

Abstract
The skeletal α-actin gene is a member of the sarcomeric contractile protein gene family and is specifically expressed in differentiated muscle. The skeletal α-actin gene is regulated efficiently by enhancer and regulatory sequences between nucleotide positions −1282 and −87. In the present study we have shown that the sequences 3′ of nucleotide position −87 can functionally interact with the SV40 enhancer in a tissue-specific manner and can restrict the ubiquitous function of the SV40 enhancer to myogenic cells. Site-specific cassette mutagenesis was used to delimit the sequences upstream of the TATA motif (−32), between nucleotide positions −64 and −37, that mediate efficient expression in myogenic cells in the presence of the SV40 enhancer. The skeletal α-actin promoter was trans-activated by the helix-loop-helix (HLH) transcription factors MyoD, MRF-4, and Myogenin, in pluripotential 10T1/2 fibroblasts and trans-repressed by the HLH protein Id (inhibitor of differentiation) in myogenic C2C12 cells. This trans-regulation required sequences upstream of −87 and occurred independently of the two consensus E boxes (CANNTG) at positions +18 and +71. The −64/−37 region interacted with purified Spl and an unidentified protein(s), proximal regulatory factor(s) I (PRF-I). We conclude that the muscle-specific expression of the skeletal α-actin promoter is not simply determined by MyoD elements and enhancer and regulatory sequences, but that the minimal promoter contains important determinants of cell-specific transcription that can function independently of the helix-loop-helix transcription factors.
A current hypothesis on muscle development is that myogenic determination appears to be controlled by the activation of one or more determination loci, probably by a hypomethylation mechanism (Konieczny and Emerson, 1984). The activation of these myogenic loci causes profound changes in gene expression by inducing a number of myogenic regulatory genes, the MyoD family: MyoDl (Davis et al., 1987), Myogenin (Edmonson and Olson, 1989; Wright et al., 1989), Myf-5 (Braun et al., 1989a; Braun et al., 1990b), MRF-4 (herculin, Myf-6; Rhodes and Konieczny, 1989; Braun et al., 1990a; Miner and Wold, 1990), and Myd (Pinney et al., 1988). The MyoD family of genes encode regulatory proteins that can direct the fate of pluripotential 10T1/2 mesodermal embryonic cells and indirectly or directly lead to the activation of muscle-specific genes in a variety of differentiated cells (Davis et al., 1987; Pinney et al., 1988; Braun et al., 1989a and 1990a; Edmonson and Olson, 1989; Rhodes and Konieczny, 1989; Weintraub et al., 1989; Wright et al., 1989; Choi et al., 1990; reviewed and summarized in Olson, 1990; Weintraub et al., 1991a). The MyoD family is expressed only in skeletal muscle (Davis et al., 1987; Braun et al., 1989a; Edmonson and Olson, 1989; Rhodes and Konieczny, 1989; Wright et al., 1989; Miner and Wold, 1990; Braun et al., 1990a). MyoD is a nuclear protein that binds to many muscle-specific enhancers (Lassar et al., 1989) that contain a consensus DNA-binding sequence that includes a CANNTG motif, present in most muscle-specific enhancers (Lassar et al., 1989; Blackwell and Weintraub, 1990; Weintraub et al., 1991a). The majority of muscle-specific enhancers contain multiple MyoD binding sites (Lassar et al., 1989; Blackwell and Weintraub, 1990; Weintraub et al., 1991). Efficient trans-activation by MyoD complexes requires two or more interaction sites (Lassar et al., 1989; Wentworth et al., 1991; Piette et al., 1990; Lin et al., 1991; Weintraub et al., 1990). Under certain circumstances, a single MyoD binding site can activate transcription if binding sites for other constitutively expressed binding sites are also present and occupied (Sartorelli et al., 1990; French et al., 1991).
All members of the MyoD family contain a 68 amino acid basic/myc-like region that is proposed to form a helix-loop-helix (HLH) motif, which is necessary and sufficient for myogenic conversion (Tapscott et al., 1988), and which mediates DNA binding and dimerization (Murre etal., 1989a,b; Sun and Baltimore, 1991). MyoDl forms regulatory heterodimers with the ubiquitously expressed and alternatively spliced products of the E2A gene that may precisely control cell determination (Murre et al., 1989a,b; Davis et al., 1990; Lassar et al., 1991). The formation of these functional regulatory heterodimers is negatively regulated by another ubiquitously expressed helix-loop-helix protein, designated Id (inhibitor of differentiation; Benezra et al., 1990; Sun and Baltimore, 1991).
The function of these regulators is modulated by environmental cues related to the concentration of growth factors/receptors and hormones. A number of growth-promoting agents inhibit myogenesis, including fibroblast growth factor (FGF), transforming growth factor-β (TGF-β), proliferin, adenovirus Ela, jun, fps, erbA, myc, fos, and the oncogenic form of rasp21 (summarized and discussed in Spizz et al., 1987; Webster et al., 1988; Vaida et al., 1989; Wilder and Linzer, 1989; Olson, 1990; Muscat et al., 1991; Weintraub et al., 1991).
We have previously demonstrated by transient transfection analyses, mutagenesis, and in vivo competition that the sequences between nucleotide positions −1282 and −87 of the human skeletal α-actin gene are sufficient and necessary for optimal muscle-specific expression and developmental regulation during myogenesis. These sequences have been shown to act as enhancers and modulators of transcription when linked to heterologous promoters (Muscat and Kedes, 1987; Muscat et al., 1992). We have now identified many different binding proteins, including MEF-2 (Gosset et al., 1989; Muscat et al., 1992), Spl (Gidoni et al., 1985; Kadonaga et al., 1986; Collie and Muscat, 1992), NF-l/CTF (Jones et al., 1987; Santoro et al., 1988; Collie and Muscat, 1992), TR, the thyroid hormone receptor (Collie and Muscat, 1992), CArG binding factor (CBF), an SRF-like protein (Muscat et al., 1988 and 1991), and two unidentified nuclear factors, DRF-1 and DRF-3 (Muscat et al., 1992). These factor-binding activities appear to be differentially expressed during myogenesis. The large number of binding sites for such ubiquitous factors (Spl, CTF, and CBF) within the α-actin promoter suggests that the complex regulation and cell-specificity of this gene during myogenesis involves combinatorial mechanisms (Muscat et al., 1988 and 1992).
To elucidate the molecular mechanisms that participate in the tissue-specific transcription of the skeletal α-actin gene, we (1) characterized the minimal cis-acting region that mediated myogenic specific expression in the presence of the SV40 enhancer; (2) examined the effects of the positive and negative helix-loop-helix trans-regulators (e.g., MyoD, Myogenin, MRF-4, and Id) on the human skeletal α-actin gene; and (3) identified proteins which interacted with the minimal cis-acting sequences required for myogenic-specific expression. In this study we show that the sequences 3′ of nucleotide position −87 can functionally interact with the SV40 enhancer in a tissue-specific manner and restrict the ubiquitous function of the SV40 enhancer to myogenic cells. Mutagenesis was used to delimit the sequences that mediate efficient expression in myogenic cells in the presence of the SV40 enhancer, upstream of the TATA motif (−32), between nucleotide positions −64 and −37. The skeletal α-actin promoter was trans-activated by MyoD, MRF-4, and Myogenin, in pluripotential 10T1/2 fibroblasts and trans-repressed by Id, in myogenic C2C12 cells. This trans-regulation required sequences upstream of −87 and occurred independently of the two consensus E-box motifs at positions +18 and +71. We conclude that the muscle-specific expression of the skeletal α-actin promoter is not simply determined by MyoD elements and enhancer and regulatory sequences and hypothesize that the cis-acting sequences involved in the regulation of tissue-specific expression and trans-activation/repression can be physically and functionally distinct in myogenic promoters.
Materials and methods
Cell culture and transfection
Mouse myogenic C2 cells (Yaffe and Saxel, 1977a; Yaffe and Saxel, 1977b; Muscat et al., 1987) were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 20% fetal calf serum (FCS) in 10% CO2 as described previously (Muscat et al., 1991). This cell line was induced to biochemically and morphologically differentiate into multinucleate myotubes by mitogen withdrawal (DMEM supplemented with 2% fetal calf serum in 10% CO2). Differentiation was essentially complete within 72–96 hours with respect to isoform switching in the actin multigene family (Bains et al., 1984). However, these cells will spontaneously differentiate at a very high confluence (100%) in the presence of mitogens. CV-1 and 10T1/2 fibroblasts were grown in DMEM supplemented with 10% FCS in 10% CO2.
Each 60 mm dish of cells was transiently transfected with 10 μg of reporter plasmid DNA expressing CAT, mixed with an additional amount of cotransfected expression vector or pUC18 DNA. The total amount of DNA in each transfection experiment (11 μg) was kept constant by the addition of pUC18 DNA. Twenty-four hours prior to transfection, the cells were placed on an appropriate number of plates. The DNA mixtures were cotransfected into C2 myoblasts, CV-1, and 10T1/2 fibroblasts by the liposome-mediated procedure. We used the cationic lipid DOTAP, N-[1-(2,3-Dioleoyloxy) propyl]-N, N, N-tri-methyl-ammonium-methyl-sulphate. Unilamellar vesicles were formed by mixing the appropriate DNAs with 30–40 μl of DOTAP and 1X HEPES buffered saline to a total volume of 200 μl. After a 10-minute incubation at room temperature, this mixture was combined with 6 ml fresh culture medium and added to the cells, which were between 50 and 70% confluence. After a period of 20–24 hours, fresh medium was added to the cells. The cells were harvested for the assay of CAT enzyme activity 48–72 hours post transfection. Each transfection experiment was performed at least twice, using two different plasmid preparations in order to overcome the variability inherent in transfections.
CAT assays
The cells were harvested and the CAT activity was measured as previously described (Muscat and Kedes, 1987; Muscat et al., 1988). Aliquots of the cell extracts were incubated at 37°C, with 0.1–0.4 μCi of 14C-chloramphenicol (Amersham) in the presence of 5 mM acetyl CoA and 0.25 M Tris-HCl pH 7.8. After a 2- to 4-hour incubation period, the reaction was stopped by the addition of 1 ml ethyl acetate, which was used to extract the chloramphenicol and its acetylated forms. The extracted materials were analyzed on silica gel thin-layer chromatography plates, as described previously (Muscat and Kedes, 1987; Muscat et al., 1988). Quantitation of CAT assays was performed by scintillation counting of the chromatograms.
Plasmids
The promoter-CAT expression plasmids used in this study, pSV2CAT (Weintraub et al., 1989); the skeletal α-actin plasmids pHSA2000CAT (Muscat and Kedes, 1987; Muscat et al., 1988) and pHSA87CAT (Muscat and Kedes, 1987; Muscat et al., 1988); and the cardiac α-actin plasmids pHCA485CAT (Minty et al., 1986; Minty and Kedes, 1986) and pHCA47CAT (Minty et al., 1986; Minty and Kedes, 1986) have been described previously. Briefly, the skeletal α-actin plasmids pHSA2000CAT and pHSA87CAT carry a 2300 base pair (bp) fragment (position −2000 to +239) and a 326 bp fragment (position −87 to +239) respectively, adjacent to the CAT gene. The cardiac α-actin plasmids pHCA485CAT and pHCA47CAT carry a 553 bp fragment (position −485 to +68) and a 115 bp fragment (position −47 and +68), respectively, adjacent to the CAT gene. These designated nucleotide (− and +) positions are all with respect to the start of transcription at +1. The plasmids expressing the helix-loop-helix transcription factors MyoD (Davis et al., 1987), Myogenin (Edmonson and Olson, 1989), MRF-4 (Edmonson and Olson, 1989), and Id (Benezra et al., 1990) driven by the Moloney sarcoma virus LTR were described previously.
Plasmid construction
In general the procedures suggested by Sambrook et al. (1989) were followed. The plasmid pCAT 3′SV was constructed (R. P. Wade, Jr.) by cloning the SV40 enhancer into the BamH I site 3′ of the CAT gene in pHCAoCAT (Minty and Kedes, 1986). The Cla I 5′ of the CAT gene was converted to Bgl II, and the BamH I/Hind III segment of the mpl8 polylinker was cloned into the Bgl II/Hind III site of pHCAoCAT The plasmid pHSA87CAT-3′SV was constructed by cloning the Xho I/Hind III fragment from pHSA87CAT containing the sequences between nucleotide positions −87/+239 into Sal I/Hind III cut pUC18. This clone was then cleaved with Xba I and Hind III, and the insert containing the α-actin promoter sequences was cloned into pCAT-3′SV. We constructed pHSA87CAT-3′SV M1, M2, and M3 and pHSA37CAT-3′SV and pHSA37CAT-3′SV M4 by PCR using the plasmid pHSA87CAT-3′SV as the template. The 5′ primers for PCR in each case were constructed to include an Xba I cloning site. The 3′ primer for all PCR reactions in this series was a CAT primer from map positions 4955–4972 in pSV2CAT (5′ CGGTGGTATATCCAGTGAT 3′). The 5′ primer sequences used in the construction of pHSA87CAT-3′SV M1, M2, and M3 and pHSA37CAT-3′SV and pHSA37CAT-3′SV M4 that carry the respective mutations were:
GGTCCTCTAGAGAAATTTCCGGCAGCGAC
GGTCCTCTAGAGTCGAGAAGTTACTTCTCATTCCTGCGGGG
GGTCCTCTAGAGTCGAGAAGGGCAGCGAGGAATTTACGGGGTGG
GGCCTCTAGAGCGGGCTATATAAAACCTGAGCAG
GGCCTCTAGATTATTTATACCTGAGCAGAGGGACAAGCGGCC
The resultant PCR products were cleaved with Xba I and Hind III and cloned into Xba I and Hind III cleaved pCAT-3′SV. We constructed pHSA87/30CAT-3′SV M1, pHSA87/30CAT-3′SV M1 and M5, pHSA37/30CAT-3′SV, and pHSA37/ 30CAT-3′SV M5 by PCR using the plasmid pHSA87CAT-3′SV as the template. The 5′ primers for PCR in each case were constructed to include an Xba I cloning site. The 3′ primers for all PCR reactions in this series were constructed to contain a Hind III cloning site. The 5′ and 3′ primers for the construction of each clone are listed as consecutive pairs.
-
pHSA87/30CAT-3′SV Ml
GGTCCTCTAGAGAAATTTCCGGCAGCGAC
GGAAGCTTGCGAGGCTTCACTTGGCGCTGTCC
-
pH5A87/30CAT-3′SV Ml, M5
GGTCCTCTAGAGAAATTTCCGGCAGCGAC
GGAAGCTTGCGAGGCTTCCCGGGGCGCTGTCC
-
pHSA37/30CAT-3′SV
GGCCTCTAGAGCGGGCTATATAAAACCTGAGCAG
GGAAGCTTGCGAGGCTTCACTTGGCGCTGTCC
-
pHSA37/30CAT-3′SV M5
GGCCTCTAGAGCGGGCTATATAAAACCTGAGCAG
GGAAGCTTGCGAGGCTTCCCGGGGCGCTGTCC
The resultant PCR product in each case was cleaved with Xba I and Hind III and cloned into Xba I and Hind III cleaved pCAT-3′SV. All clones were sequenced by double-stranded plasmid sequencing on one strand.
Nuclear extracts and gel mobility shift assays
Nuclear extracts were prepared by the method of Schreiber et al. (1989), with the addition of 1 mM phenylmethylsulfonyl fluoride (PMSF) and 2 μg/ml of aprotinin and leupeptin to all solutions (Boehringer Mannheim). Nuclear proteins were extracted with 0.4 M NaCl, 20 mM HEPES(N-2-hydroxyethylpiperazine-N′-2-ethane-sulfonic acid, pH 7.9), 20 (v/v)% glycerol, 100 mM KC1,0.2 mM EDTA (ethylenediaminetetraacetic-acid), 1 mM EDTA, 1 mM EGTA, 1 mM dithio-threitol, 1 mM PMSF, and 2 μg/ml aprotinin and leupeptin. Each binding mixture (25 μl) contained 1–2 μg of a T4 polynucleotide kinase-labeled DNA fragment, 5–10 μg of protein, 5 μg of BSA, and 3 μg of pUC18 plasmid DNA digested with Msp I or poly(dI-dC)poly(dI-dC) as a non-specific competitor (buffer C in Dignam et al., 1983). The assays were incubated at room temperature for 20 minutes and electrophoresed through a 4–6% (20:1 polyacrylamide:bisacrylamide) gel in 80 mM Tris borate and 2 mM EDTA, pH 8.3. Gels were briefly soaked in 10% acetic acid, dried, and autoradiographed (Dignam et al., 1983).
Results
The sequences downstream of positions −87 and −47 of the skeletal and cardiac α-actin promoters, respectively, restrict the activity of the SV40 enhancer to muscle
The cis-acting sequences between nucleotide positions −1282 and −87 have been shown to interact with a variety of sequence-specific DNA-binding proteins. These factor-binding activities are differentially expressed during myogenesis, and the functional role of these sites was verified by site-specific mutagenesis. Unfortunately, in vitro binding activities indistinguishable from those found in muscle cells were also detected in HeLa cells, although the relative ratios were different. To elucidate the role of the skeletal α-actin sequences between nucleotide positions −87/+239 in myogenic-specific expression, we sub-cloned this region into a promoterless CAT construct, pCAT-3′SV, containing the SV40 enhancer in the plasmid backbone, 3′ of the CAT gene linked to the SV40 splicing and polyadenylation signals (Fig. 1A). The sequence of the cis-acting region cloned into this vector is shown in Figure 1B. This plasmid allowed verification and testing of putative promoter sequences, since the presence of the enhancer in many cases will augment promoter mediated transcription. We similarly cloned the cardiac α-actin promoter between nucleotide positions −47/+68. The skeletal and cardiac clones were known as pHSA87CAT-3′SV and pHCA47CAT-3′SV, respectively. These constructs were then transfected into C2C12 myogenic cells (Fig. 2) and non-muscle CV-1 fibroblasts (Fig. 3). We also transfected additional plasmids as controls for relative expression in both cells: pSV2CAT, containing the SV40 early promoter/enhancer linked to CAT; pCAT-3′SV, the promoterless test vector; pHSA2000CAT, the native skeletal α-actin promoter (containing sequences between nucleotide positions −2000/+239) linked to CAT; pHCA485CAT, the native cardiac α-actin promoter (containing sequences between nucleotide positions −485/+68) linked to CAT; pHSA87CAT, the deleted skeletal α-actin promoter (containing sequences between nucleotide positions −87/+239) linked to CAT; and pHCA47CAT, the deleted cardiac α-actin promoter (containing sequences between nucleotide positions −47/+68) linked to CAT.
Figure 1.
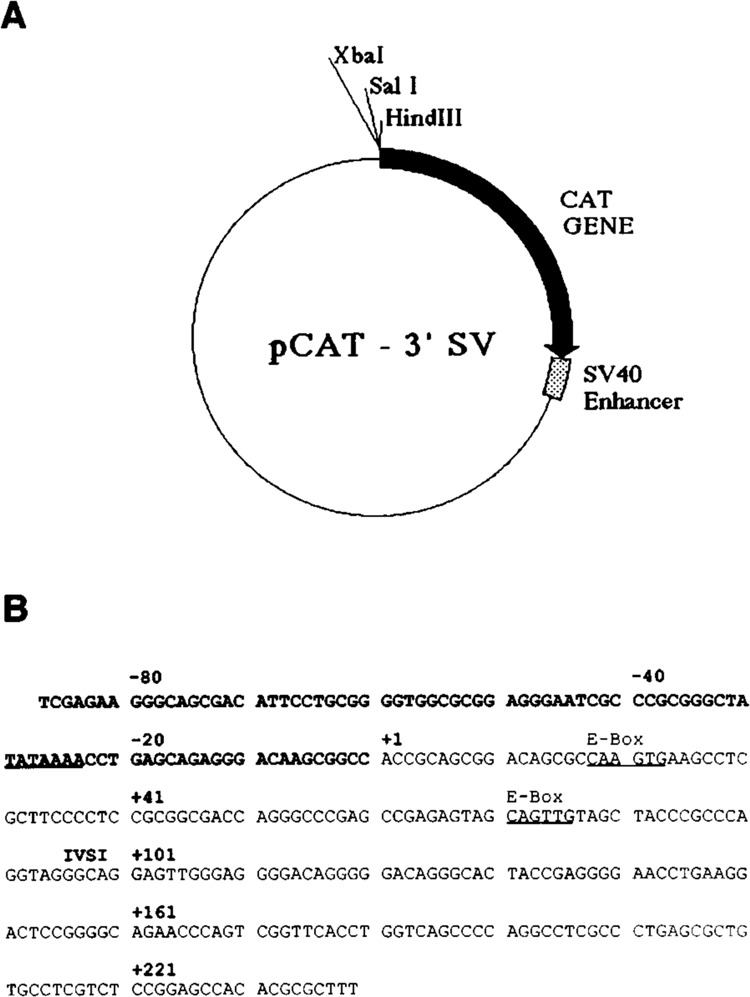
A. Prototype recombinant plasmid pCAT-3′SV, used for the construction of the various promoter mutants. This vector contains a promoterless CAT gene with the SV40 enhancer situated at the 3′ end of the CAT reporter gene. B. The nucleotide sequence of the human skeletal α-actin gene between nucleotide positions −87 and +239. The TATA and E-box motifs are underlined.
Figure 2.
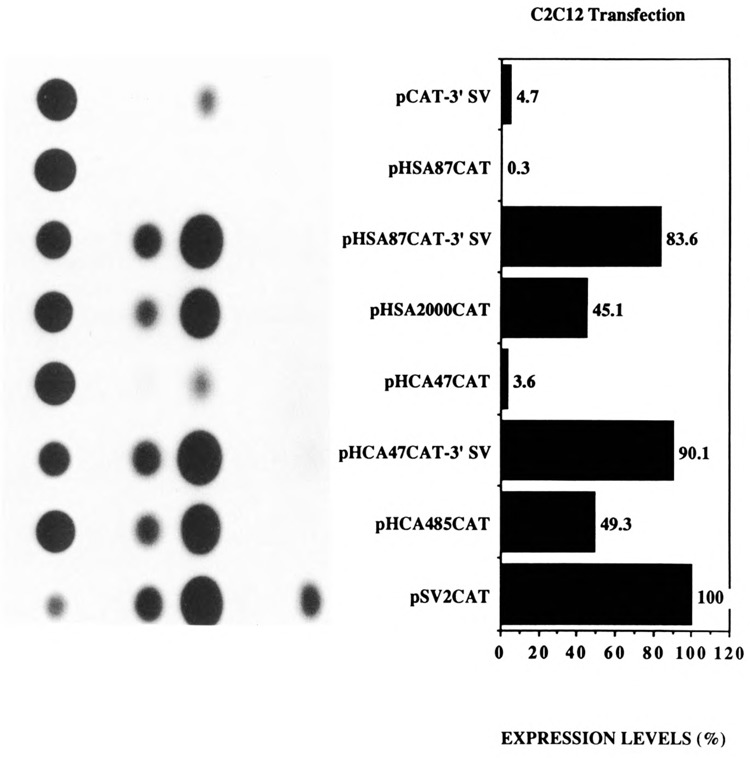
Transient CAT assays in myogenic C2C12 cells demonstrating the cooperative interaction between the myogenic basal promoter sequences and the SV40 enhancer in muscle cells. Expression levels are shown as a percentage relative to pSV2CAT, set at 100%. The plasmids pCAT-3′SV, pHSA87CAT, pHSA87CAT-3′SV, pHSA2000CAT, pHCA47CAT, pHCA47CAT-3′SV, pHCA485CAT, and pSV2CAT are described in Materials and Methods.
Figure 3.
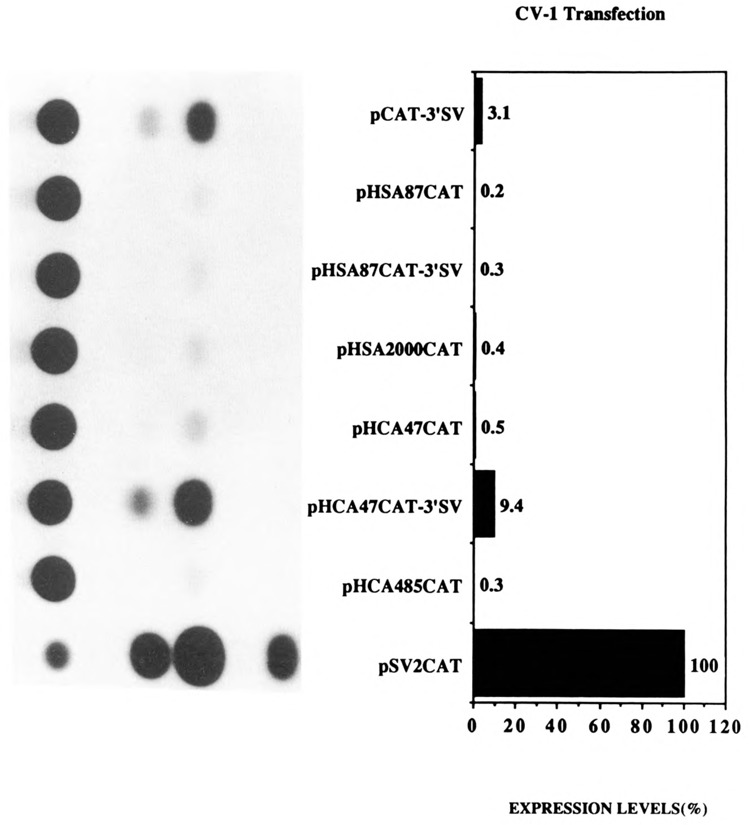
Transient CAT assays in CV-l non-muscle cells demonstrating that the myogenic basal promoter sequences restrict the function of the SV40 enhancer to myogenic cells. Expression levels are shown as a percentage relative to pSV2CAT, set at 100%. The plasmids pCAT-3′SV, pHSA87-CAT pHSA87CAT-3′SV, pHSA-2000CAT, pHCA47CAT, pHCA-47CAT-3′SV, pHCA485CAT, and pSV2CAT are described in Materials and Methods.
As expected, and as previously published, the plasmids pHSA2000CAT and pHCA485CAT expressed high levels of CAT relative to pSV2CAT in myogenic C2C12 cells (Fig. 2; Muscat and Kedes, 1987; Muscat et al., 1988; Minty et al., 1986; Minty and Kedes, 1989). Furthermore, in agreement with the published data, pHSA87CAT (Muscat and Kedes, 1987; Muscat et al., 1988) and pHCA47CAT (Minty et al., 1986; Minty and Kedes, 1986) expressed poorly (30–100 fold < native promoters; Fig. 2). As previously reported, these myogenic promoter constructs were tissue-specific (Muscat and Kedes, 1987; Muscat et al., 1988; Minty et al., 1986; Minty and Kedes, 1989) and did not function in CV-1 fibroblasts relative to the SV40 promoter pSV2CAT (Fig. 3). Interestingly, the presence of the SV40 enhancer, 3′ of the CAT gene in plasmids pHSA87CAT-3′SV and pHCA47CAT-3′SV, restored the expression of the basal α-actin sequences in muscle cells to native levels (Fig. 2). The plasmids pHSA87CAT-3′SV and pHCA47CAT-3′SV expressed 16- to 20-fold more efficiently than the basal vector pCAT-3′SV in C2C12 cells (Fig. 2). These levels of expression in myogenic cells were mediated by the basal promoter regions of the α-actin genes, since pCAT-3′SV expressed poorly in muscle cells (Fig. 2). Unexpectedly, pHSA87CAT-3′SV and pHCA47CAT-3′SV functioned poorly in CV-1 fibroblasts relative to the basal vector pCAT-3′SV (1- to 3-fold; Fig. 3). These data indicate that the sequences 3′ of nucleotide positions −87 and −47 for the skeletal and cardiac α-actin genes, respectively, can functionally interact with the SV40 enhancer in a tissue-specific manner and can restrict the ubiquitous function of the SV40 enhancer to myogenic cells. The data are in agreement with work from Bouvagnet et al. (1987), who showed that the introduction of the SV40 enhancer into embryonic skeletal myosin heavy chain gene CAT constructs in which all the upstream sequences were deleted strongly increased the level of expression of such truncated constructs without changing their muscle specificity.
Cis-acting sequences between nucleotide positions −64 and −37 are required for the efficient expression of the skeletal α-actin gene in muscle
We then used mutagenesis to delimit further the skeletal α-actin sequences between nucleotide positions −87/+239 that functioned preferentially in muscle cells. We constructed three mutations of the parent plasmid pHSA87CAT-3′SV (which expressed 17-fold more efficiently than the basal vector in C2C12 cells; Fig. 2). These plasmids, called pHSA87CAT-3′SV Ml, pHSA87CAT-3′SV M2, and pHSA87CAT-3′SV M3, had cassettes of 8 nucleotides mutated (as shown in Fig. 4) between nucleotide positions −87/−80, −79/−71, and −70/−63, respectively. These constructs functioned far more efficiently in C2C12 myogenic cells versus pluripotential 10T1/2 cells relative to the control pCAT-3′SV, arbitrarily set to 1: 12- to 20-fold in muscle versus 1- to 3-fold in non-muscle cells (Fig. 5). These data indicated that sequences between nucleotide positions −87/−63 were not significantly involved in either tissue-specific activity or in restricting the expression of the SV40 promoter to myogenic cells. The M2 and M3 mutations had minor effects on the tissue-specific expression of the gene. Hence, the sequences downstream of nucleotide position −63 were involved in the tissue-specific expression of the skeletal α-actin gene. We noted that the M3 mutation knocks out the M-CAT motif [CCATTCCT] described by Mar and Ordahl (1990).
Figure 4.
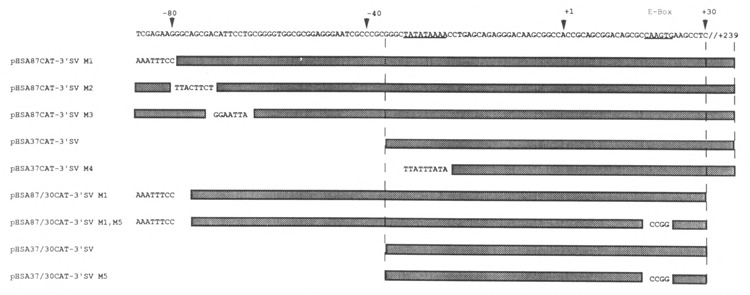
Schematic representation of the cis-acting native human skeletal α-actin sequences and respective mutations cloned into the Xba I/Hind III cloning sites of the pCAT-3′SV vector.
Figure 5.
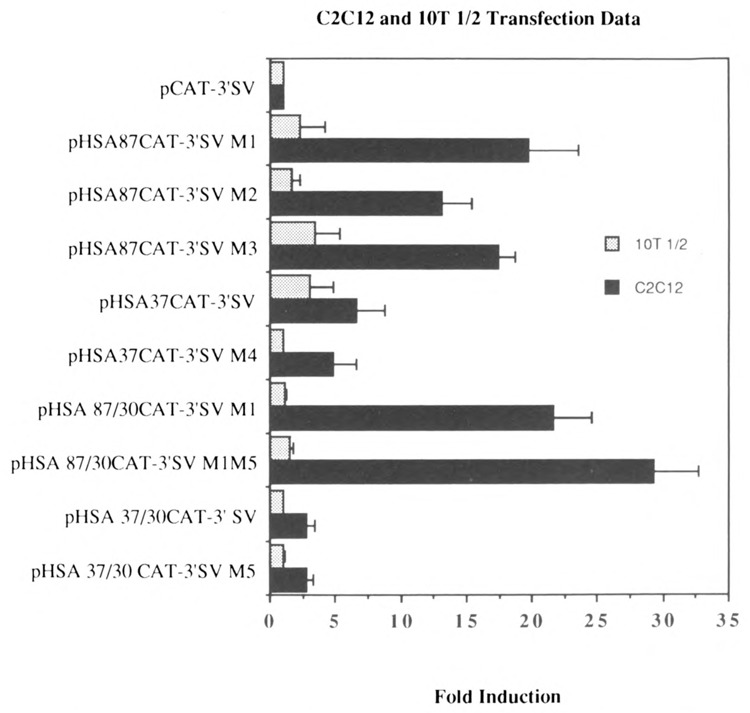
Transient CAT assays demonstrating the effect of various deletions and mutations on the transcriptional interaction between myogenic sequences and the SV40 enhancer in muscle and non-muscle cells. Shown here are the levels of expression of plasmids in myogenic C2 and pluripotential 10T1/2 cells relative to the basal control vector pCAT-3′SV, arbitrarily set at 1. The plasmids pHSA87CAT-3′SV Ml, pHSA87CAT-3′SV M2, and pHSA87CAT-3′SV M3 have cassettes of 8 nucleotides mutated between nucleotide positions −87/−80, −79/−71, and −70/−63 respectively in the cis-acting region between nucleotide positions −87/+239 (shown in Figure 4). The plasmids pHSA37CAT-3′SV and pHSA37CAT-3′SV M4 contained the sequences between nucleotide positions −37/+239, with M4 harboring an SV40 TATA motif rather than the native myogenic TATA box. The plasmid pHSA87/30CAT-3′SV M1 contained the sequences between −87 and +30 carrying the M1 mutation into the pCAT-3′SV vector and did not contain the E box at nucleotide position +71. The plasmid pHSA87/30CAT-3′SV M1 M5 was identical to the former plasmid, except that the E box at position +18 was mutated, as shown in Figure 4. The plasmid pHSA37/30CAT-3′SV contained the sequences between −37 and +30 cloned into the pCAT-3′SV vector and did not contain the E box at nucleotide position +71. This plasmid was then modified to construct the plasmid pHSA37/30CAT-3′SV M5, which was identical to the former plasmid except that the E box at position +18 was mutated, as shown in Figure 4. The results shown represent the mean ± SD of 3–5 independent experiments.
We then sub-cloned the sequences between nucleotide positions −37/+239 into the pCAT-3′SV vector. The plasmid was termed pHSA37CAT-3′SV. We constructed a mutation of this clone, pHSA37CAT-3′SV M4, which contained an SV40 TATA box in place of the native α-actin TATA box, as indicated in Figure 4. These plasmids did not express CAT efficiently in either myogenic (Fig. 5; 4- to 6-fold) or pluripotential 10T1/2 cells (Fig. 5; 1- to 3-fold) relative to the basal vector pCAT-3′SV. This indicated that the sequences between −63/−37 were necessary for the efficient expression of the α-actin gene in myogenic cells, and that the sequences between nucleotide positions −37 and +239 influence transcription minimally in muscle cells (∼3- to 5-fold) and non-muscle cells (<3-fold) in the presence of the SV40 enhancer.
We observed that two E boxes were located at nucleotide positions +18 and +71 in exon 1 of the skeletal α-actin gene. E boxes have been shown to interact with MyoD and other myogenic helix-loop-helix transcription factors and are necessary and sufficient for the tissue-specific trans-activation of a number of muscle-specific genes. We then investigated whether these E boxes were necessary for the efficient expression of the α-actin gene in myogenic cells. We cloned the sequences between −87 and +30 carrying the M1 mutation into the pCAT-3′SV vector; the resultant plasmid, pHSA87/30CAT-3′SV M1, did not contain the E box at nucleotide position +71.
We then constructed the plasmid pHSA87/30CAT-3′SV M1 M5, which was identical to the former plasmid except that the E box at position +18 was mutated, as shown in Figure 4. These constructs functioned far more efficiently in C2C12 myogenic cells versus pluripotential 10T1/2 cells relative to the control pCAT-3′SV (Fig. 5; 20- to 27-fold in myogenic cells versus 1- to 2-fold in non-muscle cells). These data indicated that sequences downstream of +18, including the E boxes at +18 and +71, were not involved in either tissue-specific expression or in restricting the influence of the SV40 promoter to myogenic cells. Hence, the sequences between nucleotide position −87 and +18 were involved in the tissue specific expression of the skeletal α-actin gene.
We then cloned the sequences between −37 and +30 into the pCAT-3′SV vector; the resultant plasmid, pHSA37/30CAT-3′SV, did not contain the E box at nucleotide position +71. This plasmid was then modified to construct the plasmid pHSA37/30CAT-3′SV M5, which was identical to the former plasmid except that the E box at position +18 was mutated, as shown in Figure 4. These plasmids did not express CAT efficiently in either myogenic or pluripotential 10T1/2 cells relative to the basal vector pCAT-3′SV (Fig. 5; 1- to 2-fold). This indicated that the sequences between −87/−37 were necessary to mediate efficient expression of the basal skeletal α-actin promoter in myogenic cells in the presence of the SV40 enhancer. The sequences between nucleotide positions −37 and +30 did not mediate efficient transcription relative to the basal vector (Fig. 5; <2-fold) in myogenic and non-muscle cells in the presence of the SV40 enhancer.
Trans-activation and repression of α-actin gene expression by the MyoD family and Id, respectively, required sequences upstream of −87
We then investigated whether the MyoD family of helix-loop-helix (HLH) proteins, including MyoD, Myogenin, and MRF-4, could transactivate the expression of the native human skeletal α-actin promoter (pHSA2000CAT) in pluripotential 10T1/2 cells. Figure 6, lanes 3–14 (relative to lanes 1 and 2) shows the trans-activation of pHSA2000CAT in 10T1/2 cells by increasing amounts of co-transfected MyoD, Myogenin, and MRF-4. Similarly, we investigated the effects of E12 and Id, anti-sense, and sense expression vectors, respectively, on the expression of pHSA2000CAT in myogenic C2C12 cells. E12 anti-sense expression did not inhibit the expression of pHSA2000CAT (Fig. 7, lanes 3–5 versus lanes 1 and 2). These data also indicated that the Moloney sarcoma virus LTR promoter driving the expression of the co-transfected E12 and Id was not non-specifically competing for general transcription factors in this assay system. However, co-transfected Id expression repressed the expression of pHSA2000CAT in myogenic cells (Fig. 7, lanes 6–8 versus lanes 1 and 2). We tested the ability of a wide variety of other plasmids with sequences downstream of nucleotide position −87 which contained consensus E boxes at nucleotide positions +18 and +71 in the human skeletal α-actin gene to be trans-activated by the MyoD family in 10T1/2 cells (Figs. 8A and B) or trans-repressed by Id (Figs. 9A, B, C, and D) in C2C12 cells. Trans-activation by the MyoD family required sequences upstream of −87 and occurred independently of two consensus E boxes (CANNTG) at +18 and +71 in 10T1/2 cells. Analogously, trans-repression by Id required sequences upstream of −87 and occurred independently of the two consensus E boxes (CANNTG) at +18 and +71 in myogenic C2C12 cells.
Figure 6.
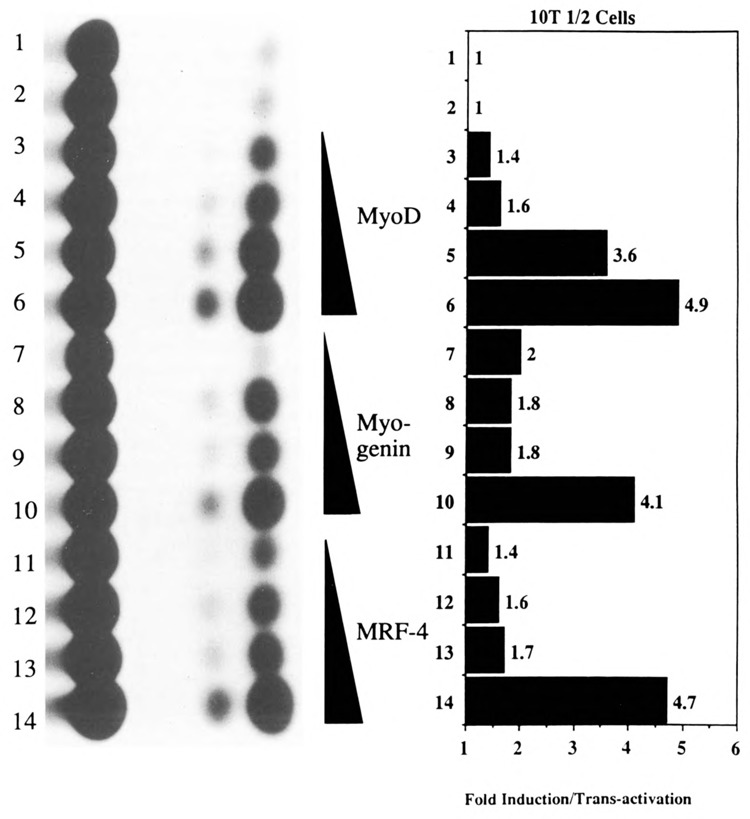
Transient CAT assays in 10T1/2 cells demonstrating the ability of the myogenic specific helix-loop-helix proteins to transactivate the human skeletal α-actin promoter. Lanes 1–12 contained the plasmid pHSA2000CAT (6 μg), which included the full-length native human skeletal α-actin promoter. Lanes 3–6 also contained 0.5, 1, 2, and 4.0 μg of co-transfected pEMSV-MyoD. Lanes 7–10 also contained 0.5, 1, 2, and 4.0 μg of co-transfected pEMSV-myogenin. Lanes 11–14 also contained 0.5, 1, 2, and 4.0 μg of co-transfected pEMSV-MRE-4. The total amount of DNA in each transfection was kept at a constant 10 μg with the carrier plasmid, pUC18.
Figure 7.
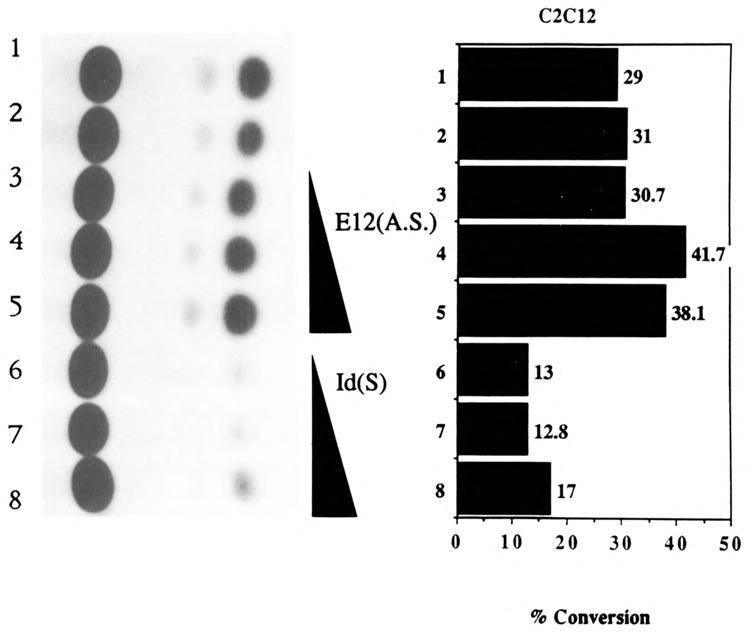
Transient CAT assays in C2C12 cells demonstrating the ability of the Id (inhibitor of differentiation) helix-loop-helix protein to trans-repress the human skeletal α-actin promoter. Lanes 1–8 contained the plasmid pHSA2000CAT (6 μg), which included the full-length native human skeletal α-actin promoter. Lanes 3–5 also contained 0.5, 1, and 2 μg of co-transfected pEMSV-E12(A.S.). Lanes 6–8 also contained 0.5, 1, and 2 μg of co-transfected pEMSV-Id. The total amount of DNA in each transfection was kept at a constant 10 μg with the carrier plasmid, pUC18.
Figure 8.
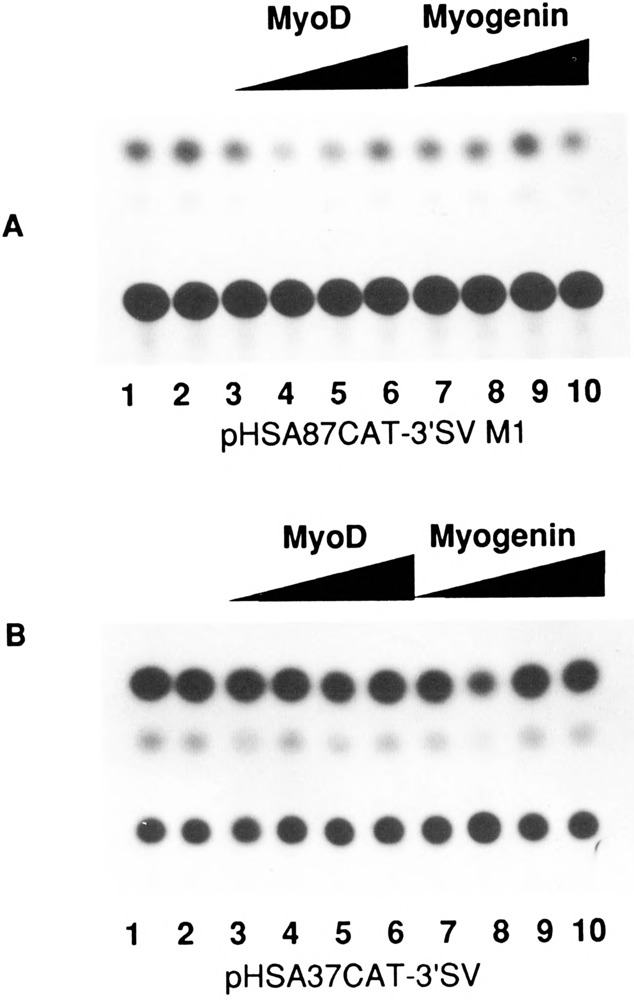
Transient CAT assays in 10T1/2 cells demonstrating the effect of the myogenic specific helix-loop-helix proteins on deleted skeletal α-actin promoters. A. Lanes 1–10 contained the plasmid pHSA87CAT-3′SV M1 (6 μg); Lanes 3–6 also contained 0.5, 1, 2, and 4.0 μg of co-transfected pEMSV-MyoD. Lanes 7–10 also contained 0.5, 1, 2, and 4.0 μg of co-transfected pEMSV-myogenin. The total amount of DNA in each transfection was kept at a constant 10 μg with the carrier plasmid pUC18. B. Lanes 1–10 contained the plasmid pHSA37CAT-3′SV, and lanes 3–10 were transfected as shown in Figure 9A.
Figure 9.
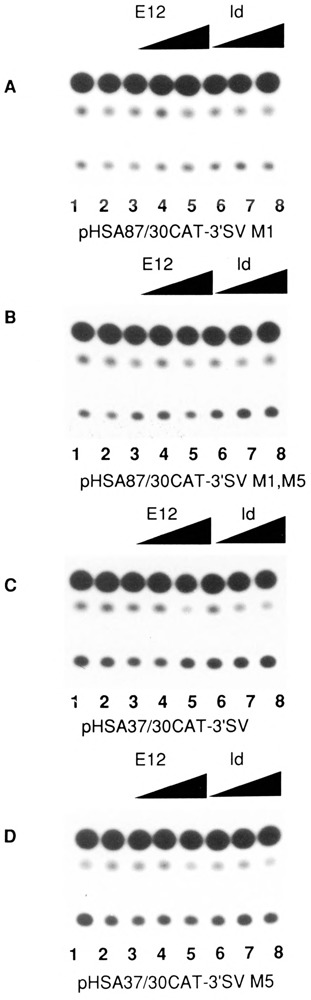
Transient CAT assays in C2C12 cells demonstrating the effect of the Id (inhibitor of differentiation) helix-loop-helix protein on deleted α-actin promoters. Lanes 1–8 of each assay contained the following plasmids: A, pHSA87CAT-3′SV M1; B, pHSA87CAT-3′SV M1,M5; C, pHSA37/30CAT-3′SV; and D, pHSA37/30CAT-3′SV M5 (6 μg). All plasmids included the full-length native human skeletal α-actin promoter. Lanes 3–5 also contained 0.5, 1, and 2 μg of co-transfected pEMSV-E12(A.S.). Lanes 6–8 also contained 0.5, 1, and 2 μg of co-transfected pEMSV-Id. The total amount of DNA in each transfection was kept at a constant 10 μg with the carrier plasmid pUC18.
The sequences between nucleotide positions −64 and −37 interact with Sp1 and an unidentified proximal regulatory factor
We then used EMSA to determine whether the sequences between nucleotide positions −67/−37 (5′CCTGCGGGGTGGCGCGGAGGGAATCGCCCGC3′) interacted with nuclear factors in vitro. After noticing that this sequence contained a putative Spl consensus sequence motif, we scanned the promoter sequences between nucleotides −87/+239 for putative Spl consensus sequence motifs. The trans-acting factor Spl has been shown to interact with the degenerate binding sites conforming to the following consensus, as described by Evans et al. (1988; the numbers are percentages of each base at that position):
| G64 | G89 | G100 | G100 | C82 | G96 | G89 | G68 | G68 | C58 |
| T21 | A11 | A14 | A4 | T11 | A21 | A18 | T21 | ||
| C11 | T4 | C11 | C7 | G14 | |||||
| A4 | T7 | A7 |
Gustafson and Kedes (1989) and Collie and Muscat (1992) have defined Spl binding sites in the human cardiac and skeletal α-actin genes that have the following consensus sequence (the numbers represent occurrences out of seven):
| G7 | G7 | G7 | G5 | A5 | G7 | G7 | G7 | G5 | G4 |
| C1 | C1 | C1 | C2 | ||||||
| T1 | T1 | T1 | T1 |
In the case of the human skeletal α-actin gene, we found at least three other putative Spl binding sites in the promoter between nucleotide positions −87/+239. We synthesized oligonucleotide probes for these putative Spl binding sites that were present between nucleotide positions
+29/+57 (5′TCGCTTCCCCTCCGCGGCGACCAGGGCCC 3′),
+74/+102 (5′ TTGTAGCTACCCGCCCAGGTAGGGCAGAG 3′) and
+94/+123 (5′ AGGGCAGGAGTTGGGAGGGGACAGGGGGAC 3′).
These putative Spl sites were then incubated with purified Spl protein (Promega). These four Spl binding sites, termed HSA (human skeletal actin) −67/−37, HSA +29/+57, HSA +74/+102, and HSA +94/+123, interacted with purified Spl in gel retardation assays, but with variable degrees of affinity (data not shown). EMSA was then used to determine whether the −67/−37 region required for efficient myogenic specific expression interacted with a myogenic nuclear factor in vitro. Nuclear extracts were prepared from mouse myogenic C2C12 myoblasts and myotubes in different stages of differentiation to assay the developmental regulation of −67/−37-bound proteins in muscle. Specifically, extracts were isolated from proliferating myoblasts (PMB), confluent myoblasts (CMB), and myotubes (MT3 and MT4; 3 and 4 days after mitogen withdrawal, respectively). Nuclear extracts were also isolated from non-muscle HeLa fibroblast cells. Figure 10 shows that this region (HSA −67/−37) interacted with a protein (proximal regulatory factor-1 or PRF-1) in myogenic cells that did not appear to be Spl. To elucidate this further we conducted in vitro competitions with a defined Spl consensus sequence (Promega, 5′ ATTCGATCGGGGCGGGGCGAGC 3′). We incubated the HSA −67/−37 region with purified Spl and competed with a 10-, 20-, 40-, and 80-fold molar excess of an Spl consensus sequence (Fig. 11, lanes 2–5). The Spl consensus sequence competed very efficiently for binding to Spl. However, as seen in lanes 8–11 (Fig. 11), the Spl consensus did not compete for binding to the myogenic nuclear factor PRF-1. This clearly indicated that PRF-1 was a distinct nuclear factor. Figure 12 shows that PRF-1 binding is very efficiently competed by a 40- and 80-fold molar excess of HSA −67/−37 (lanes 2 and 3) but not by the Spl consensus (lanes 4 and 5).
Figure 10.
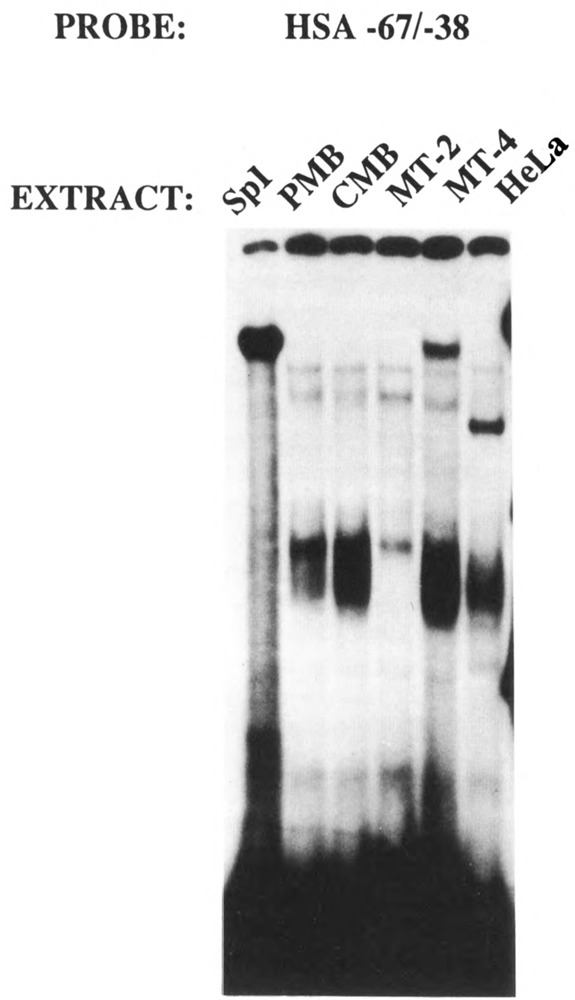
The cis-acting region between nucleotide positions −67/−38 interacts with Spl and an unidentified factor, PRF-1, in myogenic nuclear extracts. The HSA −67/−38 probe was incubated with nuclear extracts derived from proliferating (PMB) and confluent (CMB) myoblasts and myotubes after 2 (MT-2) and 4 (MT-4) days of mitogen withdrawal (see Materials and Methods).
Figure 11.
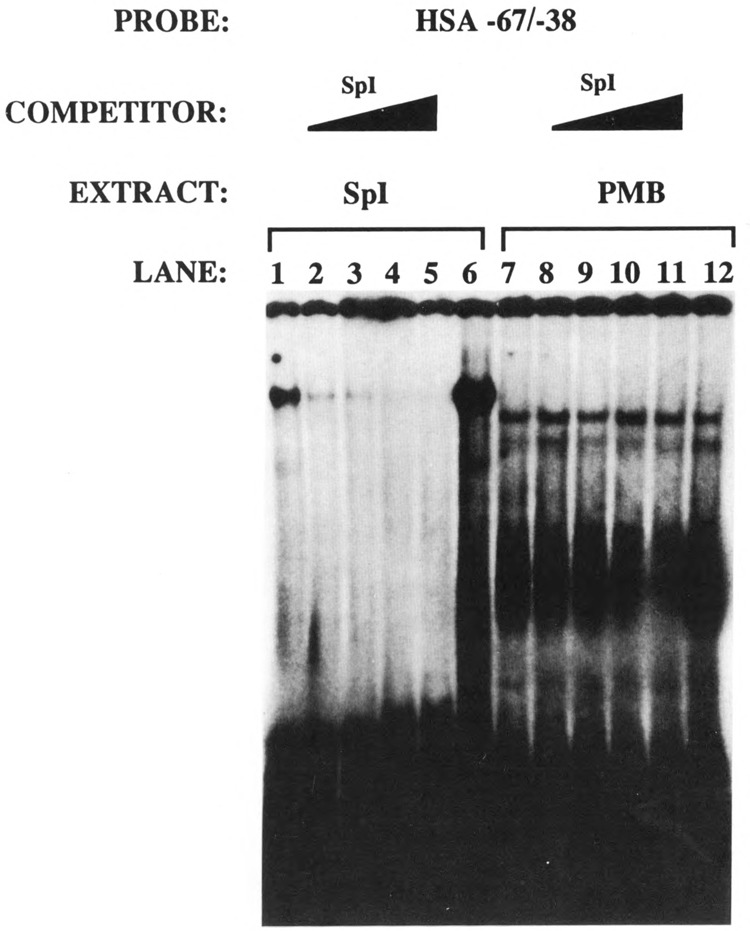
Spl and PRF-1 are distinct nuclear factors. The HSA −67/−38 probe was incubated with purified Spl (lanes 1–6) and PMB nuclear extract (lanes 7–12). Lanes 1, 6, 7, and 12 were control electrophorec tic mobility shift assays that did not contain any competitor DNAs. Lanes 2–5 and 8–11 contained 10-, 20-, 40-, and 80-fold molar excess of an Spl consensus sequence.
Figure 12.
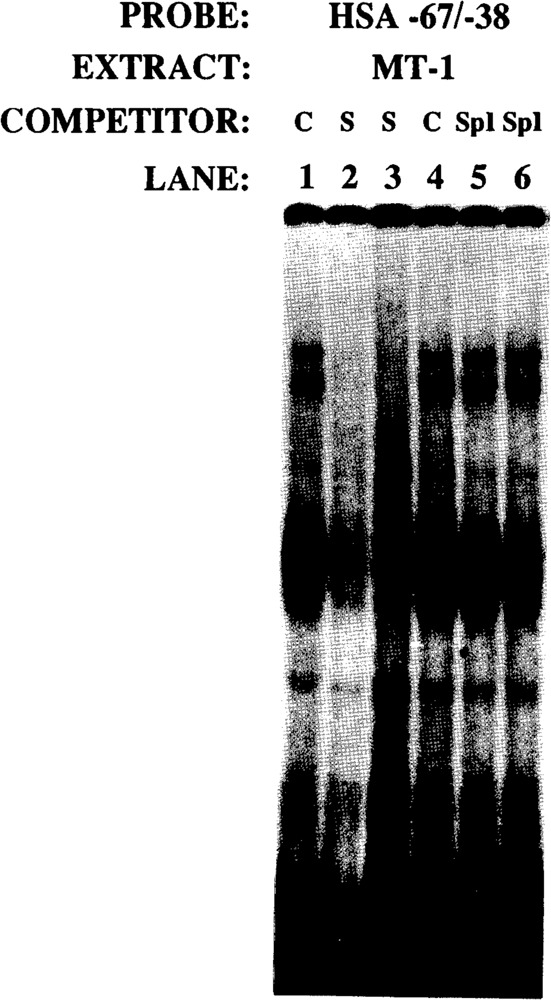
PRF-1 complex is specifically competed by HSA −67/−38, but not by Spl consensus sequences. The HSA −67/−38 probe was incubated with MT-1 nuclear extract; the control reactions are shown in lanes 1 and 4. Lanes 2 and 3, and 5 and 6, show competition with 40- and 80-fold molar excesses of self(S) and Spl consensus, respectively.
Discussion
We undertook the present investigation to define the minimal sequence requirements for the differential transcription/cell-specific expression of the skeletal α-actin gene. We have shown that the sequences 3′ of nucleotide position −87 can functionally interact with the SV40 enhancer in a tissue-specific manner. Efficient myogenic expression in the presence of the SV40 enhancer requires the sequences between nucleotide positions −64/−37, which have the potential to interact with Spl and PRF-1. These data correspond to the study on the immunoglobulin (Ig) μ heavy chain promoter, in which the sequences between nucleotide positions −154 and +57 were found to direct lymphocyte-specific transcription in the presence of a viral enhancer (Grosschedl and Baltimore, 1985). Similarly, in the pituitary-specific regulatory gene GHF 1, it was found that a 15 bp region surrounding the TATA element acted as a cell-specific promoter element (McCormick et al., 1991). The sequences 3′ of nucleotide position −87 serve as a muscle-specific basal promoter that can mediate efficient myogenic transcription in the presence of a variety of enhancer and regulatory sequences. Furthermore, the sequences between nucleotide positions −37 and +30 do not support significant transcription (∼<3-fold) in myogenic and non-muscle cells in the presence of the SV40 enhancer.
The native skeletal α-actin promoter was trans-activated by the HLH transcription factors MyoD, MRF-4, and Myogenin in pluripotential 10T1/2 fibroblasts and repressed by the HLH factor Id in myogenic C2C12 cells. However, the sequences 3′ of −87 that functionally interacted with the SV40 enhancer in a tissue specific manner could not be trans-activated in pluripotential 10T1/2 cells or trans-repressed in C2C12 cells.
We conclude from this and our previous studies that the sequences 5′ of nucleotide position −87 in the skeletal α-actin gene serve as myogenic enhancers and modulators of transcription and trans-activation and repression. However, the basal promoter 3′ of −87 plays a role in tissue-specific expression that can function independently of myogenic enhancer/regulatory sequences and the HLH family of transcription factors. Furthermore, the data indicate that the skeletal α-actin basal promoter sequences, which can suppress the activity of the SV40 enhancer in non-muscle cells, may be one of the primary determinant(s) of myogenic-specific expression. The present study in the context of previous studies by us and others on the skeletal α-actin gene suggests that trans-activation and repression, transcriptional modulation, and tissue specificity are the primary requirements of a cell-specific promoter. Myogenic expression is a consequence of a combinatorial interplay of many cis-acting regions that independently, cannot support all aspects or parameters of cell-specific expression, although they may have some overlapping functions with respect to these parameters (e.g., trans-activation/modulation or tissue-specificity/modulation).
In our previously published work we showed that −87/+239 region linked to CAT, functioned 100-fold less efficiently than the native promoter. Furthermore, these data were in agreement with the in vivo competition experiments, which established that this region did not interact with rate-limiting trans-acting factors. There appears to be a discrepancy with respect to the ability of this region to restrict the activity of the SV40 enhancer to muscle and the inability of this region to function in the in vivo competition assay developed by Scholer and Gruss (1985). It should be noted that only the cis-acting sequences that interact with rate-limiting factors (i.e., elements such as enhancers that function independently to modulate transcription) are detected in this type of in vivo competition assay (Scholer and Gruss, 1985; Scholer et al., 1986; Mercola et al., 1985). The sequences 3′ of −87 do not function independently to modulate transcription in the absence of enhancer sequences. Furthermore, sequences that have known functions in transcription, e.g., Spl and proximal promoters, do not show any activity in this assay system (Scholer and Gruss, 1985; Scholer et al., 1986; Jameson et al., 1987). We hypothesize that this region functions by tethering the enhancer regions via DNA looping mechanisms to produce stable transcription complexes in a tissue- or muscle-specific manner (Su et al., 1992).
Sartorelli et al. (1990) and French et al. (1991) found that the human and chicken cardiac α-actin genes could be trans-activated via a single rather than multiple MyoD binding site(s), if adjacent sites for Spl and a serum response factor-like protein known as the CArG binding factor (CBF) are occupied. These and other experiments have reinforced the critical role of CBF (which interacts with the CArG motif CC[A + T-rich]6GG) in myogenic-specific expression (Miwa and Kedes, 1987; Walsh and Schimmel, 1988; Boxer et al., 1989; Walsh, 1989; Chow and Schwartz, 1990; Tuil et al., 1990; Muscat et al., 1991). In support of our studies, French et al. (1991) and Sartorelli et al. (1990) found that the chicken and human cardiac α-actin basal promoters containing sequences 3′ of nucleotide positions −58 and −47 respectively could not be trans-activated by myogenin or MyoD. These studies collectively reinforce our hypothesis, since we found that the human cardiac and skeletal α-actin sequences 3′ of nucleotide positions −47 and −87, respectively, can functionally interact with the SV40 enhancer in a muscle-specific manner, in spite of their inability to be trans-activated by HUH factors in 10T1/2 cells. Furthermore, French et al. (1991) performed mutagenic analyses of the cardiac α-actin promoter. Their data suggested that an E box alone does not confer muscle-specific expression. They also constructed a mutation (cardM2) that reduced promoter activity in muscle to 2% of wild-type but retained the potential to be trans-activated in 10T1/2 cells by myogenin.
In conclusion, we hypothesize that the present study and the work of French et al. (1991) suggest that cis-acting sequences involved in the regulation of tissue-specific expression and trans-activation and repression can be physically and functionally distinct in myogenic promoters.
Acknowledgments
We sincerely thank Andrew Lassar, Harold Weintraub, Eric Olson, and Steve Konieczny for generously providing expression vectors encoding MyoD, Id, Myogenin, and MRF-4.
This work was supported by the Australian Research Council. G. E. O. Muscat is currently an R. D. Wright Fellow of the National Health and Medical Research Council.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Bains W., Ponte P., Blau H., and Kedes L. (1984), Mol Cell Biol 4, 1449–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benezra R., Davis R. L., Lockshon D., Turner D. L., and Weintraub H. (1990), Cell 61, 49–59. [DOI] [PubMed] [Google Scholar]
- Blackwell T. K. and Weintraub H. (1990), Science 250, 1104–1110. [DOI] [PubMed] [Google Scholar]
- Bouvagnet P. F., Strehler E. E., White G. E., Strehler-Page M.-A., Nadal-Ginard B., and Mahdavi V. (1987), Mol Cell Biol 7, 4377–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer L. M., Prywes R., Roeder R. G., and Kedes L. (1989), Mol Cell Biol 9, 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Buschhausen-Denker G., Bober E., Tannich E., and Arnold H. H. (1989a), EMBO J 8, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Bober E., Buschhausen-Denker G., Kohtz S., Grzeschik K.-H., and Arnold H. H. (1989b), EMBO J 8, 3617–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Bober E., Winter B., Rosenthal N., and Arnold H. H. (1990a), EMBO J 9, 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Winter B., Bober E., and Arnold H. H. (1990b), Nature 346, 663–665. [DOI] [PubMed] [Google Scholar]
- Brennan T. J. and Olson E. N. (1990), Genes Dev 4, 582–595. [DOI] [PubMed] [Google Scholar]
- Choi J., Costa M. L., Mermelstein C. S., Chagas C., Holtzer S., and Holtzer H. (1990), Proc Natl Acad Sci USA 87, 7988–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow K. L. and Schwartz R. J. (1990), Mol Cell Biol 10, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collie E. S. R. and Muscat G. E. O. (1992), Cell Growth Differ 3, 31–42. [PubMed] [Google Scholar]
- Davis R. L., Weintraub H., and Lassar A. B. (1987), Cell 51, 987–1000. [DOI] [PubMed] [Google Scholar]
- Davis R. L., Cheng P.-F., Lassar A. B., and Weintraub H. (1990), Cell 60, 733–746. [DOI] [PubMed] [Google Scholar]
- Dignam J. D., Lebovitz R. M., and Roeder R. G. (1983), Nucl Acids Res 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonson D. G. and Olson E. A. (1989), Genes Dev 2, 1779–1790. [Google Scholar]
- Evans T., DeChiara T., and Efstratiadis A. (1988), J Mol Biol 199, 61–81. [DOI] [PubMed] [Google Scholar]
- French B. A., Chow K. L., Olson E. N., and Schwartz R. J. (1991), Mol Cell Biol 11, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried M. and Crothers D. M. (1981), Nucl Acids Res 9, 6505–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidoni P., Kadonaga J. T., Barrera–Saldana H., Takahashi K., Chambon P., and Tjian R. (1985), Science 230, 511–517. [DOI] [PubMed] [Google Scholar]
- Gosset L. A., Kelvin D. J., Sternberg E. A., and Olson E. (1989), Mol Cell Biol 9, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosschedl R. and Baltimore D. (1985), Cell 41, 885–897. [DOI] [PubMed] [Google Scholar]
- Gustafson T. A. and Kedes L. (1989), Mol Cell Biol 9, 3269–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson J. L., Deutsch P. J., Gallagher G., Jaffe R. C., and Habener J. F. (1987), Mol Cell Biol 7, 3032–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. A., Kadonaga J. T., Rosenfield P., Kelly T., and Tjian R. (1987), Cell 48, 79–81. [DOI] [PubMed] [Google Scholar]
- Kadonaga J. T., Jones K. A., and Tjian R. (1986), Trends Biochem Sci 11, 20–23. [Google Scholar]
- Konieczny S. F. and Emerson C. P. (1984), Cell 38, 791–800. [DOI] [PubMed] [Google Scholar]
- Lassar A. B., Buskin J. N., Lockshon D., Davis R. L., Apone S., Hauschka S. D., and Weintraub H. (1989), Cell 58, 823–831. [DOI] [PubMed] [Google Scholar]
- Lassar A. B., Davis R. L., Wright W E., Kadesch T., Murre C., Voronova A., Baltimore D., and Weintraub H. (1991), Cell 66, 305–316. [DOI] [PubMed] [Google Scholar]
- Lin H., Yutsey K. E., and Konieczny S. F. (1991), Mol Cell Biol 11, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar J. H. and Ordahl C. P. (1990), Mol Cell Biol 10, 4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick A., Brady H., Fukushims J., and Karin M. (1991), Genes Dev 5, 1490–1503. [DOI] [PubMed] [Google Scholar]
- Mercola M., Goverman J., Mirell C., and Calame K. (1985), Science 227, 266–270. [DOI] [PubMed] [Google Scholar]
- Miner J. H. and Wold B. (1990), Proc Natl Acad Sci USA 87, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minty A., Blau H., and Kedes L. (1986), Mol Cell Biol 6, 2137–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minty A. and Kedes L. (1986), Mol Cell Biol 6, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa T. and Kedes L. (1987), Mol Cell Biol 7, 2803–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C., McCaw P. S., Vaessin H., Caudy M., Jan L. Y., Jan Y. N., Cabrerra C. V., Buskin J. N., Hauschka S. D., Lassar A. B., Weintraub H., and Baltimore D. (1989), Cell 58, 537–544. [DOI] [PubMed] [Google Scholar]
- Murre C., McCaw P. S., and Baltimore D. (1989), Cell 56, 777–783. [DOI] [PubMed] [Google Scholar]
- Muscat G. E. O. and Kedes L. (1987), Mol Cell Biol 7, 4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat G. E. O., Gustafson T. A., and Kedes L. (1988), Mol Cell Biol 8, 4120–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat G. E. O., Gobius K., and Emery J. (1991), Mol Endocrinol 5, 802–814. [DOI] [PubMed] [Google Scholar]
- Muscat G. E. O., Perry S., Prentice H., and Kedes L. (1992), Gene Expr 2, 111–126. [PMC free article] [PubMed] [Google Scholar]
- Olson E. N. (1990), Genes Dev 4, 1454–1456. [DOI] [PubMed] [Google Scholar]
- Piette J., Bessereau J. L., Huchet M., and Changeau J. P. (1990), Nature 345, 353–355. [DOI] [PubMed] [Google Scholar]
- Pinney D., Pearson-White S. H., Koniesczny S. F., Latham K. E., and Emerson C. P. (1988), Cell 53, 781–793. [DOI] [PubMed] [Google Scholar]
- Rhodes S. and Konieczny S. F. (1989), Genes Dev 3, 2050–2061. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritch E. F., and Maniatis T. (1989), Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Santoro C., Mermod N., Andrews P. C., and Tjian R. (1988), Nature 334, 218–224. [DOI] [PubMed] [Google Scholar]
- Sartorelli V., Webster K., and Kedes L. (1990), Genes Dev 4, 1811–1822. [DOI] [PubMed] [Google Scholar]
- Scholer H. R. and Grss P. (1984), Cell 36, 403–411. [DOI] [PubMed] [Google Scholar]
- Scholer H., Haslinger A., Heguy A., Holtgreve H., and Karin M. (1986), Science 232, 76–80. [DOI] [PubMed] [Google Scholar]
- Schreiber E., Mathias P., Muller M., and Schaffner W. (1989), Nucl Acids Res 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spizz G., Hu J. H., and Olson E. (1987), Dev Biol 123, 500–507. [DOI] [PubMed] [Google Scholar]
- Su W, Jackson S., Tjian R., and Echols H. (1992), Genes Dev 5, 820–826. [DOI] [PubMed] [Google Scholar]
- Sun X. H. and Baltimore D. (1991), Cell 64, 459–470. [DOI] [PubMed] [Google Scholar]
- Tapscott S. J., Davis R. L., Thayer M. J., Cheng P.-F., Weintraub H., and Lassar A. B. (1988), Science 242, 405–411. [DOI] [PubMed] [Google Scholar]
- Thayer M. J., Tapscott S. J., Davis R. L., Wright W. E., Lassar A. B., and Weintraub H. (1988), Cell 58, 241–248. [DOI] [PubMed] [Google Scholar]
- Tuil D., Clerque N., Montarass D., Pinset C., Kahn A., and Phan-Dinh-Tuy F. (1990), J Mol Biol 213, 677–686. [DOI] [PubMed] [Google Scholar]
- Vaida T. B., Rhodes S. J., Taparowsky E. J., and Konieczny S. F. (1989), Mol Cell Biol 9, 3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K. (1989), Mol Cell Biol 9, 2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh K. and Schimmel P. (1988), Mol Cell Biol 8, 1880–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster K. A., Muscat G. E. O., and Kedes L. (1988), Nature 332, 553–561. [DOI] [PubMed] [Google Scholar]
- Wefald F. C., Devlin B. H., and Sanders W. R. (1990), Nature 344, 260–262. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Davis R. L., Lockshon D., and Lassar A. B. (1990), Proc Natl Acad Sci USA 87, 5623–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H., Davis R. L., Tapscott S. J., Thayer M., Krause M., Benezra R., Blackwell T. K., Turner D., Rupp R., Hollenberg S., Zhuang Y., and Lasser A. B. (1991), Science 251, 761–766. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Dwarki V. J., Verma I., Davis R. L., Hollenberg S., Snider L., Lassar A. B., and Tapscott S. J. (1991), Genes Dev 5, 1377–1386. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Tapscott S. J., Davis R. L., Thayer M. J., Adam M. A., Lassar A. B., and Miller A. D. (1989), Proc Natl Acad Sci USA 86, 5434–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentworth B. M., Donoghue M., Engert J. C., Berglund E. B., and Rosenthal N. (1991), Proc Natl Acad Sci USA 88, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder E. and Linzer D. I. H. (1989), Mol Cell Biol 9, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright W., Sassoon D., and Lin V. (1989), Cell 56, 607–617. [DOI] [PubMed] [Google Scholar]
- Yaffe D. and Saxel O. (1977a), Differentiation 7, 159–166. [DOI] [PubMed] [Google Scholar]
- Yaffe D. and Saxel O. (1977b), Nature 270, 725–727. [DOI] [PubMed] [Google Scholar]