°C. Standard ITC experiments were performed in 50 mM HEPES, 5 mM MgCl2, and 2.5% DMSO. The iTC200 Microcalorimeter was set to deliver 19 × 2 μl injections of 500 μM ligand at 150 s intervals into 200 μl of 25 μM protein solution. All experiments were completed in triplicate. Data were analyzed using MicroCal Origin 7.0 software using a one-site binding model with n values fixed at 1.
Allelic replacement to generate isogenic USA300 AH1263 foLP mutants
The gene encoding S. aureus folP from wild-type strain NCTC 8325 was cloned into the PCR2.1TOPO vector. Site-directed mutagenesis was employed to generate eight variants of this gene with confirmed sulfonamide-resistance mutations present singly or in their observed clinical combination which included F17L, S18L, T51M, E208K, KE257_dup, F17L/E208K, F17L/KE257_dup, and T51M/E208K. These genes were sub-cloned into the shuttle vector pJB38, which has a temperature-sensitive S. aureus origin of replication. After transformation of S. aureus USA300 AH1263 with the pJB38 plasmids, growth at 43°C in the presence of chloramphenicol (CAM) allowed for plasmid integration, after which the bacteria were sub-cultured and allowed to grow at 30°C. Growth on anhydrous tetracycline allowed for the killing of cells that still contained the pJB38 plasmid. Absence of the plasmid was further confirmed by testing for sensitivity to CAM followed by sequencing of the folP gene, which was PCR amplified from the genomic DNA of each mutant. All of the isogenic mutants were successfully generated with the exception of T51M/E208K.
Minimum inhibitory concentration assay (MIC)
MIC testing was carried out in SSM9PR media (Reed et al., 2015). When necessary, pABA was added to achieve a final concentration of 5 μg/ml. Antifolates were serially diluted in 100 μl/well 2-fold across 96 well plates. Colonies of each S. aureus wild type or mutant strain to be tested were resuspended in media to a mid-log OD600 between 0.3 and 0.6, after which they were further diluted to an OD600 of 0.001, which allowed for 100 ul of bacterial suspension containing 105 CFU to be added to each well. After 16 h of incubation at 37°C, the concentration at which 80% growth inhibition had occurred was determined visually.
Growth curve analysis
Each strain studied was streaked onto LB agar and grown overnight. The overnight cultures were further diluted 1:100 in LB and the OD600 was read every 30 min. Doubling times were calculated using the linear range of the growth curve of each mutant using the following equation (Reeve et al., 2016):
The growth curve experiment was performed with three biological replicates for each mutant. The calculated doubling times were compared using One-Way ANOVA Dunnett's Multiple Comparisons Test.
Wax moth larvae infection study
Galleria mellonella larvae were purchased from Fisher Scientific (14-726-369) in their final instar stage. Larvae were stored at 13°C prior to experimentation and used within seven days of receipt. The larvae used in each experiment were obtained in a single batch and normalized for size. Each sample group contained 32 subjects. Each experiment had at least two control groups including a group that was maintained with no manipulation and a group that was injected with the vehicle solution 10 μl PBS/5% DMSO. For the rescue experiments, larvae were injected with SMX at 100 mg/kg daily in PBS/5% DMSO solution. To prepare the inoculum, S. aureus strains with chromosomal DHPS mutations of interest were streaked onto LB agar and incubated at 37°C overnight. A single colony was then used to inoculate 5 ml LB broth which was brought to mid-log at 37°C with shaking at 225 RPM. Cells were collected via centrifugation and suspended in PBS. Cell density was normalized to yield a 10 μl injection volume containing 5.9 × 105 CFU, which has an LD50 at approximately 72 h for the wild type strain. During the course of the experiment, larvae were contained in petri dishes at 37°C. Over a period of 72 h, sample groups were checked for mortality, which was defined by lack of motility and alteration in color. Mortality data were analyzed using GraphPad Prism, with the Log-rank (Mantel-Cox) test used to determine statistically significant changes in mortality for each mutant compared to the wild type strain. The same statistical test was used to determine significant alterations in mortality due to SMX treatment for each strain tested.
Crystallography
SaDHPS-F17L/E208K was brought to a concentration of 20 mg/ml in crystallization buffer (50 mM HEPES pH 7.6, 150 mM NaCl, and 1 mM DTT). Initial crystallization conditions were determined using the JCSG I-IV screens wherein 200 nl of SaDHPS were added to 200 nl of well solution in a sitting drop plate and incubated at 8°, 18°, and 23°C. Initial crystals were optimized to produce the final conditions of 0.2 M sodium nitrate, 20% PEG 3350, 75 mM NaCl, 25 mM HEPES, pH 7.6 at 18°C. Crystals were soaked overnight in compound 1530 at a 3:1 molar ratio to generate the complex structure. Twenty-five percent glycerol was added to the crystallization buffer as a cryoprotectant, and crystals were flash frozen in liquid nitrogen prior to data collection at 100°K. X-ray data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Diffraction images were indexed and integrated with XDS (Kabsch, 2010), and scaled and merged with aimless (CCP4) in space group P43. The structure was solved with PHASER (McCoy et al., 2007) using the published SaDHPS coordinates (PDB 1AD4) as the search model. Reciprocal space refinement was carried out in PHENIX (Afonine et al., 2012) using twin law h,-k,-l, NCS restraints, and TLS definitions in the final cycles. Ligand restraints were created with eLBOW (Moriarty et al., 2009) and real space refinement was performed using COOT (Emsley and Cowtan, 2004). Both structures have been deposited in Protein Data Bank with accession codes 6CLU and 6CLV. Data collection and refinement statistics are reported in Supplementary Table 2.
Modeling and computational studies
The SaDHPS transition state complexes with pABA and sulfamethoxazole were modeled based on the crystal structures of Yersinia pestis DHPS in the near transition state complex with pABA (3TYZ) and sulfamethoxazole (3TZF) using the program COOT (Emsley and Cowtan, 2004) and energy minimized using the program MOE (Molecular Operating Environment [MOE], 2018). The Amber ff12 force field was used for the protein and OPLS-AA for the small molecules. The system was energy minimized to 0.1 kcal/mol/A2.
Author contributions
RL, SW, and CR oversaw and directed the research. They also contributed to the writing of the manuscript. EG performed the kinetic experiments, isothermal calorimetry, genetic survey of folP genes in S. aureus, and crystallized as well as collected the X-ray data. She also contributed to writing the manuscript. MW participated in generating the isogenic USA300 mutant panel, performed the minimal inhibitory concentration testing, and fitness studies. She also contributed to writing the manuscript. YW and GK processed the X-ray crystallography data and refined the structures. SG performed the modeling studies and contributed to writing the manuscript. PJ participated in generating the isogenic USA300 mutant panel. ZZ performed the initial bioinformatics analysis. GP participated in the wax moth larvae fitness study and helped to analyze the data.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the Lead Discovery Informatics team at St. Jude Children's Research Hospital for their assistance in computational analysis of sulfonamide resistance mutations in DHPS. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Funding was provided by NIH grants AI070721 and GMO34496, and the American Lebanese Syrian Associated Charities (ALSAC).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01369/full#supplementary-material
References
- Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., et al. (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367. 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Wheeler D. L. (2005). GenBank. Nucleic Acids Res. 33, D34–D38. 10.1093/nar/gki063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham A., Derrick J. P. (2002). The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. Bioessays 24, 637–648. 10.1002/bies.10114 [DOI] [PubMed] [Google Scholar]
- Boratyn G. M., Schäffer A. A., Agarwala R., Altschul S. F., Lipman D. J., Madden T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct 7:12. 10.1186/1745-6150-7-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher H. W., Talbot G. H., Bradley J. S., Edwards J. E., Gilbert D., Rice L. B., et al. (2009). Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. 10.1086/595011 [DOI] [PubMed] [Google Scholar]
- Dallas W. S., Gowen J. E., Ray P. H., Cox M. J., Dev I. K. (1992). Cloning, sequencing, and enhanced expression of the dihydropteroate synthase gene of Escherichia coli MC4100. J. Bacteriol. 174, 5961–5970. 10.1128/jb.174.18.5961-5970.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis M. L., Pitcher N. P., Lee M. D., Debono A. J., Wang Z. C., Harjani J. R., et al. (2016). Structural basis for the selective binding of inhibitors to 6-Hydroxymethyl-7,8-dihydropterin Pyrophosphokinase from Staphylococcus aureus and Escherichia coli. J. Med. Chem. 59, 5248–5263. 10.1021/acs.jmedchem.6b00002 [DOI] [PubMed] [Google Scholar]
- Domagk G. (1935). Chemotherapie der bakteriellen Infektionen. Angewandte Chemie 48, 657–667. 10.1002/ange.19350484202 [DOI] [Google Scholar]
- Elena S. F., Ekunwe L., Hajela N., Oden S. A., Lenski R. E. (1998). Distribution of fitness effects caused by random insertion mutations in Escherichia coli. Genetica 102-103, 349–358. 10.1023/A:1017031008316 [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60(Pt 12 Pt 1), 2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- Fermer C., Kristiansen B. E., Sköld O., Swedberg G. (1995). Sulfonamide resistance in Neisseria meningitidis as defined by site-directed mutagenesis could have its origin in other species. J. Bacteriol. 177, 4669–4675. 10.1128/jb.177.16.4669-4675.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermer C., Swedberg G. (1997). Adaptation to sulfonamide resistance in Neisseria meningitidis may have required compensatory changes to retain enzyme function: kinetic analysis of dihydropteroate synthases from N. meningitidis expressed in a knockout mutant of Escherichia coli. J. Bacteriol. 179, 831–837. 10.1128/jb.179.3.831-837.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibreel A., Sköold O. (1999). Sulfonamide resistance in clinical isolates of Campylobacter jejuni: mutational changes in the chromosomal dihydropteroate synthase. Antimicrob. Agents Chemother. 43, 2156–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M., Mcwilliam H., Li W., Valentin F., Squizzato S., Paern J., et al. (2010). A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699. 10.1093/nar/gkq313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood D. (2010). Sulfonamides, in Antibiotic and Chemotherapy. Nottingham, UK: Elsevier B.V. [Google Scholar]
- Haasum Y., Strom K., Wehelie R., Luna V., Roberts M. C., Maskell J. P., et al. (2001). Amino acid repetitions in the dihydropteroate synthase of Streptococcus pneumoniae lead to sulfonamide resistance with limited effects on substrate K(m). Antimicrob. Agents Chemother. 45, 805–809. 10.1128/AAC.45.3.805-809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoudeh D. I., Date M., Yun M. K., Zhang W., Boyd V. A., Viacava Follis A., et al. (2014). Identification and characterization of an allosteric inhibitory site on dihydropteroate synthase. ACS Chem. Biol. 9, 1294–1302. 10.1021/cb500038g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampele I. C., D'arcy A., Dale G. E., Kostrewa D., Nielsen J., Oefner C., et al. (1997). Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J. Mol. Biol. 268, 21–30. 10.1006/jmbi.1997.0944 [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010). XDS. Acta Cryst. D 66, 125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M., Matsuoka M., Nakata N., Maeda S., Gidoh M., Maeda Y., et al. (1999). Diaminodiphenylsulfone resistance of Mycobacterium leprae due to mutations in the dihydropteroate synthase gene. FEMS Microbiol. Lett. 177, 231–235. 10.1111/j.1574-6968.1999.tb13737.x [DOI] [PubMed] [Google Scholar]
- Kazanjian P., Locke A. B., Hossler P. A., Lane B. R., Bartlett M. S., Smith J. W., et al. (1998). Pneumocystis carinii mutations associated with sulfa and sulfone prophylaxis failures in AIDS patients. AIDS 12, 873–878. 10.1097/00002030-199808000-00009 [DOI] [PubMed] [Google Scholar]
- Lane B. R., Ast J. C., Hossler P. A., Mindell D. P., Bartlett M. S., Smith J. W., et al. (1997). Dihydropteroate synthase polymorphisms in Pneumocystis carinii. J. Infect. Dis. 175, 482–485. 10.1093/infdis/175.2.482 [DOI] [PubMed] [Google Scholar]
- Levy C., Minnis D., Derrick J. P. (2008). Dihydropteroate synthase from Streptococcus pneumoniae: structure, ligand recognition and mechanism of sulfonamide resistance. Biochem. J. 412, 379–388. 10.1042/BJ20071598 [DOI] [PubMed] [Google Scholar]
- Liu C., Bayer A., Cosgrove S. E., Daum R. S., Fridkin S. K., Gorwitz R. J., et al. (2011). Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin. Infect. Dis. 52, 285–292. 10.1093/cid/cir034 [DOI] [PubMed] [Google Scholar]
- Lombardo M. N., G-Davanandan M., Wright D. L., Anderson A. C. (2016). Crystal structures of trimethoprim-resistant DfrA1 rationalizes potent inhibition by propargyl-linked antifolates. ACS Infect. Dis. 2, 149–156. 10.1021/acsinfecdis.5b00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskell J. P., Sefton A. M., Hall L. M. (1997). Mechanism of sulfonamide resistance in clinical isolates of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 41, 2121–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M. (2010). Drug resistance in leprosy. Jpn. J. Infect. Dis. 63, 1–7. [PubMed] [Google Scholar]
- McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007). Phaser crystallographic software. J. Appl. Cryst. 40, 658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Q., Gurunathan S., Masur H., Kovacs J. A. (1998). Failure of co-trimoxazole in Pneumocystis carinii infection and mutations in dihydropteroate synthase gene. Lancet 351, 1631–1632. 10.1016/S0140-6736(05)77687-X [DOI] [PubMed] [Google Scholar]
- Minato Y., Dawadi S., Kordus S. L., Sivanandam A., Aldrich C. C., Baughn A. D. (2018). Mutual potentiation drives synergy between trimethoprim and sulfamethoxazole. Nat. Commun. 9:1003. 10.1038/s41467-018-03447-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment [MOE] (2018). Molecular Operating Environment [MOE]2016.08. Montreal, QC: Computational Chemistry Group ULC. [Google Scholar]
- Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D. (2009). Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080. 10.1107/S0907444909029436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornthanakasem W., Riangrungroj P., Chitnumsub P., Ittarat W., Kongkasuriyachai D., Uthaipibull C., et al. (2016). Role of plasmodium vivax dihydropteroate synthase polymorphisms in sulfa drug resistance. Antimicrob. Agents Chemother. 60, 4453–4463. 10.1128/AAC.01835-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K. D., Tatusova T., Maglott D. R. (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–504. 10.1093/nar/gki025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed P., Atilano M. L., Alves R., Hoiczyk E., Sher X., Reichmann N. T., et al. (2015). Staphylococcus aureus survives with a minimal peptidoglycan synthesis machine but sacrifices virulence and antibiotic resistance. PLoS Pathog. 11:e1004891. 10.1371/journal.ppat.1004891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve S. M., Scocchera E. W., N G.D., Keshipeddy S., Krucinska J., Hajian B., et al. (2016). MRSA isolates from united states hospitals carry dfrg and dfrk resistance genes and succumb to propargyl-linked antifolates. Cell Chem. Biol. 23, 1458–1467. 10.1016/j.chembiol.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland S., Ferone R., Harvey R. J., Styles V. L., Morrison R. W. (1979). The characteristics and significance of sulfonamides as substrates for Escherichia coli dihydropteroate synthase. J. Biol. Chem. 254, 10337–10345. [PubMed] [Google Scholar]
- Romano K. P., Ali A., Royer W. E., Schiffer C. A. (2010). Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U.S.A. 49, 20986–20991. 10.1073/pnas.1006370107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Shaw G., Liang Y. H., Subburaman P., Li Y., Wu Y., et al. (2012). Bisubstrate analogue inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: new design with improved properties. Bioorg. Med. Chem. 20, 47–57. 10.1016/j.bmc.2011.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver L. L. (2011). Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109. 10.1128/CMR.00030-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantam G., Nichols B. P. (1998). Characterization of a mutationally altered dihydropteroate synthase contributing to sulfathiazole resistance in Escherichia coli. Microb. Drug Resist. 4, 91–97. 10.1089/mdr.1998.4.91 [DOI] [PubMed] [Google Scholar]
- Wang P., Lee C. S., Bayoumi R., Djimde A., Doumbo O., Swedberg G., et al. (1997a). Resistance to antifolates in Plasmodium falciparum monitored by sequence analysis of dihydropteroate synthetase and dihydrofolate reductase alleles in a large number of field samples of diverse origins. Mol. Biochem. Parasitol. 89, 161–177. 10.1016/S0166-6851(97)00114-X [DOI] [PubMed] [Google Scholar]
- Wang P., Sims P. F., Hyde J. E. (1997b). A modified in vitro sulfadoxine susceptibility assay for Plasmodium falciparum suitable for investigating Fansidar resistance. Parasitology 115 (Pt 3), 223–230. 10.1017/S0031182097001431 [DOI] [PubMed] [Google Scholar]
- Williams D. L., Spring L., Harris E., Roche P., Gillis T. P. (2000). Dihydropteroate synthase of Mycobacterium leprae and dapsone resistance. Antimicrob. Agents Chemother. 44, 1530–1537. 10.1128/AAC.44.6.1530-1537.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M. K., Hoagland D., Kumar G., Waddell M. B., Rock C. O., Lee R. E., et al. (2014). The identification, analysis and structure-based development of novel inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase. Bioorg. Med. Chem. 22, 2157–2165. 10.1016/j.bmc.2014.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M. K., Wu Y., Li Z., Zhao Y., Waddell M. B., Ferreira A. M., et al. (2012). Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 335, 1110–1114. 10.1126/science.1214641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Hammoudeh D., Yun M. K., Qi J., White S. W., Lee R. E. (2012). Structure-based design of novel pyrimido[4,5-c]pyridazine derivatives as dihydropteroate synthase inhibitors with increased affinity. ChemMedChem 7, 861–870. 10.1002/cmdc.201200049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Shadrick W. R., Wallace M. J., Wu Y., Griffith E. C., Qi J., et al. (2016). Pterin-sulfa conjugates as dihydropteroate synthase inhibitors and antibacterial agents. Bioorg. Med. Chem. Lett. 26, 3950–3954. 10.1016/j.bmcl.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlitni S., Ferruccio L. F., Brown E. D. (2013). Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat. Chem. Biol. 9, 796–804. 10.1038/nchembio.1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.