

Abstract
Retinoic acid-induced differentiation of mouse P19 teratocarcinoma cells is accompanied by alterations in the level of E2F transcription factor. P19 stem cells contain free, uncomplexed E2F and an E2F complex termed E2F/stem. This stem cell complex is a heterotrimeric protein aggregate consisting of E2F transcription factor, E2F-binding protein (E2F/bpl), and cyclin A. Retinoic acid treatment converts P19 stem cells into differentiated neurons, glial cells, and fibroblasts. The presented experiments clearly show that the level of uncomplexed E2F gradually decreases upon differentiation, and fully differentiated cells do not contain free E2F. In addition, the stem cell-specific E2F aggregate is converted into a smaller complex, termed E2F/diff. This smaller complex, which is specific for differentiated cells, does not contain cyclin A and consists of E2F transcription factor associated with E2F/bpl. Finally, the role of E2F complexes in the cessation of cell proliferation, which accompanies P19 cell differentiation, is discussed.
Embryonal carcinoma (EC) cells, which are cultured lines of the pluripotent stem cells of teratocarcinomas, provide an attractive model system for investigating mechanisms controlling mammalian differentiation and development (Martin, 1980; Silver et al., 1983). Incubation of EC cells with retinoic acid causes their differentiation into a wide spectrum of distinct tissue types. The formation of differentiated cells is often coupled with cessation of cell growth, and retinoic acid treatment converts rapidly proliferating EC stem cells into post-mitotic, differentiated cells. The link between acquisition of the differentiated phenotype and growth arrest is only poorly understood. Retinoic acid-induced differentiation of embryonal carcinoma cells is accompanied by numerous changes in gene expression. In order to elucidate the mechanism of differentiation and coupled growth arrest, it is therefore important to investigate the molecular details of alterations in the gene expression pattern. The recent identification of retinoic acid receptors has facilitated studies characterizing the initial steps of retinoic acid-induced differentiation (Benbrook et al., 1988; Brand et al., 1988; Giguere et al., 1987; Krust et al., 1989; Petkovich et al., 1987). By analogy to structurally related steroid and thyroid hormone receptors, retinoic acid receptors, upon binding to retinoic acid, function as dimeric transcription factors for a specific set of genes (Evans, 1988; Wahli and Martinez, 1991). Some genes respond rapidly to the action of retinoic acid, indicating that they are direct targets for retinoic acid receptors (LaRosa and Gudas, 1988; Murphy et al, 1988). This has been corroborated by the identification of retinoic acid response elements in a number of different promoters (deThe et al., 1990; Umesono et al., 1991; Vasios et al., 1991). It has been suggested that at least some of these primary response (early) genes regulate the expression of secondary (late) genes, implying that a hierarchy of steps is involved in embryonal carcinoma cell differentiation (Chiocca et al., 1988). The result is the differentiated phenotype, with genes being activated and repressed during the process (Croce et al., 1981; LaRosa and Gudas, 1988; Rickies et al., 1989; Vogt et al., 1988). Most changes in gene expression that take place upon EC cell differentiation occur at the level of transcriptional initiation and are mediated by promoter-specific transcription factors (Darrow et al., 1990; Harada et al., 1990; Lüscher et al., 1989; Yang-Yen et al., 1990). In order to identify the steps that lead to growth arrest and differentiation, it is therefore crucial to study the mechanism of transcription factor fluctuation.
A well-documented example of transcription factor regulation upon retinoic acid-induced differentiation of EC cells is that of the E2F activity. E2F is downregulated during F9 embryonal carcinoma cell differentiation, and it has been suggested that a cellular ElA-like activity is involved in the regulation process (LaThangue and Rigby, 1987; Reichel et al., 1987). The cellular E2F transcription factor was originally identified in HeLa cells and is essential for the adeno-virus ElA-mediated stimulation of the early adenovirus E2 gene (Kovesdi et al., 1986). In addition, E2F is also important for the transcriptional induction of several cellular genes (Hiebert et al., 1991; Mudryj et al., 1990). The DNA-binding activity of E2F is increased upon adenovirus infection, and two viral gene products, E1A and E4, are involved in this process (Babiss, 1989; Kovesdi et al., 1986; Reichel et al., 1989). As is evident from the above, E2F is controlled by viral as well as cellular mechanisms.
We have continued our studies on the cellular control of E2F with the help of P19 embryonal carcinoma cells. In contrast to F9 cells, initiation of P19 cell differentiation by retinoic acid leads to the emergence of neurons, glial cells, and fibroblasts (Jones-Villeneuve et al., 1982). According to our experiments, E2F is able to form protein aggregates with two cellular activities, cyclin A and a peptide–until now uncharacterized-which we term E2F-binding protein. Evidence is presented demonstrating that the E2F-containing complexes are affected by retinoic acid treatment and fluctuate upon P19 cell differentiation.
Materials and methods
Cell culture
P19 embryonal carcinoma cells (ATCC # CRL 1825) were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) + 7.5% ultra calf serum/2.5% fetal calf serum (Inovar). Differentiation of P19 cells with all-trans retinoic acid (Sigma) was initiated as described elsewhere (Rudnicki and McBurney, 1987).
Extract preparation
P19 cells were removed from tissue culture plates by scraping. Subsequently, the cells were washed with PBS (8 mM Na2HPCO4, 1.5 mM KH2PO4, 135 mM NaCl, 2.5 mM KCl) and pelleted by centrifugation (1,000 × g, 4°C, 5 minutes, Beckman AccuSpin table-top centrifuge). The cell pellet was lysed by the addition of an equal volume of lysis buffer (20 mM Tris [7.5], 2 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT [dithiothreitol], 0.5 mM PMSF [phenylmethanesulfo-nyl fluoride]). The chromatin fraction was removed by ultracentrifugation (350,000 × g,4°C, 60 minutes, SW55 Rotor), and the supernatant was dialyzed overnight at 4°C against an excess of dialysis buffer (20 mM Tris [7.5], 50 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 0.5 mM PMSF). Insoluble materials were removed by centrifugation (1,000 × g4°C, 5 minutes, Beckman AccuSpin table-top centrifuge), and the dialyzed extract was stored in aliquots at −70°C.
Glycerol gradients
P19 cell extract (3 ml of a 5 mg/ml concentration) was layered onto 35 ml of 10–30% glycerol gradients (20 mM Tris [7.5], 100 mM NaCl, 1 mM EDTA, 10-30% glycerol, 1 mM DTT, 0.5 mM PMSF). Gradients were centrifuged in an SW28 rotor at 72,000 × g for 85 hours (4°C). At the end of the run, tubes were punctured, and 1.3 ml fractions were collected from the bottom. Individual fractions were analyzed by gel shift as described below.
Gel shift assay and oligonucleotides
Binding of E2F to DNA oligonucleotides was initiated by mixing the following components: 1 ng of kinased double-stranded oligonucleotide, binding buffer (20 mM Tris [7.5], 5% glycerol, 40 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 1 mM EDTA), 1 μg of sonicated salmon sperm DNA, and 10–30 μg of extract. The total reaction mixture was 30 μl. After 30 minutes at room temperature, the reaction mixture was loaded onto a 4% polyacrylamide gel (acrylamide/bis 29:1). Electrophoresis was performed for 60–90 minutes at 150V (room temperature). Subsequently, the gel was dried and subjected to autoradiography. The double-stranded DNA oligonucleotides employed are shown in Table 1.
Table 1.
Gel shift oligonucleotides
| E2F, single site |
| 5′ACTAGTTTCGCGCCCTTTCT3′ |
| 3′TGATCAAAGCGCGGGAAAGA5′ |
| E2F, double site |
| 5′GTTTCGCGCCCTTTCTCAAATTTAAGCGCGAAAA3′ |
| 3′CAAAGCGCGGGAAAGAGTTTAAATTCGCGCTTTT5′ |
| E2F, mutated |
| 5′CTAGATTTCGAGC3′ |
| 3′TAAAGCTCGCTAG5′ |
| ATF |
| 5′GCTGGAGATGACGTAGTTTTC3′ |
| 3′CGACCTCTACTGCATCAAAAG5′ |
Antiserum
Preparation of cyclin A antiserum has been detailed elsewhere (Pines and Hunter, 1990).
Plasmids
A Hind III/Pst I DNA fragment of a cDNA encoding wild-type sequences of the 13S gene product of E1A was cloned into the Hind III/Pst I sites of pGEMl (Promega). For in vitro transcription, the plasmid was cut with EcoR I and transcribed in the presence of SP6 RNA polymerase. A cDNA for human cyclin A was cloned into the EcoR I site of pGEM 4Z (Promega). After linearizing with Hind III, cyclin A RNA was synthesized in the presence of SP6 RNA polymerase.
In vitro transcription and translation
Plasmids containing El A and cyclin A sequences were linearized, and capped RNA was synthesized by combining the following: 5 μg of linearized plasmid, 40 mM Tris (8.0), 6 mM MgCl2, 5 mM NaCl, 10 mM DTT, 0.1 mg/ml BSA (bovine serum albumin), 80 U RNAsin (Promega), 0.5 mM ATP, 0.5 mM CTP, 0.5 mM UTP, 0.05 mM GTP, 0.5 mM GpppG, 40 U SP6 or T7 RNA polymerase. After 60 minutes at 37°C, the RNA was purified by phenol extraction and precipitated by ethanol. Approximately 2 μg of in vitro transcribed RNA was translated into protein with the help of reticulocyte lysates, following the protocol of the manufacturer (Promega Biotechnology).
Results
Identification of uncomplexed E2F in P19 cells
In order to elucidate the mechanism that controls regulation of E2F during differentiation, whole cell extracts were prepared from mouse P19 stem cells and differentiated P19 cells. E2F protein was detected by gel retardation assay using a DNA oligonucleotide with a single E2F binding site (cf. Materials and Methods). P19 stem cell extract gave rise to two shifted bands (E2F and E2F/stem; Fig. 1, lane 8). In contrast, employment of differentiated P19 cell extract led to a single band (Fig. 1, lane 5) that migrated between the two stem cell bands. This band was provisionally termed E2F/diff (see Discussion, last section). Lanes 3 and 5 were slightly overloaded, and faint E2F bands, which are present in extremely low amounts in differentiated cells, were therefore detected. However, we would like to emphasize that comparisons between equal amounts of stem and differentiated cell extract clearly demonstrate that the level of the E2F band is substantially higher in stem cells (see also Fig. 8, lanes 1 and 5). For comparison, the DNA binding activity of ATF transcription factor, which interacts with an oligonucleotide derived from the −80/−70 region of the adenovirus E2 promoter, was not altered during P19 cell differentiation (Fig. 1, lanes 1 and 2). All three E2F bands were eliminated by competition with an excess of unlabeled E2F oligonucleotide (Fig. 1, lanes 7 and 4). On the other hand, addition of an excess of unlabeled mutated E2F oligonucleotide, which does not interact with E2F protein, had no effect (Fig. 1, lanes 6 and 3). These competition studies suggest that the DNA binding protein E2F is part of all three observed bands.
Figure 1.
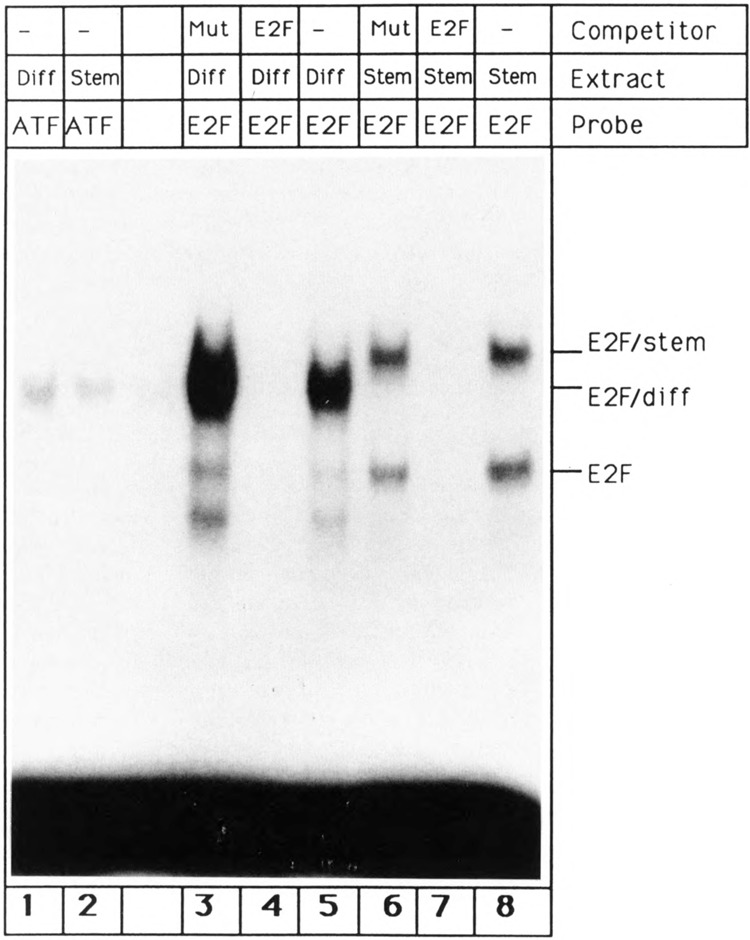
Regulation of E2Fupon differentiation. Gel shift reactions were performed with a single-site E2F oligonucleotide (lanes 3–8) and an oligonucleotide containing an ATF recognition site (lanes 1,2). The oligonucleotides were incubated with P19 stem cell extracts (lanes 2 and 6–8) or extracts prepared from P19 cells 9 days after retinoic acid-induced differentiation (lanes 1 and 3–5). 30 μg of extract were used for lanes 1 and 2 and 6–8. Lanes 3–5 were slightly overloaded and contain more that 30 μg of extract. Competitions were done with a 100-fold excess of unlabeled single site E2F oligonucleotide (lanes 4,7) or a 100-fold excess of unlabeled mutated E2F oligonucleotide (lanes 3, 6).
Figure 8.
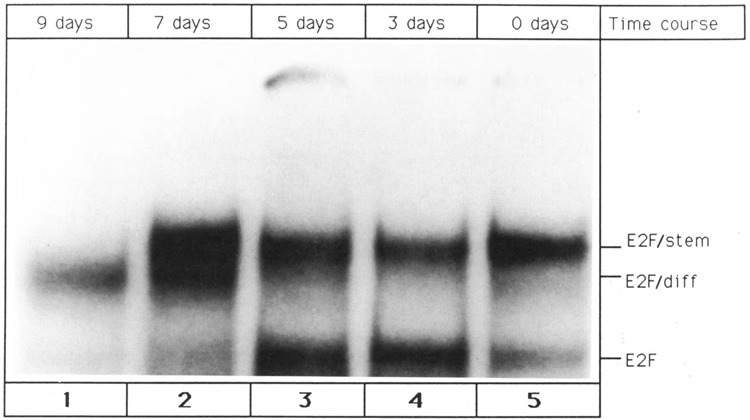
Temporal pattern of E2F fluctuation upon P19 cell differentiation. Differentiation of P19 stem cells was triggered by the addition of retinoic acid, as described in Materials and Methods. Whole cell extracts were prepared 0 days (lane 5), 3 days (lane 4), 5 days (lane 3), 7 days (lane 2), and 9 days (lane 1) after elicitation of differentiation. The extracts were used to perform gel shift reactions with single site E2F oligonucleotide. Each reaction contained 30 μg of whole cell extract. The positions of the three E2F/DNA complexes are indicated at the right.
What is the composition of the three DNA-protein complexes? The fastest migrating band, termed E2F, comigrated with a band that was observed when Hela cell extract was incubated with the single site E2F oligonucleotide (data not shown). It has been reported that this HeLa cell band represents monomeric E2F bound to the DNA oligonucleotide (Bagchi et al., 1990). It is therefore likely that the faster migrating band detected with P19 stem cell extract consists of E2F monomer bound to DNA oligonucleotide. In order to corroborate this conjecture, the molecular weight of the protein component of the E2F band was determined. Glycerol gradient centrifugation of P19 stem cell extract identified the protein part of the E2F band in fraction 20 (Fig. 2a). Comparison with the positions of protein markers suggested a molecular weight of ~50 kDa. This agrees with the molecular weight of murine E2F (P19 cells are of murine origin) which has been determined to be 54 kDa (Bagchi et al., 1990). Furthermore, another known feature of E2F was utilized to demonstrate that the faster migrating P19 stem cell band contains murine E2F monomer. Several laboratories have reported that E2F transcription factor engages in complex formation with the adenovirus E4-6/7 17 kDa protein (Huang and Hearing, 1989; Marton et al., 1990; Neill et al., 1990). This E2F/E4 aggregate can be detected by gel shift and requires a DNA probe with two adjacent E2F binding sites. In order to assay for E4 binding, protein material that gave rise to the E2F band (Fig. 1, lane 8) was isolated by glycerol gradient centrifugation of P19 stem cell extract. Incubation of this material with a double site E2F oligonucleotide yielded a single band (Fig. 3, lane 2). Preincubation of the fractionated glycerol gradient material with E4-6/7 protein that had been purified from adenovirus-infected cells (generous gift of Pradip Raychaudhuri, University of Illinois-Chicago) resulted in the detection of an additional shifted band (E2F/E4, Fig. 3, lane 1). In contrast, incubation of the E2F oligonucleotide with E4 protein alone did not lead to any complex formation (Fig. 3, lane 3). The results presented in Figure 3 demonstrate that E4-6/7 is able to interact with the protein component of the E2F band. Taking into account the DNA binding specificity, the molecular weight, and the interaction with E4-6/7, we conclude that the E2F band is a result of a specific interaction between oligonucleotide and monomeric murine E2F protein.
Figure 2.
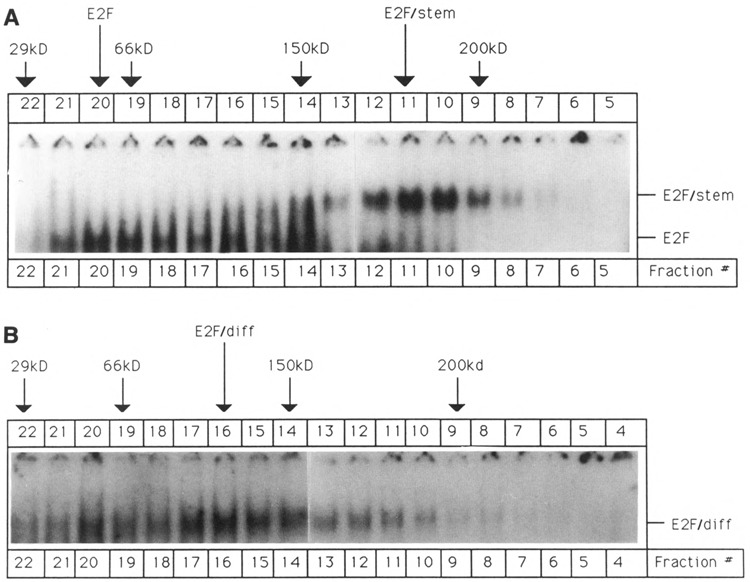
Glycerol gradient fractionation of P19 cell extracts. P19 cell extracts were fractionated by glycerol gradient centrifugation, as described in Materials and Methods. After centrifugation, 10 μl aliquots of individual fractions were assayed by gel shift using the single-site E2F oligonucleotide. The positions of protein markers that were run in parallel are indicated by arrows. The following markers were used: Carbonic anhydrase (29 kDa), Bovine serum albumin (55 kDa), Alcohol dehydrogenase (150 kDa), and β-Amylase (200 kDa). A. Sedimentation analysis of P19 stem cell extract. B. Sedimentation analysis of differentiated P19 cell extract (prepared 9 days after retinoic acid-induced differentiation).
Figure 3.
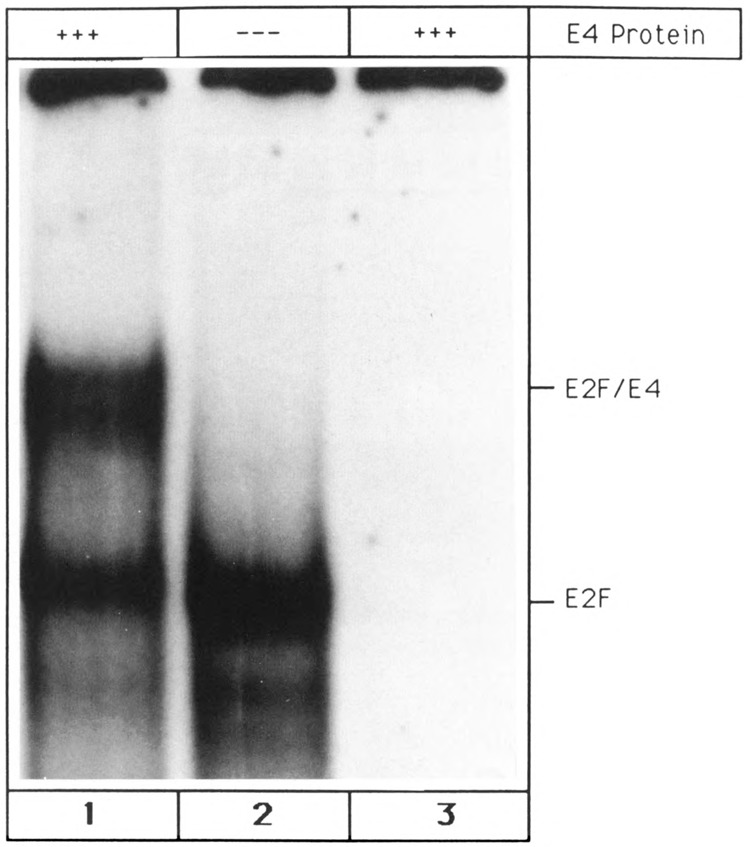
Interaction between E2F and the adenoviral E4-6/7 protein. Interaction between the double site E2F oligonucleotide (cf. Materials and Methods) and E2F-containing glycerol fractions was measured alone (lane 2) and after addition of purified adenovirus E4-6/7 protein (lane 1). Lane 3 shows the interaction between the double site E2F probe and adenovirus E4-6/7 protein alone.
Identification of complexes formed between E2F and cellular activities
Several explanations may account for the presence of the two more slowly migrating bands, termed E2F/stem and E2F/diff (Fig. 1). It is conceivable that these bands are due to the interaction of posttranslationally modified E2F proteins with DNA oligonucleotide. However, glycerol gradient fractionation of P19 cell extracts (Fig. 2, panels A and B) suggest that the protein components of E2F/stem and E2F/diff have an estimated molecular weight of 180 kDa and 120 kDa, respectively. Murine E2F has a molecular weight of 54 kDa, and it is therefore highly unlikely that association of modified E2F forms with oligonucleotides is involved in the formation of the two slower migrating bands. Furthermore, it is possible that the peptide moieties of E2F/stem and E2F/diff are large molecules that exhibit the same DNA-binding specificity as E2F However, there is no reported evidence for such molecules.
The most likely explanation for the two slower migrating aggregates is the interaction between DNA oligonucleotide and E2F complexes. Recently, it has been reported that E2F interacts with cellular activities in mouse L cells (Bagchi et al., 1990), and these complexes can be dissociated by sodium deoxycholate (DOC). Therefore, we investigated the effect of DOC on E2F/stem and E2F/diff. The protein components of E2F/stem and E2F/diff were isolated by glycerol gradient centrifugation, and DNA-binding reactions were initiated (Fig. 4, lanes 1 and 6). Addition of 0.07% sodium deoxycholate to the gel shift reactions eliminated the two more slowly migrating complexes and yielded a band that migrated at the same position as E2F (Fig. 4, lanes 3 and 8). We interpret this result as follows: 0.07% DOC disrupts complexes between E2F and cellular proteins; however, E2F protein remains stably bound to the DNA oligonucleotide, and the E2F band emerges. (Addition of even higher concentrations of DOC prevent binding of E2F to the oligonucleotide, and the E2F band disappears [Fig. 4, lanes 4, 5, 9, 10].)
Figure 4.
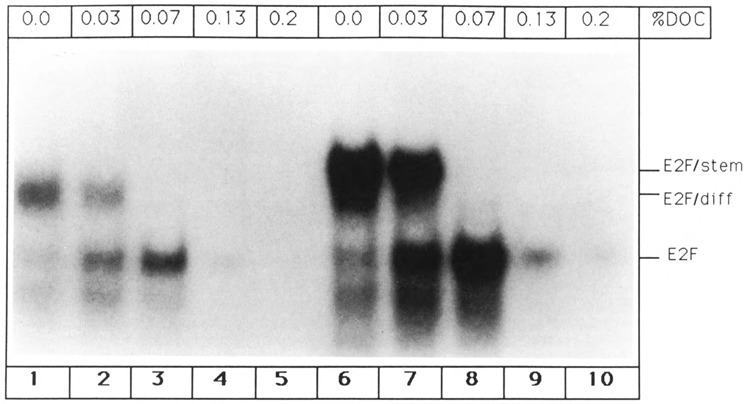
Dissociation of E2F complexes by sodium deoxycholate (DOC). E2F/stem and E2F/diff aggregates were isolated by glycerol gradient fractionation, and gel shift reactions were initiated using the single site E2F oligo-nucleotide and the indicated concentrations of DOC. Lanes 1–5: Dissociation profile of E2F/diff complex. Lanes 6–10: Dissociation profile of E2F/stem complex.
In order to test our interpretation, an additional means for the detection of E2F complexes was employed. It has been demonstrated that the adenovirus E1A protein is able to dissociate aggregates between E2F and L cell proteins (Bagchi et al., 1990; Raychaudhuri et al., 1991). Therefore, it was examined whether the viral protein is capable of disrupting the E2F/stem and E2F/diff aggregates. Indeed, gel shift reactions performed in the presence of in vitro translated 13S E1A protein resulted in the destruction of E2F/stem and E2F/diff and gave rise to the E2F band (Fig. 5, lanes 1 and 4). In contrast, reticulocyte lysate primed with brome mosaic virus (BMV) RNA did not alter the gel shift pattern (Fig. 5, lanes 2 and 5). The DOC studies combined with the above described E1A experiment strongly substantiate our notion that the protein moieties of the E2F/stem and E2F/diff bands are E2F aggregates.
Figure 5.
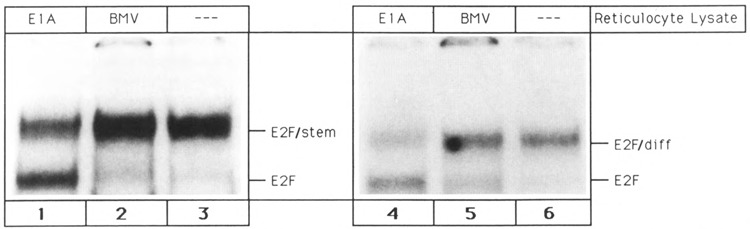
Disruption of E2F aggregates by the adenovirus 13S E1A protein. E2F/diff and E2F/stem complexes, obtained by glycerol gradient fractionation, were incubated with single site E2F oligonucleotide alone or in the presence of reticulocyte lysate primed with brome mosaic virus mRNA or 13S E1A mRNA. Lane 1: E2F/stem complex + 13S E1A mRNA primed lysate. Lane 2: E2F/stem complex + brome mosaic virus mRNA primed lysate. Lane 3: E2F/stem complex alone. Lane 4: E2F/diff complex + 13S E1A mRNA primed lysate. Lane 5: E2F/diff complex + brome mosaic virus mRNA primed lysate. Lane 6: E2F/diff complex alone.
Identity of proteins that interact with E2F
In order to examine the interactions between E2F and its associated activities in the described complexes, it is important to determine the identities of the non-E2F proteins. As shown in Figure 5, E2F/stem as well as E2F/diff can be dissociated by the adenovirus 13S E1A protein. It has been proposed that E1A disrupts protein complexes by directly interacting with components of the sensitive aggregates. Since the adenoviral E1A protein interacts with cyclin A (Pines and Hunter, 1990), this protein is a likely candidate for an E2F-associated activity. In order to test for the presence of cyclin A, all three E2F bands were incubated with polyclonal cyclin A antibodies. As is evident from Figure 6, only E2F/stem reacted with the antibody (lane 7). As a control, preimmune serum did not change the E2F/stem shift pattern (Fig. 6, lane 8). E2F/diff and E2F were not affected by the antibody (Fig. 6, lanes 1 and 4). Interaction between cyclin A antibody and E2F/stem resulted in the destruction of the E2F/stem complex and led to the emergence of a band that comigrated with E2F/diff. This result is clearly reproducible, and treatment of the E2F/stem complex with cyclin A antibodies in four independent experiments always resulted in the loss of E2F/stem and generation of E2F/diff complex. This experiment suggests that removal of cyclin A (by the antibody) from E2F/stem gives rise to E2F/diff. In addition, it indicates that E2F/diff and E2F/stem are related complexes. In other words, E2F/diff may be an integral part of E2F/stem, and addition of cyclin A to E2F/diff may result in the emergence of E2F/stem.
Figure 6.
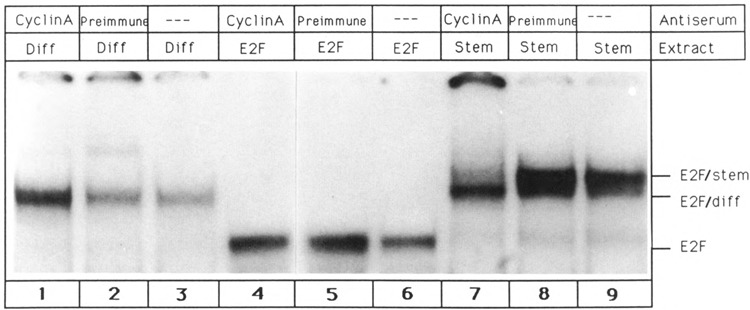
E2F/stem complex contains cyclin A. Single-site E2F oligonucleotide was incubated with glycerol fractions containing E2F/diff complex (lanes 1–3), E2F transcription factor (lanes 4–5), and E2F/stem complex (lanes 7–9). All three DNA-protein complexes were then treated with cyclin A-specific antibodies (lanes 1, 4, 7) or preimmune serum (lanes 2, 5, 8). Lanes 3, 6, and 9 show complex formation in the absence of any serum.
In order to test this hypothesis, experiments depicted in Figure 7 were performed. Cyclin A protein, which had been generated by in vitro transcription/translation of a cyclin A cDNA, was added to gel shift reactions that gave rise to all three E2F complexes. Only E2F/diff reacted with cyclin A, yielding a band that exhibited the same mobility as E2F/stem (Fig. 7, lane 7). This result is consistent with the conclusion drawn from Figure 6. That is, detachment of cyclin A from E2F/stem yields the E2F/diff complex. Furthermore, it is important to note that cyclin A did not bind to E2F (Fig. 7, lane 4). Cyclin A is therefore not an E2F-binding protein. Rather, the activity that associates with the E2F transcription factor in the E2F/diff complex is likely to fulfill this function. Therefore, this activity has been termed E2F-binding protein (E2F/bpl). As expected, cyclin A did not associate with the cyclin A-containing E2F/stem complex (Fig. 7, lane 1). In addition, reticulocyte lysate primed with brome mosaic virus RNA did not alter the E2F/diff shift pattern, strongly suggesting that the observed changes are specific for cyclin A (Fig. 7, lane 8). Given the above, the following picture emerges: using the gel shift assay, three E2F-DNA complexes can be detected in P19 cells. The smallest complex, termed E2F, is due to an interaction between the E2F transcription factor and its cognate binding site. The protein moiety of E2F/diff is the result of an interaction between E2F transcription factor and E2F-binding protein, E2F/bpl. Addition of cyclin A to the E2F/diff complex yields E2F/stem.
Figure 7.
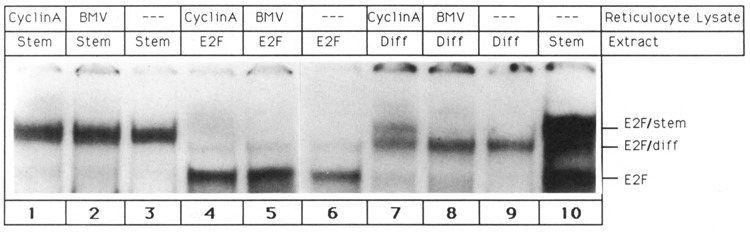
Binding of cyclin A to E2F/diff yields E2F/stem. Gel shift reactions were initiated by mixing single-site E2F oligonucleotide with glycerol fractions containing E2F/stem complex (lanes 1–3), E2F transcription factor (lanes 4–6), and E2F/diff complex (lanes 7–9). Gel shift experiments were performed in the presence of 10 μl of reticulocyte lysate primed with brome mosaic virus mRNA (lanes 2, 5, 8) or 10 μl of reticulocyte lysate primed with cyclin A mRNA (lanes 1, 4, 7). Reactions 3, 6, and 9 were performed in the absence of any reticulocyte lysate. For comparison, reaction 10 shows the shift pattern obtained with P19 stem cell extract.
Temporal pattern of E2F complex regulation during retinoic acid-induced differentiation
P19 stem cells contain free E2F transcription factor (E2F band) and E2F complexed to E2F-binding protein and cyclin A (E2F/stem band). In contrast, virtually all E2F molecules in differentiated P19 cells interact with E2F-binding protein, resulting in the appearance of E2F/diff. In order to study the fluctuation of E2F complexes during differentiation, a time course experiment was performed. P19 cell differentiation was elicited by the addition of retinoic acid, and whole cell extracts were prepared at various times thereafter. Monomeric E2F and E2F-containing complexes were detected by gel shift assay. As can be seen in Figure 8, free E2F protein (E2F band) gradually decreased upon differentiation and could not be detected in fully differentiated cells (9 day point). Between 7 and 9 days after initiation of differentiation, E2F/ stem disappeared and was replaced by E2F/diff (Fig. 8). P19 cell differentiation, elicited by retinoic acid, is therefore characterized by two regulatory events: a decrease in the level of E2F transcription factor and conversion of E2F/stem into E2F/diff. Finally, it is important to note that 7 days after induction of P19 cell differentiation (lane 2), both E2F/stem and E2F/diff can be detected simultaneously. This is consistent with our hypothesis that a precursor/product relationship exists between E2F/stem and E2F/diff.
Discussion
Regulation of E2F complexes upon differentiation
P19 teratocarcinoma cells contain E2F in three distinct forms: free, uncomplexed E2F; E2F/stem; and E2F/diff. The E2F/diff aggregate consists of E2F protein associated with E2F-binding protein (E2F/bpl). Addition of cyclin A transforms E2F/diff into E2F/stem. P19 stem cells contain E2F and E2F/stem. Since cyclin A does not bind to E2F itself, it is likely that it directly interacts with E2F-binding protein. Addition of retinoic acid triggers differentiation, which is accompanied by two regulatory events: (1) a decrease in the level of E2F transcription factor and (2) conversion of E2F/stem into E2F/diff. Figure 9 summarizes the events that occur upon retinoic acid-induced P19 cell differentiation and depicts the structure of all mentioned E2F aggregates.
Figure 9.
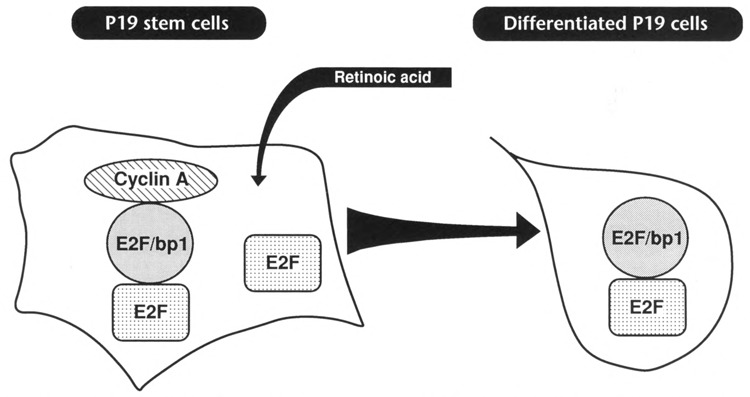
Regulation of E2F complexes upon P19 cell differentiation. Undifferentiated P19 stem cells contain free, uncomplexed E2F transcription factor and E2F/stem complex, which is a heterotrimeric complex consisting of E2F, E2F binding protein (E2F/bpl), and cyclin A. Addition of retinoic acid triggers differentiation, which is accompanied by a dramatic decrease in the level of E2E In addition, the E2F/stem complex is being converted into E2F/diff which consists of E2F associated with E2F-binding protein.
Our experiments suggest that the two regulatory events outlined above are not due to fluctuation of E2F transcription factor itself but involve changes in the levels of E2F/bpl and cyclin A. According to our studies, virtually all E2F molecules in differentiated P19 cells are associated with E2F-binding protein (E2F/bpl). The absence of uncomplexed E2F suggests that E2F-binding protein is present in excess. In contrast, P19 stem cells do contain uncomplexed E2F protein, suggesting that they do not harbor an abundance of E2F-binding protein. (The relatively low amount of E2F-binding protein in stem cells is found as part of the E2F/stem complex.) It is therefore likely that the level of E2F/bpl increases upon P19 cell differentiation. This would explain the loss of the E2F band upon differentiation (all E2F molecules in differentiated cells are complexed to E2F/bpl). On the other hand, it is conceivable that the conversion of E2F/stem into E2F/diff complex is due to a retinoic acid-induced reduction of the level of cyclin A. This is consistent with our antibody studies that clearly show that cyclin A is present in the stem cell-specific E2F/stem complex but absent from the E2F/diff complex. In addition, the absence of E2F/diff complex from stem cells also indicates that cyclin A is abundant in these cells, since cyclin A can interact with E2F/diff to form E2F/stem. Furthermore, the detection of high levels of E2F/diff in differentiated cells, combined with the absence of E2F/stem, lends further support to our hypothesis that cyclin A levels are low in differentiated cells.
Finally, the described results now enable us to examine the link between retinoic acid treatment and fluctuation of E2F aggregates. Our studies suggest that E2F/bpl and cyclin A activities are altered upon retinoic acid treatment. We have initiated experiments to determine the level(s) at which E2F/bpl and cyclin A are regulated. We anticipate that these studies will eventually reveal all the steps of the retinoic acid signal transduction pathway and bridge the gap between retinoic acid action and E2F complex regulation.
Identity of E2F/bp1
According to our experiments, E2F transcription factor interacts with an activity, termed E2F-binding protein (E2F/bpl). What is the identity of this factor? Recently, several groups have reported the presence of the retinoblastoma (Rb) protein in E2F-containing complexes (Bagchi et al., 1991; Chellappan et al., 1991; Chittenden et al., 1991). It is therefore possible that E2F/bpl is identical to the Rb protein. However, using various antibody probes specific for the retinoblastoma protein, we were not able to detect Rb in any of the E2F-containing complexes in P19 cells. Furthermore, glycerol gradient centrifugations suggest a molecular weight of approximately 60 kDa for the E2F binding protein. Since the molecular weight of the Rb protein is 105 kDa, it is unlikely that E2F/bpl and Rb are related. Finally, DNA-binding complexes containing E2F and Rb protein were detected in two leukemia cell lines but not in teratocarcinoma cells, indicating that E2F-Rb interaction is not prevalent in teratocarcinoma cells (Bandara and LaThangue, 1991; Chellappan et al., 1991). Taken together, it appears unlikely that Rb protein interacts with E2F in P19 teratocarcinoma cells.
As outlined earlier, our studies strongly suggest that E2F/bpl not only interacts with E2F transcription factor but also associates with cyclin A. It has been documented that cyclin A can form a complex with p34cdc2 kinase (Draetta et al., 1989; Minshull et al., 1990). However, recent experiments indicate that cyclin A is predominantly associated with the related p33cdk2 kinase rather than p34cdc2 (Pines and Hunter, 1990; Tsai et al., 1991). Therefore, p34cdc2 kinase or the related cdk2 gene product are possible candidates for E2F/bpl. However, antibodies that recognize both candidates (Pines and Hunter, 1990) do not affect E2F complexes (R. Reichel, unpublished results), suggesting that these kinases are not part of the E2F/diff complex and not identical with E2F/bp1. Given the association of E2F/diff with an A-type cyclin, it remains conceivable that E2F/bpl itself is a member of the cyclin family. Since cyclin B could not be detected in E2F-containing complexes (Mudryj et al., 1991), it is possible that E2F/bpl belongs to the Gl/D cyclins, members of which have recently been identified in mammalian cells (Matsushime et al., 1991; Xiong et al., 1991).
Role of E2F-containing complexes in cell growth control
According to our unpublished results, dimethyl sulfoxide (DMSO) treatment of P19 stem cells results in the same changes of the E2F complex pattern as retinoic acid treatment. In contrast to retinoic acid, DMSO converts P19 stem cells into skeletal and cardiac muscle (Edwards et al., 1983), indicating that the E2F complex alterations are not specific for a certain differentiation pathway but are a more general phenomenon. Recently, it has been reported that E2F-containing aggregates are regulated during the cell cycle of mouse L cells in a fashion that is reminiscent of the E2F complex regulation upon differentiation (Mudryj et al., 1991). Specifically, the authors describe an E2F complex, termed E2Fc2, which, according to gel shift mobilities, is equivalent to our E2F/diff complex. The E2Fc2 complex is specific for G1 phase and cannot be detected in unsynchronized cells. Since our E2F/diff complex is specific for differentiated cells that are thought to be arrested in GO, it is conceivable that E2F/diff and E2Fc2 are related and that the appearance of E2F/diff is due to a cell cycle block in G1/GO. Furthermore, this would imply that E2F/diff is already present in undifferentiated cells but was not observed by us because our cells were not synchronized. However, it is presently not clear whether E2F/diff is related to d-specific complexes such as E2Fc2. Indeed, circumstantial evidence suggests that E2F cell cycle regulation is quite distinct from E2F developmental regulation. In any case, the regulation of E2F complexes during the cell cycle point toward an important role of E2F in cell proliferation control. It is therefore equally possible that the alterations of E2F-containing aggregates that occur upon P19 cell differentiation play a role in the induction of cell growth cessation that accompanies the differentiation process. The association of E2F-containing protein aggregates with the known cell growth regulator cyclin A is consistent with our hypothesis. Our experiments indicate that cyclin A is downregulated upon P19 cell differentiation, and it is therefore possible that this downregulation participates in the growth arrest of differentiated cells. This agrees with a previous report demonstrating that cyclin A expression is diminished upon terminal differentiation of neurons (Hayes et al., 1991). How could the interaction of cyclin A with E2F-containing aggregates influence cell growth? It is conceivable that the cyclin A-containing E2F/stem complex, which is present in rapidly growing stem cells, is involved in the transcriptional activation of certain stem cell-specific genes. On the other hand, the cyclin A-less E2F/diff complex may exhibit a low affinity for these genes but is required for the induction of certain genes that participate in cessation of cell growth. This is substantiated by several studies demonstrating that E2F is crucial for the transcriptional activation of various genes that are linked to cell proliferation control (Hiebert et al., 1991; Mudryj et al., 1990). Specifically, it has been demonstrated that one of the known E2F target genes, c-myc, is regulated upon P19 cell differentiation (Thalmeier et al., 1989; St-Arnaud et al., 1988). St-Arnaud et al. showed that c-myc gene expression is increased upon retinoic acid-induced P19 cell differentiation. Furthermore, preliminary results indicate that the transcription rate of another E2F target gene, the adenoviral E2 gene, is also increased upon P19 cell differentiation (R. Reichel, unpublished data). Because transcriptional activation of these two E2F-dependent genes correlates with fluctuation of E2F complexes, it appears likely that different E2F complexes exhibit different transcriptional potentials. Alternatively, it is possible that cyclin A acts as a replication factor and affects cell growth by controlling DNA replication. According to this model, the cyclin A-containing E2F/stem complex, which is present in rapidly proliferating cells, interacts with origins of replication that contain E2F recognition sites. This would place cyclin A into the vicinity of DNA, where it can participate in the initiation of DNA synthesis. In contrast, in post-mitotic differentiated cells cyclin A does not interact with E2F-containing complexes and cannot participate in DNA replication. This attractive hypothesis is corroborated by a recent publication that suggests a role for cyclin A in the onset of DNA replication in mammalian fibroblasts (Girard et al., 1991).
Acknowledgments
We thank Pradip Raychaudhuri for E1A cDNA and purified adenoviral E4 protein. We also thank Jonathan Pines and Tony Hunter for generous gifts of cyclin A antibody and cyclin A cDNA. The technical assistance of Janet Yu during the early phase of the described experiments is acknowledged. This work was supported by a grant from the American Cancer Society, Illinois Division (90–72).
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Babiss L. (1989), J Virol 63, 2709–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi S., Raychaudhuri P., and Nevins J. R. (1990), Cell 62, 659–669. [DOI] [PubMed] [Google Scholar]
- Bagchi S., Weinmann R., and Raychaudhuri P. (1991), Cell 65, 1063–1072. [DOI] [PubMed] [Google Scholar]
- Bandara L. R. and LaThangue N. B. (1991), Nature 351, 494–497. [DOI] [PubMed] [Google Scholar]
- Benbrook D., Lernhardt E., and Pfahl M. (1988), Nature 333, 669–672. [DOI] [PubMed] [Google Scholar]
- Brand N. M., Petkovich M., Krust A., Chambon P., deThe H., Marchio A., Tiollais P., and Dejean A. (1988), Nature 332, 850–853. [DOI] [PubMed] [Google Scholar]
- Chellappan S. P., Hiebert S., Mudryj M., Horowitz J. M., and Nevins J. R. (1991), Cell 65, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Chiocca E. A., Davies P. J. A., and Stein J. P. (1988), J Biol Chem 263, 11584–11589. [PubMed] [Google Scholar]
- Chittenden T., Livingston D. M., and Kaelin W. G. Jr. (1991), Cell 65, 1073–1082. [DOI] [PubMed] [Google Scholar]
- Croce C. M., Linnenbach A., Huebner K., Parnes J. R., Margulies D. H., Appella E., and Seidman J. G. (1981), Proc Natl Acad Sci 78, 5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow A. L., Rickles R. J., Pecorino L. T., and Strickland S. (1990), Mol Cell Biol 10, 5883–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deThe H., del Mar Vivanco-Ruiz M., Tiollais P., Stunnenberg H., and Dejean A. (1990), Nature 343, 177–180. [DOI] [PubMed] [Google Scholar]
- Draetta G., Luca F., Westendorf J., Brizuela L., Ruderman J., and Beach D. (1989), Cell 56, 829–838. [DOI] [PubMed] [Google Scholar]
- Edwards M. K. S., Harris J. F., and McBurney M. W. (1983), Mol Cell Biol 3, 2280–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R. M. (1988), Science 240, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V., Ong E. S., Sequi P., and Evans R. M. (1987), Nature 330, 624–629. [DOI] [PubMed] [Google Scholar]
- Girard F., Strausfeld U., Fernandez A., and Lamb N. J. C. (1991), Cell 67, 1169–1179. [DOI] [PubMed] [Google Scholar]
- Harada H., Willison K., Sakakibara J., Miyamoto M., Fujita T., and Taniguchi T. (1990), Cell 63, 303–312. [DOI] [PubMed] [Google Scholar]
- Hayes T. E., Valtz N. L. M., and McKay R. D. G. (1991), New Biologist 3, 259–269. [PubMed] [Google Scholar]
- Hiebert S. W., Blake M., Azizkhan J., and Nevins J. R. (1991), J Virol 65, 3547–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.-M. and Hearing P. (1989), Genes Dev 3, 1699–1710. [DOI] [PubMed] [Google Scholar]
- Jones-Villeneuve E. M. V., McBurney M. W., Rogers K. A., and Kalnins V. I. (1982), J Cell Biol 94, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovesdi I., Reichel R., and Nevins J. R. (1986), Cell 45, 219–228. [DOI] [PubMed] [Google Scholar]
- Krust A., Kastner P., Petkovich M., Zelent A., and Chambon P. (1989), Proc Natl Acad Sci 86, 5310–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRosa G. J. and Gudas L. J. (1988), Proc Natl Acad Sci 85, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaThangue N. B. and Rigby P. W. J. (1987), Cell 49, 507–513. [DOI] [PubMed] [Google Scholar]
- Lüscher B., Mitchell P. J., Williams T., and Tjian R. (1989), Genes Dev 3, 1507–1517. [DOI] [PubMed] [Google Scholar]
- Martin G. R. (1980), Science 209, 768–776. [DOI] [PubMed] [Google Scholar]
- Marton M. J., Bairn S. B., Ornelles D. A., and Shenk T. (1990), J Virol 64, 2345–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushime H., Roussel M. F., Ashmun R. A., and Sherr C. J. (1991), Cell 65, 701–713. [DOI] [PubMed] [Google Scholar]
- Minshull J., Blow J. J., and Hunt T. (1990), EMBO J 9, 2865–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudryj M., Hiebert S. W., and Nevins J. R. (1990), EMBO J 9, 2179–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudryj M., Devoto S. H., Hiebert S. W., Hunter T., Pines J., and Nevins J. R. (1991), Cell 65, 1243–1253. [DOI] [PubMed] [Google Scholar]
- Murphy S. P., Garbern J., Odenwald W. F., Lazzarini R. A., and Linney E. (1988), Proc Natl Acad Sci 85, 5587–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill S. D., Hemstrom C., Virtanen A., and Nevins J. R. (1990), Proc Natl Acad Sci 87, 2008–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovich M., Brand N. J., Krust A., and Chambon P. (1987), Nature 330, 444–450. [DOI] [PubMed] [Google Scholar]
- Pines J. and Hunter T. (1990), Nature 346, 760–763. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri P., Bagchi S., Devoto S. H., Kraus V. B., Moran E., and Nevins J. R. (1991), Genes Dev 5, 1200–1211. [DOI] [PubMed] [Google Scholar]
- Reichel R., Kovesdi I., and Nevins J. R. (1987), Cell 48, 501–506. [DOI] [PubMed] [Google Scholar]
- Reichel R., Neill S. D., Kovesdi I., Simon M. C., Raychaudhuri P., and Nevins J. R. (1989), J Virol 63, 3643–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickles R. J., Darrow A. L., and Strickland S. (1989), Mol Cell Biol 9, 1691–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki M. A. and McBurney M. W. (1987), in Teratocarcinomas and Embryonic Stem Cells (Robertson E. J., ed.), IRL Press, Oxford/Washington, DC, pp. 19–49. [Google Scholar]
- Silver L. M., Martin G. R., and Strickland S. (1983), Teratocarcinoma Stem Cells, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- St-Arnaud R., Nepveu A., Marcu K. B., and McBurney M. W. (1988), Oncogene 3, 553–559. [PubMed] [Google Scholar]
- Thalmeier K., Synovzik H., Mertz R., Winnacker E.-L., and Lipp M. (1989), Genes Dev 3, 527–536. [DOI] [PubMed] [Google Scholar]
- Tsai L.-H., Harlow E., and Meyerson M. (1991), Nature 353, 174–177. [DOI] [PubMed] [Google Scholar]
- Umesono K., Murakami K. K., Thompson C. C., and Evans R. M. (1991), Cell 65, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasios G., Mader S., Gold J. D., Leid M., Lutz Y., Gaub M.-P., Chambon P., and Gudas L. (1991), EMBO J 10, 1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt T. F., Compton R. S., Scott R. W., and Tilghman S. M. (1988), Nucl Acids Res 16, 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahli W. and Martinez E. (1991), FASEB J 5, 2243–2249. [DOI] [PubMed] [Google Scholar]
- Xiong Y., Connolly T., Futcher B., and Beach D. (1991), Cell 65, 691–699. [DOI] [PubMed] [Google Scholar]
- Yang-Yen H.-F., Chiu R., and Karin M. (1990), New Biol 2, 351–361. [PubMed] [Google Scholar]