

Abstract
We report that three orphan receptors, hERRl, hERR2, and hTR2, members of the steroid/thyroid receptor (SR/TR) superfamily, can be activated by different ligand-independent pathways. hERRl and hERR2 exhibit constitutive activity in the absence of exogenously added ligands. Furthermore, this constitutive activity is localized in the carboxy terminal domain of both receptors and can be transferred to other members of this superfamily using domain switch strategies. In addition, we show that hERRl can remain constitutively active in the less evolved eukaryotic cell Saccharomyces cerevisiae. In contrast, hTR2 is not constitutively active. However, a chimera of hTR2 can be activated in a ligand-independent manner through a signal transduction pathway initiated at the cell membrane by the neurotransmitter dopamine. Like hERRl and hERR2, hTR2 is ligandindependently activated through its carboxy terminal domain. Together, these results suggest the existence of emerging subgroups w ithin the SR/TR superfamily that can regulate gene expression through different modes of activation.
Members of the SR/TR superfamily are ligand-inducible transcription factors that regulate some of the pivotal gene networks responsible for eukaryotic cell growth, development, and homeostasis (Evans, 1988; O’Malley, 1990). In the past, the prevailing view of how steroid hormone receptors exert their effects on gene transcription has been that lipophilic hormones and vitamins passively diffuse through the cell membrane of a target cell and are specifically and tightly bound by the appropriate ligand inducible receptor. As a consequence of this ligand-receptor interaction, the transformed receptor modulates RNA polymerase II activity on a responsive gene through a specific distal enhancer element. Implicit in this concept is that the receptor binds its ligand with high affinity and alters gene expression as a result of this direct ligand interaction.
Molecular cloning of the glucocorticoid, estrogen, progesterone, and vitamin D3 receptors helped to define biochemically the functional domain structure of these receptors (Hollenberg et al., 1985; Green et al., 1986; Conneely et al., 1986; Jeltsch et al., 1986; McDonnell et al., 1987). The DNA and ligand-binding domains show a general positional conservation among all of the receptors. The ligand-binding domain is located in the carboxy terminal region of the proteins. In addition to its ligand-binding functions, this domain also contains sequences required for receptor dimerization (Fawell et al., 1990) and target gene transacttvatton (Dobson et al., 1989; Webster et al., 1988; Carson et al., 1990). The ligand-binding domain is flanked on its N-terminal side by a short variable hinge region that contains an additional transactivation function together with sequences required for stabilization of interaction of inactive receptors with heat shock proteins (Dobson et al., 1989; Carson et al., 1990). The highly conserved DNA-binding domain is located N-terminal to the hinge region. This domain contains two zinc finger structural motifs that contain sequences responsible for specific recognition of hormone response elements (Umesono et al., 1989; Mader et al., 1989), and for receptor dimerization (Tsai et al., 1988; Umesomo et al., 1989). The N-terminal region of the receptors is hypervariable in terms of both size and amino acid sequence. This region contains transactivation functions that modulate both the level and promoter specificity of target gene activation by the receptors (Kumar et al., 1987; Tora et al., 1988). Finally, the functional domains outlined above are modular in structure (Picard et al., 1988). The domains can be interchanged between family members to generate functional chimeric receptors with altered specificities (Green et al., 1987; Giguere et al., 1987), or they can replace functional domains of unrelated transcription factors to modulate their function (Tasset et al., 1990).
The high degree of conservation between the DNA-binding domains of these receptors has been exploited to generate DNA probes to isolate genes encoding numerous additional members of the receptor superfamily using low stringency hybridization techniques. Activating ligands have been identified for many of these receptors which bind thyroids, mineralocorticoids, androgens, and the retinoids–including 9-cis retinoic acid, the ligand for the retinoid X receptor (RXR; Sap et al., 1986; Weinberger et al., 1986; Arriza et al., 1987; Lubahan et al., 1988; Chang et al., 1988a; Petkovich et al., 1987; Giguere et al., 1987; Levin et al., 1992; Heyman et al., 1992). However, there exists a growing family of receptors that have not been matched with their corresponding ligand (Tsai et al., 1991) and are collectively termed orphan receptors. Because orphan receptors possess a putative ligand-binding domain, a major effort is underway to identify their respective ligands. Identification of a ligand for any of these orphan receptors promises to uncover previously unknown hormonally or nutritionally controlled gene networks.
Over the past two years we have developed mammalian and yeast cell culture assays to identify ligands or chemical signals that activate a number of orphan receptors. These studies have led to the identification of a novel ligand-independent pathway of activation of several steroid receptors and the orphan receptor, COUP-TF (Power et al., 1991a,b). We have shown that several members of the SR/TR superfamily can be activated by an intracellular pathway which presumably alters phosphorylation and is stimulated by the neurotransmitter dopamine (Denner et al., 1990; Power et al., 1991b). The observation that such activation occurs in the absence of hormonal ligand provided evidence of crosstalk between two very different signal transduction pathways and prompted a revaluation of our current concepts of steroid hormone action.
In the present paper we extend these observations to address the mode of activation of three additional orphan receptors. The orphan receptors tested were human estrogen-related receptors 1 and 2 (hERRl and hERR2; Giguere et al., 1988) and hTR2 (Chang et al., 1988b).
Among the first orphan receptors to be identified were hERRl and hERR2 (Giguere et al., 1988). Both receptors were isolated using the DNA-binding domain of the human estrogen receptor as a hybridization probe under reduced stringency conditions. Based on sequence analysis of both receptors, it was inferred that each is an authentic member of the SR/TR super-family. In addition, both receptors shared a significant overall homology with each other, but despite the terminology, hERRl and hERR2 were found to be marginally related to the human estrogen receptor. Northern analysis revealed that the hERRl transcript had a general tissue distribution with particular abundance in the central nervous system. In contrast, the hERR2 mRNA expression pattern was more restricted and much less abundant, which may reflect divergent functions for these receptors in vivo. Although discovered over four years ago, a physiological function has yet to be ascribed to these two receptors.
The third orphan receptor examined in this report was hTR2. A previous study described the isolation of hTR2 from a human testis cDNA library using an oligonucleotide probe containing a consensus sequence of a conserved region within the DNA-binding domains of previously cloned steroid receptors (Chang et al., 1988b). Although sequence analysis revealed that hTR2 contained the typical modular domain structure of other members of the SR/TR superfamily, hTR2 does not share a close homology with any of the previously isolated members. Analysis of the tissue distribution of the hTR2 mRNA transcript revealed that expression of this receptor was concentrated in androgen-sensitive tissues such as the ventral prostate, seminal vesicle, and testis. In addition, preliminary results suggested that hTR2 mRNA induction was negatively controlled by androgen action in the prostate. Like hERRl and hERR2, a role for hTR2 function in cellular physiology has yet to be assigned.
The results of this study suggest that these orphan receptors may mediate their actions in the absence of direct classical ligand binding. Furthermore, the transactivational properties of these orphan receptors reveal emerging subgroups within the SR/TR superfamily with regard to the mechanism of ligand-independent activation.
Materials and methods
Sources of materials
Restriction and DNA modifying enzymes were obtained from Promega, Boehringer Mannheim, and Pharmacia. [125I]protein A was obtained from ICN. L-dopa, dopamine, and polybrene were purchased from Sigma. Nutridoma SR was obtained from Boehringer Mannheim.
Construction of PR.hERRl, PR.hERR2 and PR.hTR2
The cPRb portion of the chimeric constructs was obtained from PAD86ΔK (Conneely et al., 1989) by polymerase chain reactton (PCR) mediated amplification (Saiki et al., 1988) of the cDNA encoding amino acids 1–495:
5′-primer, 5′-GGCGAATTCCAGCGCTGCTCCGGGCACCATCGCCGAGGTGAAGAGCAAG
3′-primer, 5′-GTTAGGATCaTAAATTTTCGACCTCCCAGGACCATTCC
The 5′-primer incorporates an EcoR 1 and Nco 1 restriction site. The 3′-primer contains a BamH I site. The resulting EcoR I and BamH I digested cPRb PCR product was cloned into pSP72 (Promega). pSP72-cPRB was cut with BamH I and Xba I to introduce a polylinker containing restriction sites for Kpn I and EcoR I.
The putative orphan receptor carboxy terminal domains were generated using PCR amplification. The 5′-primer was modified to contain a BamH I site, and the 3′-primer incorporated a Kpn I restriction site. The carboxy terminal domain of hERRl encoding amino acids 246–521 was obtained from the plasmid pRShERRl using the following primers:
5′-primer,5′-AAGATCCACTGCGCCTGGACCGCGTCCGGGGTGGGCGG
3′-primer, 5′-CCCGGTACCTCAGTCCATCATGGCCTCGAGCATCTCCAA
For hERR2, the following primers were used to amplify the carboxy terminal domain encoding amino acids 173–433 from the plasmid pRShERR2:
5′-primer, 5′-AAGGATCCCGTGCGCCTTGACCGGGTGCGAGGAGGC
3′-primer, 5′-TGGATCGGTACCATCCGTCTGCATGCGGGGCCATCACACC
The 5′-primer,
5′-GAAAGGGATCCCATTGAAGTATCACGAGAAAAATCTTCC
and 3′-primer,
5′-TTTrGGGTACCCATAAGCCATTCTATAGTTAAGCATTTA
were used to amplify the carboxy terminal domain encoding amino acids 189–483 of hTR2 from the plasmid pGEM3Z hTR2. Each of the PCR-generated orphan receptor carboxy terminal domains was digested with BamH I and Kpn I and cloned into pSP72-cPRB. The overall translation reading frame of the resultant hybrid receptor was maintained as judged by sequencing, immunoblot analysis, and transcriptional activation using PREtkCAT. The hybrid expression constructs PR.hERRl, PR.hERR2, and PR.hTR2 were made by inserting the respective chimeric cDNAs into the EcoR I site of the eukaryotic expression vector p91023(B) (Wong et al., 1985).
Orphan receptor cDNA templates
The receptor expression constructs, pRShERRl and pRShERR2 were kindly provided by Ronald Evans, The Salk Institute for Biological Studies, San Diego, California, and pGEM3ZhTR2 was a gift from Chawnshang Chang, The Ben May Institute, Chicago, Illinois. The above plas-mids were used as templates for PCR. The yeast expression construct YEpPR.hERRl was made by excising the human estrogen receptor from YEPE2 (Pham et al., 1991) using Nco I and Kpn I and replacing it with the chimeric PR.hERRl cDNA.
Reporter plasmids
PREtkCAT and YRpPl have been described previously (Jantzen et al., 1987; Mak et al., 1989).
Cell transfections
CV-1 cells at a plate density of approximately 5 × 105 cells per 100 mm plate were transfected with 5 μg of both expression and reporter plasmids using the polybrene method (Chaney et al., 1989). Transfection studies using serum-free media (Dulbecco’s modified Eagle’s medium) supplemented with Nutridoma were as described by Power et al. (1991a). All transfection experiments were performed at least 6 times in duplicate, and the variation in signal between duplicate points in any one experiment was not more than 5%. For Western immunoblot analysis COSM-6 monkey kidney cells cultured in Nutridoma-supplemented media were transfected with 5 μg of the relevant hybrid receptor expression construct. Cell culture conditions were the same as for CV-1 cells.
Western immunoblot analysis
Mammalian and yeast cellular extracts equivalent to 150 μg of protein was resolved on 10% SDS-polyacrylamide gels. Standard procedures for both protein transfer and immunoblotting were followed (Power et al., 1991a). The monoclonal antibody PR22, the epitope of which resides in both cPRA and cPRB, was used as the first antibody. After consecutive incubations with rabbit antimouse IgG (Zymed) and [125I] protein A, the blot was exposed overnight to X-AR film (Kodak) at −70°C.
Transcriptional assays in Saccharomyces cerevisiae
The lithium acetate transformation procedure was used to introduce the yeast expression plasmids and reporter plasmid into the protease deficient yeast strain BJ3505 (Ito et al., 1983). At midlog phase, progesterone (10−7 M) was added to the appropriate yeast cultures grown in selective media in the presence or absence of 0.1 M copper sulphate. After a further 4–6 hour incubation in the presence of hormone, cells were harvested and assayed for β-galactosidase activity. Each test point was performed in triplicate, and each experiment was repeated 4 times. Transcriptional activity was expressed in Miller units (Miller, 1972).
Results
Transactivational studies of hERRI, hERR2, and hTR2
The challenges to understanding the role of orphan receptors such as hERRI, hERR2, and hTR2 in normal cellular physiological processes, is the identification of their respective ligands and the genes that they regulate. The overall approach we have adopted to identify potential ligands and chemical signals that lead either directly or indirectly to the activation of these orphan receptors is based on a tissue culture assay system. An expression plasmid containing the orphan receptor cDNA is transiently co-transfected into the monkey kidney cell line CV-1 with a specific receptor responsive target gene. Using this assay system, induction of transcription of the target gene can only occur through an activated orphan receptor. In these experiments, transfected CV-1 cells were grown in serum-free media. For these transactivation studies it was important to use serum-free media to avoid the apparent activating effects of “serum born factors” which have been shown to mediate their action through ligand-independent routes such as phosphorylation (Power et al., 1991b).
Tissue extracts or specific chemical ligands were added to the medium in an attempt to activate the orphan receptor, resulting in an induction of expression of the reporter gene, chloramphenicol acetyl transferase (CAT; Luckow et al., 1987). In order to prevent competition from endogenous orphan receptors in the assay, we have exploited the modular nature of the functional domains of these receptors to replace the ligand-binding domain of the chicken progesterone receptor (cPR) with the corresponding region of the orphan receptors (Fig. 1). The resulting chimeric receptors contain the N-terminal and DNA-binding domain of cPR under the control of the putative ligand-binding domain of the orphan receptor. The activity of these chimeras was assayed by co-transfection of the chimeric receptor expression plasmids with the progesterone receptor responsive target gene, PREtkCAT (Jantzen et al., 1987). This target gene contains two copies of a progesterone response element (PRE) located upstream of the herpes simplex virus thymidine kinase promoter linked to the CAT gene (Strahle et al., 1987). This domain switch strategy provides three immediate advantages in these trans-activation assays. First, it allows us to report orphan receptor activity using PREtkCAT without knowing the authentic orphan receptor responsive gene. Second, the use of the PREtkCAT gene as a reporter prevents competition with endogenous orphan receptors in the transactivation assay. Third, since specific antibodies for these orphan receptors are presently unavailable, this receptor design allows for the monitoring of chimeric receptor expression by Western immunoblot using the monoclonal antibody PR22 specific to the N-terminus of cPR (Sullivan et al., 1986).
Figure 1.
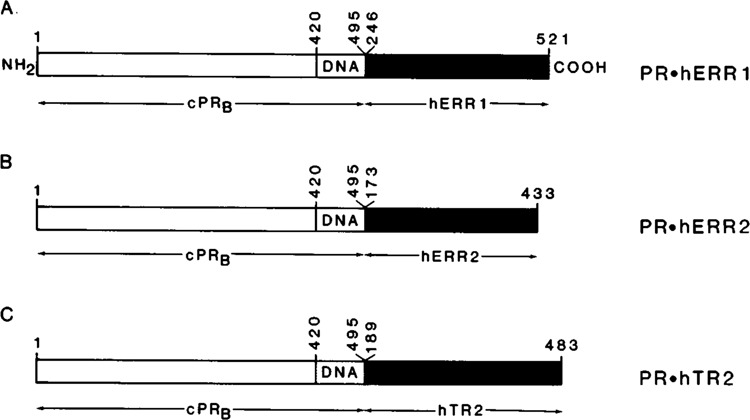
Orphan chimeric receptor constructs used for transactivation assays. The chimeric receptor constructs contained the N-terminal region (amino acids [aa] 1-420) and DNA-binding domain (DNA) (aa420-495) of the chicken progesterone receptor B subtype (cPRB) fused in frame to the carboxy terminal domain of the relevant orphan receptors. The regions of orphan receptors used to generate the chimeric receptor constructs (A) PR.hERRl, (B) PR.hERR2, and (C) PR.hTR2 included the aa246-521 of hERRl, aal73-433 of hERR2, and aal89-483 of hTR2 respectively. The chimeric receptor cDNAs were cloned into the unique EcoR I site of the eukaryotic expression plasmid p91023(B) and constitutively expressed under the control of the adenoviral major late promoter (see Materials and Methods).
Using the above approach, we examined the transactivational activities of hERRl, hERR2, and hTR2 (Fig. 2A). When the cPR expression plasmid PAD86ΔK (Conneely et al., 1989) was co-transfected with PREtkCAT into PR negative CV-1 cells cultured as described, induction of CAT activity was not detected in the absence of its ligand, progesterone. However, addition of progesterone resulted in a significant transactivation of the reporter gene. In contrast, PR.KTR2 induced undetectable levels of CAT activity. Both PR.hERRl and PR.hERR2 elicited a similar and significant CAT response in the absence of added ligands or chemical signals. This result reveals that hERRl and hERR2 exhibit constitutive transcriptional activity, a functional property also found with the orphan receptor and immediate early gene member Nur77 (Hazel et al., 1988; Davis et al., 1991). These data also show that the inherent constitutive activation function resides in the putative ligand-binding domain of hERRl and hERR2, and that this activity is transferable to other members of the SR/TR superfamily, such as the progesterone receptor. Finally, examination of the expressed chimeras using Western immunoblot analysis (Fig. 2B) with αcPR IgG, PR22, was performed in the SV40 transformed monkey kidney cell line COSM-6. Because CV-1 cells produce sub-physiological levels of receptor that are undetectable by Western analysis, COSM-6 cells were used to allow expression plasmids that contain the SV40 origin of replication to replicate to a high copy number, resulting in the production of receptor levels that could be detected by Western immunoblot. The results shown in Figure 2B confirm that PR.hERRl (87 kDa), PR.hERR2 (85 kDa), and PR.hTR2 (89 kDa) were expressed intact, in equivalent amounts, and migrated to the correct molecular mass as predicted from their amino acid sequences.
Figure 2.
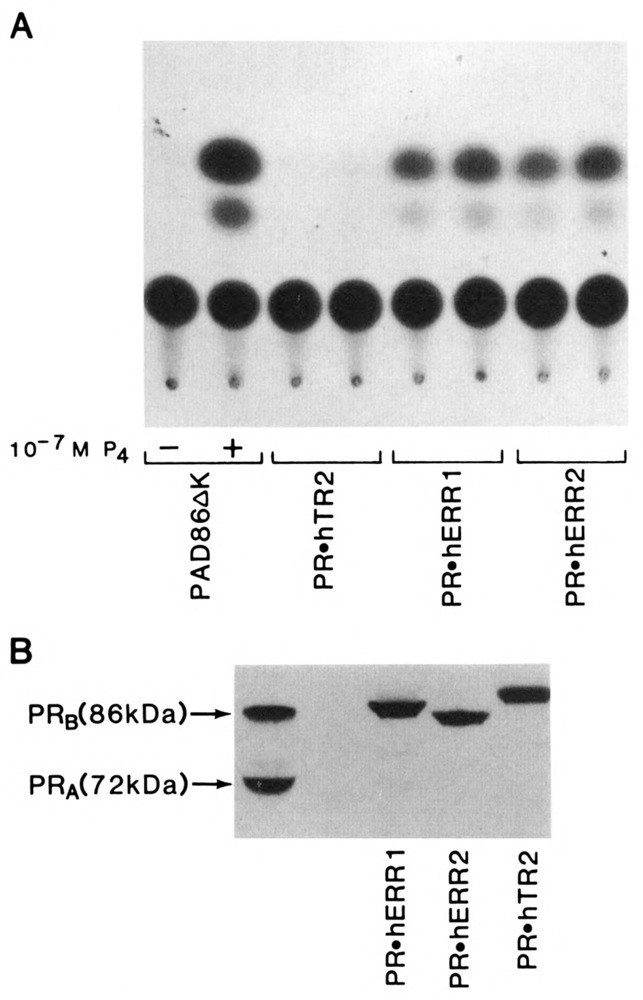
Transactivational assays of PR.hERRl, PR.hERR2, and PR.hTR2. A. The expression vectors (5 μg) encoding cPRB (PAD86ΔK) and the orphan receptor chimeras PR.hERRl, PR.hERR2, and PR.hTR2 were co-transfected with the reporter plasmid PREtkCAT (5 μg) in the monkey kidney cell line CV-1 by the polybrene method. CV-1 cells were cultured for 48 hours in serum-free media supplemented with Nutridoma-SR. The results of the CAT assay using PAD86ΔK in the absence or presence of progesterone (10−7M) are indicated. Duplicate CAT assays for each of the orphan receptor chimeras are shown. The experiment was performed 6 times, and the variation in duplicate signals between experiments did not exceed 5%. B. COS M6 monkey kidney cells cultured in nutridoma-supplemented media were transfected with the chimeric receptor constructs PRhERRl, PR.hERR2, and PR.hTR2 (5 μg), as described in A. After 48 hours of culture, high-salt extracts were prepared and analyzed by immunoblotting with the monoclonal antibody PR22 specific to the N-terminus of cPR (see Materials and Methods). The left lane indicates the molecular sizes of the progesterone receptor subtypes A and B in 100 μg of chicken oviduct cytosolic protein.
hERR1 is constitutively active in yeast
To determine whether the constitutive trans-activation function imparted by the carboxy terminal domain of hERRI required endogenous factors that were specific to mammalian cells, we employed the more basic eukaryotic assay system of Saccharomyces cerevisiae to test the activity of PR.hERRl using a similar cotransfection assay. Using this system, the ligand-dependent functional activities of a number of steroid receptors have been successfully reconstituted (Mak et al., 1989; Pham et al., 1991). In the present study, the full-length chicken progesterone (cPR) and the PR.hERRl chimeric cDNA were ligated to the yeast expression vector, YEp, as previously described (Mak et al., 1989). Receptor expression was driven by the copper-inducible yeast metallothionein (CUP1) promoter (Butt et al., 1988). The target plasmid, YRpPl, contained two copies of the PRE tandemly inserted upstream of the yeast CYC-1 (iso-cytochrome C) promoter fused to the E. coli lacZ gene encoding β-galactosidase (Mak et al., 1989). The cPR or PR.hERRl expression plasmid (YEpP2 and YEpPR.hERRI respectively) was used together with the target plasmid to transform Saccharomyces cerevisiae auxotrophic strain BJ3505 (Mak et al., 1989), and the cells were grown in defined medium. Receptor activation of the target gene was measured by assaying β-galactosidase activity in the presence or absence of copper sulphate to regulate receptor expression. The results are shown in Figure 3. The β-galactosidase activity observed is expressed in Miller units of activity.
Figure 3.
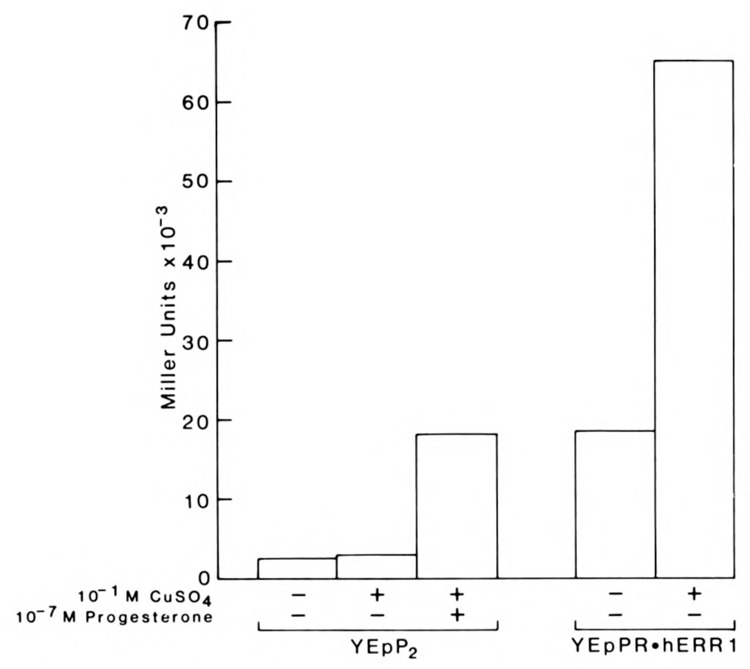
The orphan receptor chimera PR.hERRl is constitutively active in Saccharomyces cerevisiae. The yeast expression constructs YEpP2 and YEpPR.hERRl encoding the chicken progesterone receptor (cPRB) and PR.hERRl respectively, were expressed and assayed with the reporter plasmid YRpPl in the protease deficient yeast strain BJ3505. The values of each test point represent the average of 4 separate experiments performed in triplicate. Under the same experimental conditions, immunoblot analysis of expressed cPRB and PR.hERRl using the monoclonal antibody PR22 revealed that each receptor was expressed intact and in equivalent amounts (data not shown), as was shown in Figure 2B.
In the absence of copper sulphate and progesterone, basal levels of cPR were sufficient to activate a low level of transcription of the reporter plasmid, YRpPl. The addition of copper did not alter these levels significantly. After the addition of progesterone, cPR elicited a significant induction of lacZ activity (>6-fold over basal levels). The result shows that the activation of the lacZ reporter gene by cPR was both progesterone- and copper-dependent. In contrast, regulation of (β-galactosidase expression by PR.hERRl appeared to be constitutive. In the absence of copper, the lacZ gene activity induced by basal levels of expressed PR.hERRl was equivalent to levels achieved by fully expressed cPR in the presence of progesterone and was further induced >4-fold by the addition of copper sulphate. The constitutive activation of the reporter YRpPl by PR.hERRl is consistent with the mammalian transfection results described above. Furthermore, for reasons that are not clear, the relative functional activity of PR.hERRl is even greater in the yeast system compared to mammalian cells.
Both the mammalian and yeast experiments support the existence of a transactivation function in the carboxy terminal domain of hERRl and hERR2 that imparts a constitutive active phenotype to both receptors. In the case of hERRl in yeast cells, this transactivation function remains active in simple defined media and is stronger than any transactivation domain of the progesterone receptor when these domains are fully activated by progesterone.
The data, taken together, reveal that the constitutive activation function of hERRl and hERR2 is located in their carboxy terminal domains. By domain switch strategies this activity is transferable to other SR/TR superfamily members and can be maintained in less evolved eukaryotic cells such as yeast.
hTR2 is activated by the neurotransmitter dopamine
In contrast to PR.hERRl and PR.hERR2, transactivation by PR.hTR2 was not observed in CV-1 cells grown in serum free medium (Fig. 4). An exhaustive ligand screen was undertaken to identify a potential activator molecule. Despite testing more than 150 candidate compounds and tissue extracts, we did not detect induction of CAT activity. This inability to activate the PR.hTR2 orphan receptor with our collection of potential ligands and tissue extracts was also observed with the previously characterized orphan receptor, COUP-TF (Power et al., 1991a). Although the physiologic ligand for hTR2 has not been identified, like the orphan receptor COUP-TF, PR.hTR2 was induced in a ligand-independent manner by the catecholamine neurotransmitter dopamine (Fig. 4, lanes 4 and 5) and by its immediate precursor, L-dopa (Fig. 4, lanes 2 and 3).
Figure 4.
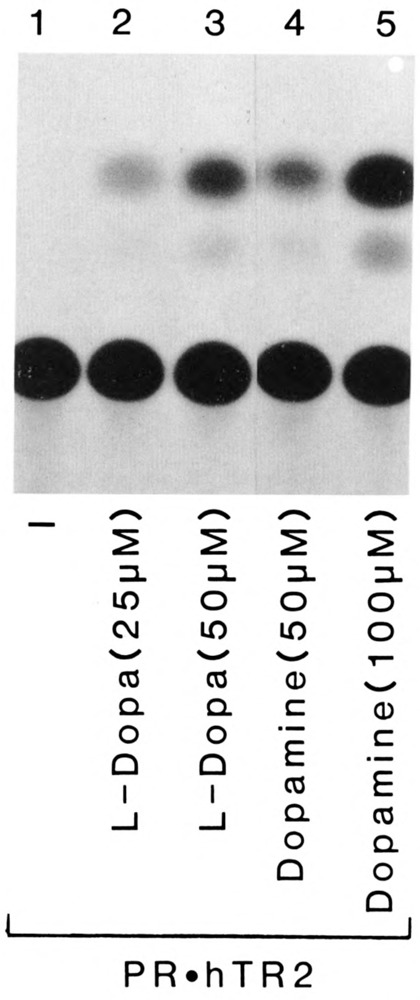
Dopamine and L-dopa induce PR.hTR2 dependent transcription of PREtkCAT. PR.hTR2 was co-transfected with PREtkCAT in CV-1 cells and cultured as described in Figure 2. Transfected cells were either untreated (lane 1) or treated with L-dopa (lanes 2 and 3) or dopamine (lanes 4 and 5) at concentrations indicated.
To confirm that dopamine mediates its action through the putative ligand-binding domain of hTR2, two progesterone gross deletion mutant receptors were examined for their ability to be activated by dopamine (Fig. 5). The first progesterone mutant receptor, C1H, described elsewhere (Carson et al., 1987), contains only the N-terminus and DNA-binding domains of cPR that were used to construct part of the PR.hTR2 chimera. The second cPR deletion mutant C1C2 (Mak et al., 1989) contains only the DNA and hormone binding domains of cPR. Analysis of dopamine effects on the activity of these mutants confirmed that the N-terminal and DNA-binding domains of cPR are unresponsive to dopamine (Panel A). The dopamine-dependent activation of C1C2 observed in these studies (Panel B) is consistent with our previous findings demonstrating that mutation of a serine residue in the carboxy terminal region of cPR results in selective loss of the ability of dopamine to activate this receptor. More importantly, in the context of these studies, the data demonstrates that PR.hTR2 is activated by the catecholamine neurotransmitter dopamine, through the putative ligand-binding domain of hTR2. It should be noted that dopamine activation of hTR2 does not exclude the possibility of modulation of hTR2 transactivation by a ligand which has yet to be identified.
Figure 5.
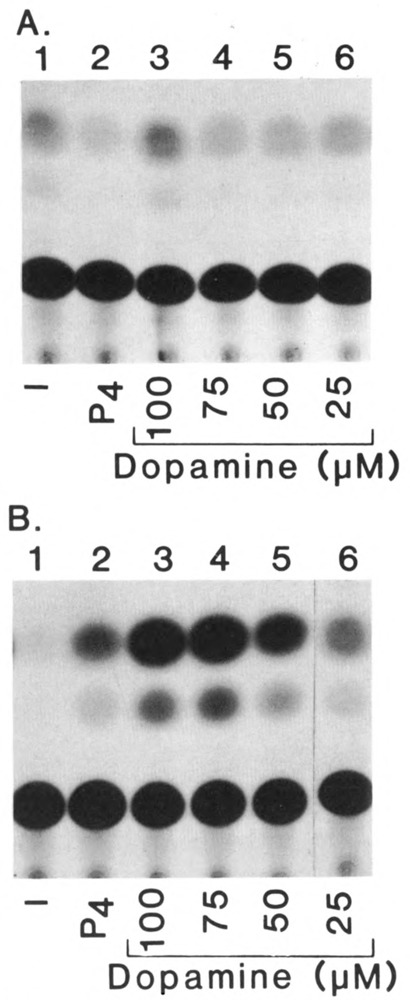
Dopamine activation of cPR mutants. CV-1 cells were cotransfected as described in Figure 2, with the receptor plasmid PREtkCAT and the expression plas mid p91023(B) containing the cPRB gross deletion mutants C1H and C1C2. The results of the transactivation studies on C1H and C1C2 are shown in A and B respectively. For both receptor mutants, transfected cells were untreated (lane 1) or treated with progesterone (P4) (10−7M) (lane 2) or dopamine (lanes 3–6) as described in Figure 4.
Discussion
The activation of the orphan receptors described in these studies supports the emergence of at least three subclasses within the SR/TR superfamily based on selective modes of activation. The three subclasses include receptors that are constitutively activated in that they do not require the addition of ligand or chemical signals to activate them, receptors that are activated by signal transduction pathways through cell membrane receptors, and receptors that are activated by a classical ligand-binding event.
Constitutive activation of a member of the SR/TR superfamily was first reported for the orphan receptor Nur77, which is a growth factor-inducible immediate early gene. It was hypothesized that because Nur77 is synthesized transiently upon stimulation, it may possess a unique transactivation function. Our studies show that this activity can be exhibited by other orphan receptors, such as hERRl and hERR2, for which membership to the immediate early gene family has yet to be established. Hence, a subgroup exists within the SR/TR superfamily that activates gene transcription through an alternative pathway to that used by the previously characterized receptors. Although hypothetical, we propose two explanations for this divergent transactivation mechanism. First, the results do not disprove the concept that these orphan receptors, like the founding members of the SR/TR superfamily, are activated by a ligand via their putative ligand-binding domain. In this case, the intracellular ligand could mediate its action through one of two pathways, termed either autocrine or intracrine. In the autocrine pathway, we propose that the ligand is synthesized in target cells, exits these cells, and then reenters to interact with its intracellular receptor. Using the hypothetical intracrine pathway, the ligand is predicted to have a permanent intracellular existence whereby the ligand is synthesized and acts within the cell without exit and reentry. Both proposed gene regulatory pathways have been reviewed recently (O’Malley et al., 1989). If they exist, we would guess that many of these putative ligands would be ubiquitous hydrophobic molecules, nutritionally or metabolically derived and involved in feedback regulation of intracellular anabolic or catabolic biochemical pathways in both mammalian and yeast cells.
Our second model suggests that hERRl and hERR2 do not require a classical ligand for activation but mediate their actions through ligand-independent routes which maintain the receptors in the constitutively active state. How this constitutive activity is controlled is a matter of speculation. It is quite possible that, like Nur77, the activities of hERRl and hERR2 are modulated at the level of transcription of their gene products by other transacting factors which are regulated in turn by external environmental cues. Alternatively, the constitutive activity might be modulated through their putative ligand binding domains by a “repressor” ligand, in the absence of which these transcription factors remain active.
The second activation subclass exemplified by hTR2 includes COUPTF, PR, and many other steroid receptors (Power et al., 1991a,b) that can be modulated by a signal transduction pathway initiated at the cell membrane- in this case, by dopamine. Neither hTR2 nor the other steroid receptors tested bind directly to [3H]-L-dopa or [3H] dopamine (data not shown). We have previously reported that the dopamine effect is mediated through the dopamine D1 subtype membrane receptor (Power et al., 1991b). In the case of one member of this class (cPR), for which mutants are available, we have identified a serine in the carboxy terminus which is required for activation by dopamine, suggesting the end point of this activation mechanism is receptor phosphorylation. The activation pathway of hTR2 and other receptors by dopamine could involve a preferential intracellular phosphorylation route, since β-adrenergic agonists shown to be active in CV-1 cells do not mimic this effect. Since members of this subclass include established ligand-inducible receptors, it is quite possible that a ligand may also exist for hTR2. However, our studies have shown that unlike the steroid receptors, hTR2 was not activated by a range of both natural and synthetic steroids, isoprenoids, prostaglandins, tri-iodothyronine (T3), thyroxine (T4), or fat soluble vitamins, or by lipid and water soluble tissue extracts. The failure to activate hTR2 by an extensive ligand screen leaves open the question of whether a ligand exists for this orphan receptor.
Until recently, the third mode of activation has been the prevailing concept on how members of the SR/TR superfamily exert their effect on gene activity. Implicit in this concept is that the receptor binds its cognate ligand with high affinity and alters gene expression as a result of this directligand interaction. In view of these recent studies, the above concept of gene regulation by this superfamily is clearly an oversimplification. The data reported herein do not invalidate this concept but help to extend it by suggesting alternative mechanisms by which this superfamily of transcription factors regulates gene activity.
Acknowledgments
We thank Deanne Gallup for technical assistance and Lisa Gamble for typing the manuscript.
This research was supported in part by NIH grant RR06499.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Arriza J. L., Weinberger C., Cerelli G., Glasser T. M., Handelin B. L., Housman D. E., and Evans R. M. (1987), Science 237, 268–275. [DOI] [PubMed] [Google Scholar]
- Butt T. R., Khan M. I., Marsh J., Ecker D. J., and Crooke S. T. (1988), J Biol Chem 263, 16364–16371. [PubMed] [Google Scholar]
- Carson M. A., Tsai M.-J., Conneely O. M., Maxwell B. L., Clark J. H., Dobson A. D. W., Elbrecht A., Toft D. O., Schrader W. T., and O’Malley B. W. (1987), Mol Endocrinol 1, 791–801. [DOI] [PubMed] [Google Scholar]
- Carson M. A., Lee A. T., Dobson A. D. W., Conneely O. M., Schrader W. T., and O’Malley B. W. (1990), J Steroid Biochem 34, 1–9. [DOI] [PubMed] [Google Scholar]
- Chaney W. G., Howard D. R., Pollard J. W., Sallustio S., and Stanley P. (1986), Somat Cell Mol Genet 12, 237–244. [DOI] [PubMed] [Google Scholar]
- Chang C., Kokontis J., and Liao S. (1988a), Science 240, 324–326. [DOI] [PubMed] [Google Scholar]
- Chang C. and Kokontis J. (1988b), Biochem Biophys Res Commun 155, 971–977. [DOI] [PubMed] [Google Scholar]
- Conneely O. M., Sullivan W. P., Toft D. O., Birnbaumer M., Cook R. G., Maxwell B. L., Zarucki-Schultz T., Greene G. L., Schrader W. T., and O’Malley B. W. (1986), Science 233, 767–770. [DOI] [PubMed] [Google Scholar]
- Conneely O. M., Kettelberger D. M., Tsai M.-J., Schrader W. T., and O’Malley B. W. (1989), J Biol Chem 264, 14062–14064. [PubMed] [Google Scholar]
- Davis I. J., Hazel T. G., and Lau L. F. (1991), Mol Endocrinol 5, 854–859. [DOI] [PubMed] [Google Scholar]
- Denner L. A., Weigel N. L., Maxwell B. L., Schrader W. T., and O’Malley B. W. (1990), Science 250, 1740–1743. [DOI] [PubMed] [Google Scholar]
- Dobson A. D. W., Conneely O. M., Beattie W., Maxwell B. L., Mak P., Tsai M.-J., Schrader W. T., and O’Malley B. W. (1989), J Biol Chem 264, 4207–4211. [PubMed] [Google Scholar]
- Evans R. M. (1988), Science 240, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawell S. E., Lees J. A., White R., and Parker M. G. (1990), Cell 60, 953–962. [DOI] [PubMed] [Google Scholar]
- Giguere V., Ong E. S., Sequi P., and Evans R. M. (1987), Nature 330, 624–629. [DOI] [PubMed] [Google Scholar]
- Giguere V., Yang N., Sequi P., and Evans R. M. (1988), Nature 331, 91–94. [DOI] [PubMed] [Google Scholar]
- Green S., Walter P., Kemar V., Krust A., Bornet J. M., Argos P., and Chambon P. (1986), Nature 320, 134–139. [DOI] [PubMed] [Google Scholar]
- Green S. and Chambon P. (1987), Nature 325, 75–78. [DOI] [PubMed] [Google Scholar]
- Hazel T. G., Nathans D., and Lau L. F. (1988), Proc Natl Acad Sci USA 85, 8444–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman R. A., Mangelsdorf D. J., Dyck J. A., Stein R. B., Eichele G., Evans R. M., and Thaller C. (1992), Cell 68, 397–406. [DOI] [PubMed] [Google Scholar]
- Hollenberg S. M., Weinberger C., Ong E. S., Cerelli G., Oro A. E., Leb R., Thompson E. B., Rosenfeld M. G., and Evans R. M. (1985), Nature 318, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Fukuda Y., Murata K., and Kimura A. (1983), J Bacteriol 153, 163–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzen H-M., Strahle U., Gloss B., Stewart F., Schmid W, Boshart M., Miksicek R., and Schutz G. (1987), Cell 49, 29–38. [DOI] [PubMed] [Google Scholar]
- Jeltsch J. M., Krozowski Z., Quirin-Stricker C., Gronemyer H., Simpson R.J., Garnier J. M., Krust A., Jacob F., and Chambon P. (1986), Proc Natl Acad Sci USA 83, 5424–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Green S., Stack S., Berry M., Jin J.-R., and Chambon P. (1987), Cell 51, 941–951. [DOI] [PubMed] [Google Scholar]
- Levin A. A., Sturzenbecker L. J., Kazmer S., Bosakowski T., Huselton C., Allenby G., Speck J., Kratzeisen C. I., Rosenberger M., Lovey A., and Grippo J. F. (1992), Nature 355, 359–361. [DOI] [PubMed] [Google Scholar]
- Lubahn D. B., Joseph D. R., Sullivan P. M., Willard H. F., French F. S., and Wilson E. M. (1988), Science 240, 327–330. [DOI] [PubMed] [Google Scholar]
- Luckow B. and Schutz G. (1987), Nucl Acid Res 15, 5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader S., Kumar V., de Verneuil H., and Chambon P. (1989), Nature 338, 271–274. [DOI] [PubMed] [Google Scholar]
- Mak P., McDonnell D. P., Weigel N. L., Schrader W. T., and O’Malley B. W. (1989), J Biol Chem 264, 21613–21618. [PubMed] [Google Scholar]
- McDonnell D. P., Mangelsdorf D. J., Pike J. W., Haussler M. R., and O’Malley B. W. (1987), Science 235, 1214–1217. [DOI] [PubMed] [Google Scholar]
- Miller J. M. (1972), in Experiments in Molecular Genetics (Miller J. M., ed.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 352–355. [Google Scholar]
- O’Malley B. W. (1989), Endocrinology 12, 1119–1120. [DOI] [PubMed] [Google Scholar]
- O’Malley B. W. (1990), Mol Endocrinol 4, 363–369. [DOI] [PubMed] [Google Scholar]
- Petkovich M., Brand N. J., Krust A., and Chambon P. (1987), Nature 330, 444–450. [DOI] [PubMed] [Google Scholar]
- Pham T. A., Elliston J. F., Nawaz Z., McDonnell D. P., Tsai M.-J., and O’Malley B. W. (1991), Proc Natl Acad Sci USA 88, 3125–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard D., Salser S. J., and Yamamoto K. R. (1988), Cell 54, 1073–1080. [DOI] [PubMed] [Google Scholar]
- Power R. F., Lydon J. P., Conneely O. M., and O’Malley B. W. (1991a), Science 252, 1546–1548. [DOI] [PubMed] [Google Scholar]
- Power R. F., Mani S. K., Codina J., Conneely O. M., and O’Malley B. W. (1991b), Science 254, 1636–1639. [DOI] [PubMed] [Google Scholar]
- Saiki R. K., Gelfand D. H., Stoffel S., Scharf S. J., Higuchi R., Horn G. T., Mullis K. B., and Erlich H. A. (1988), Science 239, 487–491. [DOI] [PubMed] [Google Scholar]
- Sap J., Munoz A., Damm K., Goldberg Y., Ghysadael J., Leutz A., Beug H., and Vennstrom B. (1986), Nature 324, 641–646. [DOI] [PubMed] [Google Scholar]
- Strahle U., Klock G., and Schutz G. (1987), Proc Natl Acad Sci USA 84, 7871–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan W. P., Beito T. G., Proper J., Krco C. J., and Toft D. O. (1986), Endocrinology 119, 1549–1557. [DOI] [PubMed] [Google Scholar]
- Tasset D, Tora L., Fromental C., Scheer E., and Chambon P. (1990), Cell 62, 1177–1187. [DOI] [PubMed] [Google Scholar]
- Tora L., Gronemyer H., Turcotte B., Gaub M-P., and Chambon P. (1988), Nature 333, 185–188. [DOI] [PubMed] [Google Scholar]
- Tsai S. Y., Carlstedt-Duke J., Weigel N. L., Dahlman K., Gustafsson J., Tsai M.-J., and O’Malley B. W. (1988), Cell 55, 361–369. [DOI] [PubMed] [Google Scholar]
- Tsai S. Y., Tsai M.-J., and O’Malley B. W. (1991), in Nuclear Hormone Receptors, Molecular Mechanisms, Cellular Functions, Clinical Abnormalities (Parker M. G., ed.), Academic Press, New York, pp. 103–124 and references therein. [Google Scholar]
- Umesono K. and Evans R. M. (1989), Cell 57, 1139–1146. [DOI] [PubMed] [Google Scholar]
- Webster N. J. G., Green S., Jin J. R., and Chambon P. (1988), Cell 54, 199–207. [DOI] [PubMed] [Google Scholar]
- Weinberger C., Thompson C. C., Ong E. S., Lebo R., Gruol D. J., and Evans R. M. (1986), Nature 324, 641–646. [DOI] [PubMed] [Google Scholar]
- Wong G. G., Witek J. S., Temple P. A., Wilkens K. M., Leary A. C., Luxenberg D. P., Jones S. S., Brown E. L., Kay R. M., Orr E. C., Shoemaker C., Golde D. W., Kaufmann R. J., Hewick R. M., Wang E. A., and Clark S. C. (1985), Science 228, 810–815. [DOI] [PubMed] [Google Scholar]