

Abstract
Yeast RNA polymerases A, B, and C share five small subunits, two of which, ABC10α and ABC10β, comigrate on SDS polyacrylamide gels. The gene encoding ABC1Oα, RPC10, was isolated based on microsequence data. RPC10 is a single copy gene localized on chromosome VIII. It codes for a very basic protein of only 70 amino acids, which contains a zinc binding domain of the form CX2CX13CX2C. Deletion of its gene indicated that, despite its very small size, the ABC10α subunit is essential for yeast cell viability. ABC10α and ABC10β have little sequence similarity.
Saccharomyces cerevisiae contains three nuclear RNA polymerases A(I), B(II), and C(III) which are responsible for the synthesis of rRNA, mRNA, and small stable RNAs, respectively. Each enzyme is composed of two large subunits homologous to bacterial RNA polymerase sub-units β′ (A190, B220, C160) and β (A135, B150, C128), and of subunits related to α (AC40, AC19, and B44.5). 1 In addition, the three enzymes share common subunits and contain a variable set of enzyme-specific polypeptides (for a review see Sentenac, 1985).
To understand the structure and function of yeast RNA polymerase subunits, the cloning of their structural genes was undertaken. Most of the genes were cloned, in particular the large subunit genes (Allison et al., 1985; Mémet et al., 1988; Sweetser et al., 1987; Yano and Nomura, 1991; James et al., 1991), three genes encoding subunits shared by the three enzymes (ABC27, ABC23, and ABC14.5) (Woychik et al., 1990), and the genes encoding the two subunits common to enzymes A and C (AC40 and AC19) (Mann et al., 1987; Dequard-Chablatetal., 1991). We recently showed that the smallest component of the three RNA polymerases, the ≈10 kDa subunit, can be resolved by reverse phase chromatography into two distinct polypeptides (Carles et al., 1991). These two polypeptides comigrate on SDS-PAGE, and both bind zinc in vitro. However, they were shown to be structurally different on the basis of several criteria, including partial microsequence data. The presence of the two polypeptides (called ABC10α and ABC10β) in the three forms of RNA polymerases brings to five the number of shared subunits (ABC27, ABC23, ABC14.5, ABC10α, and ABC10β) (Carles et al., 1991). The gene coding for ABC10β has been cloned recently (Woychick and Young, 1990). In this work we have isolated and characterized the gene RPC10 encoding ABC10α.
Materials and methods
Yeast strains
Yeast strain CMY214 (a/α trp1-Δ1/trp1-Δ1 his3Δ200/his3Δ200 ura3-52/ura3-52 ade2-101/ade2-101 lys2-801/lys2-801 can1/CAN1) was obtained from C. Mann (Mann et al., 1987). Yeast strains YNN295 (α ura3-52 lys2 ade1 ade2 his7 trp1-Δ1), YNN281 (a trp1-Δ1 his3-Δ200 ura3-52 lys2-801 ade2-1 gal mal CUPr), and S288C (a SUC2 mal mel gal2 CUP1) were from the yeast genetic stock center (Berkeley).
ABC10α purification and amino acid sequence determination
Yeast RNA polymerases A(I), B(II), and C(III) were prepared according to Buhler et al. (1974), Dezélée et al. (1972), and Huet et al. (1985), respectively. The A10α, B10α, and C10α subunits were purified, digested with trypsin, and micro-sequenced as described by Carles et al. (1991). The three subunits contained the following tryptic polypeptides: peptide I: EGFQIPTNLDAA (A) (A) GTSQA; peptide II: LVQFEA (uncertain aminoacids are within parenthesis).
Oligonucleotides
Oligonucleotides were synthesized by the phosphoramidite method using an Applied Biosystems 380B DNA synthesizer. Three degenerate oligonucleotides were synthesized based on the amino acid sequence of the N-terminal and C-terminal part of peptide I: oligonucleotide A (GGNTTYCARATHCC) encoding GFQIP; oligonucleotides B (GCYTGNGANGTNCC) and B′ (GCYTGRCTNGTNCC), corresponding to GTSQA (N stands for A, C, G, or T; R for purine; Y for pyrimidine; and H for A, C, or T).
Following gel electrophoresis to control their size, they were used directly in the polymerase chain reaction (PCR). For the screening of the λEMBL3a yeast genomic library, the 30-mer oligonucleotide CCAACAAATTTAGACGCCGCAGCTGCAGGT was synthesized, 32P end-labeled with T4 polynucleotide kinase, and purified on a denaturing 20% polyacrylamide gel before use.
Cloning of the RPC10 gene
Oligonucleotides A, B, and B′ were used to amplify a genomic DNA fragment from yeast S288C in a polymerase chain reaction. The mixture contained 100 pmol of oligonucleotide A, 50 pmol each of B and B′ oligonucleotides, 100 ng of yeast genomic DNA, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 200 μM each dATP, dCTP, dGTP, dTTP, 1 mg/ml of gelatin, and 2.5 units of Ampli Taq polymerase (United States Biochemical) in a total volume of 50 μl. After an initial denaturation step at 92°C for 4 minutes, the reaction was carried out for 30 cycles of 1 minute denaturation at 92°C, 1 minute hybridization at 50°C, and 1 minute polymerization at 72°C. The reaction mixture was fractionated on a 15% native polyacrylamide gel, and a fragment of 53 bp was isolated and used as a template for a second amplification reaction under the same conditions. The ends of the DNA fragments were made blunt by Klenow polymerase I and ligated into the EcoR V site of the pBluescript SK+ plasmid (Stratagene). Several clones were sequenced, and one of them (pLS190) was found to contain an insert that encoded the peptide sequence GFQIPTNLDAAAAG. The genomic DNA sequence of the internal peptide PTNLDAAAAG was used to design a 30-mer non-degenerate oligonucleotide which was 32P-end labeled to probe a Southern blot of yeast genomic DNA digested with different restriction enzymes (Maniatis et al., 1982). After washing the membrane at 55°C in 5 × SSC (750 mM NaCl, 75 mM sodium citrate pH 7.0), the probe remained hybridized to single EcoR I and Hind III genomic fragments of 2.1 and 7 kb, respectively, and to a single EcoR I-Hind III fragment of 1.8 kb. The same oligonucleotide probe was used to screen a yeast genomic λEMBL3a library (a gift from M. Snyder). Filters were hybridized and washed under the same conditions as for the Southern hybridization. Six positive clones were found by Southern analysis to contain a 2.1 kb EcoR I insert that hybridized with the 30-mer oligonucleotide. The 2.1 kb EcoR I fragment was subcloned into the EcoR I site of pBluescript SK+ to give pLS193.
Sequence analysis of RPC10
The sequence of RPC10 was determined on both strands of pLS193 DNA, around a unique Pvu II restriction site present within the cloned 53 bp fragment, using the dideoxynucleotide chain-termination method (Sanger et al., 1977). Ten oligonucleotides were used as primers for this purpose. Sequence analysis was done with the “DNA strider” program (Marck, 1988). The homology search of databases was performed on GenPro (release 68) and NBRF Protein data banks (release 29) with the Kanehisa program (Kanehisa, 1984), or on Swiss Prot (release 18) and EMBL (release 27) data banks with the Fasta program.
Gene copy number and chromosomal localization
Yeast genomic DNA (1 μg) from YNN281 was digested separately with five restriction enzymes or some combinations of them, electrophoresed on a 0.7% agarose gel, and transferred onto a nitrocellulose membrane. The blot was pre-hybridized for 1 hour at 65°C in 10 ml of 6X SSC, 5X Denhardt’s solution, 0.5% SDS, and 20 μg/ml denatured salmon sperm (Maniatis et al., 1982). After hybridization with the 30-mer oligonucleotide for 4 hours at 65°C, the membrane was washed at 65°C in 0.1X SSC, 0.1 % SDS for 10 minutes and autoradiographed.
The chromosomes of YNN295 yeast strain were separated by pulse field electrophoresis as described by Carle and Olson (1985). The chromosomal DNA bands were visualized by ethidium bromide staining and transferred onto a nitrocellulose membrane. Southern analysis of blotted chromosomal DNA was performed as above, using the Nru I-EcoN I 32P-labeled fragment as hybridization probe.
Disruption of the RPC10 gene
To construct the rpc10 ∷ HIS3 allele, the Nru I-EcoN I fragment of pLS193 was replaced by the BamH I fragment from pSZ63 that contains the yeast HIS3 gene (Parent et al., 1985). The resulting plasmid, pLS195, was cleaved with EcoR I, and the DNA was used to transform the yeast diploid strain CMY214. Genomic DNA from eight HIS+ transformants was digested with EcoR I and analyzed by Southern hybridization with the 2.1 kb EcoR I DNA probe of pLS193. In each case, two bands corresponding to the wild-type and the disrupted gene were detected. The HIS+ diploid cells were sporulated and subjected to tetrad analysis. The auxotrophy for histidine was tested on minimal medium with or without histidine.
Results
Cloning of the RPC10 gene
Yeast RNA polymerases A(I), B(II), and C(III) were shown to share two additional common subunits of ≈10 kDa, called ABC10α and ABC10β (Carles et al., 1991). The amino-terminal sequence of ABC10β was found to correspond to the recently isolated RPB10 gene (Woychick and Young, 1990). Since the N-terminus of ABC10α was blocked, internal protein sequence information was sought in order to clone the corresponding gene. To obtain peptide sequence information, subunits A10α, B10α, and C10α were purified from the three enzymes by RP-HPLC and digested with trypsin. The sequence of a 19 amino acid long tryptic peptide was found to be identical in the three proteins (peptide I, EGFQIPTNLDAA(A) (A)GTSQA), confirming the identity of A10α, B10α, and C10α. Based on this peptide sequence, three degenerate oligonucleotides were made and used as primer to amplify a predicted 53 bp genomic DNA fragment which was cloned and sequenced (see Materials and Methods). The sequence of the cloned DNA was found to encode the internal part of the peptide not encoded by the priming oligonucleotides. The genomic DNA sequence coding for the central part of the peptide (PTNLDAAAAG) was then used to design a 30-mer non-degenerate oligonucleotide. In Southern experiments, this oligonucleotide hybridized strongly to unique EcoR I, Hind III, and EcoR I-Hind III fragments of 2.1, 7, and 1.8 kb respectively (data not shown). The same probe was used to screen a λEMBL3a library of S. cerevisiae S288C genomic DNA. λEMBL3a recombinant clones which contained a 2.1 kb EcoR I fragment hybridizing with the oligonucleotide were selected. This 2.1 kb EcoR I fragment was subcloned into the pBluescript SK+ vector and sequenced.
Sequence analysis
The nucleotide sequence, shown in Figure 1, revealed an open reading frame of 210 nucleotides encoding a 70 amino acid polypeptide which contained the two tryptic peptides previously described. This identified the gene encoding ABC10α, which was called RPC10, following our nomenclature for common subunits (Riva et al., 1986). From its amino acid composition, ABC10α is a very basic protein with a pi of 10.06. A computer analysis with protein sequences in the GenPro, NBRF, and Swiss-Prot data bases did not reveal any protein with significant sequence similarity. A potential zinc binding motif of structure CX2CX13CX2C is located in the middle of the protein. The presence of a metal binding motif was expected, since we had previously found that ABC10α subunit binds zinc (Carles et al., 1991). The protein does not contain any consensus nuclear localiation signal of the form [(R/K/T/A)KK(R/Q/N/T/S/G)K] (Gomez-Marques and Segade, 1988) or [K(K/R)X(K/R)] (Chelsky et al., 1989). Nevertheless, we noticed the presence of an hexapeptide rich in arginine and lysine (KARTKR) near the C-terminal part of ABC10α.
Figure 1.

Nucleotide sequence of RPC10 coding and flanking regions. The uninterrupted open reading frame of 210 bp encodes a sequence of 70 amino acids which contains the two sequenced tryptic peptides (underlined). The four cysteines of the putative zinc binding domain are circled. The basic heptapeptide discussed in the text is underlined (dotted line). In the upstream region, a consensus ABF1 binding site and a T-rich element are boxed. The position of the Nru I and EcoN I restriction sites used for gene disruption are indicated.
The open reading frame is immediately preceded by a sequence that contains 11 of 12 nucleotides that match the yeast optimal translation initiation sequence (A/T)A(A/C)A(A/C)AATGTC(T/C) (Hamilton et al., 1987). The 5′ untranslated region contains an ABF1 binding site, at position – 158 (consensus RTCRYBN4ACG, Della Seta et al., 1990), followed by a T-rich cluster. ABF1 sites were found in front of many RNA polymerase genes (Dequard-Chablat et al., 1991). T-rich elements have been shown to activate transcription in conjunction with ABF1 sites (Lue et al., 1989). The recently described PAC box (RNA Polymerase A and C box) is found in the promoters of most of the genes coding for RNA polymerase A and C subunits (Dequard-Chablat et al., 1991). This sequence is not present in the RPC10 promoter or in the reported upstream sequence of the other subunits common to all three nuclear RNA polymerases.
ABC10α is encoded by a single gene located on chromosome VIII
To determine whether ABC10α is encoded by a single gene, a Southern blot analysis was performed with YNN281 DNA digested with five restriction endonucleases, separately or in various combinations, and probed with the 30-mer oligonucleotide encoding an internal peptide sequence of ABC10α. In each case, the probe hybridized strongly to a single DNA fragment indicating that only one gene encodes ABC10α (Fig. 2). A unique DNA fragment also hybridized to the 2.1 kb EcoR I probe that encompasses the RPC10 gene (Fig. 4B). The possibility of a tandem repetition of the gene was also excluded by cleaving genomic DNA with EcoR I and EcoN I. EcoN I restriction enzyme cleaves within the RPC10 gene once, next to the sequence complementary to the oligonucleotide probe. Again, a unique 0.65 kb fragment hybridized to the probe (Fig. 2, lane E + En). RPC10 was localized to chromosome VIII by probing a Southern blot of chromosomal DNA from YNN295 strain separated by pulse field electrophoresis (Fig. 3). We noted that the probe (the Nru I-EcoN I gene internal fragment) also hybridized faintly to chromosome IV, which suggested a weak homology of part of the probe with another DNA fragment.
Figure 2.
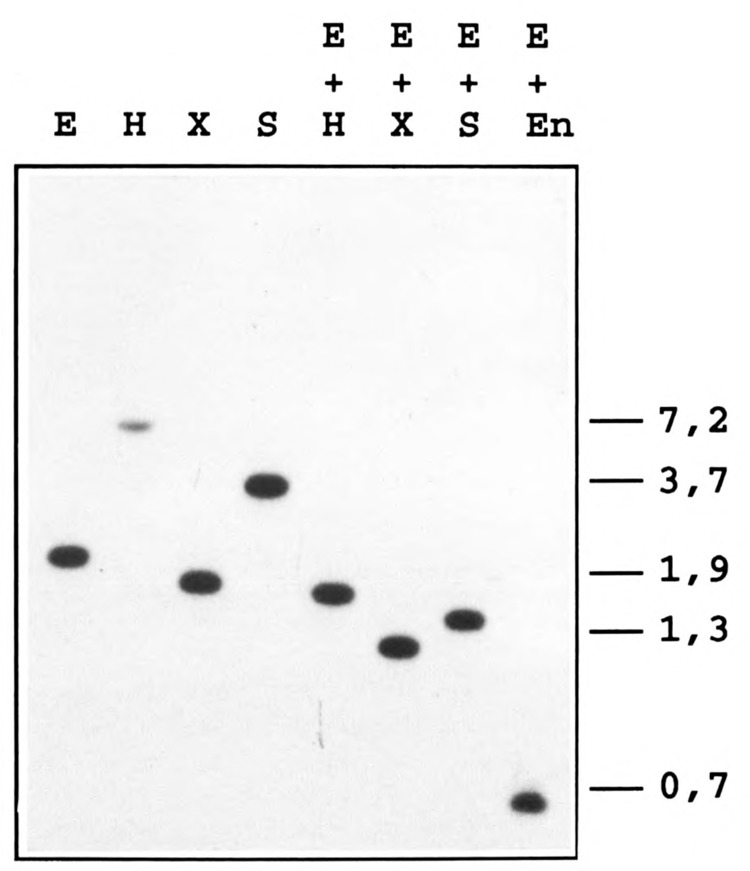
RPC10 is a single copy gene. Yeast genomic DNA from YNN281 was digested with the indicated restriction enzymes (E, EcoR I; H, Hind III; X, Xba I; S, Stu I; En, EcoN I), electrophoresed on a 0.7% agarose gel, transferred onto a nitrocellulose membrane, and probed with the 32P-end labeled 30-mer oligonucleotide as described in Materials and Methods. The size of marker DNA fragments is given in kilobases.
Figure 4.

Chromosomal disruption of RPC10. A. Restriction map of RPC10 chromosomal locus and replacement of the Nru I-EcoN I internal fragment by HIS3 gene. Restriction sites are: H, Hind III; E, EcoR I; N, Nru I; En, EcoN I; B, BamH I. The arrows indicate the size of relevant restriction fragments. B. Southern analysis of the gene disruption. The diploid strain CMY214 (his−) was transformed with the 3.7 kb EcoR I fragment containing RPC10 disrupted by the HIS3 gene. The genomic DNA of several HIS+ transformants was isolated, restricted with EcoR I, and subjected to Southern analysis by hybridization with the 2.1 kb EcoR I fragment. Lanes 1, 2, and 3 show the Southern blot from three HIS+ transformants; wt, control hybridization with DNA from the original CMY214 strain. C. Tetrad analysis from one HIS+ disrupted diploid transformant containing one wild-type and one disrupted copy of RPC10. All dissected tetrads contained only two viable spores on YPD medium which were always his−.
Figure 3.
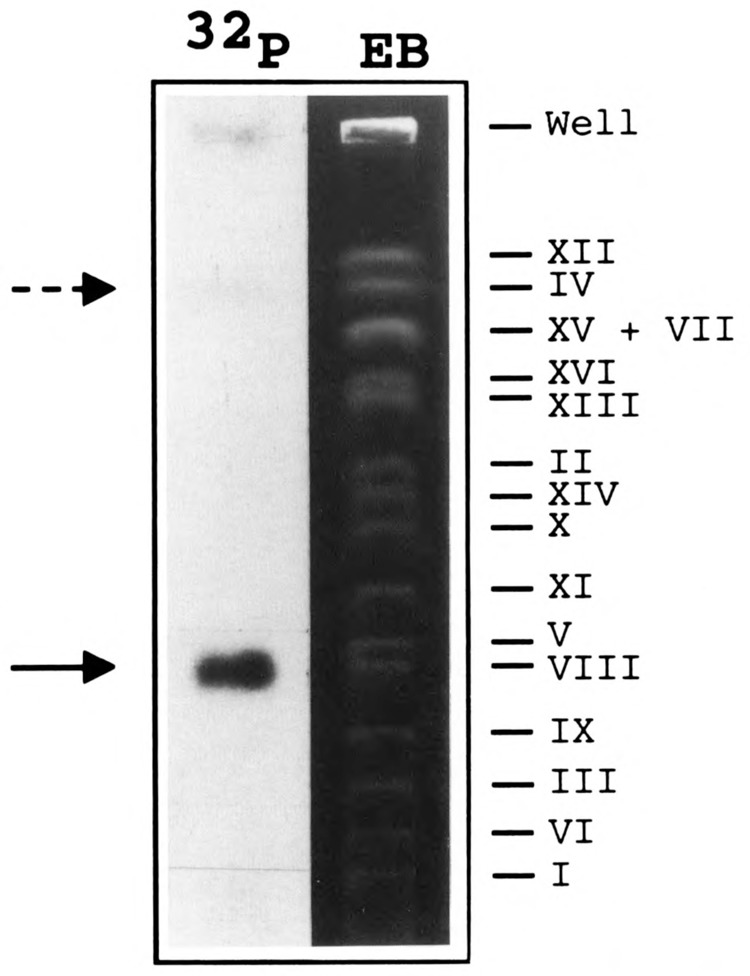
Chromosomal localization of RPC10. Chromosomal DNA of YNN295 yeast strain was separated by pulse field electrophoresis and stained with ethidium bromide (EB). After transfer onto a nitrocellulose membrane, the blotted DNA was hybridized with the Nru I-EcoN I 32P-labeled fragment. The labeled chromosomal DNA band was identified by auto radiography (32P). The probe hybridized strongly to chromosome VIII (arrow). The dotted arrow points to the faint hybridization signal on chromosome IV.
RPC10 is an essential gene
To test whether the RPC10 gene is essential for growth, the chromosomal coding sequence was disrupted by the method of Rothstein (1983). To do this, the Nru I-EcoN I fragment from RPC10 gene was replaced by a BamH I fragment carrying the yeast HIS3 gene (Fig. 4A). The resulting plasmid (pLS195) was cut with EcoR I and used to transform the his− homozygous diploid strain CMY214. A Southern blot experiment was done on the genomic DNA of 8 transformants and confirmed that, in each case, one of the two chromosomal copies had been replaced by the HIS3 disrupted construct (Fig. 4B). Two transformants were sporulated, and all asci analyzed (of 30) gave rise to only two viable spores (Fig. 4C). As expected, the two resulting colonies from each tetrad were always his−, which shows that the RPC10 gene is an essential gene. The two nonviable spores from each tetrad ceased to divide after one or two divisions.
Discussion
RPC10 codes for an additional yeast RNA polymerase common subunit. Like all the common subunit genes, it is unique and essential. It encodes a very basic protein (pI 10.06) of 70 amino acids which contains a zinc binding motif of the form CX2CX13CX2C.
ABC10α and ABC10β are intriguingly similar. These two small basic polypeptides comigrate on SDS-gel electrophoresis, and both bind zinc in vitro (Carles et al., 1991). It now appears that they are unrelated in sequence. In addition their size is different, and ABC10α is modified at its amino-terminus in the three forms of nuclear RNA polymerase, whereas the amino-terminus of ABC10β is unmodified (Carles et al., 1991; Woychick and Young, 1990).
As the genes for ABC10α and ABC10β are essential for cell growth (Woychick and Young, 1990; this work), these two subunits must carry out different, but possibly related, functions. The sequence of ABC10α and ABC10β reveal that these two basic proteins contain a putative metal-binding motif. The significance of these motifs is strengthened by the recent observation that both proteins bind zinc (Carles etal., 1991). In view of the small size of ABC10α and β, the metal-binding domain encompasses about one-third of the whole polypeptide. The role of these zinc-binding domains in enzyme structure or function remains to be explored.
The identification of ABC10α and ABC10β as additional common subunits brings to five the number of subunits shared by the three RNA polymerases, and to seven the subunits common to RNA polymerases A and C (Table 1). In the case of AC40 and AC19 shared by A and C enzymes, there is a relationship to the bacterial a subunit (Dequard-Chablat et al., 1991). The abundance of small subunits shared by the three enzymes is more puzzling, particularly when considering that none of them has an equivalent in eubacterial RNA polymerases. Why are there so many common subunits, and what are their roles? One could speculate that the common subunits carry out functions shared by all three RNA polymerases that are specific for eukaryotic cells. They may perform functions involving interaction with chromatin proteins, assembly or nuclear localization of RNA polymerase subunits, or they might be involved in some coordination of rRNA, tRNA, and mRNA synthesis. In at least two cases, however, homologues of shared subunits were identified in archaebacterial RNA polymerase with sequence similarity to ABC27 (Sentenac et al., 1992) and to ABC14.5 (Zillig, personal communication). Therefore, a general role should be anticipated for these two common subunits that is unrelated to the multiplicity of eukaryotic RNA polymerases or to their localization in the nucleus.
Table 1.
Shared subunits of yeast RNA polymerases.
| Subunit | Gene | Calculatedmass | Chromosomal location | pl | Particularities |
|---|---|---|---|---|---|
| ABC27 | RPB5 | 25 | II | 10.2 | Two copies |
| ABC23 | RPB6 | 18 | XVI | 5.2 | Phosphorylated |
| ABC14.5 | RPB8 | 17 | XV | 4.3 | |
| ABC10α | RPC10 | 7.7 | VIII | 10.1 | Zinc binding; N-terminus blocked |
| ABC10β | RPB10 | 5.4 | XV | 9.6 | Zinc binding |
| AC40 | RPC40 | 38 | XVI | 5.6 | Related to α |
| AC19 | RPCI9 | 16 | II | 4.5 | Related to α; phosphorylated |
References for gene cloning are: ABC27, ABC23 and ABC14.5, Woychik et al., 1990; ABC10α, this work; ABC10β, Woychik and Young, 1990; AC40, Mann et al., 1987; AC19, Dequard-Chablat et al., 1991.
Acknowledgments
We are grateful to D. Averbeck and M. Dardahlon (I.B.P.C., Paris) for providing us with a yeast chromosome blot, to Catherine Doira for oligonucleotide synthesis, to Françoise Bouet for peptide sequencing, and to Christian Marck for assistance in preparing Figure 1. We thank Alain Vincent (C.N.R.S., Toulouse) for his comments on zinc finger, Carl Mann for discussions and for correcting the manuscript, and W. Zillig for sharing his information prior to publication.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Footnotes
RNA polymerase subunits are referred to by a letter indicating the form of enzyme from which they are isolated, followed by their apparent size (in kDa). Shared sub-units are indicated by two or three letters.
References
- Allison L. A., Moyle M., Shales M., and Ingles C. J. (1985), Cell 42, 599–610. [DOI] [PubMed] [Google Scholar]
- Buhler J. M., Sentenac A., and Fromageot P. (1974), J Biol Chem 249, 5963–5970. [PubMed] [Google Scholar]
- Carle G. and Olson M. V. (1985), Proc Natl Acad Sci USA 82, 3756–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carles C., Treich I., Bouet F., Riva M., and Sentenac A. (1991), J Biol Chem 266, 24092–24096. [PubMed] [Google Scholar]
- Chelsky D., Ralph R., and Jonak G. (1989), Mol Cell Biol 9, 2487–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Seta F., Treich I., Buhler J. M., and Sentenac A. (1990), J Biol Chem 265, 15168–15175. [PubMed] [Google Scholar]
- Dequard-Chablat M., Riva M., Carles C., and Sentenac A. (1991), J Biol Chem 266, 15300–15307. [PubMed] [Google Scholar]
- Dezélée S., Sentenac A., and Fromageot P. (1972), FEBS Lett 21, 1–6. [DOI] [PubMed] [Google Scholar]
- Gomez-Marques J. and Segade F. (1988), FEBS Lett 226, 217–219. [DOI] [PubMed] [Google Scholar]
- Hamilton R., Watanabe C. K., and de Boer H. A. (1987), Nucl Acids Res 15, 3581–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet J., Riva M., Sentenac A., and Fromageot P. (1985), J Biol Chem 260, 15304–15310. [PubMed] [Google Scholar]
- James P., Whelen S., and Hall B. D. (1991), J Biol Chem 266, 5616–5624. [PubMed] [Google Scholar]
- Lue N. F., Buchman A. R., and Koonberg R. D. (1989), Proc Natl Acad Sci USA 86, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E. F., and Sambrook J. (1982), Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Mann C., Buhler J.-M., Treich I., and Sentenac A. (1987), Cell 48, 627–637. [DOI] [PubMed] [Google Scholar]
- Marck C. (1988), Nucl Acids Res 16, 1829–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mémet S., Gouy M., Marck C., Sentenac A., and Buhler J.-M. (1988), J Biol Chem 263, 2830–2839. [PubMed] [Google Scholar]
- Parent S. A., Fenimore C. M., and Bostian K. A. (1985), Yeast 1, 83–138. [DOI] [PubMed] [Google Scholar]
- Riva M., Mémet S., Micouin J. Y., Huet J., Treich I., Dassa J., Young R. A., Buhler J.-M., Sentenac A., and Fromageot P. (1986), Proc Natl Acad Sci USA 83, 1554–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R. J. (1983), Methods Enzymol 101, 202–211. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., and Coulson A. R. (1977), Proc Natl Acad Sci USA 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentenac A. (1985), CRC Crit Rev Biochem 18, 31–90. [DOI] [PubMed] [Google Scholar]
- Sentenac A., Riva M., Thuriaux P., Buhler J.-M., Treich I., Carles C., Werner M., Ruet A., Huet J., Mann C., Chiannilkulchai N., Stettler S., and Mariotte-Labarre S. (1992), in Transcriptional Regulation (Yamamoto K. R., and McKnight S. L., eds.), Cold Spring Harbor Laboratory Monograph Series, Cold Spring Harbor, NY, in press. [Google Scholar]
- Sweetser D., Nonet M., and Young R. A. (1987), Proc Natl Acad Sci USA 84, 1192–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woychik N. A. and Young R. A. (1990), J Biol Chem 265, 17816–17819. [PubMed] [Google Scholar]
- Woychik N. A., Liao S.-M., Kolodziej P. A., and Young R. A. (1990), Genes Dev 4, 313–323. [DOI] [PubMed] [Google Scholar]
- Yano R. and Nomura M. (1991), Mol Cell Biol 11, 754–764. [DOI] [PMC free article] [PubMed] [Google Scholar]