

Abstract
About 3 kb of the promoter region of the gene encoding cytochrome P-450 2B2 (CYP2B2) in the rat were sequenced and searched for potential cis-acting elements. Apart from putative binding sites for (liver-specific) protein factors, a region showing homology with the LINE 1 retrotransposon element was also found. Three proximal promoter fragments, encompassing nucleotides −579 to −372, −372 to −211, and −211 to +1, respectively, were shown to contain binding sites for multiple protein factors by bandshift analyses. The strongest protein-binding element, designated BRE (basic regulatory element), occurs between −103 to −66. Its structure is very similar to a negative control element in the murine cmyc promoter and displays a composite feature having a tandemly repeated sequence homology with the BTE (basic transcription element; Yanagida et al., 1990) separated by a CCAAA-box. The use of a deletion series of this template in in vitro transcription assays, provided evidence that the BRE serves as a major cis-acting element in the (regulated) transcription activation of the CYP2B2 gene.
The cytochrome P-450 enzyme family comprises more than 60 isoenzymes, occurring mainly in the eneoplasmatic reticulum of liver cells, which are involved in the oxidative metabolism of endogenous as well as exogenous substrates (see for reviews Gonzalez and Nebert, 1990; Nebert et al., 1989; Okey, 1990; Ryan and Wayne, 1990). These proteins serve predominantly as detoxifying enzymes, but they can, in some cases, also convert certain substrates to very toxic intermediates.
Synthesis of most isoenzymes occurs at detectable amounts only after exposure of the organism to inducing compounds. In many cases these substances themselves are substrates for the enzymes. Cytochrome P-450c (1A1) activity, for instance, is effectively induced by some aromatic carbohydrates like 3-methylcholanthrene (3MC) and tetrachlorodibenzo-p-dioxin (TCDD; Kawajiri et al., 1984; Thomas et al., 1983). CytP450b (2B1) and the related P450e (2B2) are induced by phenobarbital (PB) and some polychlorinated biphenyls (PCB; Scholte et al., 1985; Vlasuk et al., 1982; Waxman and Walsh, 1982; recently reviewed by Waxman and Azaroff, 1992). CytP450b and e belong to the CYP2B subfamily consisting of at least 10 members (Ryan and Wayne, 1990). Induction causes an approximately 40-fold rise in the respective cellular enzyme concentrations (Phillips et al., 1983a,b). For CYP1A1 and 2B1 and 2, this elevated gene expression is accomplished by enhanced transcription (Hardwick et al., 1983; Kawajiri et al., 1984; Phillips et al., 1983b).
Transcription activation of the CYP1A1 gene is highly stimulated by a receptor protein bound to the xenobiotic substance (3MC or TCDD; Sogawa et al., 1986). This receptor–inducer complex interacts with a specific site (XRE) in the promoter region of the respective genes, and acts synergistically with other trans-acting factors in the formation of an efficient transcription initiation complex (Fujisawa-Sehara et al., 1988; Nebert et al., 1984; Poland et al., 1980; Poland and Glover, 1976; Wen et al., 1990; Whitlock and Galeazzi, 1984).
Most likely induction of the CYP2B1 and 2 activity also occurs by specifically enhancing the transcription of the respective genes. Previously, evidence has been obtained that PB can induce transcription of these genes (Hardwick et al., 1983; Phillips et al., 1983b); in the present paper these findings are extended for the inducer 2,4,5,2′,4′5′-hexachlorobiphenyl (2,4,5-HCB). So far, no PB- or HCB-receptor could be identified. The genes for CYP2B1 and 2 are highly similar with respect to their nucleotide sequence (Okey, 1990; Rampersaud and Walz, Jr., 1987a,b). The same holds for the corresponding promoter regions up to −1406 (Jaiswal et al., 1987). On the other hand, the expression of both genes appeared to display distinct developmental and tissue-specific patterns (Okey, 1990). This paper deals with the functional elements constituting the promoter of the rat cytochrome P-450 2B2 gene. We have identified the upstream region between −103 and −66 as a major activating element implicated in the (regulated) transcription of this gene.
Materials and methods
Plasmid construction
A DNA fragment containing the cytochrome P-450 2B2 promoter sequence was isolated by screening a cosmid bank of rat genomic DNA using the CYP2B2 cDNA clone published previously (Scholte et al., 1985) as a probe. A 3.8 kb Bgl II-generated fragment from this cosmid clone, which contains the 5′-flanking region of the gene, was subcloned in pIC20R. The sequence of the 5′-flanking region up to position −2919 was determined using the method of Sanger et al. (1977). The constructs used in the in vitro transcription experiments are based on the plasmid p(C2AT)[380], a generous gift from Dr. A. Monaci (Monaci et al., 1988). A synthetic oligonucleotide inserted in the EcoR V site immediately upstream of the G-less cassette created a unique Sph I site, which made it possible to fuse promoter fragments with the G-less cassette at position +1, comparable to the in vivo situation. The 5′-deletions of the CYP2B2 promoter were generated either by BAL 31 digestion or by using unique restriction sites. Plasmid AdML50[180], used as a control, was a generous gift from Dr. A. Monaci (Monaci et al., 1988).
Analysis of RNA
Total RNA was isolated from the livers of control rats as well as rats PB-treated for 2, 4, 8, 12, and 24 hours, using the AGPC extraction procedure described by Chomszynski and Sacchi (1987). Slot blot analysis was performed using a 2.6 kb CYP2B2 cDNA fragment (Scholte et al., 1985) as a probe for CYP2B1 and 2B2 mRNAs.
Mapping of the transcription start sites was performed by an RNase A protection assay. Total liver RNA was isolated from PB- and 2,4,5 HCB-treated as well as control rats and subsequently hybridized to labeled antisense RNA. This riboprobe was based upon a genomic CYP2B2 DNA fragment encompassing the region from −1406 to +173. After RNase A treatment, the length of the protected fragment was analyzed on a denaturing polyacrylamide gel.
Preparation of rat liver nuclear extracts
Liver nuclear extracts from three-month-old Wistar rats were prepared essentially as described by Gorski et al. (1986) and Graves et al. (1986). All experiments were carried out at 0° to 4°C. Usually 30 g of minced liver was suspended in homogenization buffer (10 mM HEPES-KOH [pH 7.6], 25 mM KC1, 0.15 mM spermine, 0.5 mM spermidine, 1 mM EDTA, 2 M sucrose, 10% glycerol) up to a final volume of 30 ml. It was then homogenized in the absence of air in a modified Waring blender. The homogenate was diluted 3 times in homogenization buffer, layered in 25 ml portions on 10 ml cushions of the homogenization buffer, and centrifuged at 24,000 rpm for 45 minutes at 0°C in a SW28 rotor (100,000 × g; Ueno and Gonzalez, 1990).
This centrifugation step was repeated after resuspending the combined nuclear pellets in 50 ml homogenization buffer, layered on two 10 ml cushions of the homogenization buffer. The pellets were washed twice with nuclear lysis buffer (10 mM HEPES-KOH [pH 7.6], 100 mM KC1, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 10% glycerol) and resuspended in 20 ml of nuclear lysis buffer, using an all-glass Dounce homogenizer (Wheaton pestle B). An aliquot was diluted 50 times in 0.5% SDS, and the A260nm was measured. The nuclear suspension was diluted to approximately 10 A260nm units per ml and extracted essentially as described by Parker and Topol (1984). One-tenth volume of 4 M (NH4)2SO4 (pH 7.9) was added directly, and the extract was gently shaken and left on ice for 30 minutes. The viscous lysate was then centrifuged at 45,000 rpm for 90 minutes in a 50.2 Ti rotor at 0°C to pellet the chromatin (200,000 × g). Subsequently solid (NH4)2SO4 (0.3 g/ml) was added to the supernatant and allowed to dissolve by gentle agitation. After an additional 30 minutes on ice, the precipitated proteins were pelleted at 26,000 rpm in a SW28 rotor at 0°C. The protein pellet was stored overnight on ice, and suspended in dialysis buffer (25 mM HEPES-KOH [pH 7.6], 40 mM KC1, 0.1 mM EDTA, 1 mM DTT, 10% glycerol, and 0.5 mM PMSF) at 1 ml/400 A260nm units of nuclear lysate. The protein extract was dialyzed against dialysis buffer. After dialysis the protein extract was centrifuged for 5 minutes in a microfuge to remove unsoluble material. The protein concentration usually amounted to 4–8 mg/ml. The protein extract was stored in aliquots under liquid nitrogen.
In vitro transcription assay
In vitro transcription reactions were carried out essentially as described by Gorski et al. (1986). The transcription reaction mixtures (20 μl) contained 60 ng/μl circular template DNA and 4 (μg/μl nuclear protein extract in a buffer containing 25 mM HEPES-KOH (pH 7.6), 50 mM KCl, 6 mM MgCl2, 0.6 mM each of ATP and CTP, 35 μM UTP, 7 μCi [α-32P]UTP (Amersham; 3000 Ci/mmol), 0.1 mM 3′-O-Methyl GTP (Pharmacia), 12% glycerol, 1 μl RNAsin (∼30 units; Promega). After 45 minutes of incubation at 30°C the reactions were terminated by the addition of 20 μl stop mix (50 mM Tris.HCl [pH 8.0], 50 mM EDTA, 1.2% SDS).
Proteins were removed by a proteinase K treatment (10 μl of 20 mg/ml proteinase K, 0.5 mg/ml tRNA) for 20 minutes at 55°C. RNA was precipitated by the addition of 350 μl 86% EtOH, 0.3M NH4AC. The RNA pellets were rinsed with 80% EtOH and dried for 5 minutes in a speed vac. The dried pellets were resuspended in 15 μl RNA loading dye (10% glycerol, 0.1% xylene cyanol FF, 0.1 % bromophenol blue, 6 M urea in TBE) and loaded on a 6% polyacrylamide, 7 M urea sequencing gel. Autoradiography was carried out at −80°C with intensifying screen.
In vitro protein–DNA binding assays
Binding assays were performed essentially as described by Monaci et al. (1988). Gel retardation experiments were carried out either with a labeled double-stranded oligonucleotide (5 to 10 fmol) or with a suitable labeled DNA fragment (10 to 20 fmol). The DNA was incubated with 5 to 20 μg of nuclear proteins in a buffer containing 25 mM HEPES-KOH (pH 7.6), 7.5% glycerol, 60 mM KC1, 4 mM spermidine, 0.75 mM DTT, 0.1 mM EDTA for 15 minutes at room temperature. After this incubation 2 μl of 20% Ficoll, 0.1% xylene cyanol FF, 0.1% bromophenol blue were added, and the samples were loaded onto a 5% polyacrylamide gel in 0.5 × TBE buffer and subjected to electrophoresis at 10 V/cm for 2–3 hours at 4°C.
Results
Sequence of the rat CYP2B2 promoter region
Transcription of the gene encoding CYP2B2 (cytP450e) in the rat is induced upon treatment with PB (Hardwick et al., 1983) or 2,4,5-HCB (this laboratory, unpublished results).
In order to investigate the transcriptional activation of the CYP2B2 gene, a cosmid bank of rat genomic DNA was screened using a previously described CYP2B2-cDNA (Scholte et al., 1985) as a probe. A DNA fragment carrying the CYP2B2 gene including about 3 kb of its 5′-flanking region was isolated and subcloned. The physical map of this region is depicted in Figure 1B. Sequence analysis of 2919 bp upstream of the transcriptional start site—see Figure 1A—showed, for the first 1406 bp, almost identity with the sequence previously reported by others (Jaiswal et al., 1987; Suwa et al, 1985). In Figure 1 the location of the transcriptional start site +1 is indicated; by RNase A-mapping (see Fig. 2A) it was found to be identical in liver cells of both PB- and 2,4,5 HCB-treated animals.
Figure 1.

Structure of the rat CYP2B2 promoter region. In A the sequence of 2919 bp of the promoter is shown. The location of putative protein factor binding sites is indicated, as well as the region showing homology with the LINE 1 element (discussed in the text). In B the physical map of the 5′-flanking region of the CYP2B2 gene is given, ranging from the transcription initiation point (+1) to 2919 bp upstream. B = BamH I, Bg = Bgl II, H = Hind III, Hi = Hinc II, N = Nco I, P = Pst I, S = Sma I, Sp = Sph I, St = Stu I, X = Xba I, EV = EcoR V, Xo = Xho I. Fragments used as probes in bandshift analyses are boxed. The location of the sequence from −98 to −77 of the proximal part of the CYP2B2 promoter showing homology with a c-myc regulatory element (see text) is indicated.
Figure 2.
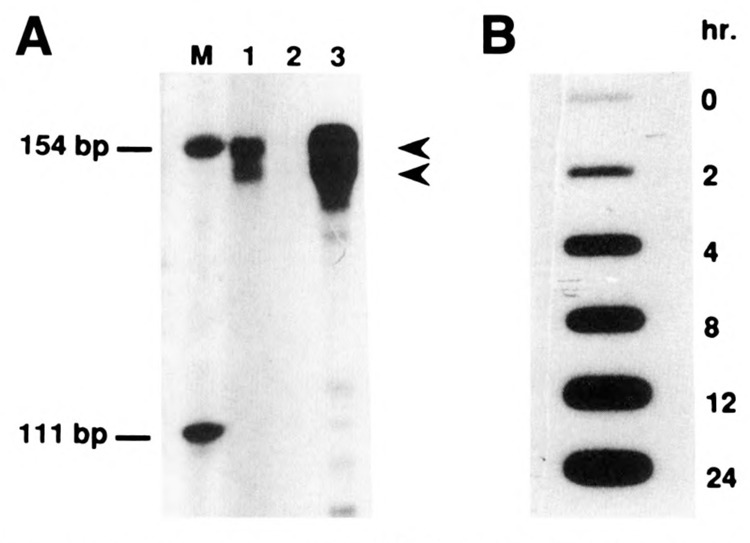
Mapping of the CYP2B2 transcription start site and slot blot analysis of CYP2B-mRNAs. A. RNase A protection assay of total liver RNA from 2,4,5 HCB-treated rats (lane 1), from control rats (lane 2), and from PB-treated rats (lane 3). The length of the RNase A protected riboprobe fragment (see Materials and Methods) was estimated relative to size markers (M). Arrows correspond to the mapped transcription start sites. B. Total liver RNA from rats treated for different time periods with PB were used for slot blot hybridization with a CYP2B gene-specific probe. A rat ribosomal protein L12 gene-specific probe was used as a loading control (result not shown).
A computer-aided search revealed a region extending from −2440 to further upstream that displayed a strong homology with a so-called LINE 1 retrotransposon element (Soares et al., 1985). Whether the presence of this element has any relevance for the transcription activity of the CYP2B2 gene in vivo, is as yet unknown. In addition, the presence of several putative protein factor binding sites was elucidated, which may serve as cis-acting elements for transcription (Fig. 1A). The most notable elements are two potential glucocorticoid-responsive elements at −1341 (Jaiswal et al., 1990) and at −2665, and two dioxin-responsive elements (Denison et al., 1988) at −821 and −1257, respectively, both types of motifs acting as regulatory elements in the CYP1A1 promoter. Moreover, several putative binding sites for liver-specific factors are present: e.g., for LF-A1 (Hardon et al., 1988) at −1772, −1127, and −599; for ATIII-H [Ochoa et al., 1989] at −1213; for HP-1 (Kugler et al., 1988) repeatedly over the entire promoter region; for AP-1 at −1441; and for NF-1 (Cereghini et al., 1987; Lichtsteiner et al., 1989) at −2205 and −121 (half site; also alb DE-II element).
Finally, in the region between −98 and −77 a striking homology is present with a repressor element located upstream of the murine c-myc gene (Corcoran et al., 1985; Remmers et al., 1986). Part of the latter element also turned out to share homology with a so-called basic transcription element (BTE) recently reported to be functional in the CYP1Al promoter (Yanagida et al., 1990; see below). To establish the occurrence of actual (in vitro) protein-binding sites in the proximal part of the CYP2B2 promoter that might play a part in transcription activation in vivo, band shift analyses were performed.
The CYP2B2 promoter contains multiple in vitro protein-binding sites
For the band shift analyses we made use of liver nuclear extracts prepared from both PB-treated and control organisms (to be designated PB- and control extracts). For the former, at first the required induction period was determined. Consistent with previous reports (Hardwick et al., 1983), our slot blot analysis (see Fig. 2B) demonstrated that in control liver cytP450 2B (1 and 2) mRNA levels are very low, whereas a strong induction can be observed after several hours of treatment with PB. Since, obviously, at 4 hours after PB addition the transcriptional activity is already high, we have chosen this induction period for preparing PB extracts.
For the analysis of in vitro protein-binding sites, the proximal promoter fragment of the CYP2B2 gene was divided into three parts: a Pst I-Xba I-generated fragment extending from −579 to −372, an Xba I-Hinc II-generated fragment from −372 to −211, and a Hinc II-Sph I-generated fragment from −211 to +1 (see Fig. 1B). With all three fragments complex formation occurred, as shown in Figure 3A–C.
Figure 3.
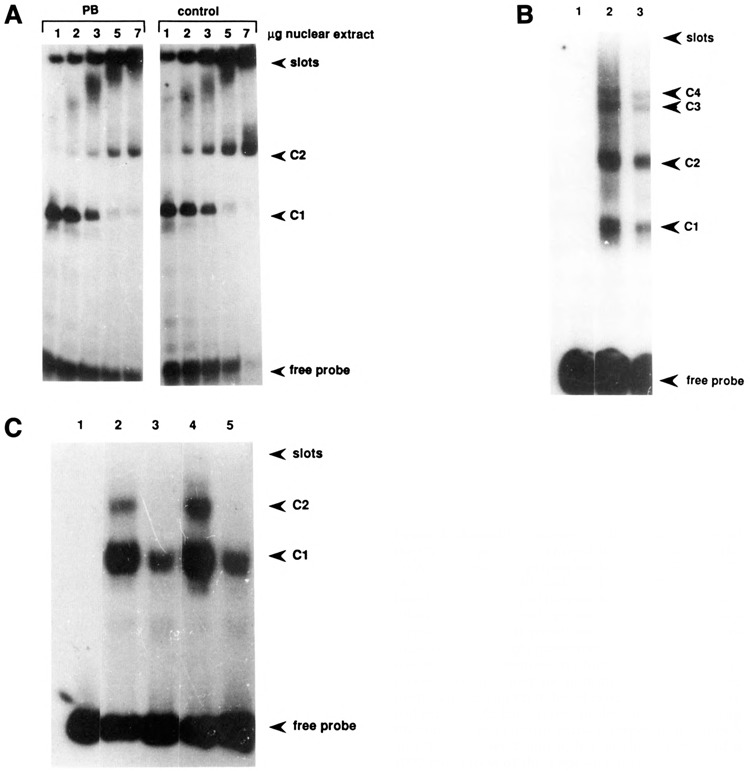
Bandshift analyses of the proximal part of the CYP2B2 promoter. Three different probes, viz. the DNA fragments comprising nucleotides −579 to −372 (A), −372 to −211 (B), and −211 to +` (C) were incubated in the presence of liver nuclear ext acts from both PB-treated and control rats and analyzed for complex formation. A. Left panel: increasing amount of PB-induced extract; right panel: increasing amount of control extract. The positions at which free probe and complexes have migrated are indicated. B. Lane 1: free probe; lane 2: 2 μg PB-induced extract; lane 3: 2 μg control extract. C. Lane 1: free probe; lanes 2 and 4: 5 μg PB-induced and control extract, respectively; lanes 3 and 5: like lanes 2 and 4, but in the presence of a 1000-fold excess of the c-myc oligomer.
Panel A shows the band shift assay with the most distal fragment. Control and PB-induced extracts indistinguishably gave rise to complex formation, though with PB-induced extracts the complexes tended to be more intense. In both cases, increasing the amount of added protein led to further retardation of the probe fragment, which may indicate either that multiple protein-binding sites are occupied or that multimerization of the protein(s) occurs.
A similar result was obtained with the fragment encompassing the nucleotide sequence from −372 to −211 (Fig. 3B). Both control and PB-induced extracts gave rise to two major complexes of different mobility and two minor complexes, which may be due to the simultaneous binding of different protein factors to the same DNA molecule.
The most proximal DNA fragment, which contains the putative (modified) TAATA and CAAT boxes, characteristic of cytP450 genes (Okey, 1990), forms two major complexes with both control and PB-induced extracts. Since this fragment contains the above-mentioned sequence element showing strong homology with the c-myc repressor element, we wished to examine whether this sequence element could be responsible for the observed retention. Therefore, a synthetic oligomer encompassing the promoter sequences from −104 to −71 was made (see also Fig. 5) and then used as a competitor or probe in a band shift analysis. Indeed, the complexes formed with this oligomer (Fig. 4) correspond to the prominent complexes formed with the proximal promoter fragment, since competition occurred first with C2 and, using a higher excess of oligomer, also with C1 (Fig. 3C, lanes 3 and 5). Apparently, a large excess of oligomer is needed to compete fully for binding of factors to the proximal promoter fragment. This finding suggests that the oligomer may not encompass the entire or optimal protein-binding element located in this promoter region. Both control and PB-induced extracts gave rise to complex formation with the oligomer (Fig. 4A). Again, increasing the amount of protein added caused a shift of the complex to a slower mobility. This phenomenon also occurred using partially purified protein factors (Fig. 4B), suggesting that multimerization of binding proteins results in the progressively increasing retardation of the protein-DNA band in the gel.
Figure 5.
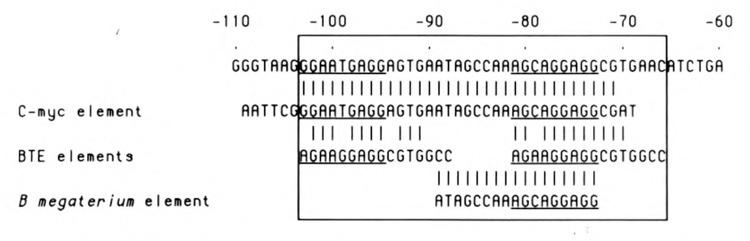
Comparison of sequence elements present in the proximal part of the CYP2B2 promoter. The nucleotide sequence of the region from −110 to −60 is aligned with the sequence of the “c-myc repressor oligomer” (Remmers et al., 1986), containing nucleotides −104 to −71, the BTE elements (Yanagida et al., 1990), and the element displaying homology with a sequence in the promoter of the B. megaterium cytP450 gene (He and Fulco, 1991). Oligomers encompassing the indicated sequences were used for the band shift analyses presented in Figures 4 and 6. The composite sequence element from −66 to −103 is designated BRE (basic regulatory element)—see text. The repeated protein factor binding motif RGNANGAGG is underlined.
Figure 4.
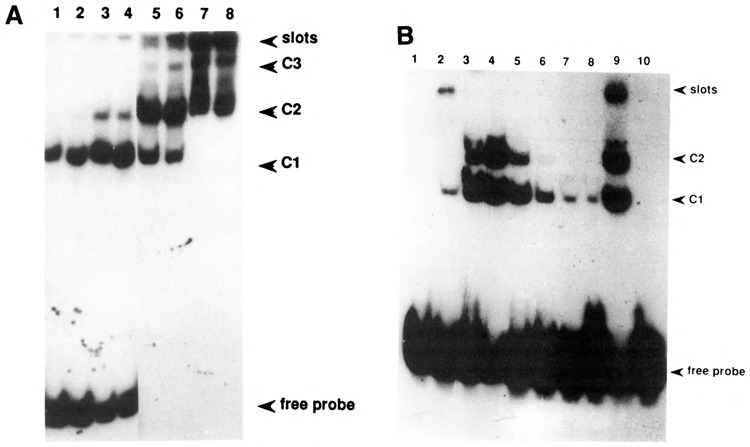
Bandshift analysis of the −104 to −71 oligomer. A. Lanes 1, 3, 5, and 7: 1.2, 2, 3.5, and 10 μg, respectively, of crude control extract; lanes 2, 4, 6, and 8: 1.2, 2, 3.5, and 10 μg, respectively of crude PB-induced extract. B. Crude nuclear PB-extract was chromatographed on a superose 12HR column (Pharmacia; result not shown), and consecutive fractions were then used for a band shift analysis of the oligomer. Numbers 1–8 correspond to these consecutive fractions, of which 15 μl were used in a total assay volume of 30 μl; lane 9: crude PB-extract; lane 10: free probe.
A closer inspection of the primary structure of the pertinent proximal promoter element indicated a tandemly repeated homology with the previously identified BTE, present in the promoter of several liver-specific genes (Yanagida et al., 1990), as well as an homology with a sequence motif identified by He and Fulco (1991) in the promoter of a barbiturate-inducible P450 gene from Bacillus megaterium. The comparison of the respective sequences is shown in Figure 5. To further establish the presumed composite character of the CYP2B2 proximal promoter region, we decided to use short oligomers encompassing the two different homologous sequences in a band shift analysis. The results, shown in Figure 6, clearly indicate (1) that both oligomers contain a binding site for a protein factor; and (2) that the complexes formed using the oligonucleotide showing homology with the c-myc repressor element (which was used for the experiment presented in Figure 4) are competed by adding excess amounts of both short oligomers. Obviously, two equivalent binding sites, sharing the motif RGNANG AGG, are present in this part of the CYP2B2 promoter. That the protein-binding sites on both short oligomers are identical was further proven by the fact that mutual competition occurred (not shown). On the basis of these analyses, it is also likely that the additional complexes formed using the extended oligomer (Fig. 4) are due to the binding of multiple proteins. Because low-mobility complexes were not found using the short oligomers—not even when high protein concentrations were used (Fig. 6)—the complexes showing a lower mobility than C2 (Fig. 4) may thus be due to interactions with other, as yet unidentified, factors.
Figure 6.
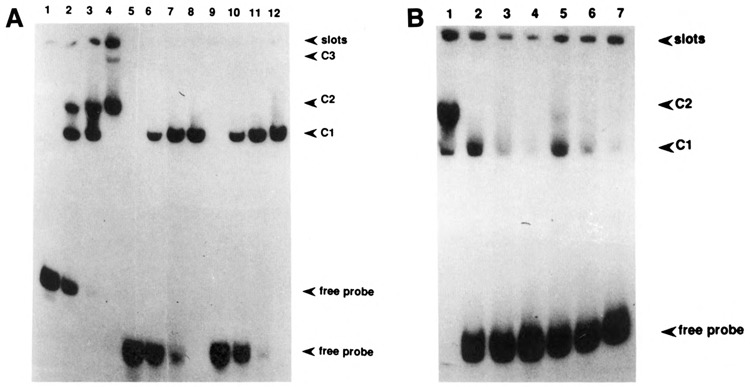
Band shift analysis of the CYP2B2-oligomers indicated in Figure 5. A. Increasing protein concentrations were used: 0, 2.5, 5, and 10 μg of crude control extract for lanes 1–4, 5–8, and 9–12. Lanes 1–4: using the −104 to −71 (c-myc) oligomer as a probe; lanes 5–8: using the BTE-oligomer as a probe; and lanes 9–12: using the B. megaterium oligomer as a probe. B. Competition experiment: the −104 to −71 oligomer was used as a probe for a band shift analysis using 5 μg of crude control extract (lane 1). Lanes 2, 3, and 4: competition with 10, 20, and 50×, respectively, molar excess of BTE oligomer; lanes 5, 6, and 7: competition with 10, 20, and 50×, respectively, molar excess of the B. megaterium oligomer.
In none of the band shift analyses performed so far we were able to detect qualitative differences between control and PB-induced extracts. This might be due to the fact that presumed PB-responsive elements are located elsewhere, or that interactions occurring in vivo under induction conditions escape this in vitro analysis.
Demarcation of functional sites in the proximal part of the CYP2B2 promoter
To study the possible functional role of DNA elements in the proximal part of the CYP2B2 promoter, we applied in vitro transcription using rat liver nuclear extracts. To this end we made a series of constructs that contain different parts of the promoter sequence fused to a G-less cassette—see Materials and Methods (Monaci et al., 1988). Proper transcription of these templates leads to the formation of a 380-nt transcript. We made use of restriction sites to generate constructs −806, −579, −372, and −177. In addition, using BAL 31 treatment, 5′ deletions were carried out, starting from the Hinc II site at −211 up to position −23. The adenovirus 2 major late promoter cloned in front of an 180 bp G-less cassette was used as an internal control. This promoter is known to be active in most cell types (Monaci et al., 1988) and was shown to act efficiently in both PB-induced and control extracts (see Fig. 7).
Figure 7.
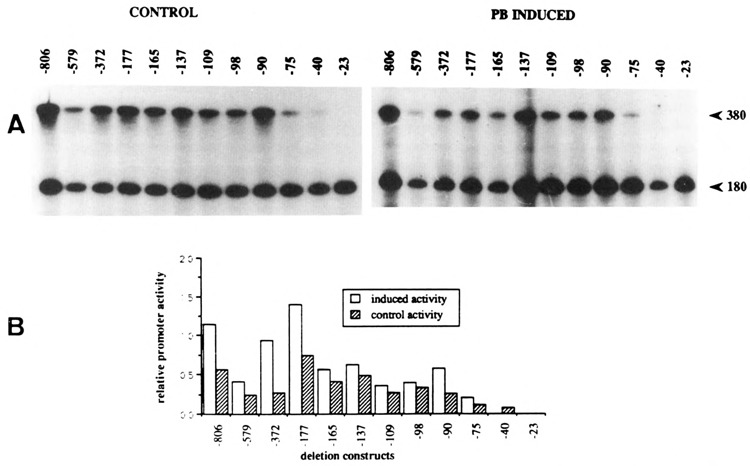
In vitro transcription on CYP2B2 promoter G-less templates. Deletion fragments of the CYP2B2 promoter cloned in the G-less cassette vector (see Materials and Methods) were used as templates for in vitro transcription, both in control and PB-induced nuclear extracts. The adenomajor late promoter fused to a 180 bp G-less fragment was used as internal reference. A. Left panel: control extracts; right panel: PB-induced extracts. B. Graphical representations. The autoradiographs shown in A were scanned using an LKB UltroScan Densitometer.
Analysis of the in vitro synthesized transcripts was performed on sequencing gels, the results of which are presented in Figure 7. The in vitro transcription assays have been repeated several times, using both the same and independently prepared extracts, and are highly reproducible; Figure 7A shows the result of a typical experiment. Different exposures of the autoradiorams were subsequently scanned in order to quantitate the differences in signal intensity; the results of the densitometric scans are shown in Figure 7B.
Using PB-induced extracts, deletion of the DNA region between −806 and −579 appeared to result in a twofold loss of transcription activity, which is, however, restored by a further deletion down to −372. This data may indicate the combinatorial action of positive as well as negative cis-acting elements in this part of the CYP2B2 promoter region. Deletions extending from −372 to −109 give rise to a gradual decrease in the level of transcription, indicating that this DNA region contributes to the strength of the promoter in vitro. A putative repressing element is present between nucleotides −98 and −90, since transcription increases by removal of this element. Clearly, an essential positive cis-acting element is destroyed by extending the deletion downstream of −90. A fivefold reduction in in vitro transcription activity occurs as a consequence of deleting 15 bp between −90 and −75. The remaining transcription activity is close to the low basal level exerted by a promoter containing just the TATA-element: construct −40. Apparently, the most important transcription-activating element in the proximal part of the CYP2B2 promoter is located directly upstream of −75.
Remarkably, extracts from control cells—which show no endogenous CYP2B2 gene expression—also display an in vitro transcription activity which is qualitatively quite similar to that obtained with PB-induced extracts. In general, however, the relative levels are lower compared to those obtained using PB-induced extracts (see Fig. 7).
Discussion
Expression of the CYP2B2 gene in rat liver is transcriptionally induced upon exposure of the organism to drugs like phenobarbital (PB) and certain PCB’s. In this paper we propose a model in which a proximal promoter element, encompassing nucleotides −103 to −66, plays an important part in this control process. This element is therefore designated BRE, for basic regulatory element.
Several lines of evidence support this model. First of all, the pertinent nucleotide element shows a composite structure consisting of two tandemly arranged homologous segments separated by a CCAAA-box sequence (summarized in Fig. 5). The conserved sequence element was first identified by us on the basis of homology with an upstream repressor site present in the murine c-myc promoter (Corcoran et al., 1985; Remmers et al., 1986) and was later demonstrated to be part of other liver-specific promoters as well (Yanagida et al., 1990). Recently, He and Fulco (1991) reported that barbiturate-inducible P450-genes in the rat and Bacillus megaterium share a common promoter element. On the basis of band-shift analysis, these authors suggested that this sequence element could mediate the transcriptional induction by PB. This sequence is part of the BRE, described in this paper, viz. nucleotides −89 to −73 (see Fig. 5).
Band shift analysis of the proximal part of the CYP2B2 promoter revealed a major composite protein binding site in the region up to −211 which appeared to coincide with the −104 to −71 element. The results of gel retardation experiments using synthetic oligomers encompassing different sub-segments of this element (Fig. 6) strongly suggest that binding of more than one protein to this sequence element occurs. On the basis of sequence comparisons we propose that the sequence motif RGNANGAGG represents the actual factor-binding site. This sequence is tandemly repeated in the promoter element from −104 to −71, which could explain that we were unable to demonstrate the involvement of specific nucleotides in the interaction with the factor by a methylation interference analysis using complex C1 shown in Figure 4 (result not shown). Apart from the factors interacting with the two equivalent protein-DNA binding sites, additional proteins are likely to bind to this part of the CYP2B2 promoter, since low-mobility complexes were found using the −104 to −71 oligomer (cf. Figs. 4 and 6). No qualitative differences between control and PB-induced extracts could be detected, however.
The results of our in vitro transcription experiments revealed the functional importance of the −90 to −75 region for transcription activation of the CYP2B2 gene. Deletion of this element leads to a fivefold loss in transcription activity in vitro (Fig. 7). Taken together, therefore, the results support the hypothesis that the complex sequence element present between −103 and −66 mediates the transcription activation of the CYP2B2 gene (BRE in Fig. 5).
Unlike the situation in vivo, where no transcription of CYP2B under non-induced conditions can be detected (see Fig. 2), control extracts appeared unexpectedly active in vitro. As a matter of fact, PB-induced extracts displayed only a slightly elevated CYP2B2 gene transcription as compared with control extracts. We favor the interpretation that this is due to the absence of the natural chromatin structure in the pertinent part of the promoter. The transcriptional induction of the CYP2B2 gene may thus involve a derepression mechanism which renders this promoter region accessible for the assembly of an active transcription–initiation complex. Probably the required trans-acting protein factors are present at non-induced conditions—as is also suggested by the failure to find differences in the band-shift assays—while some modification may be responsible for the actual transcriptional induction.
Of course the experiments described in this paper do not exclude the involvement in transcription activation of (PB-responsive) nucleotide elements located further upstream. In vitro transcription assays are not suitable for identifying such elements. So far, however, functional analyses of the CYP2B2 promoter by transient expression assays are hampered by the fact that isolated hepatocytes very rapidly lose the ability to express this gene (see also Waxman and Azaroff, 1992).
When this manuscript was ready to submit, we learned the results obtained by Upadhya et al., which are in agreement with ours (Upadhya et al., 1992).
Acknowledgments
We express our gratitude to Rutger Meinsma, Etienne Bido, and Gerard Griffioen for their contribution in the sequencing work; to Martine Smit for performing the band shift analyses using purified protein fractions; to Mrs. P. G. Brink for carefully preparing this manuscript; and to Dr. H. van Heerikhuizen for useful discussions.
References
- Cereghini S., Raymondjean M., Carranca A. G., Herbomel M., and Yaniv M. (1987), Cell 50, 627–638. [DOI] [PubMed] [Google Scholar]
- Chomczynski P. and Sacchi N. (1987) Anal Biochem 64, 156–159. [DOI] [PubMed] [Google Scholar]
- Corcoran L. M., Cory S., and Adams J. M. (1985), Cell 40, 71–79. [DOI] [PubMed] [Google Scholar]
- Denison M. S., Fisher J. M., and Whitlock J. P. Jr. (1988), J Biol Chem 263, 17221–17224. [PubMed] [Google Scholar]
- Fujisawa-Sehara A., Yamane M., and Fujii-Kuriyama Y. (1988), Proc Natl Acad Sci USA 85, 5859–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F. J. and Nebert D. W. (1990), Trends Genet 6, 182–186. [DOI] [PubMed] [Google Scholar]
- Gorski K., Carneiro M., and Schibler U. (1986), Cell 47, 767–776. [DOI] [PubMed] [Google Scholar]
- Graves B. J., Johnson P. E., and McKnight S. L. (1986), Cell 44, 565–576. [DOI] [PubMed] [Google Scholar]
- Hardon E. M., Frain M., Paonessa G., and Cortese R. (1988), EMBO J 7, 1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick J. P., Gonzalez F. J., and Kasper C. B. (1983), J Biol Chem 258, 8081–8085. [PubMed] [Google Scholar]
- He J-S. and Fulco A. J. (1991), J Biol Chem 266, 7864–7869. [PubMed] [Google Scholar]
- Jaiswal A. K., Rivkin E., and Adesnik M. (1987), Nucleic Acids Res 15, 6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal A. K., Haaparante T., Luc P., Schembri J., and Adesnik M. (1990), Nucleic Acids Res 18, 4237–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawajiri K., Gotoh O., and Tagashira Y. (1984), J Biol Chem 259, 10145–10149. [PubMed] [Google Scholar]
- Kugler W., Wagner U., and Ryffel G. U. (1988), Nucleic Acids Res 16, 3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtsteiner S., Wuarin J., and Schibler U. (1989), Cell 51, 963–973. [DOI] [PubMed] [Google Scholar]
- Monaci P., Nicosia A., and Cortese R. (1988), EMBO J 7, 2075–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert D. W., Eisen H. J., and Hankinson O. (1984), Biochem Pharmacol 33, 917–924. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Nelson D. R., Adesnik M., Coon M. J., Estabrook F. J., Gonzalez R. W., Guengerich F. P., Gunsalus I. C., Johnson E. F., Kemper B., Levin W., Phillips I. R., Sato R., and Waterman R. (1989), DNA 8, 1–13. [DOI] [PubMed] [Google Scholar]
- Ochoa A., Brunel F., Mendelzon D., Cohen G. N., and Zakin M. M. (1989), Nucl Acids Res 17, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okey A. B. (1990), Pharmacol Ther 45, 241–289. [DOI] [PubMed] [Google Scholar]
- Parker C. S. and Topol J. (1984), Cell 36, 357–369. [DOI] [PubMed] [Google Scholar]
- Phillips I. R., Shephard E. A., Bayney R. M., Pike S. F., Rabin B. R., Heath R., and Carter N. (1983a), Biochem J 212, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips I. R., Shephard E. A., Ashworth A., and Rabin B. R. (1983b), Gene 24, 41–52. [DOI] [PubMed] [Google Scholar]
- Poland A. and Glover E. (1976), J Biol Chem 251, 4936–4946. [PubMed] [Google Scholar]
- Poland A., Mak I., Glover E., Boatman R. J., Ebetino F. H., and Kende A. S. (1980), Mol Pharmacol 18, 571–580. [PubMed] [Google Scholar]
- Rampersaud A. and Walz F. G. Jr. (1987a), Biochem Genet 25, 527–534. [DOI] [PubMed] [Google Scholar]
- Rampersaud A. and Walz F. G. Jr. (1987b), J Biol Chem 262, 5649–5653. [PubMed] [Google Scholar]
- Remmers E. E., Yang J-Q., and Marcu K. B. (1986), EMBO J 5, 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D. E. and Wayne L. (1990), Pharmacol Ther 45, 153–239. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., and Coulson A. R. (1977), Proc Natl Acad Sci USA 83, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholte B. J., te Velde B., Groot G. S. P., and de Vries J. (1985), Eur J Biochem 151, 67–73. [DOI] [PubMed] [Google Scholar]
- Sinclair P. R., Schuetz E. G., Bement W. J., Haugen S. A., Sinclair J. F, May B. K., Li D., and Guzelian P. S. (1990), Arch Biochem Biophys 282, 386–392. [DOI] [PubMed] [Google Scholar]
- Soares M. B., Schon E., and Efstratiadis A. (1985), J Mol Evol 22, 117–133. [DOI] [PubMed] [Google Scholar]
- Sogawa K., Fujisawa-Sehara A., Yamane M., and Fujii-Kuriyama M. (1986), Proc Natl Acad Sci USA 83, 8044–8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwa Y., Mizukami Y., Sogawa K., and Fujii-Kuriyama Y. (1985), J Biol Chem 260, 7980–7984. [PubMed] [Google Scholar]
- Thomas P. E., Reik L. M., Ryan D. E., and Levin W. (1983), J Biol Chem 258, 4590–4598. [PubMed] [Google Scholar]
- Ueno T. and Gonzalez F. J. (1990), Mol Cell Biol 10, 4495–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya P., Venkateswara-Rao P. M., Venkateswar V., and Padmanaban G. (1992), Nucleic Acids Res 20, 557–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasuk G. P., Ghrayeb J., Ryan D. E., Reik L., Thomas P. E., Levin W., and Walz F. G. Jr. (1982), Biochemistry 21, 789–798. [DOI] [PubMed] [Google Scholar]
- Waxman D. J. and Walsh C. (1982), J Biol Chem 257, 10446–10457. [PubMed] [Google Scholar]
- Waxman D. J. and Azaroff L. (1992), Biochem J 281, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L-P., Koeiman N., and Whitlock J. P. Jr. (1990), Proc Natl Acad Sci USA 87, 8545–8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock J. P. Jr. and Galeazzi D. R. (1984), J Biol Chem 259, 980–985. [PubMed] [Google Scholar]
- Yanagida A., Sogawa K., Yasumoto K-I., and Fujii-Kuriyama Y. (1990), Mol Cell Biol 10, 1470–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]