

The 5′-flanking regions of protein encoding genes in yeast contain specific sequences which are involved in the regulation of transcription of these genes. These sequences, called upstream activations sites (UASs) or upstream repression sites (URSs), represent specific binding sites for protein factors that can stimulate or inhibit the formation of an active transcription initiation complex. Some of these trans-acting proteins are present in the yeast cell in great abundance, and their respective binding sites are found not only within gene promoter regions, but also at numerous locations on the genome. These trans-acting factors play a role in various nuclear processes, such as transcriptional activation, transcriptional silencing, chromosome segregation during mitosis, initiation of DNA-replication, and regulation of telomere stability. In this review we will present the currently available, most relevant data concerning the abundant, multifunctional DNA-binding proteins ABF1, RAP1, REB1, and CPF1 from yeast, with the major emphasis on the former two. Some characteristics of these proteins are summarized in Table 1.
Table 1.
Characteristics of multifunctional DNA-binding yeast factors.
| Protein | Other designationsa , b | Molecular weightc | Amino acids | Consensus binding site | Putative function |
|---|---|---|---|---|---|
| ABF1 | SBF-B,2 BAF1,3 OBF1,4 SUF,5 TAF,6 GFI,7 TyBF,8 Yprotein9 | 81.7 | 731 | RTCRY YYNNNACG | Transcriptional activation |
| Transcriptional silencing | |||||
| DNA replication | |||||
| RAP110 | SBF-E,11 TUF,12 GRF1,13 TBA14 | 92.5 | 827 | ACACCCATACATTT | Transcriptional activation |
| Transcriptional silencing | |||||
| Telomere control | |||||
| Attachment to nuclear scaffold | |||||
| REB115 | Factor Y,16 GRF2,17 RBP118 | 92.1 | 809 | CCGGGTRR | Transcriptional activation? |
| Nucleosome positioning | |||||
| CPF119 | αΠ×,20 CBP1,21 CBF1,22 GFII23 | 39.4 | 351 | RTCACGTG | Chromosome separation during mitosis |
| Expression MET genes |
Names in bold: identity has been established by nucleotide sequencing.
References: 1 Buchman et al., 1988a; 2 Shore et al., 1987b; 3 Halfter et al., 1989; 4Biswas et al., 1987; 5 Herruer et al., 1989; 6 Hamil etal., 1988; 7 Dorsmanetal., 1988; 8 Goel and Perlman, 1988; 9 Chambers et al., 1990; 10 Brand et al., 1987; 11 Shore etal., 1987b; 12 Huet et al., 1985; 13 Buchman et al., 1988a; 14 Berman et al., 1986; 15 Ju et al., 1990; 16 Fedor et al., 1988; 17 Chasman et al., 1990; 18 Kulkens et al., 1989; 19 Mellor et al., 1991; 20 Bram and Kornberg, 1987; 21 Cai and Davis, 1989; 22 Cai and Davis, 1990; 23 Dorsman et al., 1988.
On the basis of the sequence deduced from the DNA-sequence.
ABF1, RAP1, and REB1 are indispensable for growth, since disruptions of these genes appear to be lethal (Rhode et al., 1989; Shore and Nasmyth, 1987a; Ju et al., 1990). On the other hand, null mutants for CPF1 were shown to be viable. These mutants, however, display chromosome loss and nondisjunction, as well as—remarkably—methionine auxotrophy (Cai and Davis, 1990; Baker and Masison, 1990; Mellor et al., 1990; Mellor et al., 1991).
The abundance of these protein factors in the yeast cell, at least for ABF1 and RAP1, is high: estimates vary from several hundred to a few thousand molecules per nucleus (Buchman et al., 1988a; Sweder et al., 1988; Morrow et al., 1991). Overproduction of ABF1 does not give rise to an abnormal phenotype; in contrast, excess synthesis of RAP1 severely affects growth (Rhode et al., 1989; Francesconi and Eisenberg, 1991; Conrad et al., 1990).
ABF1-binding sites have been found near many ARS elements (domain B), in the promoter regions of many nuclear genes (amongst others, those encoding components of the transcription and translation machinery, glycolytic enzymes, and mitochondrial proteins), as well as in silencer elements of the silent mating type loci (Dorsman et al., 1988; Eisenberg et al., 1988; Goel and Pearlman, 1988; Hamil et al., 1988; Kimmerly et al., 1988; Sweder et al., 1988; Francesconi and Eisenberg, 1989; Halfter et al., 1989b; Herruer et al., 1989; Mahoney and Broach, 1989; Rhode et al., 1989; Biswas et al., 1990; Brindle et al., 1990; Chambers et al., 1990; Della Seta et al., 1990a and b). In agreement with their various locations, ABF1-sites have been demonstrated to serve as cis-acting elements involved in transcriptional activation, transcriptional silencing, and DNA-replication. ABF1 binds to its cognate sequence through a DNA-binding domain encompassing an atypical Zn-finger structure (residues 57–71; Rhode et al., 1989; Diffley and Stillman, 1989), while a second domain might determine the specificity of DNA-binding.
RAP1-binding sites (also designated as RPG-boxes; cf. Leer et al., 1985) occur in the promoter regions of a large number of genes encoding components of the translational and transcriptional machinery, as well as most glycolytic enzymes and several other proteins. RAP1-binding sites are also present in silencer elements of the silent mating-type loci and in telomere repeats (Shore et al., 1987b; Buchman et al., 1988a and b; Mager and Planta, 1991; Moore et al., 1991; Giesman et al., 1991; Kurtz and Shore, 1991; Longtine et al., 1989). In addition, RAP1 has been implicated in the attachment of chromosomal DNA to the nuclear scaffold. A domain extending from amino acids 361 to 596, whose structural motif has not yet been described, is required for binding to the pertinent DNA element (Henry et al., 1990; Hofmann et al., 1989).
REB1-binding sites occur in the promoter regions of several protein-encoding genes, near the promoter and in the enhancer of the rRNA operon and in telomeres (Fedor et al., 1988; Kulkens et al., 1989; Chasman et al., 1990; Ju et al., 1990; Wang et al., 1990). Its actual cellular function is still not clear. It has been suggested that binding of REB1 to the DNA makes that stretch of DNA nucleosome-free, and, in this way, promotes the binding of other transcriptional activators to their respective sites in this region (Chasman et al., 1990).
Finally, CPF1-binding sites have been found in both centromere regions (CDE1) and in several promoter regions. However, so far no evidence has been found that these sites actually function as transcriptional activation sites (Bram and Kornberg, 1987; Cai and Davis, 1989; Mellor et al., 1991; Dorsman et al., 1988; Kraakman et al., 1991; Thomas et al., 1989). CPF1 binds to its recognition sequence as a dimer, probably through a helix-loop-helix motif (Cai and Davis, 1990; Mellor et al., 1990).
The multifunctional nature of these DNA-binding proteins might, on the one hand, enable the yeast cell to control different nuclear processes — most of them, in one way or another, related to cellular growth — with a limited set of regulatory factors. On the other hand, the intriguing question arises as to how the desired specific action of such proteins at a given location is accomplished. It is likely that their actual cellular activity depends upon the context of the respective binding sites, presumably through interaction with additional protein factors (see Fig. 1). Indeed, evidence obtained by both genetic and molecular biological approaches supports the idea that additional proteins are involved. These additional proteins may be other protein factors binding to adjacent nucleotide elements, protein-binding factors, or (structural) nuclear components.
Figure 1.
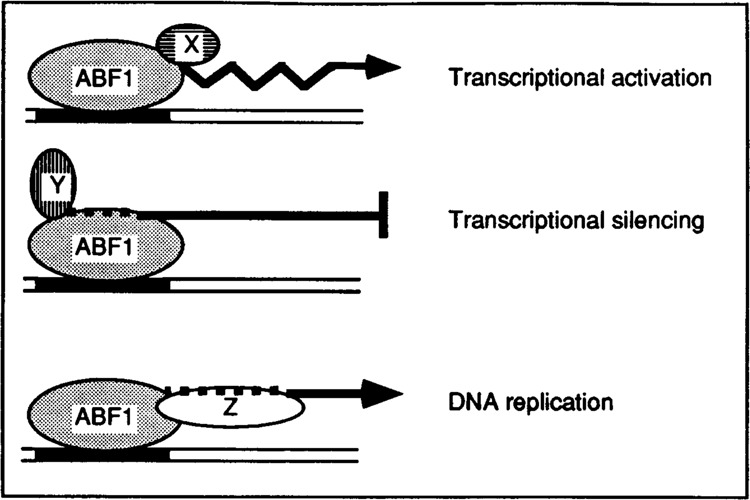
Model representing the different functions of a multifunctional protein, e.g., ABF1. Additional proteins X, Y, and Z, interacting with different putative domains of ABF1, may be other protein factors binding to adjacent nucleotide elements, protein-binding factors, or (structural) nuclear components.
A prime example of a complex molecular interplay of several regulatory proteins is provided by the complicated regulation at the silent mating-type loci. For reviews concerning the mating-type switch in yeast, see Herskowitz (1981) and Spraque et al. (1983). Multifunctional proteins RAP1 and ABF1 both play a part in the complex that keeps the mating-type gene-copies at the HMR and HML locus in a transcriptionally repressed state. The regulatory regions of the silent mating-type genes HML and HMR were found to be located both upstream (E-region) and downstream (I-region) of each gene (Brand et al., 1987; also see Fig. 2). Each of these regions, except HMR-I, is functional as a silencer element on its own (Mahoney and Broach, 1989), implying that transcription of HMR and HML is repressed by one and two independent silencer elements, respectively.
Figure 2.
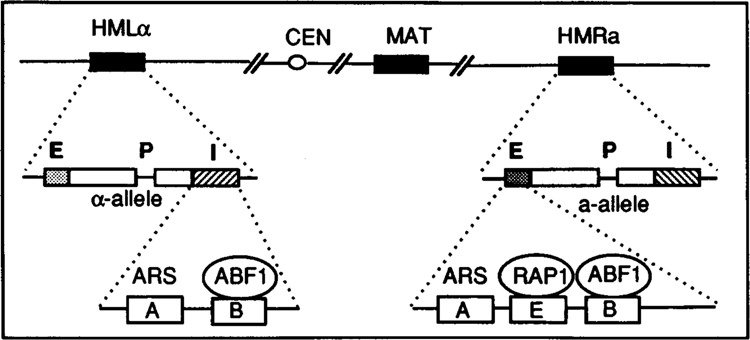
Schematic representation of mating-type elements on Chromosome III (not drawn to scale). E and I (in bold) indicate the silencer regions flanking the silent loci. P represents the promoter in the silent loci.
The best studied silencer is the HMR-E region, in which three functional elements, A, E, and B, have been identified (Brand et al., 1987). The A-element contains an 11 bp ARS-consensus sequence and is able to serve as an origin of DNA-replication when it is cloned into a plasmid. The B-element is the binding site for ABF1, and the E-element harbors a RAP1-binding site (Brand et al., 1987). Any combination of two of these elements possesses silencer activity: elements E + A and E + B are fully functional as a silencer, whereas elements A + B also repress transcription at HMR, but not completely. These results suggest that the E-element, harboring the RAP l-binding site, is necessary for full transcriptional repression (Brand et al., 1987). On the other hand, the HML-I region is composed of only an A and a B element, and therefore lacks a RAP1-binding site. Yet this region is able to repress HML completely in the absence of a functional HML-E region (Mahoney and Broach, 1989). Therefore, in addition to the sequences necessary for ARS-function or for binding of RAP1 or ABF1, these elements are likely to harbor other functional sequences, involved in repression of transcription, which vary among silencer regions (see also Kimmerly et al., 1988).
In addition to the above-mentioned proteins, the gene-products of SIR1, SIR2, SIR3, and SIR4 are also essential for silencer function (Herskowitz, 1981). Since DNA binding of these proteins has not yet been established, they probably interact with the silencer indirectly via protein–protein contacts (Shore and Nasmyth, 1987a; Buchman et al., 1988a). Recently, Sussel and Shore (1991) showed that mutations in the RAP1 protein, which cause a silencer-defective phenotype, can be overcome by overproduction of SIR1 or SIR4. However, in wild-type yeast cells, elevated levels of SIR4 result in a loss of repression at the mating-type locus (Marshall et al., 1987). These results suggest that balanced levels of RAP1 and SIR4 are required for transcriptional repression.
Histone protein H4 is also essential for proper function of the silencer regions (Kayne et al., 1988). Substitution experiments have shown that four adjacent basic amino acid residues in the N-terminal part of the H4 protein — which make direct contact with DNA (Ebralidse et al., 1988) — are involved in repression of the silent mating-type loci (Johnson et al., 1990; Megee et al., 1990; Park and Szostak, 1990). Among the four important residues, only acetylation of lysine causes derepression of HMR and HML (Johnson et al., 1990). Extragenic mutations that suppress the effect of the acetylated lysine of H4 have been isolated. All suppressor mutations were found to be located in the SIR3-gene. However, these sir3-mutations cannot overcome the derepression of the silent mating-type gene caused by complete removal of the N-terminal part of histone H4. It is likely, therefore, that SIR3, by direct or indirect interaction with histone H4, stabilizes nucleosomes at the silencer region, hence preventing the formation of a transcription pre-initiation complex (Johnson et al., 1990). This view is consistent with the observations of Nasmyth (1982), who showed that the accessibility of the DNA at the HMR-E locus for the endonuclease DNAse I is enhanced in sir −-mutants.
In this respect it is relevant to note that RAP1 has been implicated in the association of DNA with the nuclear matrix. Gasser and coworkers (Hofmann et al., 1989) have shown that RAP1 copurifies with nuclear scaffold preparations and is necessary (and probably sufficient) to reconstitute DNA loop-formation in in vitro assays. The E- and I-regions of the HML silent mating-type locus form the bases of a DNA loop that is dependent on the presence of RAP1. Also the promoter region, harboring a RAP1-binding site, can interact with both the E- and I-region at this locus in a RAP1-dependent manner. These loop-formations fit well with the premise that the silencers may function by locking the pertinent DNA region into a chromatin state that renders the DNA inaccessible for components of the transcription machinery (Nasmyth, 1982; Johnson et al., 1990). This model suggests a complex interaction of DNA (silencer, promoter), RAP1, ARS-binding proteins, histone proteins (H4), SIR proteins, and components of the nuclear scaffold. However, though HML-I does not contain a RAP1-binding site, it nevertheless serves as a base for DNA-loop formation (Mahoney and Broach, 1989). Therefore, in this model, the nuclear scaffold interaction with either ARS core binding proteins or ABF1 is also a prerequisite (see below).
Binding sites for the multifunctional proteins RAP1 and ABF1 are also in proximity to each other in the promoters of glycolytic enzyme-genes, e.g., PGK and PYK (Stanway et al., 1989; Chambers et al., 1990). In these promoters, the ABF1-site is located upstream of the RAP1-site. If tested on multicopy plasmids, the ABF1-site is not involved in transcriptional regulation of these genes. Rather, control of the transcriptional activation of PGK and PYK seems to depend primarily on RAP1, with elements consisting of the sequence 5′ CTTCC 3′ acting as auxiliary enhancing elements (Ogden et al., 1988; Chambers et al., 1990). Possibly the actual function of an ABF1-binding site in these promoters can only be revealed in the proper chromosomal context.
In the promoter of the enolase 2- (ENO2-) gene (Cohen et al., 1986; Brindle et al., 1990), two cis-acting elements, UAS1 and UAS2, have been found, which also include binding sites for both RAP1 and ABF1, respectively. In contrast to the situation described above, in this case both proteins seem to be involved in transcriptional activation. Deletion of either the RAP1- or ABF1-binding site does not interfere with glucose-dependent expression of the ENO2-gene (Cohen et al., 1986). In addition to RAP1 and ABF1, a third protein, named GCR1 — found to be involved in the regulation of many genes coding for glycolytic enzymes — is needed for transcription activation of the ENO2-gene (Holland et al., 1987). A non-functional GCR1-gene causes a severe reduction in transcription levels of the ENO2-gene and of other glycolytic enzyme-genes (Santangelo and Tornow, 1990).
Interestingly, removing the ABF1-binding site from the ENO2 promoter allows RAP1 to induce wild-type transcription levels in a gcr1-null mutant, suggesting that ABF1 represses RAP1-induced transcription in a gcr1-background (Holland et al., 1990). This repression may be relieved by the GCRl-protein in wild-type cells. Arguing against this view is the finding that an oligomer containing the region with both the RAP1- and ABF1-binding site is capable of activating transcription from UAS-less ENO1 and ENO2 cassettes in a GCRl-independent manner (Holland et al., 1990; Brindle et al., 1990). Since it has been suggested that RAP1, ABF1, and GCR1 influence the chromatin structure (Buchman et al., 1988a; Kayne et al., 1988; Pavlovic and Hörz, 1988), it is not unlikely that these conflicting results can be explained by a difference in nucleosome positioning between wild-type and mutant ENO2-gene promoters. Very recently, evidence has been presented that GCR1 is a DNA-binding protein whose activity is affected by mutations in the CTTCC-motif (Baker, 1991).
GCR1 is not the only protein factor supposed to be involved in RAP1/ABF1-mediated transcriptional activation. Nishizawa and coworkers (1990) have published data suggesting that the transcriptional-activating function of RAP1 might be mediated via the GAL11-gene product. GAL11 had previously been described to be required for maximal levels of GAL4-activated transcription (Nogi and Fukasawa, 1980). A gal11-null mutant displays growth defects on non-fermentable carbon-sources, as well as in mating and sporulation (Fassler and Winston, 1989; Nishizawa et al., 1990). Northern blot analysis revealed that the mating-type abnormalities were caused by a severely reduced level of MATα-gene expression. The transcription of the MATα-gene is regulated by RAP1. To test whether the reduction of transcription of the MATα-gene in the gal11-mutant was caused by an inability of RAP1 to activate transcription, the transcriptional activity of RAP1 in gal11-null mutant was examined at the PYK locus. Interestingly, Nishizawa and coworkers could suppress the gal11-effect on RAP1-regulated transcription of the PYK-gene by placing the UAS close to the TATA-box. They explain these results by assuming a function for GAL11 in transmitting the activating signal of RAP1 to the general transcription machinery. By placing the UAS in proximity to the TATA-box, RAP1 is probably able to contact TFIID or other components of the transcription initiation complex directly by protein–protein interactions. Relevant to this view are observations of Hoffmann and coworkers (1990), who noticed homologous repetitive stretches of serine, threonine, and proline residues (STP-regions) in GAL11 and TFIID. However, the GAL11-model of modulating RAP1-mediated transcription cannot be a general model applicable to all transcriptional activities of RAP1, since the transcription of several essential genes, e.g., ribosomal protein genes, depends upon the transcription activation of RAP1. If a mutation in the GAL11-gene would cause a similar reduction in the transcription of these essential genes, as in the case of the MATα-gene, this mutation would be lethal under all growth conditions.
The promoters of many nuclear genes encoding mitochondrial proteins represent another example of adjacent binding sites for multifunctional proteins. The promoter of the gene coding for complex III subunit VIII of the respiratory chain, for instance, harbors the binding sites for ABF1 and CPF1 (Dorsman et al., 1988). These binding sites even overlap each other, suggesting that a mutually exclusive binding of the two factors occurs. The transcription of this gene is regulated in essentially similar fashion as other nuclear encoded mitochondrial genes, i.e., through the HAP2/3/4/-system (Maarse et al., 1988). The ABF1/CPF1-binding site is not a part of the UAS and is only marginally involved in establishing a basal transcription level (Dorsman et al., 1990).
A combination of ABF1- and REB1-sites also occurs in the yeast genome, particularly in the enhancer region of the rRNA operon. Surprisingly, deletion of the REB1-site from the ribosomal enhancer (as well as from the second site near the promoter region) has no effect on the transcription activation of the rRNA operon or a rRNA minigene when present in a plasmid (Kulkens et al., 1989; Ju et al., 1990). Chasman and coworkers (1990) suggest an interplay between the REB1- and the ABF1-sites with a third T-rich element.
T-stretches have been implicated in the promoter function of many protein-encoding genes as well. Several authors reported that the binding sites of multifunctional factors in promoter regions — for instance of ribosomal protein genes — are often located close to DNA-stretches rich in thymine-residues (Rotenberg and Woolford, 1986; Lue et al., 1989; Buchman and Kornberg, 1990; Chasman et al., 1990). In those cases examined, T-rich elements can activate transcription independently (Kelleher et al., 1990; Buchman and Kornberg, 1990; Munholland et al., 1990), and can even augment considerably the effect of nearby located transcriptional activation sites (Rotenberg and Woolford, 1986; Lue et al., 1989; Buchman and Kornberg, 1990). Since T-elements can form specific DNA structures, which may be unable to become assembled into nucleosomes, they may render the promoter accessible to components of the transcription machinery. On the other hand, the effect of a T-rich element may be mediated by a protein binding to the dA:dT sequence. Buchman and coworkers have presented evidence for the occurrence of such a T-rich binding factor in yeast (Buchman and Kornberg, 1990). A well-studied promoter-region harboring a powerful T-rich element is the promoter-region of the constitutively expressed DED1-gene. At first, the T-rich element was thought to be the only cis-acting element responsible for the transcriptional activation of the DED1-gene (Struhl, 1985). However, it has recently been found that two ABF1-binding sites flanking this T-rich region, also play an important role in the activation of the DED1-gene (Buchman and Kornberg, 1990). ABF1-sites function only weakly on their own in heterologous promoters in vivo, but can establish a synergism with other elements, such as a T-rich element, to become a powerful transcription-activating element. Furthermore, both RAP1 and REB1 show a synergistic effect on transcription in a test promoter if the T-rich element derived from the DED1-promoter is cloned near their relevant binding sites (Buchman and Kornberg, 1990; Chasman et al., 1990). Therefore, the presence of T-rich elements in the relevant promoters might enhance the transcription activation effect of all these multifunctional factors.
Functional dissection of the promoters of some ABF1-regulated ribosomal protein genes also revealed the involvement of auxiliary activating elements (Hamil et al., 1988; Herruer et al., 1989; Kraakman et al., 1991). Strikingly enough, ABF1 is not able to function as a transcriptional activator in vitro, whereas many of the known transcriptional activators, such as GAL4, GCN4, and RAP1, can (Lue et al., 1989; Buchman and Kornberg, 1990). This result may indicate that, at least in the case of transcriptional activation by ABF1, higher-order structures not present in in vitro assays are required for ABF1-function.
The possible involvement of chromosomal structures in determining the cellular function of the multifunctional DNA-binding proteins has been suggested above. Indeed, DNA in the nucleus is attached to the nuclear scaffold at numerous sites (Gasser and Laemmli, 1987; Amati and Gasser, 1990b). These attachment sites (scaffold attachment regions or SAR) on the DNA are characterized by a high dA:dT content (Amati et al., 1990a). In yeast, SARs are primarily found at ARS and CEN elements (Amati and Gasser, 1988). Although nuclear attachment is a structural feature of ARS elements, it is not essential for ARS function (Amati et al., 1990a). It is not known whether ABF1 or CPF1 is involved in interactions with the nuclear scaffold at ARS and CEN elements, respectively. However, it is not likely that ABF1, with a binding site in the B-domain of ARS elements, mediates nuclear scaffold contact at ARS-SAR elements, since the attachment site is mapped to the ARS core consensus. The protein that binds to this core sequence, named ACBF (for ARS core binding factor), was found to be enriched in nuclear scaffold preparations (Hofmann and Gasser, 1991), whereas ABF1 could not be found in this type of protein preparation (Rhode et al., 1989).
Though the evidence summarized so far strongly suggests the combined action of the multifunctional DNA-binding factors with additional proteins, modifications of these proteins may also play a part in determining their actual activity. Since these multifunctional proteins mediate the regulation of processes that are essential for the growth of the yeast cell, rapid responses to changing physiological demands are required. Posttranslational modifications of these factors may provide the mechanism for such responses.
Several recent reports provide evidence that ABF1, RAP1, and REB1 harbor multiple phosphorylated amino acid residues in vivo (Chambers et al., 1989; Henry et al., 1990; Ju et al., 1990; Tsang et al., 1990; Morrow et al., 1991; Francesconi and Eisenberg, 1991). The potential phosphorylation sites of RAP1 are located mainly in and around the DNA-binding domain, suggesting a role for phosphorylation in the formation of a stable protein–DNA complex. Indeed, altering the phosphorylation state of full-length RAP1 or its minimal DNA-binding domain, results in changes in the affinity of this protein fragment for a RAP1-binding site in the PGK-gene promoter (Tsang et al., 1990). Interestingly, the nature of these changes depends upon sequences flanking the RAP1-binding site on the DNA. If a DNA-binding domain fragment of RAP1 is phosphatase-treated and used in binding assays, an increase of complex formation can be observed with the genuine PGK-RAP1 binding site. However, if the 5′-flanking sequences of the binding site are absent from the DNA-probe or substituted by plasmid DNA, the binding ability of this phosphatase-treated protein fragment is abolished (Tsang et al., 1990). Although it is not known whether this difference also occurs in vivo, these results indicate that a change in the phosphorylation state of RAP1 might induce a redistribution of the protein among its various binding sites on the genome.
ABF1 and REB1 have also been reported to be phosphoproteins. However, phosphorylated amino acid residues appear to be more equally distributed over these proteins than in the case of RAP1 (Ju et al., 1990; Francesconi and Eisenberg, 1991). For ABF1 and REB1, it has not yet been documented whether an altered phosphorylation state affects the ability to bind DNA, or whether phosphorylation influences, directly or indirectly, the activation of a particular function of the protein.
Insight into the actual mechanism of action of the multifunctional DNA-binding proteins will greatly benefit from a detailed knowledge of their domain structure. On the basis of presently available evidence, it seems likely that different parts of the proteins have distinctive functions. Data supporting this idea are beginning to emerge Recently, Sussel and Shore (1991) have introduced mutagenized RAP1 into rapl-null mutants. This strategy yielded several temperature-sensitive yeast strains, which at the restrictive temperature are defective in repression of transcription at the silent mating type loci. Interestingly, these mutant strains grow normally, indicating that the transcription of essential genes is not impaired. In addition, some — but not all — of the mutant strains have elongated telomeres. These results clearly show that the three functions of RAP1, viz., transcriptional activation, transcriptional repression, and telomere length control, reside in three physically separable domains of the protein. Indirectly, Kurtz and Shore (1991) provided evidence supporting the same conclusion by showing that some of their temperature-sensitive rapl-mutants grow equally well but differ in their extent of derepression at the HMR-locus. It will be interesting to see whether the active domains of other multifunctional DNA-binding proteins can also be physically uncoupled from each other and from the DNA-binding domain.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Amati B., Pick L., Laroche T., and Gasser S. M. (1990a) EMBO J 9, 4007–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B. and Gasser S. M. (1990b) Mol Cell Biol 10, 5442–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B. B. and Gasser S. M. (1988), Cell 54, 967–978. [DOI] [PubMed] [Google Scholar]
- Baker R. E. and Masison D. C. (1990), Mol Cell Biol 10, 2458–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker H. V. (1991), Proc Natl Acad Sci USA 88, 9443–9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman J., Tachinaba C. Y., and Tye B. K. (1986), Proc Natl Acad Sci USA 83, 3713–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas E. E., Stefanec M. J., and Biswas S. B. (1990), Proc Natl Acad Sci USA 87, 6689–6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bram R. J. and Kornberg R. D. (1987), Mol Cell Biol 7, 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H., Micklem G., and Nasmyth K. (1987), Cell 51, 709–719. [DOI] [PubMed] [Google Scholar]
- Brindle P. K., Holland J. P., Willet C. E., Innis M. A., and Holland M. J. (1990), Mol Cell Biol 10, 4872–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman A. R., Kimmerly W. J., Rine J., and Kornberg R. D. (1988a), Mol Cell Biol 8, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman A. R., Lue N. F., and Kornberg R. D. (1988b), Mol Cell Biol 8, 5086–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman A. R. and Kornberg R. D. (1990), Mol Cell Biol 10, 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M. and Davis R. W. (1989), Mol Cell Biol 9, 2544–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M. and Davis R. W. (1990), Cell 61, 437–446. [DOI] [PubMed] [Google Scholar]
- Chambers A., Tsang J. S. H., Stanway C., Kingsman A. J., and Kingsman S. J. (1989), Mol Cell Biol 9, 5516–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers A., Stanway C., Tsang J. S. H., Henry Y., Kingsman A. J., and Kingsman S. M. (1990), Nucl Acids Res 18, 5353–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasman D. I., Lue N. F., Buchman A. R., LaPointe J. W., Lorche Y., and Kornberg R. D. (1990), Genes Dev 4, 503–514. [DOI] [PubMed] [Google Scholar]
- Cohen R., Holland J. P., Yokoi T., and Holland M. J. (1986), Mol Cell Biol 6, 2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M. N., Wright J. H., Wolf A. J., and Zakian V. A. (1990), Cell 63, 739–750. [DOI] [PubMed] [Google Scholar]
- Della Seta F., Ciafre S. A., Mark C., Santoro B., Sentenac A., and Bozzoni I. (1990a), Mol Cell Biol 10, 2437–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Seta F., Treich I., Buhler J. M., and Sentenac A. (1990b), J Biol Chem 265, 15168–15175. [PubMed] [Google Scholar]
- Diffley J. F. X. and Stillman B. (1989), Science 246, 1034–1038. [DOI] [PubMed] [Google Scholar]
- Dorsman J. C., van Heeswijk W. C., and Grivell L. A. (1988), Nucl Acids Res 16, 7287–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsman J. C., van Heeswijk W. C., and Grivell L. A. (1990), Nucl Acids Res 18, 2769–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebralidse K. K., Grachev S. A., and Mirzabekov A. (1988), Nature 331, 365–367. [DOI] [PubMed] [Google Scholar]
- Eisenberg S., Civalier C., and Tye B. K. (1988), Proc Natl Acad Sci USA 85, 743–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler J. S. and Winston F. (1989), Mol Cell Biol 9, 5602–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedor M. J., Lue N. F., and Kornberg R. D. (1988), J Mol Biol 204, 109–127. [DOI] [PubMed]
- Francesconi S. C. and Eisenberg S. (1989), Mol Cell Biol 9, 2906–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi S. C. and Eisenberg S. (1991), Proc Natl Acad Sci USA 88, 4089–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser S. M. and Laemmli U. K. (1987), Trends Genet 3, 16–22. [Google Scholar]
- Giesman D., Best L., and Tatchell K. (1991), Mol Cell Biol 11, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A. and Perlman R. E. (1988), Mol Cell Biol 8, 2572–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfter H., Kavety B., Vandekerckhove J., Kiefer F., and Gallwitz D. (1989), EMBO J 8, 4265–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamil K. G., Nam H. G., and Fried H. M. (1988), Mol Cell Biol 8, 4328–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Y. A. L., Chambers A., Tsang J. S. H., Kingsman A. J., and Kingsman S. M. (1990), Nucl Acids Res 18, 2617–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herruer M. H., Mager W. H., Doorenbosch T. M., Wessels P. L. M., Wassenaar T. M., and Planta R. J. (1989), Nucl Acids Res 17, 7427–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. and Oshima Y. (1981), in Molecular Biology of the Yeast Saccharomyces-Life Cycle and Inheritance (Strathern J. N., Jones W. W., and Broach J., eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 181–209. [Google Scholar]
- Hoffmann A., Sinn E., Yamamoto T., Wang J., Roy A., Horikoshi M., and Roeder R. G. (1990), Nature 346, 387–390. [DOI] [PubMed] [Google Scholar]
- Hofmann J. F.-X., Laroche T., Brand A. H., and Gasser S. M. (1989), Cell 57, 725–737. [DOI] [PubMed] [Google Scholar]
- Hofmann J. F.-X. and Gasser S. M. (1991), Cell 64, 951–960. [DOI] [PubMed] [Google Scholar]
- Holland J. P., Brindle P. K., and Holland M. J. (1990), Mol Cell Biol 10, 4863–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland M. J., Yokoi T., Holland J. P., Myambo K., and Innis M. A. (1987), Mol Cell Biol 7, 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet J., Cotrelle P., Cool M., Vignais M.-L, Thiele D., Marck C., Buhler J. M., Sentenac A., and Fromageot P. (1985), EMBO J 4, 3539–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. M., Kayne P. S., Kahn E. S., and Grunstein M. (1990), Proc Natl Acad Sci USA 87, 6286–8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Q., Morrow B. E., and Warner J. R. (1990), Mol Cell Biol 10, 5226–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayne P. S., Kim U. J., Han M., Mullen J. R., Yoshizaki F., and Grunstein M. (1988), Cell 55, 27–39. [DOI] [PubMed] [Google Scholar]
- Kelleher R. J. III, Flanagan P. M., and Kornberg R. D. (1990), Cell 61, 1209–1215. [DOI] [PubMed] [Google Scholar]
- Kimmerly W., Buchman A., Kornberg R., and Rine J. (1988), EMBO J 7, 2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraakman L. S., Mager W H., Grootjans J. J., and Planta R. J. (1991), Biochim Biophys Acta 1090, 204–210. [DOI] [PubMed] [Google Scholar]
- Kulkens T., VanHeerikhuizen H., Klootwijk J., Oliemans J., and Planta R. J. (1989), Curr Genet 16, 351–359. [DOI] [PubMed] [Google Scholar]
- Kurtz S. and Shore D. (1991), Genes Dev 5, 616–628. [DOI] [PubMed] [Google Scholar]
- Leer R. J., van Raamsdonk-Duin M. M. C., Mager W. H., and Planta R. J. (1985), Curr Genet 9, 273–277. [DOI] [PubMed] [Google Scholar]
- Longtine M. S., Maxfield Wilson N., Petracek M. E., and Berman J. (1989), Curr Genet 16, 225–239. [DOI] [PubMed] [Google Scholar]
- Lue N. F., Buchman A. R., and Kornberg R. D. (1989), Proc Natl Acad Sci USA 86, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarse A. C., De Haan M., Bout A., and Grivell L. A. (1988), Nucl Acids Res 16, 5797–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager W. H. and Planta R. J. (1991), Mol Cell Biochem 104, 181–187. [DOI] [PubMed] [Google Scholar]
- Mahoney D. and Broach J. R. (1989), Mol Cell Biol 9, 4621–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall M., Mahoney D., Rose A., Hicks J. B., and Broach J. R. (1987), Mol Cell Biol 7, 4441–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megee P. C., Morgan B. A., Mittman B. A., and Smith M. M. (1990), Science 247, 841–845. [DOI] [PubMed] [Google Scholar]
- Mellor J., Funk M., Rathjen J., Barnes C. A., Hinz T., Hegemann J. H., and Phillipsen P. (1990), EMBO J 9, 4017–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J., Rathjen J. R., Jiang W., and Dowell S. J. (1991), Nucl Acids Res 19, 2961–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore P. A., Sagliocco F. A., Wood R. M. C., and Brown A. J. P. (1991), Mol Cell Biol 11, 5330–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow B. E., Ju Q., and Warner J. R. (1991), J Biol Chem 265, 20778–20783. [PubMed] [Google Scholar]
- Munholland J. M., Kelly J. K., and Wildeman G. (1990), Nucl Acids Res 18, 6061–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. (1982), Cell 30, 567–578. [DOI] [PubMed] [Google Scholar]
- Nishizawa M., Suzuki Y., Nogi Y., Matsumoto K., and Fukasawa T. (1990), Proc Natl Acad Sci USA 87, 5373–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi Y. and Fukasawa T. (1980), Curr Genet 2, 115–120. [DOI] [PubMed] [Google Scholar]
- Ogden J. E., Stanway C., Kim S., Mellor J., Kingsman A. J., Kingsman S. M. (1988), Mol Cell Biol 6, 4335–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E. and Szostak J. W. (1990), Mol Cell Biol 10, 4932–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic B. and Hörz W. (1988), Mol Cell Biol 8, 5513–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhode P. R., Sweder K. S., Oegema K. F., and Campbell J. L. (1989), Genes Dev 3, 1926–1939. [DOI] [PubMed] [Google Scholar]
- Rotenberg M. O. and Woolford J. L. (1986), Mol Cell Biol 6, 674–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo G. M. and Tornow J. (1990), Mol Cell Biol 10, 859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D. and Nasmyth K. (1987a), Cell 51, 721–732. [DOI] [PubMed] [Google Scholar]
- Shore D., Stillman D. J., Brand A. H., and Nasmyth K. (1987b), EMBO J 6, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spraque G. F Jr., Blair L. C., and Thorner J. (1983), Annu Rev Microbiol 37, 623–660. [DOI] [PubMed] [Google Scholar]
- Stanway C. A., Chambers A., Kingsman A. J., and Kingsman S. M. (1989), Nucl Acids Res 17, 9205–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. (1985), Nucl Acids Res 13, 8587–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L. and Shore D. (1991), Proc Natl Acad Sci USA 88, 7749–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweder K. S., Rhode P. R., and Campbell J. L. (1988), J Biol Chem 263, 17270–17277. [PubMed] [Google Scholar]
- Thomas D., Cherest H., and Surdin-Kerjan Y. (1989), Mol Cell Biol 9, 3292–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J. S. H., Henry Y. A. L., Chambers A., Kingsman A. J., and Kingsman S. M. (1990), Nucl Acids Res 18, 7331–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Nicholson P. R., and Stillman D. J. (1990), Mol Cell Biol 10, 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]