

Abstract
Background & objectives:
Human parvovirus B19V (B19V) is known to be associated with erythema infectiosum commonly in children, aplastic crisis, especially in persons with underlying haemolytic disorders, hydrops fetalis in pregnancies and arthritis. This cross-sectional study was aimed to determine the presence of B19V infection in childhood febrile illnesses, association of B19V with arthropathies and in adult patients with end-stage renal disease (ESRD) on dialysis. The genetic diversity among the sequences was also analysed.
Methods:
A nested polymerase chain reaction (nPCR) assay was used for B19V DNA targeting VP1/VP2 region and used for testing 618 patients and 100 healthy controls. Phylogenetic analysis on nucleotide and amino acid sequences was carried out to compare our sequences with other Indian strains and global strains.
Results:
Among 618 samples tested, seven (1.13%) were found positive. The phylogenetic analysis revealed that all the seven sequences belonged to genotype 1 and showed low genetic diversity. The clustering pattern of seven sequences was similar both by nucleotide and by predicted amino acid sequences. The fixed effects likelihood analysis showed no positive or negatively selected sites.
Interpretation & conclusions:
Seven samples (4 from non-traumatic arthropathies, 2 from patients with ESRD and 1 from febrile illness patient) were found positive by nPCR. When our seven sequences were compared with global strains, the closest neighbour was other Indian strains followed by the Tunisian strains.
Keywords: Arthropathy, end-stage renal disease, febrile illness, nested polymerase chain reaction, parvovirus B19, polymerase chain reaction
Parvovirus B19 (B19V) is a single-stranded DNA virus belonging to the family Parvoviridae, the subfamily Parvovirinae, the genus Erythrovirus. B19V infection is associated with a wide spectrum of clinical manifestations. Diseases associated with B19V include erythema infectiosum commonly in children, aplastic crisis, especially in persons with underlying haemolytic disorders, hydrops fetalis in pregnancy, arthralgia and arthritis1. The virus is implicated in causing acute glomerulopathy and as a cause of anaemia in end-stage renal disease (ESRD) and recipients of kidney transplantation. It causes a variety of symptoms, ranging from mild febrile illness to life-threatening disease conditions. The virus usually distributes through respiratory droplets; recurrent blood transfusions and immunosuppression are also risk factors for B19V acquisition. A wide range of manifestations other than well-established syndromes have been reported in association with infection, but a causal role for B19V in many of these has not been conclusively established2.
The B19V viral genome encodes three proteins: non-structural protein, NS1, and two viral capsid proteins, VP1 and VP23. There is a scarcity of information on the prevalence of B19V in many clinical conditions, and the available information is based on serology alone. Screening of patients for the diagnosis of B19V infection with polymerase chain reaction (PCR) rather than with IgG and IgM antibody-based serology is recommended when infection is suspected4. Reports on the evidence of B19V infection in patients with renal disease, arthritis and febrile illness are limited worldwide. The B19V has been designated genotype 1 (B19-related viruses), genotype 2 (A6-related viruses) and genotype 3 (V9-related viruses) and two subgroups within genotypes 1 and 3. Most infections are caused by genotype 1. However, it was also found that there was no significant correlation between viral genotypes and clinical manifestations5.
A nested PCR (nPCR) assay for the detection of B19V DNA in the patients serum samples has been developed in our laboratory6. The present study was undertaken to determine the presence of B19V infection in febrile illness, non-traumatic arthropathies (inflammatory polyarthritis/reactive arthritis) and ESRD in adults and distribution of genotypes among the Indian strains. The genetic diversity among the sequences was also analyzed to observe frequency of the B19V genotype in the Indian population and understand the degree of genetic variation.
Material & Methods
This study was cross-sectional in nature and conducted from February 2013 to April 2015 in Sri Narayani Hospital and Research Centre (SNH&RC), Sripuram, Vellore, Tamil Nadu, India. The laboratory study was carried out in Sri Sakthi Amma Institute of Biomedical Research, a unit of SNH&RC.
The patient groups included those presenting with fever with or without rash (n=201), rheumatoid factor-negative non-traumatic arthropathy (n=216) and individuals on dialysis for ESRD and chronic renal disease (n=201). The study protocol was approved by the Institutional Review Board. Written informed consent was obtained for each participant.
Taking an average prevalence of 5 per cent and worst acceptable as 3 per cent the calculated sample size was 4557. A total of 618 patients and 100 healthy volunteers were included in the study by convenient sampling.
Cases were selected based on the following inclusion criteria: patients who were admitted to the medical wards and those who attended the outpatient department presenting with fever with or without rash, oedema, joint pain, arthralgia/myalgia, general fatigue, mild illness of pyrexia, malaise, rash, and chronic anaemia with ESRD and who were admitted to the dialysis unit. Patients or their attendants who did not give informed consent to participate and had conditions not of relevance to the proposed study were excluded. Control samples from healthy donors (n=100) were collected. Healthy volunteers included staff (n=30) in the range of age of 17-44 yr and students (n=70) in the age range of 17-18 yr of SNH&RC, Vellore. The clinical features and clinicopathological findings were obtained from patients medical charts.
Samples were collected and tested prospectively. A portion of blood samples collected for other routine clinical management and laboratory investigations were utilized for this study. Whole-blood samples were collected at first point of contact with the patient, and serum was used for nPCR. The serum samples were collected and stored at −20°C until testing.
DNA was extracted from the serum samples using Qiagen DNA Blood Mini Kit (Qiagen, Hilden, GmBH, Germany) as per the manufacturer's instruction.
Nested PCR (nPCR) assay for B19V detection: A panel of 30 serum samples of qualitative positive and negative controls was used (kindly provided by Dr Amita Jain, KGMC, Lucknow). A plasmid construct was used as quantitative positive control. In all PCR runs, known positive controls and negative extraction controls (no-template controls) were used. The nPCR for the detection of B19V DNA was performed using two primer pairs6 selected in the overlapping region common to the minor (VP1) and major (VP2) capsid protein genes. The outer primers were sense 5’- CAAAAGCATGTGGAGTGAGG-3’ (nucleotides 3187-3206); antisense 5’- CTACTAACATGCATAGGCGC -3’ (nucleotides 3584-3565). The inner primers were sense 5’- CCCAGAGCACCATTATAAGG-3’ (nucleotides 3271-3290); anti-sense 5’ GTGCTGTCAGTAACCTGTAC-3’ (nucleotides 3558-3539). Product sizes were 398 bp and 288 bp for the external and internal round of nPCR, respectively.
The nPCR mix (Qiagen, Hilden, Germany) contained DNAse-free water, Qiagen Buffer, deoxynucleotides (1 mM), forward and reverse primers (25 pmol) (Eurofins, Bangalore), HotStar Taq polymerase (2 U) and template (5 μl). The thermal cycling conditions for both the rounds were 95°C for 15 min, 40 cycles at 95°C for one minute, 55°C for one minute, 72°C for one minute and 72°C for five minutes for first round and 95°C for 15 min, 35 cycles of 95°C for one minute, 55°C for one minute, 72°C for one minute and 72°C for five minutes for the second round. All amplifications were carried out using thermal cyclers Eppendorf Master Cycler Personal (Hamburg, Germany). The amplified product was detected by agarose gel electrophoresis using 2 per cent agarose (Sigma, MO, USA) in 1X Tris-acetate-EDTA buffer containing 0.5 μg/ml ethidium bromide (Sigma, St. Louis, USA) at 110 V/45 min.
The validation of the nPCR was carried out by establishing the lower limit of detection using cloned plasmids. The TOPO TA Cloning Kit (Invitrogen, CA) was used to clone the nPCR product.
Phylogenetic analysis: The nPCR amplicons were sequenced, aligned and submitted in the GenBank repository. Phylogenetic analysis was carried out to compare our sequences with other Indian strains and global strains. The sequences were clustal aligned, followed by phylogenetic construction applying maximum likelihood method and Tamura-Nei model in MEGA 5.0 software programme8. Uniform rates among the sites and bootstrap analysis with 1000 replicates were used. Reference sequences from NCBI database (https://www.ncbi.nlm.nih.gov/nucleotide) of each subtype were included and compared towards scrutiny of genetic diversity. The sequences were selected from different countries from continents of Asia, Africa, Europe and South America for analysis. A dendrogram was plotted for analysis.
The amino acid sequences were identified for nPCR-positive DNA sequences using online tool (http://in-silico.net/tools/biology/sequence_conversion). The amino acid sequences were also used for phylogeny construction using the same programme and settings.
Selective pressure analysis: Evidence for strong selective pressure at the population level (i.e. along internal branches) was searched for using the codon-based maximum likelihood method in the web-based interface of the HyPhy software package (http://www.datamonkey.org). The sequences obtained in our study and other Indian strains were analyzed. Positive and negative selection based on the non-synonymous (dN) and synonymous (dS) rates of substitution per site were estimated using the internal fixed effects likelihood (IFEL) method9 and the default settings of the programme. Sites that were under selective pressure were identified using a significance level of 0.1 (www.datamonkey.org, online server program). This statistical method used fits an independent dN and dS to every site in the context of codon substitution models and test whether dN≠dS. IFEL is a codon-based maximum likelihood method that can be used to estimate the dN/dS ratio at every codon in the alignment.
Results
A total of 618 serum samples from an equal number of patients were tested. The age of the patients ranged from 6 to 92 yr (median 45 yr), 208 were females. In the healthy control group (n=100), the age ranged from 17 to 44 years (median 18 yr). Among 618 patients tested, seven were found positive for B19V by nPCR (Table I). A gel picture showing amplification for parvovirus B19 positive sample in one patient with febrile illness is shown in Fig. 1. In experiments for determination of lower limit of detection, the nPCR assay for parvovirus B19 was able to detect 31.5 plasmid copies per 5 μl reaction mixture. The reproducibility of the assay was confirmed by testing all the positive samples thrice. None of the 100 controls was positive.
Table I.
Nested polymerase chain reaction findings in different patient groups
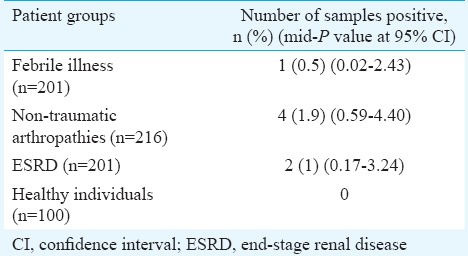
Fig. 1.
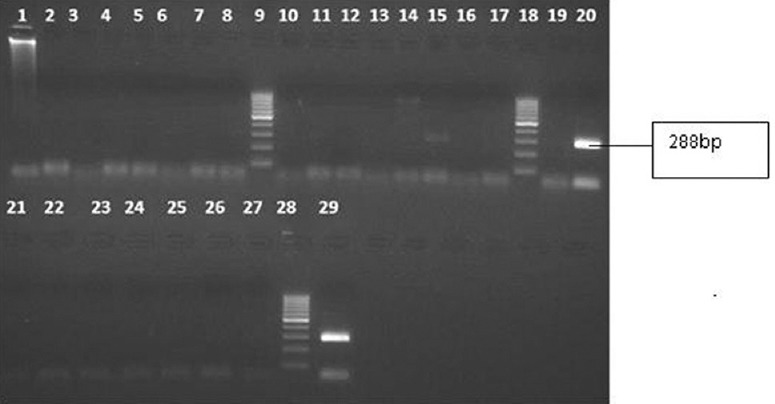
Gel picture showing amplification for parvovirus B19 positive sample in one patient with febrile illness. Lane 15, specific bands for parvovirus B19 (288 bp); lanes 20 and 29, positive control for parvovirus B19 (288 bp); lanes 9, 18 and 28, molecular weight marker; lanes 1, 2, 4, 5, 7, 8, 11, 12, 14, 17, 19, 22, 23, 25 and 26, patients samples; lanes 3, 6, 10, 13, 16, 21, 24 and 27, negative controls.
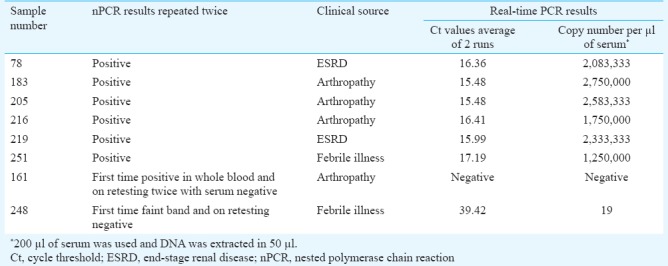
Selected serum samples were also tested by real-time PCR for validation of nPCR findings in a different laboratory. Six samples positive by nPCR were positive by quantitative real-time PCR picked up at early cycle threshold (Ct) with high virus load. One sample was negative by both nPCR and real-time PCR. One sample showed faint band in nPCR which was also weak positive in real-time PCR with low copy numbers at late Ct (Table II).
Table II.
Clinical information and real-time polymerase chain reaction findings for positive samples
The partial VP1/VP2 sequences from seven B19V-positive samples were submitted to GenBank (accession numbers are KJ856907, KJ856908, KJ856909, KJ856910, KJ856911, KM659027 and KM659028).
The comparison of alignment of nucleotide and their predicted amino acid sequences showed a low level of heterogeneity in nucleotide and amino acid sequences. It is generally agreed upon that amino acid sequences produce a tree closest to the true tree. This is due to the higher rate of conservation of amino acid sequences in the protein structure.
The FEL analysis showed three positively selected sites and 10 negatively selected sites. When our seven sequences alone were tested, there were no positive or negatively selected sites. This could be either inherent nature of the strains or result of a low number of sequences analyzed.
The seven sequences from this study were subjected to BLAST search and found to have maximum (94-100%) identity to previously reported human parvovirus B19 strains. The sequences when aligned with 24 reference sequences from GenBank representing each genotype revealed that all our seven sequences belonged to genotype 1. However, these were also distinct to genotype 1a and 1b (Fig. 2). The two of six QC strains matched with genotype 3; the other four strains matched with genotype 1 along with other strains reported from Lucknow. In India, Genbank B19V sequences (n=21) were available only from Lucknow. Sequences other than our target gene (NS1 and unverified entries) were excluded. The 21 sequences were included for our analysis to compare with the seven sequences obtained in our study.
Fig. 2.
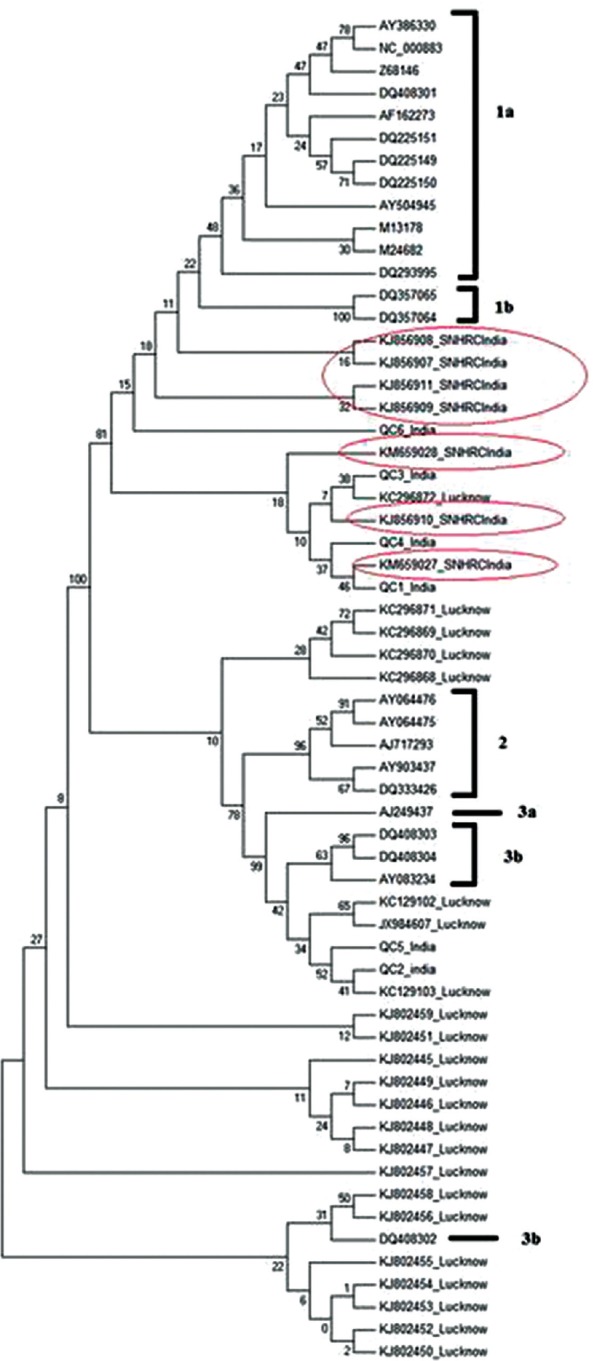
Phylogenetic tree of human B19V for genotype determination. The sequences identified in our study were circled.
A total of 95 sequences from global strains were used for phylogenetic analysis. The strains included representative strains from different geographical regions such as Poland, Germany, France, Taiwan, Brazil, USA, Côte d’Ivoire, Tunisia, India including our strains. Selection pressure analysis was carried out for all 95 sequences and also for the seven sequences identified in our study. The nucleotide comparison of 95 global partial VP1/VP2 gene sequences from GenBank that included seven sequences from our study and six QC strains revealed four major phylogenetic clusters (Fig. 3). Sequences (n=27) from European countries and Taiwan clustered separately (Cluster I). Indian strains (n=21) reported from Lucknow, India clustered separately (cluster II). Strains from Brazil and USA, and strains from Côte d’Ivoire, Tunisia, two of our QC strains and Lucknow strains grouped as three sub-clusters (cluster III). The sequences identified from our strains remained separated and formed a cluster (cluster IV), homogeneously grouped with four other QC strains and a few Indian strains reported from Lucknow.
Fig. 3.

Phylogenetic tree of human B19V compared with global strains.
The alignment of seven nucleotide sequences was similar when compared to their predicted amino acid sequences (data not shown).
Selective pressure analysis was carried out on all available sequences (n=31) reported from India including our seven sequences. The FEL analysis showed three positively selected sites and 10 negatively selected sites indicating a strong purifying selection.
Discussion
In our study, the presence of parvovirus B19 infection in childhood febrile illnesses, arthropathies (inflammatory polyarthritis/reactive arthritis) seen in adults and children and ESRD and chronic renal disease seen in adults was studied. Among the 618 patients tested, seven were positive for B19V nPCR and were subsequently confirmed by sequencing. None of the healthy individuals was tested positive by the PCR assay. Further, a case was made to show the intrinsic value of the nPCR only and identify the genetic variants if any in comparison to the Lucknow study5.
Human B19V infection has a varied spectrum of clinical manifestations, ranging from erythema infectiosum, transient aplastic crisis, foetal hydrops, cardiomyopathy and recently arthritis and hepatitis with potential fatality10. Diagnosis often is only made clinically. Serologic testing can be confirmatory.
Among the seven B19V positives, four were from non-traumatic arthropathy group. All the patients in this group were pretested for rheumatoid factor and those who were negative were recruited for further B19V PCR testing. Schmid et al11 demonstrated B19V DNA in synovial biopsies from patients with arthritis, including patients with lack of rheumatoid factor. Human B19V infection-associated joint symptoms occur most frequently in adults, presenting as a self-limited, acute symmetric polyarthritis affecting the small joints of the hands, wrists and knees. However, a small percentage of patients have persistent chronic polyarthritis that mimics rheumatoid arthritis. This indicates the role of B19 virus as a concomitant or precipitating factor in the pathogenesis of autoimmune conditions12.
The other B19V-positives samples included two from ESRD patients and one from a patient with febrile illness. However, all the seven positive patients were febrile and presented with cough, myalgia and joint pain. In Brazil, de Moraes et al13 reported 2.4 per cent of B19V in febrile-rash illness. Patients having ESRD and on haemodialysis are at high risk of B19V infection14. The maximum likelihood estimator is consistent when a sufficiently large number of observations (n) are available improving arbitrary precision, thus converging the estimate of probability with its true value. Hence, in Table I, the mid-P value at 95 per cent CI of nPCR findings in different patient groups is shown.
When analyzed with other B19V reference sequences, all our strains clustered significantly with B19V genotype 1. Thus, our results further confirm the predominance of genotype 1 in south India and clustered separately from 1a and 1b.
Jain et al5 reported the detection of 13 (5.5%) cases of B19V by PCR in children with aplastic anaemia/leukaemia and chronic haematological disorders in Lucknow, Uttar Pradesh, north India. They revealed that 84.6 per cent of sequences belonged to genotype 1a, followed by genotype 3b (15.4%). Individual cases of B19V infections have been reported in several parts of the country15,16,17,18,19. Gupta et al20 reported B19V seroprevalence of 25.8 per cent in paediatric patients with aplastic anaemia.
The comparison of alignment of nucleotide and their predicted amino acid sequences showed a low level of heterogeneity in nucleotide and amino acid sequences. It is generally agreed upon that amino acid sequences produce a tree closest to the true tree21,22. This is due to the higher rate of conservation of amino acid sequences and protein structure21. The association of high genetic diversity with low amino acid variability has been reported to be consistent with the apparent lack of difference in pathogenicity, clinical manifestations and antigenic reactivity between genotypes23. When our sequences were compared with global strains, the closest neighbour was other Indian strains followed by the Tunisian strains.
Natural selection can be detected by comparing the rate of synonymous nucleotide substitution (dS) with that of non-synonymous nucleotide substitution (dN). The relationship dN/dS<1 indicates purifying selection (negative), dN/dS=1 indicates neutral and dN/dS>1 indicates diversifying selection (positive)24. Human B19V genotypes and subtypes have a high ratio of synonymous to non-synonymous nucleotide changes per site, suggesting that NS1 and VP1/VP2 regions are under strong purifying selection25,26.
Barros et al27 investigated selection pressures in strains of parvovirus B19 causing infections in Brazilian population. They studied two different populations for evidence of positive selection among the strains examining nucleotide (nt) sequences size of 476 bp of Vp1/Vp2. The authors examined different codon sites responsible for the difference among B19V strains obtained from the two Brazilian populations. The authors indicated that under positive selection pressure, lineages of B19 virus present in one region might increase in the number of non-synonymous mutations through adaptive evolution facilitating the spread of the virus. These changes increase viral survival and replication which is an outcome of positive selection pressure. However, in our study, the FEL analysis showed three positively selected sites and 10 negatively selected sites. There were no positive or negatively selected sites in our seven sequences. This could be either inherent nature of the strains or result of low number of samples screened. However, a detailed study including several other important immunosuppressive patient groups, pregnant women and children with malignancies needs to be done.
In conclusion we recommend the use of nested PCR for the detection of active B19V infection in high risk groups especially individuals with arthropathies.
Footnotes
Financial support & sponsorship: This work was financially supported by the Indian Council of Medical Research, New Delhi, India (grant no. VIR/51/2011/ECD-I). The authors also acknowledge Sri Sakthi Amma Health Care Trust, Vellore for additional financial support
Conflicts of Interest: None.
References
- 1.Heegaard ED, Brown KE. Human parvovirus B19. Clin Microbiol Rev. 2002;15:485–505. doi: 10.1128/CMR.15.3.485-505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldman M, Kopp JB. Parvovirus B19 and the kidney. Clin J Am Soc Nephrol. 2007;2:47–56. doi: 10.2215/CJN.01060307. [DOI] [PubMed] [Google Scholar]
- 3.Tu M, Liu F, Chen S, Wang M, Cheng A. Role of capsid proteins in parvoviruses infection. Virol J. 2015;12:114. doi: 10.1186/s12985-015-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soliman Oel-S, Abd El-Aal Hegazi Hasan M, El-Ashry R, Zaghloul MH, Kora B. Parvovirus B19 infection in pediatric oncology patients: Diagnostic value of clinical and serologic parameters compared with nested PCR. J Pediatr Hematol Oncol. 2009;31:173–6. doi: 10.1097/MPH.0b013e3181983b2d. [DOI] [PubMed] [Google Scholar]
- 5.Jain P, Jain A, Prakash S, Khan DN, Singh DD, Kumar A, et al. Prevalence and genotypic characterization of human parvovirus B19 in children with hemato-oncological disorders in North India. J Med Virol. 2015;87:303–9. doi: 10.1002/jmv.24028. [DOI] [PubMed] [Google Scholar]
- 6.Yamakawa Y, Oka H, Hori S, Arai T, Izumi R. Detection of human parvovirus B19 DNA by nested polymerase chain reaction. Obstet Gynecol. 1995;86:126–9. doi: 10.1016/0029-7844(95)00092-6. [DOI] [PubMed] [Google Scholar]
- 7.Manchanda A, Datta V, Jhunjhunwala K, Saili A, Kumar A, Agarwal N. Parvovirus B19 nonimmune hydrops in a neonate. Indian J Pediatr. 2007;74:585–6. doi: 10.1007/s12098-007-0099-6. [DOI] [PubMed] [Google Scholar]
- 8.Tamura K1, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolf JB, Künstner A, Nam K, Jakobsson M, Ellegren H. Nonlinear dynamics of nonsynonymous (dN) and synonymous (dS) substitution rates affects inference of selection. Genome Biol Evol. 2009;1:308–19. doi: 10.1093/gbe/evp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perrini S, Guidi B, Torelli P, Forte A. Parvovirus B19 associated acute cholestatic hepatitis. Pediatr Med Chir. 2014;36:102. doi: 10.4081/pmc.2014.102. [DOI] [PubMed] [Google Scholar]
- 11.Schmid S, Bossart W, Michel BA, Brühlmann P. Outcome of patients with arthritis and parvovirus B19 DNA in synovial membranes. Rheumatol Int. 2007;27:747–51. doi: 10.1007/s00296-007-0337-2. [DOI] [PubMed] [Google Scholar]
- 12.Colmegna I, Alberts-Grill N. Parvovirus B19: Its role in chronic arthritis. Rheum Dis Clin North Am. 2009;35:95–110. doi: 10.1016/j.rdc.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 13.de Moraes JC, Toscano CM, de Barros EN, Kemp B, Lievano F, Jacobson S, et al. Etiologies of rash and fever illnesses in Campinas, Brazil. J Infect Dis. 2011;204(Suppl 2):S627–36. doi: 10.1093/infdis/jir490. [DOI] [PubMed] [Google Scholar]
- 14.Waldman M, Kopp JB. Parvovirus B19 and the kidney. Clin J Am Soc Nephrol. 2007;2(Suppl 1):S47–56. doi: 10.2215/CJN.01060307. [DOI] [PubMed] [Google Scholar]
- 15.Singh S, Chand G, Charan S, Arora S, Singh P. Recurrent severe anaemia: A rare presentation of parvovirus b19 infection. J Clin Diagn Res. 2014;8:MD01–2. doi: 10.7860/JCDR/2014/7840.4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharada Raju R, Nalini Vinayak K, Madhusudan Bapat V, Preeti Balkisanji A, Shaila Chandrakant P. Acute human parvovirus b19 infection: Cytologic diagnosis. Indian J Hematol Blood Transfus. 2014;30:133–4. doi: 10.1007/s12288-013-0287-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishore J, Kishor D. Can Parvovirus B19 infection be naturally oncolytic: Clinical findings raise such a possibility in leukaemic children. Indian J Med Res. 2014;139:952–3. [PMC free article] [PubMed] [Google Scholar]
- 18.Aravindh R, Saikia UN, Mishra B, Kumari V, Sarkar S, Sharma M, et al. Persistence of human parvovirus B19 in tissues from adult individuals: A comparison with serostatus and its clinical utility. Arch Virol. 2014;159:2371–6. doi: 10.1007/s00705-014-2065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bihari C, Rastogi A, Saxena P, Rangegowda D, Chowdhury A, Gupta N, et al. Parvovirus b19 associated hepatitis. Hepat Res Treat 2013. 2013 doi: 10.1155/2013/472027. 472027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta V, Saini I, Nath G. Prevalence of parvovirus B 19 infection in children with aplastic anemia. Indian Pediatr. 2013;50:489–91. doi: 10.1007/s13312-013-0149-2. [DOI] [PubMed] [Google Scholar]
- 21.Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–27. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- 22.Hershkovitz MA, Leipe DD. Phylogenetic analysis. Methods Biochem Anal. 1998;39:189–230. doi: 10.1002/9780470110607.ch9. [DOI] [PubMed] [Google Scholar]
- 23.Parsyan A, Szmaragd C, Allain JP, Candotti D. Identification and genetic diversity of two human parvovirus B19 genotype 3 subtypes. J Gen Virol. 2007;88(Pt 2):428–31. doi: 10.1099/vir.0.82496-0. [DOI] [PubMed] [Google Scholar]
- 24.Spielman SJ, Wilke CO. The relationship between dN/dS and scaled selection coefficients. Mol Biol Evol. 2015;32:1097–108. doi: 10.1093/molbev/msv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukashov VV, Goudsmit J. Evolutionary relationships among parvoviruses: Virus-host coevolution among autonomous primate parvoviruses and links between adeno-associated and avian parvoviruses. J Virol. 2001;75:2729–40. doi: 10.1128/JVI.75.6.2729-2740.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Servant A, Laperche S, Lallemand F, Marinho V, De Saint Maur G, Meritet JF, et al. Genetic diversity within human erythroviruses: Identification of three genotypes. J Virol. 2002;76:9124–34. doi: 10.1128/JVI.76.18.9124-9134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barros de Freitas R, Durigon EL, Oliveira Dde S, Romano CM, Castro de Freitas MR, Linhares Ada C, et al. The “pressure pan” evolution of human erythrovirus B19 in the Amazon, Brazil. Virology. 2007;369:281–7. doi: 10.1016/j.virol.2007.07.007. [DOI] [PubMed] [Google Scholar]