

Abstract
G1-specific temperature-sensitive (ts) mutants of the cell cycle arrest in G1 after serum stimulation at the restrictive temperature. Under these conditions, the RNA levels of late growth-regulated genes (such as DNA polymerase alpha, PCNA, thymidine kinase, and core histones) are markedly decreased or even undetectable, while early growth-regulated genes (for instance, c-myc) are normally expressed, and certain promoters are actually super-induced. We have used the human PCNA gene transfected into TK−tsl3 cells (a G1-specific ts mutant) to investigate whether the inhibition of gene expression caused by this type of growth inhibition occurs at a transcriptional or posttranscriptional level. Constructs were made in which the 5′ and 3′ flanking sequences of the human PCNA gene were replaced by the corresponding elements of the SV40 T antigen coding gene. Using these constructs and data from run-on assays and RT-PCR, we conclude that the failure of expression of the PCNA gene in G1-arrested TK−tsl3 cells occurs at the transcriptional level.
The product of the PCNA gene, as the co-factor of DNA polymerase δ (Prelich et al., 1987; Bravo et al., 1987), is necessary for DNA replication (Tsurimoto and Stillman, 1989; Prelich et al., 1987a,b; Bravo et al., 1987), and for cellular proliferation (Jaskulski et al., 1988a; Liu et al., 1989). The PCNA mRNA levels are growth-regulated (Almendral et al., 1987; Jaskulski et al., 1988b; Shipman et al., 1988). Both transcriptional and posttranscriptional mechanisms operate in the growth regulation of PCNA mRNA levels (Chang et al., 1990; Shipman-Appasamy et al., 1990). Specifically, the ability of the PCNA gene to respond to serum stimulation resides in a −73 promoter, i.e., a promoter reduced to the 73 bp immediately upstream of the capsite (Pietrzkowski et al., 1991). Elements in intron 4 (Ottavio et al., 1990) and intron 1 (Alder et al., 1992) also participate in the regulation of PCNA mRNA levels.
While the promoter and intragenic sequences necessary for a response to serum have been identified (for a review, see Baserga, 1991), G1-specific temperature-sensitive (ts) mutants of the cell cycle offer a unique system to study the regulation of genes whose expression is growth-regulated. When these ts mutants are stimulated with serum or growth factors at the permissive temperature of 34°C, all growth-regulated genes increase normally, as in other cell types (Hirschhorn et al., 1984; Jaskulski et al., 1988b; Koniecki et al., 1991; Travali et al., 1991). When the cells are serum-stimulated at the restrictive temperature, the RNA levels of early growth-regulated genes like c-myc increase normally, but genes that are expressed at the G1-S boundary, such as thymidine kinase, DNA polymerase α, and histones, are inhibited, i.e., their mRNAs cannot be detected in ordinary RNA blots (Hirschhorn et al., 1984; Liu et al., 1985; Ide et al., 1985; Jaskulski et al., 1988b; Koniecki et al., 1991; Travail et al., 1991). This is also true for PCNA, whose mRNA is easily detectable in G1 specific ts mutants stimulated at the permissive temperature of 34°C, but is undetectable, or markedly decreased, in the same cells serum-stimulated at the restrictive temperature of 39.6°C (Jaskulski et al., 1988b; Koniecki et al., 1991; Travali et al., 1991). The inhibition in the expression of late growth-regulated genes is made even more interesting by the fact that at this stage other promoters are still actively transcribed. For instance, while the RNA transcribed from a thymidine kinase (TK) gene is no longer detectable in G1-specific ts mutants serum-stimulated at the restrictive temperature, it becomes easily detectable—in fact overexpressed—when transcribed from its cDNA placed under the control of the early SV40 promoter (Lipson et al., 1989). It seems, therefore, that transcriptional mechanisms are still operating in these cells, even at the restrictive temperature, and that the inhibition affects only a subset of genes, specifically the genes that are involved in chromosomal replication (Liu et al., 1985). It is important to notice that in this model, the cells are always serum-stimulated, whether at the permissive or restrictive temperatures. The failure of late growth-regulated genes to be expressed (at the mRNA levels) is therefore related, not to serum, but to the ts block (see also Discussion). In this paper we have investigated the regulatory elements involved in the expression of the PCNA gene in a G1-specific ts mutant of the cell cycle, TK−ts13 cells (Shen et al., 1982). These results show unequivocally that at the restrictive temperature of 39.6°C the promoter is responsible for the lack of expression of the PCNA gene and that the failure to express PCNA mRNA can be fully explained by a transcriptional inhibition.
Materials and methods
Cell lines
TK−ts13 Syrian hamster fibroblasts (Shen et al., 1982), a TK-deficient ts mutant cell line derived from tsl3 cells, which arrest in the G1 phase of cell cycle at the non-permissive temperature of 39.6°C (Talavera and Basilico, 1977), were co-transfected using the calcium-phosphate precipitation procedure (Shen et al., 1982) with one of the desired constructs and pTK11 (human TK cDNA cloned into an Okayama-Berg vector; Flemington et al., 1987) as a selectable marker. Selection started 2 days after transfection, in glycine-hypoxanthine-aminopterine-thymidine (gHAT) medium supplemented with 10% calf serum (CS). Cell lines were maintained in selective medium.
Plasmid construction
Using the sequences of the human PCNA gene (Travali et al., 1989) and the human PCNA cDNA (Almendral et al., 1987; Jaskulski et al., 1988b), we have constructed the various plasmids used in these experiments.
Strategy to construct the SV40pr/PCNA gene/3′
cob 12 (PcDX, Okayama Berg vector) containing the PCNA cDNA (Okayama and Berg, 1983; Jaskulski et al., 1988b) was digested with Pvu I (nucleotide 1953 in the vector’s ampicillin resistance gene) and Nru I (nucleotide +61 in the PCNA cDNA). The fragment containing part of the ampicillin resistance gene, the SV40 early promoter and splice junction, and the first 61 bases of the PCNA cDNA were isolated.
The p3BamFullPCNA (BamH I promoter and full-length human PCNA gene cloned in pGEM 3) was digested with Pvu I (nucleotide 1543 in the vector’s ampicillin resistance gene) and Nru I (nucleotide +61 in the PCNA gene). The fragment containing part of the ampicillin resistance gene, the origin of replication, and the PCNA gene from +61 to the end was isolated.
The 2 fragments were ligated, generating a construct in which the early SV40 promoter was driving the full-length human PCNA gene with its own 3’ flanking sequence.
Strategy to construct the Bampr/cDNA/SV40 poly(A)
The p3BamFullPCNA was digested with Pvu I (nucleotide 1543 in the vector’s ampicillin resistance gene) and Nru I (nucleotide +61 in the PCNA gene). The fragment containing part of the ampicillin resistance gene, the PCNA promoter from BamH I, and the first 61 bases of the gene was isolated.
cob 12 was digested with Pvu I (nucleotide 1953 in the vector’s ampicillin resistance gene) and Nru I (nucleotide +61 in the PCNA cDNA). The fragment containing part of the ampicillin resistance gene, the origin of replication for the plasmid, the SV40 polyadenylation signal, and the PCNA cDNA from +61 to the end was isolated.
The 2 fragments were ligated to generate a construct in which the PCNA promoter was driving its own cDNA, but with an SV40 poly(A) signal.
Strategy to construct the Bampr/cDNA/3′
The pBampr/cDNA/SV40 poly(A)—see above—was digested with Hpa I (nucleotide +1150 in the PCNA cDNA) and Pvu I (nucleotide 1543 in the vector’s ampicillin resistance gene). The fragment containing part of the ampicillin resistance gene, the PCNA promoter from BamH I to the first 61 bases of the PCNA gene, and the PCNA cDNA from Nru I (+61) to Hpa I (+1150) was isolated.
The p3Bam FullPCNA was digested with Pvu I (nucleotide 1543 in the vector’s ampicillin resistance gene) and Hpa I (nucleotide +4682 in the PCNA gene), and the fragment containing part of the ampicillin resistance gene, the origin of replication for the plasmid, and the 3′ end of the PCNA gene from Hpa I (+4862) to the end was isolated.
The 2 fragments were ligated to generate a construct similar to pBampr/cDNA/SV40 poly(A), but with the 3’ flanking sequence of the PCNA gene.
The strategies used to construct the other expression plasmids were essentially similar (with minor variations in restriction sites) to generate the constructs listed in Table 1. Details for one more construct are given below.
Table 1.
TK-ts13 cells were cotransfected with one of the construct and pTKII as a selectable marker (see Materials and Methods).
| Established cell lines were made quiescent and then serum-stimulated for 24 hours. RNA blots were hybridized to the Ava ll-Pst I fragment of the human PCNA cDNA, to the histone H3 probe and to the 3A10 cDNA. The densitometry readings of the autoradiographs represent the ratios of the PCNA bands to the 3A10 bands (PCNA/3A10), and the H3 bands to the 3A10 bands (H3/3A10). | |||
| Cell Line | H3/3A10 | PCNA/3A10 | |
|---|---|---|---|
| Bam pr-PCNA gene-3′ | GO | 0.19 | 0.018 |
| 34° | 0.66 | 0.04 | |
| 39°6 | 0.06 | 0.015 | |
| SV pr-PCNA gene-SV | GO | 0.38 | 0.06 |
| 34° | 0.88 | 0.12 | |
| 39°6 | 0.21 | 0.17 | |
| Bam pr-PCNA gene-SV | GO | 0.8 | 2.74 |
| 34° | 1.9 | 2.5 | |
| 39°6 | 0.2 | 0.5 | |
| SV pr-PCNA gene-3′ | GO | – | – |
| 34° | 1.9 | 1.8 | |
| 39°6 | 0.4 | 3.1 | |
| Bam pr-PCNA cDNA-3′ | GO | – | – |
| 34° | 0.6 | 0.62 | |
| 39°6 | 0.1 | 0.2 | |
| SV pr-PCNA cDNA-SV | GO | 0.28 | 0.05 |
| 34° | 0.6 | 0.3 | |
| 39°6 | 0.05 | 0.8 | |
| Bam pr-PCNA cDNA-SV | GO | 0.04 | 0.05 |
| 34° | 1.18 | 0.2 | |
| 39°6 | – | – | |
| SV pr-PCNA cDNA-3′ | GO | 0.03 | 0.08 |
| 34° | 0.5 | 0.5 | |
| 39°6 | 0.05 | 0.9 | |
Strategy to construct SV40pr/cDNA/SV40 poly(A) without the SV40 splicing site
The SK plasmid (Stratagene) was linearized with BamH I. The overhanging end was filled with the Klenow fragment of DNA polymerase, and the plasmid was digested with Pst I.
cob 12 was digested with Pst I; the 273 bp fragment was subsequently digested with Stu I. The fragment containing the first 194 bases of the PCNA cDNA was isolated, and the two fragments were ligated.
The plasmid resulting from the first two steps was digested with Nru I (nucleotide +61) and Sca I (nucleotide 2526 in the vector’s ampicillin resistance gene). The fragment containing part of the ampicillin resistance gene and the first 61 bases of the PCNA cDNA was isolated.
cob 12 was digested with Sea I (nucleotide 2063 in the vector’s ampicillin resistance gene) and Nru I (nucleotide +61). The fragment containing part of the ampicillin resistance gene, the origin of replication of the plasmid and the PCNA cDNA from +61 to the end, was isolated and the 2 fragments were ligated.
The resulting plasmid was linearized with Sal I (674).
cob 12 was digested with Sal I and Xho I. The fragment containing the SV40 promoter without the splice junction was isolated and the 2 fragments ligated.
The Dsal-PCNA gene was previously described (Pietrzkowski et al., 1991). It is the full-length human PCNA gene with a 73 bp promoter.
RNA isolation and Northern blot analysis
RNA was isolated by the method of Chomczynski and Sacchi (1987). RNA blots were performed by standard procedures (Thomas, 1983). Radioactive probes were prepared by the random primer method (Feinberg and Vogelstein, 1984). The following probes were used:
A 186 bp Pst I-Ava II fragment corresponding to the human specific part of the human PCNA cDNA (subcloned into pGEM 3-Z; Promega).
The histone H3 probe previously described (Jaskulski et al., 1988b).
3A10 cDNA (Lin and Lee, 1984) was used as an internal control in all of the RNA hybridization measurements reported here. The 3A10 cDNA was derived from a cDNA library constructed by using poly(A)+ RNAs extracted from hamster K12 cells incubated at the restrictive temperature. 3A10 encodes an RNA species that is expressed at constant levels throughout the cell cycle and at either restrictive or permissive temperatures.
Nuclei isolation and nuclear run-on assays
For each growth condition, 3 × 107 cells were grown on Petri dishes. Cells were washed twice with ice-cold PBS and collected by scraping. After centrifugation, the cell pellet was gently resuspended in lysis buffer containing Nonidet P-40 (NP-40) (10 mM Tris-HCl, pH 7.4; 10 mM NaCl; 3 mM MgCl2; 0.5% NP-40). After 5′ incubation on ice, the lysed cells were centrifuged, the pellet resuspended in NP-40 lysis buffer, and centrifuged again. The nuclear pellet was then resuspended in glycerol storage buffer (50 mM Tris-HCl, pH 8.3; 40% glycerol; 5 mM MgCl2; 0.1 mM EDTA). The nuclei were kept at −70°C.
Nuclear run-on assays were performed as described previously (Groudine et al., 1981; Koniecki et al., 1991). The single-stranded PCNA probes were generated from fragments sub-cloned into SK (Stratagene). The subcloned fragments were: (1) exon 1, Nru I-Pst I; (2) exons 2 and 3, EcoR V-EcoR I. These plasmids were rescued as single-stranded phagmids using a helper phage (R408, Stratagene), and the inserts were excised as described (Sell et al., 1992). The actin probe was obtained by digestion of pHF-β-A-l plasmid (Gunning et al., 1983) with BamH I.
The λ DNA used as a negative control was a Hind III digest commonly used as an electrophoresis marker (Boehringer Mannheim).
Reverse transcriptase-polymerase chain reaction (RT-PCR)
The RT-PCR techniques used were similar to published methods (Rappolee et al., 1988; Rappolee et al., 1989; Saiki et al., 1988). The amplimers used for the amplification (exon 1-intron 1) and the intron 1 probe have been described previously (Chang et al., 1990).
Results
In previous experiments it was shown that the human TK gene under the control of its own promoter was not expressed when transfected into TK−ts13 cells serum-stimulated at the restrictive temperature of 39.6°C, while its mRNA was easily detectable in the same cells stimulated at the permissive temperature. However, when the human TKcDNA was placed under the control of the early SV40 promoter and transfected into the same cells, it was expressed both at the permissive and the restrictive temperatures (Lipson et al., 1989). Indeed, at the restrictive temperature the human TKcDNA, under the control of the SV40 promoter, was actually overexpressed. The same observation was found to be true for the PCNA cDNA and gene (see below). Such a discrepancy at the restrictive temperature in the expression of the TK or PCNA genes versus that of an SV40 controlled cDNA could only depend on one of three components: the 5′ flanking sequences, the presence of introns, or the 3′ untranslated sequences. We therefore proceeded to make constructs in which these three components were varied. We started from the PCNA gene, as cloned and sequenced by Travali et al. (1989). This human PCNA gene contains 2.8 kb of 5′ flanking sequence, the whole gene which consists of 6 exons and 5 introns, and about a kilobase of 3′ flanking sequences. Figure 1 shows three constructs made with the PCNA gene. The first has its own promoter, but with the SV40 3′ flanking sequence. The second has the SV40 promoter instead of the PCNA promoter, and the SV40 3′ untranslated sequence. The third construct has the SV40 promoter and the human PCNA gene with its own 3′ flanking sequence. All these constructs were cloned in pGEM 3. Similar constructs were made using–instead of the PCNA gene – the human PCNA cDNA (Jaskulski et al., 1988b).
Figure 1.
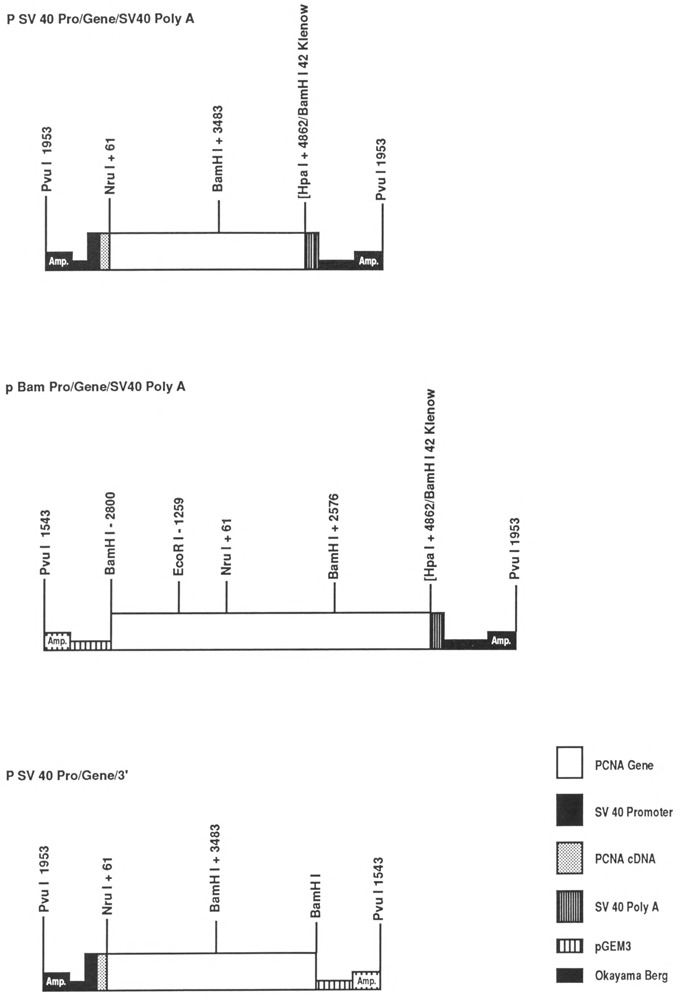
Constructs made with the human PCNA gene. The diagram shows only the constructs made with the gene, but similar constructs were also made with the human PCNA cDNA (see Materials and Methods). Empty boxes: PCNA gene; black boxes: SV40 promoter; white striped boxes: pGEM 3; black striped boxes: SV40 polyadenylation sequence; spotted boxes: PCNA cDNA; black squares: Okayama-Berg vector sequences; Amp: ampicillin resistance gene.
These various constructs were transfected into TK−tsl3 cells together with pTK11, which is a plasmid containing the human TKcDNA under the control of the SV40 promoter. Mixed populations (from 40 or more independent colonies) were selected in gHAT and used for subsequent experiments.
The type of promoter determines the expression of PCNA at the restrictive temperature
The experiments described in Figure 2 were carried out with the cells stably transfected with different constructs (see above) under three different conditions: serum deprived cells (lane 1), cells serum-stimulated for 24 hours at the permissive temperature of 34° C (lane 2), and cells stimulated for the same period of time at the restrictive temperature of 39.6°C (lane 3). RNA was extracted, and RNA blots were simultaneously hybridized to the human-specific PCNA probe (see Materials and Methods), to a human histone H3 probe, and to the 3A10 probe that is constantly expressed throughout the cell cycle (Lin and Lee, 1984). Figure 2 shows that the expression of the human PCNA (PCNA pro/ PCNA 3′) gene is completely suppressed at the restrictive temperature, although human PCNA RNA is abundantly present in cells stimulated at 34°C.
Figure 2.
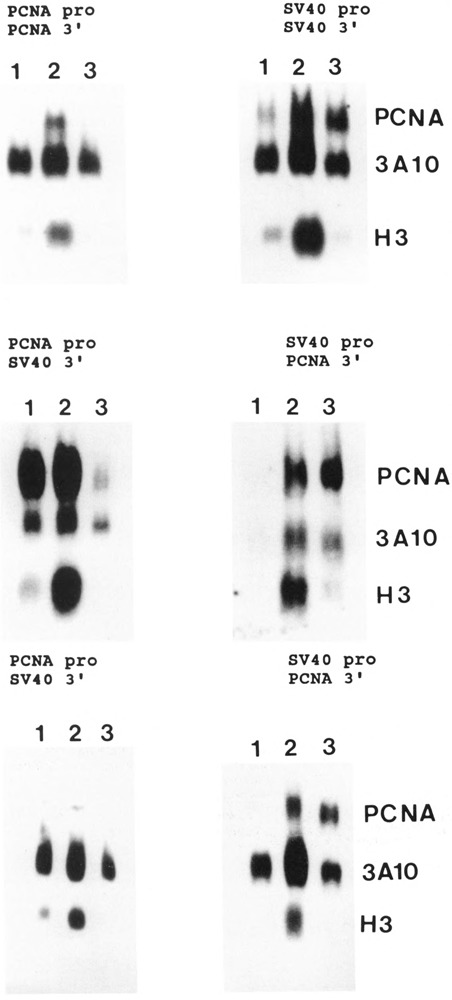
Expression of the transfected PCNA gene in TK−tsl3 cells. Experiments were carried out with cell lines obtained after transfection of TK−tsl3 cells with the constructs diagrammed in Figure 1 and in Materials and Methods. The cells were cotransfected with the selectable marker as described in Materials and Methods. Mixed populations were then selected and amplified. The cells were made quiescent and subsequently serum-stimulated for 24 hours. Total RNA was extracted and analyzed by Northern blot, the filters were hybridized to the Ava II-Pst I fragment of the human PCNA cDNA (which recognizes only the human PCNA mRNA), to the histone H3 probe, and to the 3A10 cDNA. Lane 1, quiescent cells; lane 2, serum-stimulated cells at the permissive temperature; lane 3, serum-stimulated cells at the restrictive temperature. The top four panels are from cells carrying the human PCNA gene, pro: promoter; 3′: 3′ flanking sequences.
The same result was obtained in cells carrying either (1) the construct with the PCNA promoter, the PCNA gene, and the SV40 3′ flanking sequence (PCNA pro/SV40 3′, middle panel); (2) the construct with the PCNA promoter, the PCNA cDNA, and the SV40 3′ flanking sequence (PCNA pro/SV40 3′, lower panel); or (3) the PCNA 5′ and 3′ flanking sequences and the PCNA cDNA (not shown). In all instances, the human PCNA mRNA levels were markedly decreased at the restrictive temperature, following the same pattern of histone H3 RNA levels.
The results were different when the cells carrying the constructs in which the SV40 promoter driving the human PCNA gene or cDNA were analyzed. PCNA mRNA levels were as high at the restrictive temperature as at the permissive temperature (Fig. 2; the construct with the SV40 promoter, the PCNA cDNA, and the SV40 3′ flanking sequence is not shown). Neither the presence or absence of introns, nor the type of 3′ flanking sequence has any influence on the expression of PCNA mRNA at the restrictive temperature. The absence of histone H3 in lane 3 confirms that the cells had been blocked in G1 in all instances.
The results of these experiments are summarized in Table 1, where they are presented as ratios of H3 RNA levels to 3A10 RNA levels, and of PCNA RNA levels to 3A10 RNA levels, in serum deprived G0 cells and in the same cells stimulated for 24 hours at either 34°C or 39.6°C. The H3/3A10 ratio is always markedly decreased in cells stimulated at the restrictive temperature. The PCNA/3A10 ratio is decreased at the restrictive temperature only when the transfected construct had the original PCNA promoter. When the cells carried similar constructs but with the SV40 promoter, the ratios were essentially the same at 34°C and 39.6°C, although the absolute levels may have varied.
The activity of the SV40 promoter at 39.6°C is not due to its splicing site
The SV40 promoter used in all these experiments is derived from the Okayama-Berg vector (Okayama and Berg, 1983); it contains a very small intron with the appropriate splicing signals. It is possible that the ability of this SV40 promoter to generate PCNA mRNA levels may be due to the presence of these splicing signals and the small intron. In the next construct we removed the SV40 intron and splicing sites (see Materials and Methods), and used it again to drive the human PCNA cDNA. The construct was transfected as usual in TK"tsl3 cells, together with pTK11; mixed populations were selected; and RNA was extracted. The resultant blots are shown in Figure 3. Clearly, the human PCNA cDNA is vigorously expressed at the restrictive temperature, although this SV40 promoter does not contain the splicing signals.
Figure 3.
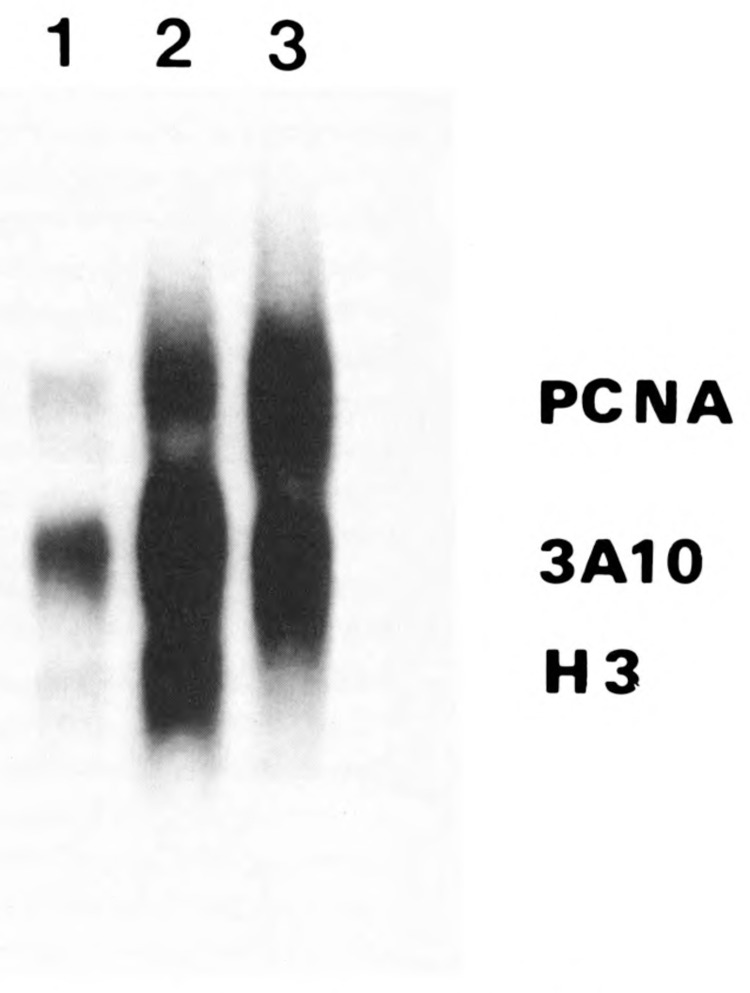
Effect of the removal of the SV40 intron and splicing sites on the ability of the SV40 promoter to generate PCNA mRNA levels. We used the construct in which the SV40 intron and splicing sites were removed from the promoter (see Materials and Methods) to cotransfect TK−tsl3 cells together with pTK 11. Total RNA was extracted and analyzed as described in Figure 2. Lane 1, quiescent cells; lane 2, serum-stimulated cells at the permissive temperature; lane 3, serum-stimulated cells at the restrictive temperature.
Transcriptional activity of the PCNA promoter and of the SV40 promoter at both permissive and restrictive temperatures
The transcriptional activity of these two promoters was analyzed by two different methods. The first method we used was that of the reverse transcriptase polymerase chain reaction (RT-PCR) as outlined by Rappolee et al. (1989), and Lipson and Baserga (1989). Only two cell lines were studied in this case. The first cell line was the one containing the human PCNA gene, with its 5′ and 3′ flanking sequences, and the second cell line was the one in which the human transfected PCNA gene had an SV40 promoter and an SV40 3′ flanking sequence. Appropriate amplimers of exon 1 and intron 1 were used to generate hnRNA, originating from the human PCNA gene. The amplification products were then transferred by a Southern blot and hybridized to a PCNA intron 1 probe. The expected size of the amplification product was 493 bp, and the results are shown in Figure 4. Very little human PCNA hnRNA is present in cells that are serum-deprived (lanes 1 and 4). In cells stimulated at 34°C, the human PCNA hnRNA is clearly detectable, both in cells carrying the human PCNA gene construct and in cells carrying the same gene but under the control of the SV40 promoter (lanes 2 and 5). At 39.6°C, however, the results are different: no transcripts are detectable in cells carrying the full-length, human PCNA gene (lane 3), while transcripts are easily detectable in cells in which the human PCNA gene was driven by the SV40 promoter (lane 6). Appropriate controls to the RT-PCR were made as follows: (1) when the RT-PCR was run in the absence of the reverse transcriptase, no amplification products were obtained, indicating that DNA had been totally excluded; and (2) two different cycles of amplification were used to ascertain that the quantitation was correct (data not shown).
Figure 4.
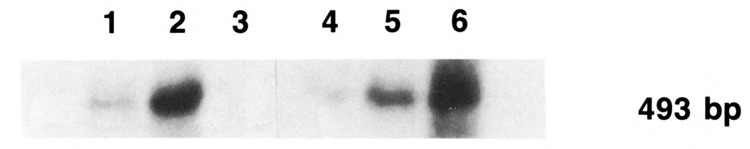
PCNA hnRNA levels in TK−tsl3 cells with either Bampr/PCNA gene or SV40pr/ PCNA gene. mRNA levels were measured by RT-PCR. One μg of total RNA was amplified with 2 amplimers from exon 1 and intron 1 of the human PCNA gene for 20 cycles. After amplification, the amplification products were blotted and hybridized to an oligonucleotide from intron 1 of the human PCNA gene. Lanes 1-3, RT-PCR performed with the cells transfected with the human PCNA gene with its own flanking sequences; lanes 4-6, RT-PCR performed with the cells transfected with the human PCNA gene under the control of the SV40 promoter and with the SV40 polyadenylation signal; lanes 1 and 4, quiescent cells; lanes 2 and 5, serum-stimulated cells at the permissive temperature; lanes 3 and 6, serum-stimulated cells at the restrictive temperature.
The second technique we used to investigate transcriptional activity of this construct was the run-on assay (Groudine et al., 1981) as described in Materials and Methods. The cells were treated as in Figure 4, except that the nuclei were prepared and analyzed for transcriptional activity. To detect PCNA transcription, we used single-stranded probes from exon 1 and from exons 2 and 3. A β-actin probe was used as the positive control, and λ DNA as the negative control. The results are shown in Figure 5. Confirming the results obtained with RT-PCR, run-on transcription shows that there is no detectable signal at 39.6°C in cells carrying the human PCNA gene with its own flanking sequences (Panel A). The run-on assay of cells–with the same gene, but under the control of the SV40 promoter–gives instead a very clear signal that is as strong at 39.6°C as at 34°C (panel B). These experiments were done with single-stranded probes to avoid the problems of antisense transcription (Sell et al., 1992).
Figure 5.
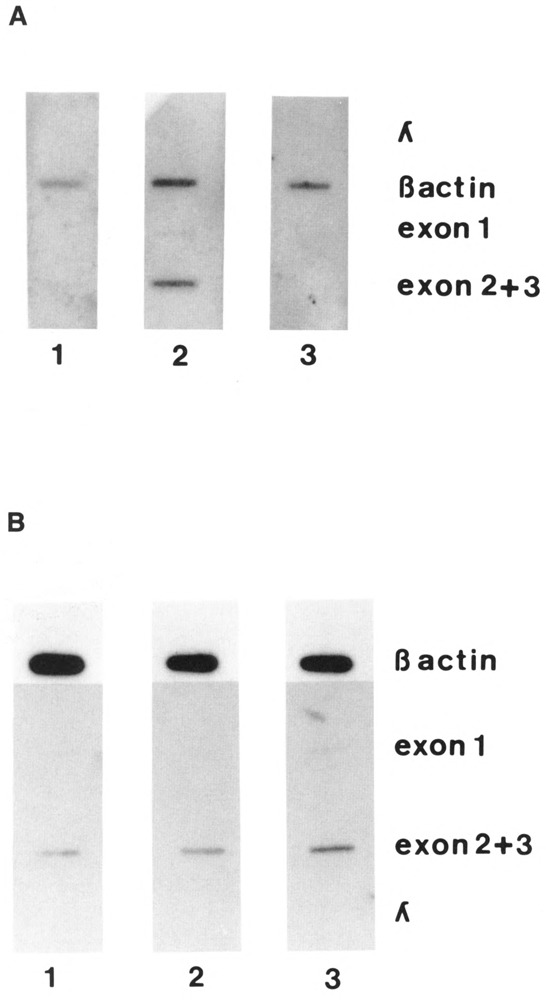
Transcriptional activity of the different constructs measured by run-on assay. The experiments were performed as described in Materials and Methods. The cells were made quiescent (lane 1), then serum-stimulated for 20 hours at either the permissive temperature (lane 2) or the restrictive temperature (lane 3). A. Cells transfected with the human PCNA gene with its own flanking sequences. B. Cells transfected with the human PCNA gene under the control of the SV40 promoter and with the SV40 polyadenylation signal. The amount of DNA/slot was 1 μg. The probes used were the Nru I-Pst I fragment of the exon 1 of the human PCNA gene, the EcoR V-EcoR I fragment containing both exon 2 and 3 of the PCNA gene, the λ DNA and the β-actin DNA (see Materials and Methods).
A 73 bp promoter of the PCNA gene is thermosensitive
In a previous paper (Pietrzkowski et al., 1991), we had shown that a human PCNA promoter consisting only of the 73 bp immediately upstream of the CAP site is fully active and is growth-regulated, i.e., its mRNA levels are markedly increased by serum stimulation. The same cell line used by Pietrzkowski et al. (1991) of transfected TK−tsl3 expressing the human PCNA gene driven by a 73 bp promoter was used again. These cells are stably transfected, and Figure 6 shows that in this cell line (1) the PCNA mRNA levels increase when the cells are stimulated at the permissive temperature; and (2) the increase is negligible when the cells are serum-stimulated at the restrictive temperature. Again, histone H3 serves as a control for both stimulations. At the restrictive temperature, the −73 bp promoter also fails to produce PCNA mRNA when driving the cDNA, instead of the gene, with the SV40 3′ end (not shown).
Figure 6.
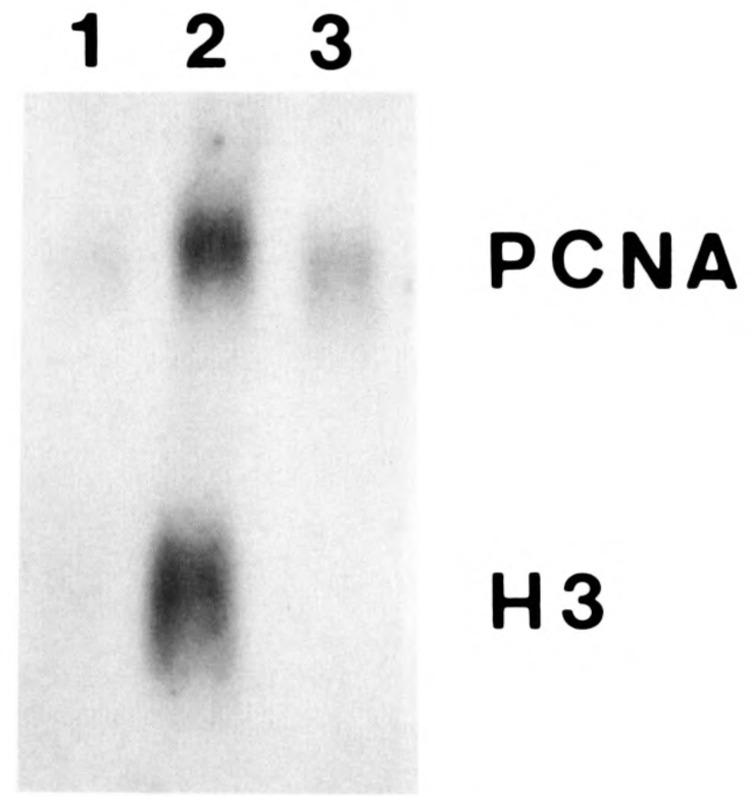
Expression of the −73bp PCNA promoter (Dsal) driving the human PCNA gene. Experiments were carried out with the transfected human PCNA gene under the control of the −73bp PCNA promoter (Pietrzkowski et al., 1991). The cells were transfected, and mixed populations were then selected and amplified. The cells were made quiescent and subsequently serum-stimulated for 24 hours. Total RNA was extracted and analyzed by Northern blot; the filters were hybridized both to the Ava II-Pst I fragment of the human PCNA cDNA that recognizes only the human PCNA mRNA and to the histone H3 probe. Lane 1, quiescent cells; lane 2, serum-stimulated cells at the permissive temperature; lane 3, serum-stimulated cells at the restrictive temperature.
Discussion
The PCNA mRNA levels are growth-regulated (see review in Baserga, 1991). Both serum- and platelet-derived growth factor increase PCNA mRNA expression in 3T3 cells (Almendral et al., 1987, Jaskulski et al., 1988b). The serum-responsive element in the PCNA promoter has been localized to the 73 bp immediately upstream of the CAP site (Pietrzkowski et al., 1991). In this paper we have addressed a different problem, namely, the element(s) in the PCNA gene responsible for its failure to generate mRNA in G1-specific ts mutants of the cell cycle stimulated at the restrictive temperature.
The rationale for this investigation is based on the fact that late growth-regulated genes, like histones and DNA synthesis genes, are not expressed in G1-specific ts mutants of the cell cycle serum-stimulated at the restrictive temperature (Hirschhorn et al., 1984b; Liu et al., 1985; Ide et al., 1895; Jaskulski et al., 1988b; Koniecki et al., 1991; Venturelli et al., 1990; Travali et al., 1991). Under the same conditions, early growth-regulated genes (like c-myc) are normally expressed (Hirschhorn et al., 1984a; Koniecki et al., 1991), and certain promoters are actually superinduced (Lipson et al., 1989; Travali et al., 1991). G1-specific ts mutants, therefore, offer a very attractive system for studying the regulation of gene expression – specifically, why the expression of a subset of genes is inhibited at the restrictive temperature, while other genes are normally expressed. The fact that some genes are expressed while others are not, rules out trivial explanations, such as a total inhibition of unique copy gene transcription. The possibility of identifying a factor or a mechanism specific to a subset of genes is clearly of interest.
We have selected for our purpose one G1 specific ts mutant, TK−tsl3 cells (Shen et al., 1982) and the human PCNA gene (Travali et al., 1989), since our laboratory has had considerable experience with both of them. In preliminary experiments based on a previous observation by Lipson et al. (1989), we had established that a human PCNA cDNA under the control of an early SV40 promoter and with an SV40 poly(A) signal, stably transfected in TK−tsl3 cells, was expressed vigorously at the restrictive temperature. Different constructs were made in which the human PCNA gene or cDNA were flanked by either PCNA or SV40 sequences. These constructs were transfected into TK−tsl3 cells to generate cell lines derived from multiple clones and expressing human PCNA mRNA, which is easily distinguishable in Northern blots from hamster PCNA mRNA (Chang et al., 1990). These cell lines were then tested for the expression of human PCNA mRNA at both permissive and restrictive temperatures. The results unequivocally show that the PCNA promoter is responsible for the failure of expression of the PCNA gene at the restrictive temperature. Presence of absence of introns, and the 3’ flanking sequence do not seem to play an appreciable role in the regulation of the PCNA gene under these conditions. We have already shown in previous papers that the endogenous hamster PCNA gene is completely inhibited at the restrictive temperature (Jaskulski et al., 1988b; Travali et al., 1991; Koniecki et al., 1991), like the transfected human PCNA gene.
By run-on transcription and by RT-PCR we show that the inhibition is at the transcriptional level. Furthermore, we show that a −73 bp PCNA promoter is fully incapable of expression at the restrictive temperature. This inhibition, therefore, must be due to a specific mechanism, specific not to PCNA but to a subset of genes like PCNA, while the SV40 promoter and the promoters of early growth-regulated genes are not affected by this mechanism. We are cognizant of the fact that PCNA mRNA levels in G0 cells vary with different constructs, and this is the subject of further investigation. In this paper, we concentrate only on the difference between cells serum-stimulated at either permissive or restrictive temperatures. As mentioned above, it is well established that the expression of the PCNA gene is growth-regulated and that this regulation occurs at both transcrip-tional and posttranscriptional levels (Ottavio et al., 1990; Chang et al., 1990; Shipman et al., 1988, 1990). Intron 4 (Chang et al., 1990) and intron 1 (Alder et al., 1992) also play a role in regulating PCNA mRNA levels. The growth-regulated expression of the PCNA gene is still evident when the gene is under the control of a promoter consisting of the first 73 bp immediately upstream of the transcription initiation site (Pietrzkowski et al., 1991). In G1-specific ts mutants whose growth is arrested at the restrictive temperature, the PCNA promoter does not direct transcription, whether full-length or reduced to 73 bp.
This 73 bp promoter contains an enhancer, located between −73 and −45 (Pietrzkowski et al., 1991). It also contains a direct repeat of the sequence ACGCGG, which matches in 5 out of 6 nucleotides a sequence present in the region immediately upstream of the CAP site in all yeast DNA synthesis genes (Lowndes et al., 1991) and is essential for cell cycle regulation (Lowndes et al., 1991; Gordon and Campbell, 1991). Even in yeast, two genes have this sequence as 5 out of 6 matches, so that the ACGCGG sequences in the human PCNA gene are reasonable candidates for a similar cell cycle role. Incidentally, the 5′ flanking regions of both the human DNA polymerase a gene (Wong et al., 1988) and the human thymidine kinase gene (Flemington et al., 1987) have similar sequences. The PCNA 73 bp promoter binds proteins by gel shift analysis, and the binding is cell cycle-dependent (Pietrzkowski et al., 1991). By methylation interference analysis, protein binding also involves the distal ACGCGG sequence (Pietrzkowski et al., 1991), suggesting that this sequence plays a role in the growth regulation of the PCNA gene. The direct repeats are actually located in the 46 bp immediately upstream of the CAP site, and it is of interest that a 45 bp promoter, although weak because of the loss of the enhancer, is still growth-regulated (Chang et al., 1990; Pietrzkowski et al., 1991).
Our results indicate that, in cells stimulated at the restrictive temperature, a factor (or factors) is missing that is necessary for the function of the −73 bp PCNA promoter. Such factors do not seem to be required for the function of the SV40 promoter. Although we have studied in this paper only these two promoters, it is possible that the promoters of early growth-regulated genes (like c-myc, expressed at the restrictive temperature) may behave like the SV40 promoter, while the G1-S boundary genes will be similar to the PCNA promoter. These findings suggest that this model can be used to isolate a factor (or factors) necessary for the transcription of DNA synthesis genes.
Acknowledgments
This work was supported by grants GM-33694 and GM-42383 from the National Institutes of Health.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Alder H., Yoshinouchi M., Prystowsky M. B., Appasamy P., and Baserga R. (1992), Nucl Acids Res 20, 1769–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almendral J. M., Huebsch D., Bluendell P. A., MacDonald-Bravo H., and Bravo R. (1987), Proc Natl Acad Sci USA 84, 1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baserga R. (1991), J Cell Sci 98, 433–436. [DOI] [PubMed] [Google Scholar]
- Bravo R., Frank R., Blundell P. A., and MacDonald-Bravo H. (1987), Nature (London) 326, 515–517. [DOI] [PubMed] [Google Scholar]
- Chang C.-D., Ottavio L., Travali S., Lipson K. E., and Baserga R. (1990), Mol Cell Biol 10, 3289–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P. and Sacchi N. (1987), Anal Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Feinberg A. P. and Vogelstein B. A. (1983), Anal Biochem 132, 6–13. [DOI] [PubMed] [Google Scholar]
- Flemington E., Bradshaw H. D., Traina-Dorge V., Slagel V., and Deininger P. A. (1987), Gene 52, 267–277. [DOI] [PubMed] [Google Scholar]
- Gordon C. B. and Campbell J. L. (1991), Proc Natl Acad Sci USA 88, 6058–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groudine M., Peretz M., and Weintraub H. (1981), Mol Cell Biol 1, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning P., Okayama E., Blau H., and Kedes L. (1983), Mol Cell Biol 3, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn R. R., Aller P., Yuan Z-A., Gibson C. W., and Baserga R. (1984a), Proc Natl Acad Sci USA 82, 6004–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn R. R., Marashi F., Baserga R., Stein J., and Stein G. (1984b), Biochemistry 23, 3731–3735. [DOI] [PubMed] [Google Scholar]
- Ide T., Ninomiya-Tsuji J., Ferrari S., Philiponis V., and Baserga R. (1986), Biochemistry 25, 7041–7046. [DOI] [PubMed] [Google Scholar]
- Jaskulski D., DeRiel J. K., Mercer W. E., Calabretta B., and Baserga R. (1988a), Science 240, 1544–1546. [DOI] [PubMed] [Google Scholar]
- Jaskulski D., Gatti G., Travali S., Calabretta B., and Baserga R. (1988b), J Biol Chem 263, 10175–10179. [PubMed] [Google Scholar]
- Koniecki J., Nugent P., Kordowska J., and Baserga R. (1991), Cancer Res 51, 1465–1471. [PubMed] [Google Scholar]
- Lipson K. E. and Baserga R. (1989), Proc Natl Acad Sci USA 86, 9774–9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson K. E., Chen S. T., Koniecki J., Ku D-H., and Baserga R. (1989), Proc Natl Acad Sci USA 86, 6848–6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A. Y. and Lee A. S. (1984), Proc Natl Acad Sci USA 81, 988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.-T., Baserga R., and Mercer W. E. (1985), Mol Cell Biol 5, 2936–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. C., Marraccino R. L., Keng P. C., Bambara R. A., Lord E. M., Chou W. G., and Zain S. B. (1989), Biochemistry 28, 2967–2974. [DOI] [PubMed] [Google Scholar]
- Lowndes N. F., Johnson A. L., and Johnston L. H. (1991), Nature (London) 350, 247–250. [DOI] [PubMed] [Google Scholar]
- Okayama H. and Berg P. A. (1983), Mol Cell Biol 3, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottavio L., Chang C. D., Rizzo M.-G., Petralia S., Travali S., and Baserga R. (1990), Biochem Biophys Res Commun 169, 509–516. [DOI] [PubMed] [Google Scholar]
- Pietrzkowski Z., Alder H., Chang C.-D., Ku D.-H., and Baserga R. (1991) Exp Cell Res 193, 283–290. [DOI] [PubMed] [Google Scholar]
- Prelich G., Tan C. K., Kostura M., Mathews M. B., So A. G., Downey K. M., and Stillman B. (1987a), Nature (London) 326, 517–520. [DOI] [PubMed] [Google Scholar]
- Prelich G., Kostura M., Marshak K. D. R., Mathews M. B., and Stillman B. (1987b), Nature (London) 326, 471–475. [DOI] [PubMed] [Google Scholar]
- Rappolee D. A., Wang A., Mark D., and Werb Z. (1988), Science 241, 708–712. [DOI] [PubMed] [Google Scholar]
- Rappolee D. A., Wang A., Mark D., and Werb Z. (1989), J Cell Biochem 39, 1–11. [DOI] [PubMed] [Google Scholar]
- Saiki R. K., Gelfand D. H., Stoffel S., Sharf S. J., Higuchi R., Horn G. T., Mullis K. B., and Erlich H. A. (1988), Science 239, 487–497. [DOI] [PubMed] [Google Scholar]
- Sell C., Chen H., and Baserga R. (1992), Biotechniques 12, 692–694. [PubMed] [Google Scholar]
- Shen Y. M., Hirschhorn R. R., Mercer W. E., Surmacz E., Tsutsui Y., Soprano K. J., and Baserga R. (1982), Mol Cell Biol 2, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman P. M., Sabath D. E., Fisher A. H., Comber P. G., Sullivan K., Tan E. M., and Prystowsky M. B. (1988), J Cell Biochem 38, 189–198. [DOI] [PubMed] [Google Scholar]
- Shipman-Appasamy P., Cohen K. S., and Prystowsky M. B. (1990), J Biol Chem 265, 19180–19184. [PubMed] [Google Scholar]
- Talavera A. and Basilico C. (1977), J Cell Physiol 92, 425–436. [DOI] [PubMed] [Google Scholar]
- Thomas P. (1983), Methods Enzymol 100, 255–266. [DOI] [PubMed] [Google Scholar]
- Travali S., Lipson K. E., Jaskulski D., Lauret E., and Baserga R. (1988), Mol Cell Biol 8, 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travali S., Ku D-H., Rizzo M-G., Ottavio L., Baserga R., and Calabretta B. (1989), J Biol Chem 264, 7466–7472. [PubMed] [Google Scholar]
- Travali S., Ferber A., Reiss K., Sell C., Koniecki J., Calabretta B., and Baserga R. (1991), Oncogene 6, 887–894. [PubMed] [Google Scholar]
- Tsurimoto T. and Stillman B. (1990), Nature 346, 534–539. [DOI] [PubMed] [Google Scholar]
- Venturelli D., Travali S., and Calabretta B. (1990), Proc Natl Acad Sci USA 87, 5963–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. W., Wahl A. F., Yuan P. M., Arai N., Pearson B. E., Arai K., Horn D., Hunkapillar M. W., and Wang T. S. F. (1988), EMBO J 7, 3767–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]