

Abstract
The HIV Tat protein is a potent transactivator of HIV transcription, increasing both RNA initiation and elongation. We now demonstrate that purified, full-length 86 amino acid Tat protein specifically transactivates the HIV LTR in vitro to a high level (25- to 60-fold). Tat transactivation was specifically blocked by anti-Tat serum, but not preimmune serum. Tat did not transactivate transcription from the control adenovirus major late promoter (AdMLP). HIV transcription was blocked at various functional steps during initiation and elongation complex formation. Similar to the control AdMLP, HIV basal initiation complex assembly was sensitive to the addition of 0.015 % sarkosyl prior to the addition of nucleoside triphosphates. Resistance to 0.05% sarkosyl required the addition of G, C, and U, which constitute the first 13 bases of the HIV RNA transcript. The addition of Tat to the in vitro transcription relieved the 0.015% sarkosyl block. These Tat-induced complexes were sensitive to 0.05 % sarkosyl, suggesting that transcriptional initiation had not occurred. Consistent with this hypothesis, the addition of G, C, and U to the Tat-induced transcription complexes allowed the rapid conversion to transcription initiation complexes. Tat also facilitated the formation of 0.015% sarkosyl-resistant complexes in a reconstituted transcription system containing partially purified transcription factors and polymerase II. Following the formation of stable initiation complexes, Tat increased the rate and efficiency of transcription elongation on the HIV but not the AdML template. Kinetic analysis of Tat transactivation suggests that approximately 30% of the Tat initiation complexes are converted to elongation complexes. We conclude that Tat, in addition to its demonstrated role in RNA elongation, facilitates transcription initiation in vitro.
The genome of human immunodeficiency virus type 1 (HIV-1; Barre-Sinousi et al., 1983; Gallo et al., 1984; Levy et al., 1984), etiological agent of acquired immune deficiency syndrome (AIDS), encodes the common retroviral structural gag, pol, and env gene products and two essential regulatory proteins, Tat and Rev (Rosen and Pavlakis, 1990). Tat is localized primarily in the nucleolus and acts as a potent activator of HIV-1 long terminal repeat (LTR)-directed gene expression to increase the steady-state levels of all HIV-1 mRNAs (Arya et al., 1985; Sodroski et al., 1985; Dayton et al., 1986; Cullen, 1986; Rosen et al., 1986; Fisher et al., 1986; Peterlin et al., 1986; Wright et al., 1986; Hauber et al., 1987; Muesing et al., 1987; Rice and Matthews, 1988; Sadaie et al., 1988). In vivo studies have suggested that Tat regulates gene expression at both transcriptional and post-transcriptional levels (Cullen, 1986; Hauber et al., 1987; Jeang et al., 1988; Parkin et al., 1988; Rice and Matthews, 1988; Edery et al., 1989; SenGupta et al., 1990; Braddock et al., 1989 and 1990), and that transcriptional activation of the HIV-1 LTR promoter by Tat is the result of increased rates of transcription initiation and elongation (Laspia et al., 1989 and 1990). Recent in vivo and in vitro studies have supported a model for Tat transactivation primarily at the level of transcription elongation (Marciniak et al., 1990a; Marciniak and Sharp, 1991; Feinberg et al., 1991). In contrast, compelling evidence has been presented which suggests that Tat may also function in the context of a DNA-binding protein (Berkhout and Jeang, 1990), and more specifically, that Tat and acidic activators may act on a similar step in the transcription process (Southgate and Green, 1991). In addition, Tat has been proposed to act as an antiterminator of transcription (Kao et al., 1987; Toohey et al., 1989; Selby et al., 1989), potentiating full-length elongation of abundant HIV-1 transcripts normally truncated in the absence of Tat.
The Tat transactivation response (TAR) element (Rosen et al., 1985) contains cis-acting sequences located between +14 and +44 relative to the start site of transcription and is necessary for Tat transactivation (Peterlin et al., 1986; Muesing et al., 1987; Hauber and Cullen, 1988; Garcia et al., 1988; Jakobovits et al., 1988; Selby et al., 1988; Feng and Holland, 1988; Garcia et al., 1989; Berkhout and Jeang, 1989; Roy et al., 1990). The 5′-untranslated leader RNA forms an energetically favorable stem-loop structure (Muesing et al., 1987) that binds cellular proteins and Tat specifically in vitro (Dingwall et al, 1989; Gaynor et al., 1989; Marciniak et al., 1990b). It has been proposed that Tat binding to TAR sequences and Tat transactivation of HIV-1 LTR-driven genes are linked coordinately to p68-TAR RNA binding (Marciniak et al., 1990a,b). Although the TAR sequences required for RNA binding to Tat are essential for Tat transactivation, the physiological significance of Tat binding to TAR RNA remains unclear. In addition to requiring TAR sequences for Tat activity, upstream LTR promoter regions inclusive of the SP1 binding and TATA elements are essential for optimum Tat transactivation (Cullen, 1986; Luciw et al., 1986; Jones et al., 1986; Wright et al., 1986; Nabel and Baltimore, 1987; Bohnlein et al., 1988; Hauber and Cullen, 1988; Wu et al., 1988; Gaynor et al., 1989; Leonard et al., 1989; Clark et al., 1990; Berkhout et al., 1990; Parrott et al., 1991). Interestingly, TAR-independent HIV-1 LTR activation by Tat occurs in vivo with LTR constructs containing heterologous RNA-binding sites and Tat fusion proteins having either the Rev or MS2 RNA binding site domains (Southgate et al., 1990; Selby and Peterlin, 1990).
Is it possible that Tat facilitates both transcription initiation and elongation? Certainly, there is precedent for eukaryotic transcription factors that provide a dual role in promoting both initiation and elongation. TFIIF (RAP 30 and RAP 74) mediates the association of RNA polymerase II with the TFIID-TFIIA-TFIIB (DAB) preinitiation complex (Flores et al., 1991). In addition, TFIIF apparently promotes a conformational change in the eukaryotic promoter DNA (Buratowski et al., 1991). This conformational change has been compared to the transition from a closed to an open complex in the prokaryotic transcription complex. The ability of Tat to regulate both transcription initiation and elongation may be due to the modular structure of Tat (Southgate et al., 1991). In the present studies, full-length 86 amino acid Tat was utilized to analyze in vitro Tat transactivation. Analysis of in vitro transcription at successive functional steps in preinitiation and initiation complex formation with the anionic detergent sarkosyl, in both unfractionated cell-free extracts and reconstituted systems using partially purified transcription factors, indicated that Tat increased HIV-1 transcription initiation and elongation.
Materials and methods
Plasmids, reagents, and DNA templates
Reagents used in the transcription assays were sarkosyl (N-lauroyl sarcosine sodium salt; Sigma Chemical Company) prepared as a 20% stock solution (wt/vol), unlabeled FPLC purified nucleoside triphosphates (Pharmacia), and [α-32P]UTP (400 Ci/mmol; Amersham). The DNAs used in transcription assays as templates were the plasmids pCD12CAT, which contains the HIV-1 LTR (−453 to +126) fused to the prokaryotic chloramphenicol acetyltransferase (CAT) gene (Ensoli et al., 1989; Okamoto et al., 1990), and pAdMLP, which contains the 430 bp Alu I fragment of pAd6 that has the major late promoter and 33 base pairs of downstream sequences. HIV-1 LTR deletion mutants have been described previously (Ensoli et al., 1989; Okamoto et al., 1990; Boris-Lawrie et al., 1992). pCD23dlS contains a deletion of HIV-1 leader sequences +34 to +39 which form the loop in the TAR RNA secondary structure. pLM2 contains HIV-1 LTR sequences (−126 to +57) fused to the CAT gene and is the parent plasmid of pTM25, which contains a four-base compensatory substitution mutation at nucleotides +25 to +28 (CTCG) and +35 to +38 (CGAG) within the HIV-1 leader sequence. The mutation changes the structure of both the TAR bulge and loop.
Templates for in vitro transcription were prepared by digesting 100 μg of plasmid DNA with a 5- to 10-fold unit excess of restriction enzyme for 1–2 hours under buffer conditions suggested by the manufacturer (New England Biolabs). After termination of reactions, DNA digests were subjected to two phenol-chloroform-isoamyl alcohol (50:50:1) extractions and subsequently precipitated by ethanol.
In vitro transcription: unfractionated extracts
For most transcription experiments, preincubation of protein extracts (40 μg) and DNA template in the absence of nucleoside triphosphates was followed by the addition of nucleoside triphosphates and a second incubation. All incubations were at 30°C. The times of these incubations varied and are presented in the legends to the figures. The volume of the preincubation reaction was 15 μl and was increased by the addition of Tat, sarkosyl, or nucleoside triphosphates to reach a final volume of 16.5 μl. The in vitro transcription buffer contained 10 mM HEPES (pH 7.9), 50 mM KCl, 0.5 mM EDTA, 1.5 mM DTT, 6.25 mM MgCl2, and 8.5% glycerol. The DNA templates pCD12, CD54, CD52, CD38, CD23, CD16, CD7, CD23dlS, TM25, and LM2 were linearized with EcoR I and added to a final concentration of 5 μg/ml (75 ng/reaction). pAdML was linearized with BamH I and added at a concentration of 5–20 μg/ml (75–350 ng/reaction). HeLa whole-cell extracts, prepared as described previously (Manley et al., 1980), were added to a final concentration of 2.4 mg/ml (40 μg/reaction). Purified Tat (in Tat storage buffer: PBS without Ca++ and Mg++, 0.1% BSA [RNase- and DNase-free], 0.1 mM DTT) was added to a final concentration of 0.4 μM unless otherwise stated. Nucleoside triphosphates in water were added to a final concentration of 500 μM unless otherwise stated. After a 30-minute preincubation period, 20 μCi (2 μl) of [α-32P]UTP (400 Ci/mmole) was added. Transcription reactions were terminated by the addition of 20 mM Tris-HCl (pH 7.8), 150 mM NaCl, and 0.2% SDS. The quenched reactions were extracted with equal volumes of phenol-chloroform and chloroform and precipitated with 2.5 volumes of ethanol and 0.1 volume of 3.0 M sodium acetate. Following centrifugation, RNA pellets were resuspended in 12 μl of formamide denaturation mix containing xylene cyanol and bromophenol blue, heated at 90°C for 3 minutes, and electrophoresed at 400 V in a 4% poly-acrylamide (19:1 acrylamide:bisacrylamide) gel containing 7 M urea (prerun at 200 V for 30 minutes) and 1X TBE. Gels were exposed to Kodak X-Omat XR-5 film at −70°C with intensifying screens for autoradiography.
In vitro transcription: reconstituted system
Transcription reactions were reconstituted with partially purified TFIIA, TFIIB, TFIID, TFIIE/F, and RNA polymerase II generously provided by Dr. Ulla Hansen, Dana-Farber Cancer Institute. The reaction buffer contained 50 mM Tris-HCl (pH 7.8), 60 mM KCl, 5% glycerol, 6 mM MgCl2, and 2 mM DTT. RNA was purified as described above.
Antibody neutralization assays
Antibody to Tat was raised against purified Tat in rabbits. The polyclonal antiserum was purified by affinity chromatography on a Tat column and tested by immunoprecipitation and by neutralization of Tat-induced AIDS-KS cell growth (Ensoli et al., 1989). Antibody at a 1:10 dilution was added to the preincubation reaction containing Tat.
Results
Purified Tat specifically transactivates HIV-1 in vitro
The 86 amino acid Tat protein used in these studies was expressed in E. coli and purified to >99% homogeneity by successive rounds of molecular sieve and HPLC reverse phase chromatography (B. Ensoli et al., unpublished data; Fig. 1A). Purified Tat (0.04–4.0 μM) was added to in vitro transcription reactions containing 75 ng of an EcoR I-linearized HIV-1 DNA template (CD12R1), 500 μM NTPs, and HeLa cell extract (40 μg protein). In the presence of optimal concentrations of Tat (0.4 μM), a reproducible 25- to 60-fold increase in the abundance of the full-length 382 nt HIV-1 run-off transcript was observed (Fig. 1B, lane 3) under conditions yielding low basal HIV-1 transcription. Transactivation was minimal (fourfold) at low Tat concentrations (0.04 μM; Fig. 1B, lane 2), and promoter activity was inhibited at high Tat concentrations (4 μM; Fig. 1B, lane 4). High concentrations of HIV-1 LTR DNA templates in in vitro transcription reactions yielded high basal transcription activity, which diminished the observable Tat transactivation effect. Tat storage buffer did not contribute to Tat transactivation, as an equal volume of Tat storage buffer added to the in vitro transcription reaction yielded basal levels of transcription (Fig. 1B, lane 1).
Figure 1.
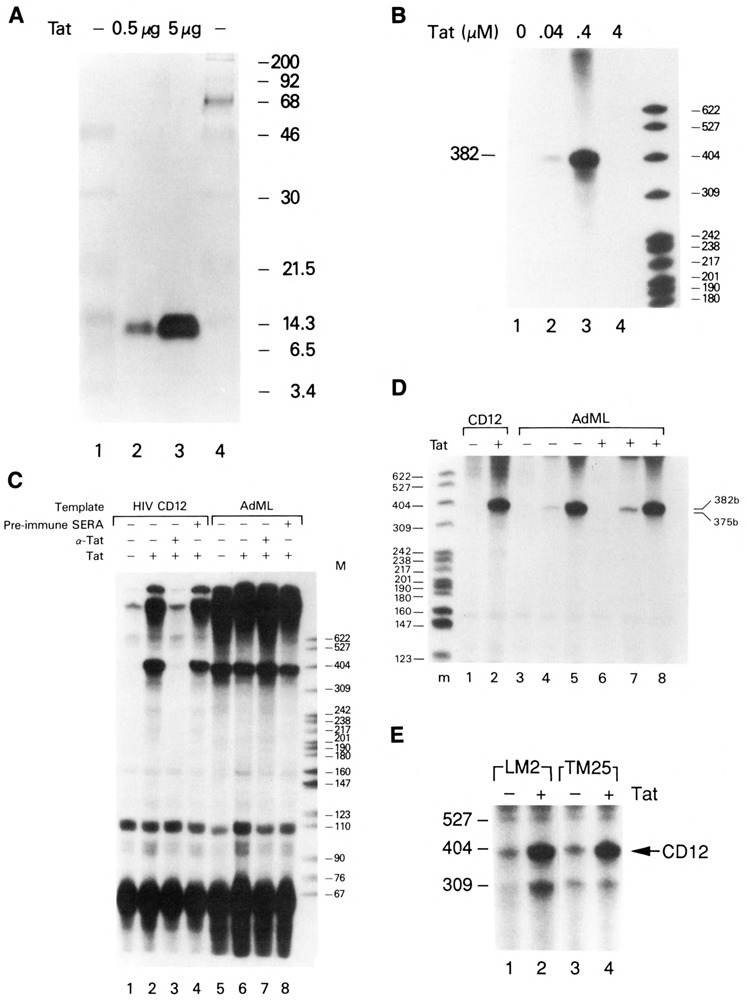
Tat-specific transactivation of HIV-1 transcription in vitro. A. Silver stain of an SDS-PAGE gel of purified Tat. Lanes 1 and 4, molecular weight markers; lane 2, 0.5 μg Tat; lane 3, 5 μg Tat. B. Tat concentration dependence for maximal transactivation in vitro. The CD12 HIV template (75 ng) was incubated with HeLa whole-cell extract in the absence (lane 1) or presence of 0.04 μM (lane 2), 0.4 μM (lane 3), or 4.0 μM (lane 4) Tat. C. Inhibition of Tat transactivation by anti-Tat antibodies in vitro. The CD12 HIV or AdML DNA template was incubated in the absence (lanes 1 and 5) or presence (lanes 2–4 and 6–8) of 0.4 μM Tat. Rabbit anti-Tat polyclonal antibody (1:10 dilution; lanes 3 and 7) or rabbit preimmune serum (1:10 dilution; lanes 4 and 8) were added to in vitro transcription reactions containing the HIV (75 ng) or AdML (250 ng) templates in the presence of 0.4 μM Tat. D. Titration of AdMLP in the presence and absence of Tat. 75 ng (lanes 3 and 6), 150 ng (lanes 4 and 7), or 350 ng (lanes 5 and 8) of the AdMLP were transcribed in the presence or absence of 0.4 μM Tat as indicated. 75 ng of the HIV CD12 template was incubated in the absence (lane 1) or presence (lane 2) of 0.4 μM Tat. E. Cotranscription of HIV-1 wild-type and TAR mutant DNA templates. Wild-type HIV templates CD12 (75 ng) and LM2 (75 ng) or CD12 (75 ng) and TAR mutant template (TM25) were coincubated in HeLa whole-cell extracts in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of 0.4 μM Tat. Full-length transcripts from CD12, LM2, and TM25 HIV-1 LTR DNA templates were 382, 313, and 309 nt, respectively.
To demonstrate that the transactivation activity was specific to Tat, affinity-purified polyclonal antibodies to Tat were tested for their ability to block transactivation (Fig. 1C). Anti-Tat antibody efficiently blocked Tat transactivation of the HIV-1 LTR in vitro (Fig. 1C, lanes 1–3), but not transcription from the control AdML template (Fig. 1C, lanes 5–7). In contrast, preimmune rabbit serum did not affect Tat transactivation (Fig. 1C, lanes 2 and 4). This experiment also demonstrated that Tat transactivation in vitro was promoter-specific, as Tat was unable to stimulate transcription from an AdML template in parallel assays (Fig. 1C, lanes 5 and 6). In addition to demonstrating the promoter specificity of the Tat protein, these control assays also show that the addition of anti-Tat antibody or preimmune serum had no effect on basal transcription (Fig. 1C, lanes 7 and 8).
It could be argued that the lack of Tat transactivation of the AdMLP was due to the high basal activity of the promoter. To address this possibility, the AdML template was transcribed, in the presence or absence of Tat, at various template concentrations (Fig. 1D); 75 ng (lanes 3 and 6), 150 ng (lanes 4 and 7), or 350 ng (lanes 5 and 8) of the AdML template were added to the transcription reaction. No significant difference in the level of transcription was observed in the presence or absence of Tat. Transactivation of the HIV CD12 template (Fig. 1D, lanes 1 and 2) demonstrated that the Tat used in these studies was active.
To demonstrate further tne promoter specificity of Tat transactivation in vitro, an internal control was added to the transcription assay. The wild-type HIV-1 LTR DNA template (CD12R1) was cotranscribed with either a Tat transactivation-competent leader deletion mutant (LM2), a Tat transactivation-defective leader substitution mutant (TM25), or the AdMLP. The DNA templates were added simultaneously to in vitro transcription reactions in the absence or presence of Tat. Consistent with the results presented above, the wild-type HIV-1 LTR DNA template was responsive to Tat (Fig. 1E; 382 nt transcript). Transcription from the HIV-1 LTR DNA template (LM2) containing leader sequences 1–57 was also responsive to Tat transactivation (Fig. 1E, lanes 1 and 2; 313 nt transcript). In contrast, the leader substitution mutant DNA template (TM25) containing a TAR region defective for Tat and p68 binding was not responsive to Tat (Fig. 1E, lanes 3 and 4; 309 nt transcript). Similarly, cotranscription of the HIV and AdML promoters resulted in transactivation of the HIV but not the AdML template (data not shown). In addition, the TAR mutant CD23dlS containing a deletion of the TAR loop region was not transactivated by Tat in vitro (Fig. 2B, lanes 1 and 2; 338 nt transcript). Wild-type and LM2 DNA templates, but not TM25 and CD12dlS DNA templates, were shown to be active for Tat transactivation in vivo (Dingwall et al., 1989; Ensoli et al., 1989; Marciniak et al., 1990a; Boris-Lawrie et al., 1992). Extension of these analyses to other TAR mutants demonstrated a positive correlation between in vivo and in vitro Tat transactivation (data not shown).
Figure 2.
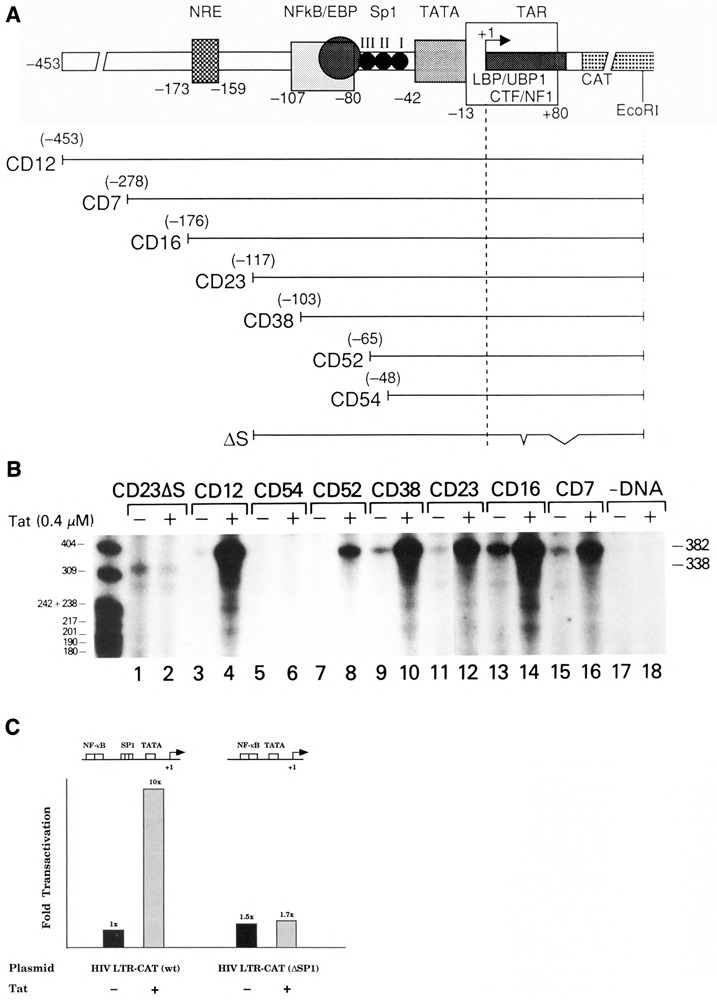
Tat-specific transactivation of transcription from 5′-deletion and TAR-deletion mutant HIV-1 LTR DNA templates in vitro. A. Diagram of HIV-1 LTR DNA templates used for in vitro transcription. B. Tat trans-activation of wild-type and mutant HIV-1 LTR transcription in vitro. Promoter templates (75 ng) were added to in vitro transcription reactions containing either Tat storage buffer (−) or 0.4 μM Tat (+). C. Effect of SP1 on Tat transactivation. In vitro transactivation was performed with approximately 100 ng of either the wild-type HIV-1 LTR or an internal SP1 deletion mutant (SPE-CAT) in the presence or absence of Tat (0.4 μM). Bar graphs indicate fold transactivation as measured by 32P cpm determination of RNA products.
Proximal LTR promoter elements and TAR are essential for Tat transactivation
Maximum transactivation of HIV-1 LTR-directed gene expression by Tat in vivo required both proximal promoter sequences (NF-kB, SP1, and TFIID binding sites) and TAR sequences (Cullen, 1986; Wright et al., 1986; Nabel and Baltimore, 1987; Hauber and Cullen, 1988; Leonard et al., 1989; Berkhout et al., 1990; Parrott et al., 1991). To determine whether similar LTR sequences were required for maximum Tat transactivation in vitro, 5′-LTR and TAR deletion mutants were used as transcription templates in cell-free extracts in the absence or presence of purified Tat (Fig. 2A and B). Tat transactivation was observed with templates CD12 (−453), CD7 (−278), CD16 (−176), CD23 (−117), CD38 (−103), and CD52 (−65) (Fig. 2B, lanes 3, 4, and 7–16). Tat transactivation was abolished upon deletion of LTR DNA sequences between −65 and −48 (CD54), which contained part of the SP1 binding sites of the HIV-1 LTR (Fig. 2B, lanes 5 and 6). Analysis of a specific internal deletion mutant within the HIV LTR that lacks the SP1 binding sites, but retains the NFkB and other regulatory sequences, demonstrated that SP1 is critical for efficient Tat transactivation in vitro (Fig. 2C). Therefore, Tat transactivation in vitro required a minimal promoter with several SP1 binding sites adjacent to a TATA box. Mutant CD23dlS (deletion of +35 to +38), inactive for Tat transactivation in vivo (Ensoli et al., 1989), was not transactivated by Tat in the in vitro transcription system (Fig. 2B, lanes 1 and 2). These mutant analyses showed that both proximal LTR promoter elements and TAR sequences were necessary for Tat transactivation in vitro.
Tat facilitates HIV-1 transcription preinitiation complex formation in the presence of sarkosyl
Tat transactivation in vitro (25- to 60-fold) was feasibly the composite of Tat effects on several steps in HIV-1 transcription. Therefore, the roles of Tat during HIV-1 transcription initiation and elongation in vitro were examined. The anionic detergent sarkosyl had been used to define discrete functional steps during RNA polymerase II transcription initiation in vitro using the AdML promoter (Cai and Luse, 1987; Reinberg and Roeder, 1987; Hawley and Roeder, 1987; Buratowski et al., 1989; Saltzman and Weinmann, 1989). A nucleotide-independent step involving the binding of TFIID and TFIIA to the promoter was sensitive to 0.015% sarkosyl (Fig. 3). Partial resistance to 0.015% sarkosyl resulted upon stable binding of TFIID and TFIIA to the promoter. Complete resistance to 0.015% sarkosyl required binding of RNA polymerase II to the DAB preinitiation complex. The assembly of preinitiation complexes remained sensitive to 0.05% sarkosyl at stages preceding transcription initiation. Productively initiated transcription complexes, formed by the incorporation of the first dinucleotide in the nascent transcript, were resistant to 0.05% sarkosyl (Luse et al., 1987).
Figure 3.
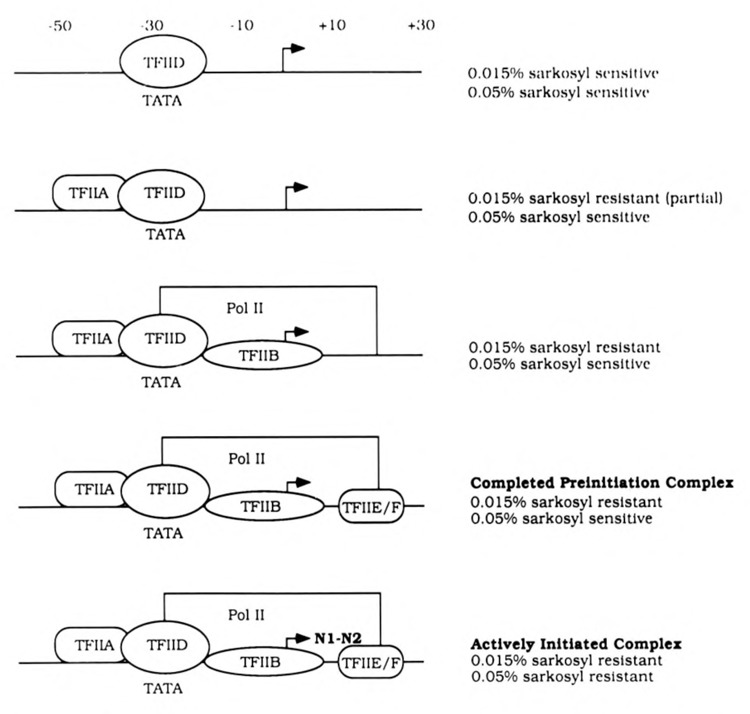
Schematic diagram of discrete functional steps during transcription initiation and their differential sensitivities to sarkosyl in vitro. Adapted from Cai and Luse, 1987; Reinberg and Roeder, 1987; Hawley and Roeder, 1987; Buratowski et al., 1989; Saltzman and Weinmann, 1989.
Experiments were first performed using the AdMLP as a control DNA template. Consistent with previous observations, the addition of sarkosyl at concentrations ⩾0.015% during the preincubation period inhibited the formation of full-length transcripts (375 nt) from the AdMLP (Fig. 4, lanes 1–8). When preinitiation complexes were permitted to form for 30 minutes prior to the addition of sarkosyl and nucleoside triphosphates, transcription was resistant to 0.015% sarkosyl but was blocked by sarkosyl concentrations ⩾0.05% (Fig. 4, lanes 9–15). These experiments demonstrated that the preincubation period allowed sufficient time for preinitiation complexes to form on the AdMLP. That complexes which assembled during the preincubation period were sensitive to 0.05% sarkosyl indicated that productively initiated complexes had not been formed prior to the addition of exogenous nucleoside triphosphates.
Figure 4.
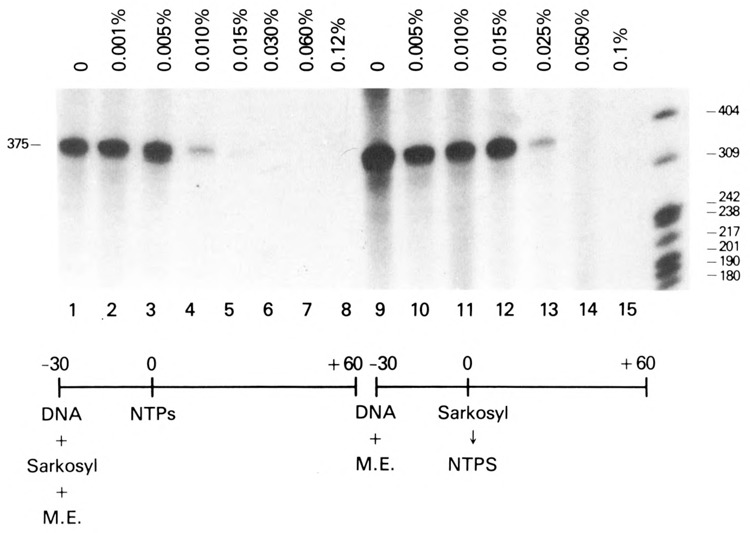
Effects of sarkosyl on in vitro transcription from the adenovirus major late promoter (AdMLP). The indicated concentrations of sarkosyl were added before (lanes 1–8) or after (lanes 9–15) a 30-minute preincubation in the absence of exogenous nucleoside triphosphates to in vitro transcription reactions containing the AdML template (300 ng).
In parallel experiments, sarkosyl (0.001%–0.25%) was added to in vitro transcription reactions containing the HIV-1 LTR promoter (CD12R1) in the absence or presence of Tat (Fig. 5). As observed with the AdMLP, HIV-1 LTR basal transcription was sensitive to the addition of 0.015% sarkosyl at the onset of the preincubation period (Fig. 5A, lanes 1–5). A tenfold decrease in the level of basal transcription was observed (Fig. 5A, lanes 1 and 4). This number was probably an underestimate of the relative effectiveness of the sarkosyl block, since no transcription was detected at sarkosyl concentrations ⩾0.015%. In contrast, when Tat was added to transcription reactions containing ⩽0.015% sarkosyl, efficient transactivation of HIV-1 RNA synthesis was observed (>25-fold; Fig. 5A, lanes 1–4 and 6–9). Less than a twofold reduction in Tat transactivation was observed in the presence of 0.015% sarkosyl (Fig. 5A, lanes 6 and 9). When the concentration of sarkosyl was >0.015%, Tat transactivation of the HIV-1 LTR DNA template was not observed (Fig. 5A, lanes 5 and 10; data not shown). That Tat transactivation was observed in the presence of 0.015% sarkosyl, a concentration of detergent that was inhibitory for basal HIV-1 transcription, suggested that Tat was able to overcome a sarkosyl block that occurred early in preinitiation complex formation, specifically at a step prior to or inclusive of the stable formation of the DAB-Pol II complex on the HIV-1 LTR promoter.
Figure 5.
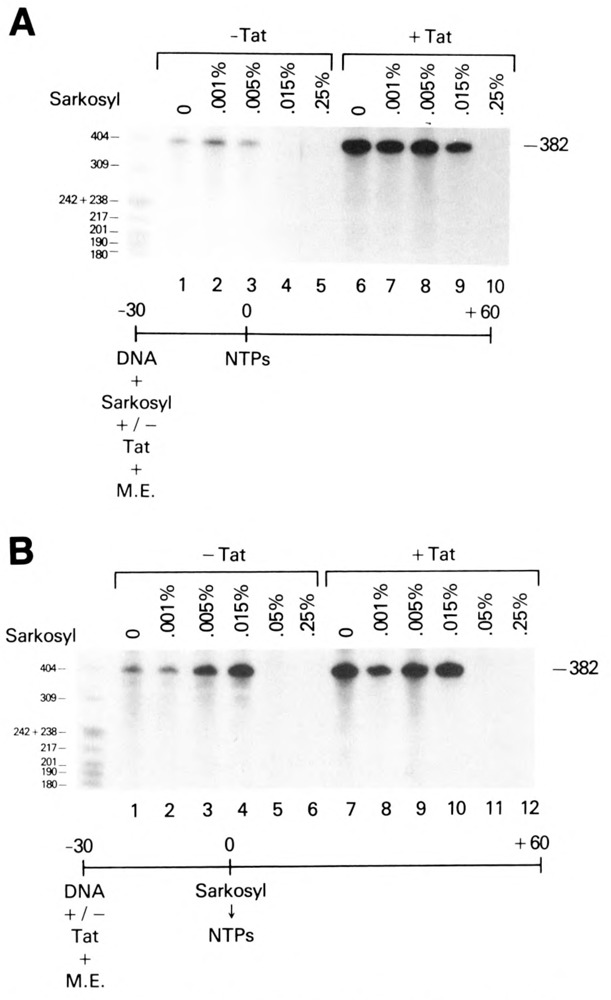
Facilitation of preinitiation complex formation on the HIV-1 LTR promoter in the presence of Tat. A. Resistance of HIV-1 transcription to 0.015% sarkosyl added at the start of preincubation in the presence of Tat. 75 ng of the CD12 template was transcribed in HeLa whole-cell extracts containing 0.0% to 0.25% sarkosyl during the preincubation period. Reactions were carried out in the absence (lanes 1–5) or presence (lanes 6–10) of 0.4 μM Tat. B. Effect of sarkosyl addition to in vitro transcription reactions after preincubation.
Preincubation of transcription reactions prior to the stepwise addition of sarkosyl and nucleoside triphosphates conferred HIV-1 transcription resistance to concentrations of sarkosyl ⩽0.015% and decreased the level of Tat transactivation from the HIV-1 LTR promoter (Fig. 5B, lanes 1–4 and 7–10). These results indicated two important points: first, that the sensitivities of basal transcription from the HIV-1 LTR and AdML promoters to sarkosyl were similar; and second, that preincubation of the HIV-1 LTR DNA template with a HeLa cell extract permitted the efficient formation of preinitiation complexes and negated the quantitative effect of Tat transactivation. Transcription from the HIV-1 LTR promoter was blocked by the addition of 0.05% sarkosyl to in vitro transcription reactions at the end of the preincubation period and prior to the addition of exogenous nucleoside triphosphates (Fig. 5B, lanes 5 and 11) and indicated that productively initiated transcription complexes were unable to form in the absence of exogenous nucleoside triphosphates.
Nucleotide requirements for HIV transcription in the presence or absence of Tat
The above studies suggested that low concentrations of nucleotide pools (<1 μM) in the HeLa cell extracts used in the in vitro transcription assays were insufficient to support productive transcription initiation. This hypothesis could be tested directly, since the nucleoside triphosphates required for initiation of RNA synthesis could be predicted from the HIV-1 nucleotide sequence. Combinations of exogenous nucleoside triphosphates lacking either ATP or GTP and CTP were added to preincubation reactions containing the HIV-1 LTR template with or without Tat. After sarkosyl addition to 0.05%, the full complement of nucleoside triphosphates were introduced, and transcripts were analyzed following elongation. In the absence of Tat, detectable levels of full-length transcripts were observed only under conditions when all nucleoside triphosphates were present prior to sarkosyl addition (Fig. 6A, lanes 1–3). Tat stimulated transcription when either all four nucleoside triphosphates (Fig. 6A, lane 4) or GTP, CTP, and UTP (Fig. 6A, lane 5) were added to the transcription reaction prior to the addition of sarkosyl. In contrast, the addition of GTP, CTP, and ATP; CTP, ATP, and UTP; GTP, ATP, and UTP; or ATP and UTP did not result in the formation of Tat-induced 0.05% sarkosyl resistant complexes (Fig. 6A, lanes 3 and 6; data not shown).
Figure 6.
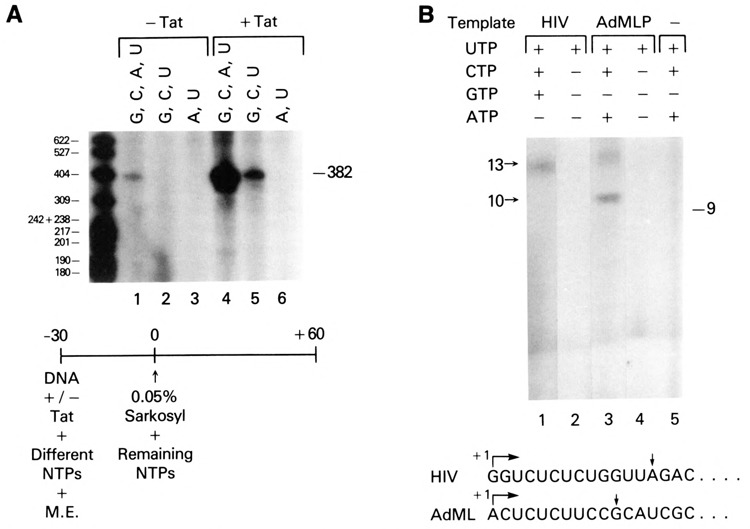
Ability of Tat to facilitate transcription preinitiation complex formation in the absence of nascent RNA synthesis. A. Effect of preincubation of HIV template and Tat in the presence of various nucleoside triphosphates. Different combinations of nucleoside triphosphates were added to in vitro transcription reactions containing the HIV CD12 promoter template (75 ng) without (lanes 1–3) or with (lanes 4–6) 0.4 μM Tat. After a 30-minute preincubation, 0.05% sarkosyl and then a full complement of nucleoside triphosphates were added to the reactions. B. Effect of limiting exogenous nucleoside triphosphates on HIV-1 transcription in vitro. Different combinations of nucleoside triphosphates were added to in vitro transcription reactions containing 300 ng of either AdMLP or HIV-1 LTR DNA templates. Transcription reactions were analyzed after a 30-minute incubation.
The level of HIV transcription in the presence of G, C, and U to the preincubation was lowered because of the ATP-dependent step in transcriptional initiation. That initiation complex formation was observed in the presence of GTP, CTP, and UTP (Fig. 6A, lane 5) was consistent with the observation that GTP, albeit at lower efficiency, was able to substitute for ATP in the catalytic conversion of RNA polymerase IIA to 110, an energy-dependent polymerase phosphorylation “activation” step occurring prior to or concomitant with the formation of the first phosphodiester bond (Payne et al., 1989; Laybourn and Dahmus, 1990).
The addition of GTP, CTP, and UTP to the preincubation reaction was expected to allow the synthesis of 13 nucleotide nascent RNA blocked in elongation due to the lack of ATP, the next base in the nascent HIV-1 RNA (Fig. 6B, bottom). To determine whether the expected short transcripts were synthesized, transcription reactions from HIV-1 LTR and AdMLP DNA templates were analyzed 30 minutes post-initiation for short transcripts. Thirteen nucleotide transcripts were synthesized from the HIV-1 LTR promoter in transcription reactions containing GTP, CTP, and UTP but lacking ATP (Fig. 6B, lane 1). When only UTP was added to the reaction, no short transcripts were detected (Fig. 6B, lane 2), which indicated that HIV-1 transcription was not initiated. As a control for these studies, short transcripts from the AdMLP were also analyzed. Exclusion of GTP from AdMLP transcription reactions yielded transcripts of 10 nucleotides, since the first guanosine in the nascent transcript from the AdMLP was positioned at +11 (Fig. 6B, lane 3). UTP alone was unable to initiate transcription from the AdMLP (Fig. 6B, lane 4). As expected, no transcripts were detected in transcription reactions containing HeLa cell extract, CTP, ATP, and UTP in the absence of promoter DNA template (Fig. 6B; lane 5).
Productive initiation requires adequate threshold concentrations of appropriate nucleoside triphosphates to allow formation of the first dinucleotide in the nascent transcript (Luse et al., 1987) and a source of energy for two distinct steps leading to initiated complexes (Payne et al., 1989; Laybourn et al., 1990). To exclude the possibility that the absence of HIV-1 transcripts in the presence of UTP alone was due to a lack of energy, the above experiments were repeated in the presence of UTP and ATP or dATP (data not shown). No HIV transcription was observed, providing further evidence that the HeLa cell extracts used in the in vitro transcription assays throughout these studies contained nucleoside triphosphate pools inadequate to support the formation of productive initiation complexes.
Tat stimulates HIV-1 transcription in a reconstituted in vitro transcription system
To discount the possibility that transcription initiation occurred during the preincubation step in HeLa whole-cell extracts, Tat transactivation was studied in in vitro transcription reactions reconstituted with partially purified transcription factors and RNA polymerase II. In this reconstituted system, transcription was totally dependent upon the addition of exogenous nucleoside triphosphates. The HIV-1 LTR DNA template (CD12R1) was preincubated with TFIIA, TFIIB, TFIID, TFIIE/F, SP1, and RNA polymerase II in the absence or presence of Tat. Sarkosyl (0.015%) was added to in vitro transcription reactions at different times during the preincubation period, prior to the addition of nucleoside triphosphates. HIV-1 LTR transcription in the absence of Tat was inhibited by 0.015% sarkosyl (Fig. 7A, lanes 1 and 3). The presence of Tat in the reconstituted system stimulated 0.015% sarkosyl-resistant transcription within 7.5 and 15 minutes of incubation (Fig. 7A, lanes 2 and 4). Transcription in the presence of Tat was blocked completely when 0.05% sarkosyl was added prior to the addition of nucleoside triphosphates (Fig. 7A, lane 5). The level of Tat transactivation with partially purified transcription factors was quantitatively less than that observed with cell extracts.
Figure 7.
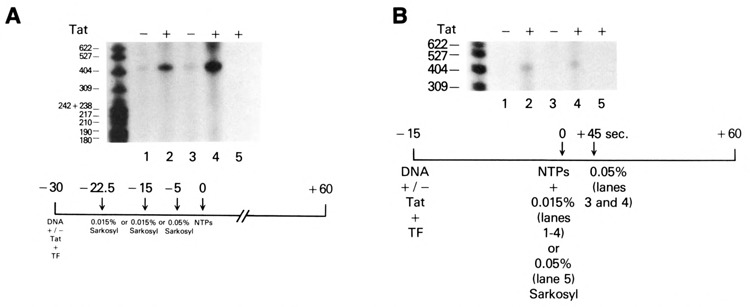
Stimulation of preinitiation complex formation by Tat in an in vitro transcription system reconstituted with partially purified transcription factors and RNA polymerase II. A. Kinetics of Tat transactivation in an in vitro transcription system reconstituted with purified transcription factors (TF) and RNA polymerase II. Transcription reactions containing a HIV-1 LTR DNA (CD12R1) template in the absence (lanes 1 and 3) or presence (lanes 2, 4, and 5) of 0.4 μM Tat were reconstituted with partially purified TFIIA, TFIIB, TFIID, TFIIE/F, Spl, and RNA polymerase II. Sarkosyl (0.015%) was added 7.5 minutes (lanes 1 and 2) or 15 minutes (lanes 3 and 4) after the start of incubation. In a separate reaction, 0.05% sarkosyl was added 5 minutes before addition of exogenous nucleoside triphosphates (lane 5). B. Tat transactivation of HIV-1 transcription initiation in an in vitro reconstituted system. Purified TFIIA, TFIIB, TFIID, TFIIE/F, Spl, and RNA polymerase II were preincubated with an HIV-1 LTR DNA template (75 ng) and 0.015% sarkosyl in the absence (lanes 1 and 3) or presence (lanes 2, 4, and 5) of 0.4 μM Tat. Sarkosyl concentration was either maintained at 0.015% throughout the 90-minute incubation (lanes 1 and 2) or raised to 0.05% 5 minutes before (lane 5) or 45 seconds after (lanes 3 and 4) exogenous nucleoside triphosphate addition.
To demonstrate that preinitiation complexes were formed in the presence of Tat but required nucleoside triphosphates to initiate transcription, in vitro reconstituted reactions were preincubated for 15 minutes in the presence of 0.015% sarkosyl, pulsed with nucleoside triphosphates for 45 seconds, and finally incubated in 0.05% sarkosyl to allow elongation of transcription complexes that were initiated stably and to block preinitiation of transcription in the reconstituted reactions. The pulse with nucleoside triphosphates allowed efficient conversion of the Tat-induced preinitiation complexes to actively initiated complexes (Fig. 7B, lanes 2 and 4). The 30–45 second pulse with nucleoside triphosphates quantitatively converted 0.05% sarkosyl-sensitive preinitiation complexes to 0.05% sarkosyl-resistant initiation complexes.
Tat stimulates transcriptional elongation of preformed HIV-1 initiation complexes
Recently, Tat was shown to stimulate HIV-1 transcription elongation in vitro (Marciniak et al., 1990a and 1992; Kato et al., 1992). To determine whether our purified Tat was able to promote transcription elongation of the HIV-1 LTR promoter using our in vitro transcription system, Tat was added to cotranscription reactions containing productively initiated HIV-1 transcription complexes formed by an initial preincubation of HeLa whole-cell extracts with wild-type and TAR mutant HIV-1 LTR DNA templates, followed by an additional incubation in the presence of optimal concentrations of nucleoside triphosphates. Tat was added to the reactions 10 minutes following transcription initiation. Transcription reactions were then terminated at various times, and full-length transcripts were analyzed. In the absence of Tat, full-length transcripts were initially detected at 30 minutes following the addition of nucleoside triphosphates (Fig. 8, lane 3). In the presence of Tat, full-length HIV-1 transcripts were observed within 15 minutes (5 minutes after the addition of Tat; Fig. 8, lanes 1 and 5). Interestingly, the TAR mutant template, CD23dlS, which was unresponsive to Tat at the level of initiation (Fig. 2B, lanes 1 and 2), was responsive to Tat at the level of elongation (338 b transcript). The stimulatory effect of Tat added to transcription reactions containing actively initiated complexes was similar to that observed when 0.05% sarkosyl was added (data not shown), which may indicate that Tat, like 0.05% sarkosyl, may potentiate the release of regulators of basal transcription.
Figure 8.
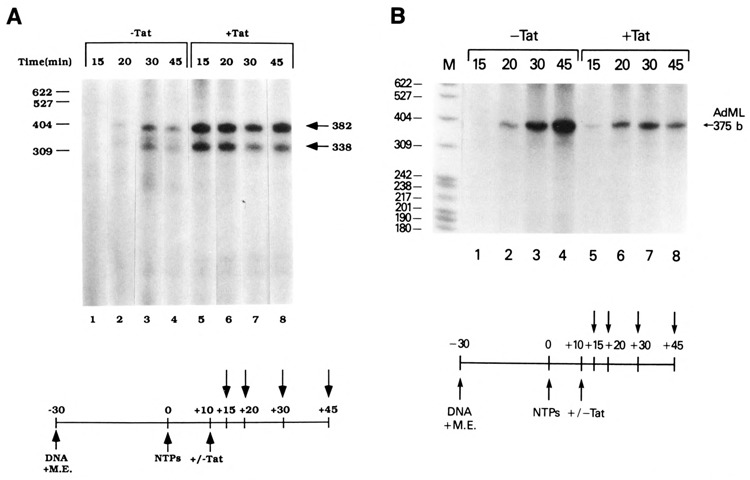
Stimulation of transcription elongation on the HIV-1 LTR promoter in the presence of Tat. Wild-type (CD12R1) and TAR mutant (CD23dlS) HIV-1 LTR DNA templates (A) or AdML template (B) in HeLa whole-cell extracts were permitted to form preinitiation complexes for 30 minutes. Subsequent addition of nucleoside triphosphates (500 μM) and [α-32P]UTP began productive initiation. Transcription was allowed to proceed for 10 minutes prior to the addition of purified Tat (0.4 μM). Following mock or Tat addition, assays were terminated at 45 minutes.
The Tat-containing extract did not increase transcription elongation from all polymerase II promoters (Fig. 8B). A similar set of elongation assays was performed with the AdMLP. The results of this experiment demonstrated that Tat does not increase elongation on the AdML template (Fig. 8B, compare lanes 1–4 and 5–8). In this particular assay, a slight decrease in the intensity of the AdML RNA was observed in the 45-minute sample (lane 8). This decrease was not observed in the 20- and 30-minute elongation samples and likely represented a difference in RNA recovery with this particular sample. These results suggested that Tat specifically increased the number and efficiency of processive HIV-1 elongation complexes.
Tat-induced transcription initiation complexes are efficiently converted to elongation complexes
Since Tat functioned at both transcription initiation and elongation, then Tat may act separately on these events and/or may facilitate the transition from transcription initiation to elongation. As an initial study to address these possibilities, the kinetics of transition from 0.015% sarkosyl-resistant preinitiation complexes to 0.05% sarkosyl-resistant elongation complexes in the absence and presence of Tat was investigated. Sarkosyl (0.015% or 0.05%) was added at 5, 10, and 30 minutes following the start of transcription in the absence or presence of Tat. In this experiment, HIV-1 basal transcription was sensitive to the addition of 0.015% sarkosyl during the first 30 minutes of incubation (Fig. 9). In the presence of Tat, HIV-1 LTR transcription was resistant to 0.015% sarkosyl after 5 minutes of incubation (Fig. 9, lane 2), which indicated a significant increase in the level of 0.015% sarkosyl-resistant preinitiation complexes. These complexes were sensitive to 0.05% sarkosyl (Fig. 9, lane 4), which suggested that only a small percentage of the formed preinitiation complexes had initiated transcription. By 10 minutes of incubation, HIV-1 LTR transcription in the presence of Tat was partially resistant to 0.05% sarkosyl (Fig. 9, lane 8), indicating that a greater number of preinitiation complexes had converted to elongation complexes. Finally, by 30 minutes of incubation, quantitative conversion of HIV-1 preinitiation complexes to elongation complexes was noted in the presence of Tat (Fig. 9, lanes 10 and 12). The time-dependent and quantitative transition of HIV-1 transcription from 0.015% sarkosyl-resistance to 0.05% sarkosyl-resistance suggested that Tat-induced preinitiation complexes were efficiently converted to elongation complexes.
Figure 9.
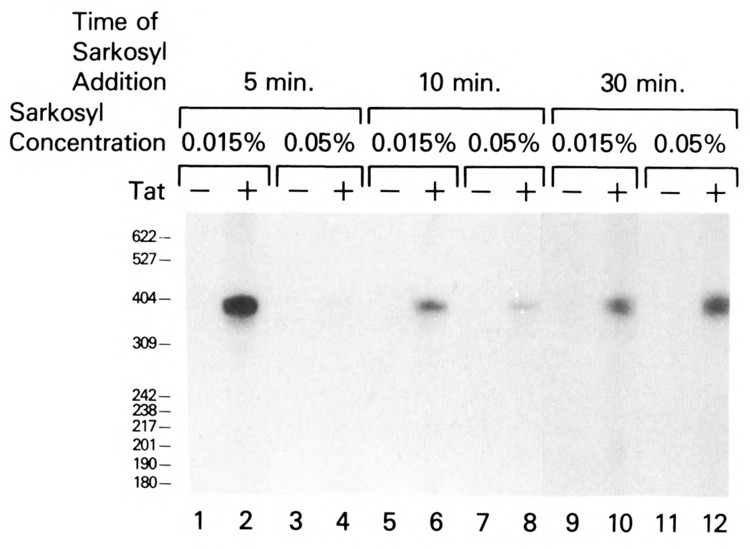
Kinetics of Tat transactivation in unfractionated HeLa cell extracts. Sarkosyl (0.015% or 0.05%) was added (5, 10, or 30 minutes) to complete in vitro transcription reactions containing the HIV CD12 template (75 ng), HeLa whole-cell extract, and exogenous nucleoside triphosphates (300 μM) in the absence (lanes 1, 3, 5, 7, 9, 11) or presence (lanes 2, 4, 6, 8, 10, 12) of 0.4 μM Tat.
Discussion
Several lines of evidence suggest that HIV Tat binds to TAR RNA and functions as an RNA binding protein (Dingwall et al., 1989). It is also evident that Tat increases the efficiency of RNA elongation, both in vivo and in vitro (Feinberg et al., 1991; Laspia et al., 1989 and 1990; Marciniak et al., 1990 and 1991). Recently, however, it has been suggested that Tat may also function in the context of a DNA-binding protein (Berkhout and Jeang, 1990), and more specifically, that Tat and acidic activators may act on a similar step in the transcription process (Southgate and Green, 1991). The sequential assembly of general transcription components, including TFIIA, TFIIB, TFIID, TFIIE/F, and RNA polymerase II, into functional preinitiation and initiation complexes has been analyzed (Luse et al., 1987; Reinberg and Roeder, 1987; Hawley et al., 1987; Nakajima et al., 1988; Van Dyke et al., 1988 and 1989; Saltzman and Weinmann, 1989; Buratowski et al., 1989; Maldonado et al., 1990). Interestingly, an initial and rate-limiting step in transcription initiation, the stable binding of TFIIA and TFIID to the promoter template, is sensitive to 0.015% sarkosyl. Resistance to 0.015% sarkosyl is conferred upon subsequent association of RNA polymerase II to the actively maturing preinitiation complex. Tat transactivates HIV-1 transcription in the presence of 0.015% sarkosyl added at the beginning of preinitiation complex formation. One interpretation of these results is that Tat stimulates RNA polymerase II transcription by facilitating the assembly and/or stabilization of basal transcription factors with the HIV-1 LTR promoter. Although one cannot rule out the possibility that the Tat effect in the presence of sarkosyl results from the Tat-activated elongation of a minor fraction of complexes that are able to form in the presence of sarkosyl, several lines of evidence suggest that this interpretation is unlikely. There is less than a twofold decrease in the level of Tat transactivation when 0.015% sarkosyl is added to the Tat containing reactions. In contrast, sarkosyl quantitatively blocks HIV basal transcription (>10-fold decrease). This would necessitate a tenfold increase in the level of reinitiation to account for the level of RNA synthesized. This seems unlikely, since the level of transcription reinitiation is extremely low in the in vitro transcription systems. In addition, using gel shift assays to analyze the binding of transcription factors to the HIV TATA sequence, we have recently demonstrated that Tat interacts with and stabilizes the interaction of TFIID with this promoter element, directly supporting a role for Tat in preinitiation complex assembly (F. Kashanchi andj. N. Brady, unpublished data). The effect of this interaction on factors such as TFIIA, TFIIB, and polymerase II with the initiation complex is presently under investigation.
Tat’s putative role in the stimulation of initiation complex formation may be similar to the functions attributed to several DNA-dependent transactivating proteins (Sawadogo and Roeder, 1985; Horikoshi et al., 1988a,b; Abmayr et al., 1988; Katagiri et al., 1990; Stringer et al., 1990; Ptashne and Gann, 1990; Yamazaki et al., 1990; Lin and Green, 1991). The pseudorabies immediate-early protein, PIE, and the tobacco mosaic virus DNA-binding protein, TGAla, activate transcription in in vitro systems by increasing the number of active preinitiation complexes (Abmayr et al., 1988; Yamazaki et al., 1990). PIE transactivates in vitro transcription by facilitating the interactions between the target promoter and TFIID. Interestingly, both PIE and Tat trans-activation may be circumvented by preincubation of the cell-free extract and the DNA template to permit the formation of preinitiation complexes. The acidic transcriptional activator VP16 stimulates transcription by increasing the number of functional preinitiation complexes (Lin and Green, 1991). It has been reported that VP16 interacts directly with TFIID (Stringer et al., 1990; Ingles et al., 1991) and TFIIB (Lin et al., 1991), and in vitro evidence suggests that VP16 could increase preinitiation complex formation by recruiting TFIIB to the promoter (Lin and Green, 1991).
Analysis by Laspia et al. (1989) of HIV-1 transcription in the presence of Tat in vivo indicates that transcription initiation from wild-type HIV-1 LTR promoters increases significantly (15-fold). In addition, both Laspia et al. (1989) and Feinberg et al. (1991) have reported that Tat-transactivated HIV transcription complexes elongate more efficiently into the distal region of the RNA transcript. Consistent with these observations are reports that Tat transactivation of the HIV-1 LTR promoter in vitro results from an increase in the efficiency of transcription elongation (Marciniak et al., 1990a; Marciniak and Sharp, 1991; Kato et al., 1992). These results suggest that transcription complexes formed in the presence of Tat are quantitatively and possibly qualitatively distinct in their competence to initiate and elongate transcription from the HIV-1 LTR promoter. Our results are not inconsistent with the model in which Tat transactivation increases the efficiency of RNA elongation. Our results extend these observations and provide evidence that Tat may also have a critical function that is prior to, but perhaps coupled with, its elongation effect. Support for the bifunctional role of Tat comes from the demonstration of differential effects of Tat on the mutant CD23dlS. This mutant deletes the sequences adjacent to the downstream DNA binding site for UBP-1. In addition, the secondary structure of the binding sites for both Tat and p68 are destroyed. Tat does not transactivate this mutant template if Tat is added prior to preinitiation complex formation. In contrast, a 10-fold stimulation in HIV-1 transcription elongation is evident upon the addition of Tat after the onset of RNA synthesis (Fig. 8, lanes 5–8). These results suggest that both downstream DNA (initiation) and RNA (elongation) sequences may be involved in Tat transactivation. Mutant CD23dlS is not transactivated by Tat in vivo (Ensoli et al., 1989). The fact that this mutant exhibits a Tat initiation-defective, but Tat elongation-competent, phenotype in our in vitro analyses may suggest that the loss of Tat transactivation by this mutant in vivo may be due to a defect in initiation complex formation. Consistent with previous studies, these results suggest that Tat binding to RNA is not sufficient to explain TAR element function (Dingwall et al., 1989; Roy et al., 1990). These results further suggest that cellular proteins distinct from the p68 loop binding protein, such as pl40 SBP (Rounseville and Kumar, 1992), may facilitate RNA elongation.
Toohey and Jones (1989) report that sarkosyl affects the ratio of full-length and short HIV-1 transcripts in vitro. Full-length HIV-1 transcripts are detected at sarkosyl concentrations ⩽0.015% but disappear upon incubation of the extract with intermediate levels of sarkosyl (0.05–0.15%), whereupon truncated leader transcripts (58–61 nt) become abundant. At higher concentrations of sarkosyl (0.25–0.40%), the appearance of the full-length HIV-1 RNA returns, while the levels of truncated leader transcripts decline. The disappearance of full-length HIV-1 transcripts at a sarkosyl concentration of 0.05% is directly relevant to the results presented in this manuscript. When 0.05% sarkosyl is added at the end of the preincubation reaction containing Tat, but before the addition of nucleoside triphosphates, a dramatic decrease in the levels of full-length HIV-1 transcripts is observed. In contrast to the results of Toohey and Jones (1989), the sensitivity of HIV-1 basal transcription to 0.05% sarkosyl in our transcription system is overcome by the addition of 300 μM nucleoside triphosphates prior to 0.05% sarkosyl addition. The reason for the differences in the results of the two in vitro transcription systems is not readily apparent. Differences in the respective analyses include (1) time of sarkosyl addition relative to transcription initiation; (2) types of extracts, i.e., Manley whole-cell vs. partially purified nuclear; (3) HIV-1 LTR DNA templates; and (4) conditions for HIV-1 basal transcription (e.g., template concentration). Most notably, the level of HIV-1 basal transcription in the analysis by Toohey and Jones (1989) is optimized for near maximal activity, which is inhibitory for Tat transactivation in vitro. Further refinement of in vitro transcription systems as afforded in our reconstituted system using purified transcription factors and RNA polymerase II should resolve differences between HIV-1 basal transcription and Tat transactivation of transcription.
Marciniak and Sharp (1991) have reported that there are two classes of elongation complexes which initiate from the HIV promoter, a less-processive and a more-processive form. Tat was found to increase the more-processive class of elongation complexes. Since no increase in the amount of transcription between nucleotides 1 and 82 was observed, the authors conclude that the total effect of Tat is due to an increase in elongation. In light of our results, it is interesting to speculate that Tat does regulate transcription initiation by specifically facilitating the assembly of these more-processive complexes. Thus, even though there would be no increase in the level of promoter proximal transcription, the level of promoter distal transcription could be increased by the HIV Tat protein.
Acknowledgments
We thank Mike Radonovich and Janet Duvall for expert technical assistance; Drs. Rebecca Sundseth and Ulla Hansen for generously providing purified transcription factors; and Drs. Elizabeth Ross and Malcolm Martin for the HIV LTR SP1 deletion mutant, SPE-CAT. We also thank Ms. Judie Ireland for secretarial assistance.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Cindy A. Bohan is currently in the School of Biology, Georgia Institute of Technology, Atlanta, GA 30332-0230.
Kathleen A. Boris-Lawrie is currently at the McArdle Laboratory #518, University of Wisconsin, 1400 University Avenue, Madison, WI 53706.
References
- Abmayr S. M., Workman J. L., and Roeder R. G. (1988), Genes Dev 2, 542–553. [DOI] [PubMed] [Google Scholar]
- Arya S. K., Guo C., Josephs S. F., and Wong-Staal F. (1985), Science 229, 69–73. [DOI] [PubMed] [Google Scholar]
- Barre-Sinoussi T., Chermann J. C., Reye F., Nugeure M. T., Charmaret S., Gruest J., Dauquet C., Aixer-Blin C., Vezinet-Brun F., Rouzious C., Rozenbaum W., and Montagnier L. (1983), Science 220, 868–871. [DOI] [PubMed] [Google Scholar]
- Berkhout B., Gatignol A., Rabson A. B., and Jeang K.-T. (1990), Cell 62, 757–767. [DOI] [PubMed] [Google Scholar]
- Berkhout B. and Jeang K.-T. (1989), J Virol 63, 5501–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnlein R., Lowenthan J. W., Siekevitz M., Ballard D. W., Franza B. R., and Greene W. C. (1988), Cell 53, 827–836. [DOI] [PubMed] [Google Scholar]
- Boris-Lawrie K. A., Brady J. N., and Kumar A. (1992), Gene Expr 2, 215–230. [PMC free article] [PubMed] [Google Scholar]
- Braddock M., Chambers A., Wilson W., Esnouf M. P., Adams A. J., Kingsman A. J., and Kingsman S. M. (1989), Cell 58, 269–279. [DOI] [PubMed] [Google Scholar]
- Braddock M., Thorburn A. M., Chambers A., Elliot G. D., Anderson G. J., Kingsman A. J., and Kingsman S. M. (1990), Cell 62, 1123–1133. [DOI] [PubMed] [Google Scholar]
- Buratowski S., Hahn S., Guarente L., and Sharp P. A. (1989), Cell 56, 546–561. [DOI] [PubMed] [Google Scholar]
- Buratowski S., Sopta M., Greenblatt J., and Sharp P. A. (1991), Proc Nad Acad Sci USA 88, 7509–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H. and Luse D. S. (1987), J Biol Chem 262, 262–304. [PubMed] [Google Scholar]
- Chowdhury M., Taylor J. P., Tada H., Rappaport J., Wong-Staal F., Amini J., and Khalili K. (1990), Oncogene 5, 1737–1742. [PubMed] [Google Scholar]
- Clark L., Matthews J. R., and May R. T. (1990), J Virol 64, 1335–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen B. (1986), Cell 46, 973–982. [DOI] [PubMed] [Google Scholar]
- Cullen B. R. (1990), Cell 63, 655–657. [DOI] [PubMed] [Google Scholar]
- Dayton A. I., Sodroski J. G., Rosen C. A., Goh W. C., and Haseltine W. A. (1986), Cell 44, 941–947. [DOI] [PubMed] [Google Scholar]
- Dingwall C., Ernberg I., Gait M. J., Green S. M., Heaphy S., Karn J., Lowe A. D., Singh M., Skinner M. A., and Vallerio R. (1989), Proc Nad Acad Sci USA 86, 6925–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery I., Petryshyn R., and Sonenberg N. (1989), Cell 56, 303–312. [DOI] [PubMed] [Google Scholar]
- Ensoli B., Lusso P., Schachter F., Josephs S. F., Rappaport J., Negro F., and Gallo R. C. (1990), EMBO J 8, 3019–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli B., Barillari G., Salahuddin S. Z., Gallo R. C., and Wong-Staal F. (1990), Nature 345, 84–86. [DOI] [PubMed] [Google Scholar]
- Feinberg M. B., Baltimore D., and Frankel A. D. (1991), Proc Natl Acad Sci USA 88, 4045–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. and Holland E. C. (1988), Nature 334, 165–167. [DOI] [PubMed] [Google Scholar]
- Fisher A. G., Feinberg M. B., Josephs S. F., Harper M. E., Marselle L. M., Reyes G., Gonda M. A., Aldovini A., Debouk C., Gallo R. C., and Wong-Staal F. (1986), Nature 320, 367–371. [DOI] [PubMed] [Google Scholar]
- Flores O., Lu H., Killeen M., Greenblatt J., Burton A. F., and Reinberg D. (1991), Proc Natl Acad Sci USA 88, 9999–10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo R. C., Salahuddin S. Z., Popovic M., Shearer G. M., Kapten M., Haynes B. F., Palker T. J., Redfield R., Oleske J., Safai B., White G., Foster P., and Markham P. D. (1984), Science 224, 500–503. [DOI] [PubMed] [Google Scholar]
- Garcia J. A., Harrich D., Pearson I., Mitsuyasu R., and Gaynor R. B. (1988), EMBO J 7, 3143–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J. A., Harrich D., Soultanaki E., Wu F., Mitsuyasu R., and Gaynor R. B. (1989), EMBO J 8, 765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor R., Soultanakis E., Kuwabara M., Garcia J., and Sigman D. S. (1989), Proc Natl Acad Sci USA 86, 4858–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber J. and Cullen B. R. (1988), J Virol 62, 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauber J., Perkins A., Heimer E. P., and Cullen B. R. (1987), Proc Natl Acad Sci USA 84, 6364–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley D. K. and Roeder R. G. (1987), J Biol Chem 262, 3452–3461. [PubMed] [Google Scholar]
- Horikoshi M., Carey M. F., Kakidani H., and Roeder R. G. (1988a), Cell 54, 665–669. [DOI] [PubMed] [Google Scholar]
- Horikoshi M., Hai T., Lin Y. S., Green M. R., and Roeder R. G. (1988b), Cell 54, 1033–1042. [DOI] [PubMed] [Google Scholar]
- Ingles C. J., Shales M., Cress W. D., Treizenburg S. J., and Greenblatt J. (1991), Nature 351, 588–590. [DOI] [PubMed] [Google Scholar]
- Jakobovits A., Smith D. H., Jakobovits E. B., and Capon D. J. (1988), Mol Cell Biol 8, 2555–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeang K.-T., Shank P. R., Rabson A. R., and Kumar A. (1988), J Virol 62, 3874–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyeapul J., Reddy M. R., and Khan S. A. (1990), Proc Natl Acad Sci USA 87, 7030–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. A., Kadonaga J. T., Luciw P. A., and Tjian R. (1986), Science 232, 755–759. [DOI] [PubMed] [Google Scholar]
- Kao S. Y., Calman A. F., Luciw P. A., and Peterlin B. M. (1987), Nature 330, 489–493. [DOI] [PubMed] [Google Scholar]
- Katagiri F., Yamazaki K., Horikoshi M., Roeder R. G., and Chua N. H. (1990), Genes Dev 4, 1899–1909. [DOI] [PubMed] [Google Scholar]
- Kato H., Sumimoto H., Pognonec P., Chein-Hwa C., Rosen C. A., and Roeder R. G. (1992), Genes Dev 6, 655–666. [DOI] [PubMed] [Google Scholar]
- Laspia M. F., Rice A. P., and Matthews M. B. (1989), Cell 59, 283–292. [DOI] [PubMed] [Google Scholar]
- Laspia M. F., Rice A. P., and Matthews M. B. (1990), Genes Dev 4, 2397–2408. [DOI] [PubMed] [Google Scholar]
- Laybourn P. J. and Dahmus M. E. (1990), J Biol Chem 265, 13165–13173. [PubMed] [Google Scholar]
- Leonard J., Parrot C., Buckler-White A. J., Turner W., Ross E. K., Martin M. A., and Rabson A. B. (1989), J Virol 63, 4919–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy J. A., Hoffman A. D., Kramer S. M., Landis J. A., Shimabukuro J. M., and Oshino L. S. (1984), Science 225, 840–842. [DOI] [PubMed] [Google Scholar]
- Lin Y. S., Ha I., Maldonado E., Reinberg D., and Green M. R. (1991), Nature 353, 569–571. [DOI] [PubMed] [Google Scholar]
- Lin Y. S. and Green M. R. (1991), Cell 64, 971–981. [DOI] [PubMed] [Google Scholar]
- Luciw P. A. and Tjian R. (1986), Science 232, 755–759. [DOI] [PubMed] [Google Scholar]
- Luse D. S., Kochel T., Kuempel E. D., Coppola J. A., and Cai H. (1987), J Biol Chem 262, 289–297. [PubMed] [Google Scholar]
- Maldonado E., Ha I., Corest P., Weis L., and Reinberg D. (1990), Mol Cell Biol 10, 6335–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley J. L., Fire A., Cano A., Sharp P. A., and Gefter M. L. (1980), Proc Nad Acad Sci USA 77, 3855–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak R. A. and Sharp P. A. (1991), EMBO J 10, 4189–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak R. A., Cainan B. J., Frankel A. D., and Sharp P. A. (1990a), Cell 63, 791–802. [DOI] [PubMed] [Google Scholar]
- Marciniak R. A., Garcia-Blanco M., and Sharp P. A. (1990b), Proc Natl Acad Sci USA 87, 3624–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muesing M. A., Smith D. H., and Capon D. J. (1987), Cell 48, 691–701 [DOI] [PubMed] [Google Scholar]
- Nabel G. and Baltimore D. (1987), Nature 426, 711–713. [DOI] [PubMed] [Google Scholar]
- Nakajima N., Horikoshi M., and Roeder R. G. (1988), Mol Cell Biol 8, 4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T., Benter T., Josephs S. F., Sadaie M. R., and Wong-Staal F. (1990), Virology 177, 606–614. [DOI] [PubMed] [Google Scholar]
- Parkin N. T., Cohen E. A., Darveau A., Rosen C. A., Haseltine W. A., and Sonenberg N. (1988), EMBO J 7, 2831–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvin J. D., Timmers H. Th. M., and Sharp P. A. (1992), Cell 68, 1135–1144. [DOI] [PubMed] [Google Scholar]
- Parrott C., Seidner T., Duh E., Leonard J., Theodore T. S., Buckler-White A., Martin M. A., and Rabson A. B. (1991), J Virol 65, 1414–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J. M., Laybourn P. J., and Dahmus M. E. (1989), J Biol Chem 264, 19621–19629. [PubMed] [Google Scholar]
- Peterlin B. M., Luciw P. A., Barr P. J., and Walker M. D. (1986), Proc Natl Acad Sci USA 83, 9734–9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. and Gann A. F. (1990), Nature 346, 329–331. [DOI] [PubMed] [Google Scholar]
- Reinberg D. and Roeder R. G. (1987), J Biol Chem 262, 3310–3321. [PubMed] [Google Scholar]
- Rice A. P. and Matthews M. B. (1988), Nature 332, 551–553. [DOI] [PubMed] [Google Scholar]
- Rosen C. A. and Pavlakis G. N. (1990), AIDS 4, 499–509. [PubMed] [Google Scholar]
- Rosen C. A., Sodroski J. G., and Haseltine W. A. (1985), Cell 41, 813–823. [DOI] [PubMed] [Google Scholar]
- Rosen C. A., Sodroski J. G., Goh W. C., Dayton A. I., Lipke J., and Haseltine W. A. (1986), Nature 319, 555–559. [DOI] [PubMed] [Google Scholar]
- Rounseville M. P. and Kumar A. (1992), J Virol 66, 1688–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Parkin N. T., Rosen C., Itovitch J., and Sonenberg N. (1990), J Virol 64, 1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M. R., Benter T., and Wong-Staal F. (1988), Science 239, 910–913. [DOI] [PubMed] [Google Scholar]
- Saltzman A. G. and Weinmann R. (1989), FASEB J 3, 1723–1733. [DOI] [PubMed] [Google Scholar]
- Sastry K. J., Reddy H. R., Pandita R., Totpal K., and Aggarwal B. B. (1990), J Biol Chem 265, 20091–20093. [PubMed] [Google Scholar]
- Sawadogo M. and Roeder R. G. (1985), Cell 43, 165–175. [DOI] [PubMed] [Google Scholar]
- Selby M. J., Baia E. S., Luciw P. A., and Peterlin B. M. (1989), Genes Dev 3, 547–558. [DOI] [PubMed] [Google Scholar]
- Selby M. J. and Peterlin B. M. (1990), Cell 62, 769–776. [DOI] [PubMed] [Google Scholar]
- SenGupta D. N., Berkhout B., Gatignol A., Zhou A., and Silverman R. H. (1990), Proc Natl Acad Sci USA 87, 7492–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodroski J. G., Patarca R., Rosen C. A., Wong-Staal F., and Haseltine W. A. (1985), Science 229, 74–77. [DOI] [PubMed] [Google Scholar]
- Southgate C. D. and Green M. R. (1991), Genes Dev 5, 2496–2507. [DOI] [PubMed] [Google Scholar]
- Southgate C. D., Zapp M. L., and Green M. R. (1990), Nature 345, 640–642. [DOI] [PubMed] [Google Scholar]
- Spencer C. A. and Groudine M. (1990), Oncogene 5, 777–785. [PubMed] [Google Scholar]
- Stringer K. F., Ingles C. J., and Greenblatt J. (1990), Nature 345, 783–786. [DOI] [PubMed] [Google Scholar]
- Toohey M. G. and Jones K. A. (1989), Genes Dev 3, 265–283. [DOI] [PubMed] [Google Scholar]
- Van Dyke M. W., Roeder R. G., and Sawadogo M. (1988), Science 241, 1335–1338. [DOI] [PubMed] [Google Scholar]
- Van Dyke M. W., Sawadogo M., and Roeder R. G. (1989), Mol Cell Biol 9, 342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C. M., Felber B. K., Paskalis H., and Pavlakis G. N. (1986), Science 234, 988–992. [DOI] [PubMed] [Google Scholar]
- Wu F. K., Garcia J. A., Harrich D., and Gaynor R. B. (1988), EMBO J 7, 2117–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki K., Katagiri F., Imaseki H., and Chua N. (1990), Proc Natl Acad Sci USA 87, 7035–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]