

Abstract
We have isolated human cDNA clones for USF2, a new member of the upstream stimulatory factor (USF) family of transcription factors. Analysis of these clones revealed the existence of highly conserved elements in the C terminal region of all USF proteins. These include the basic region, helix-loop-helix (HLH) motif, and, in the case of the human proteins, the C-terminal leucine repeat (LR). In addition, a highly conserved USF-specific domain islocated immediately upstream of the basic region. Using invitro translated proteins, we found that all members of the USF family bound DNA as dimers. The N-terminal portion of USF, including the USF-specific domain, was entirely dispensable for dimer formation and DNA-binding. However, deletion mutants of USF2 lacking the LR were deficient in DNA-binding activity. Interestingly, each of the USF proteins could form functional heterodimers with the other family members, including the sea urchin USF, which does not have a LR motif. This indicates that the conserved LR in human USF is not required for dimer formation, and influences only indirectly DNA-binding.
The upstream stimulatory factor (USF) was first identified because of its involvement in transcription from the adenovirus major late (ML) promoter (Sawadogo and Roeder, 1985; Carthew et al., 1985; Miyamoto et al., 1985). Binding of USF to the ML upstream element induces a significant stimulation of transcription from this promoter in vitro and is cooperative with the binding of the TATA box factor TFIID and the initiator binding protein (Sawadogo and Roeder, 1985; Roy et al., 1991). In the viral environment, the USF binding site was also found to play a critical role in transcription of the adenovirus ML, although its function is redundant with that of an upstream CAAT box element (Reach et al., 1991). A possible involvement of USF in the transcription of several other cellular and viral genes has also been reported (Carthew et al., 1987; Chodosh et al., 1987; Lemaigre et al., 1989; Chang et al., 1989; Scotto et al., 1989; Sato et al., 1990; Greaves et al., 1991; Potter et al., 1991).
Purification of USF from HeLa cells has revealed that at least two different polypeptides, with apparent molecular weights of 43,000 and 44,000, contribute to the USF activity detected in human cells (Sawadogo et al., 1988). A cDNA clone was isolated which encodes the human 43-kDa USF protein (Gregor et al., 1990). Analysis of this clone revealed that USF belongs to the helix-loop-helix (HLH) family of proteins, which includes a number of regulatory transcription factors and oncoproteins (Murre et al, 1989). USF is conserved during evolution, and USF cDNA clones were recently isolated from Xenopus (Kaulen et al., 1991) and sea urchin (Kozlowski et al., 1991).
The DNA-binding sites with which USF interacts are palindromic motifs most often centered on a CACGTG sequence (Sawadogo and Roeder, 1985). A core containing the nucleotides CANNTG is characteristic of the recognition sequence of all known HLH proteins (Murre et al., 1989). Interestingly, USF binding sites are also recognized by several other families of HLH proteins, namely the various Myc, Myn/Max, and TFE3/TFEB family members (Blackwell et al., 1990; Kerkhoff et al., 1991; Blackwood and Eisenmann, 1990; Prendergast et al., 1991; Beckman et al., 1990; Carr and Sharp, 1990). Vertebrate USFs, like Myc and TFE3, contain a leucine repeat (LR) contiguous with the HLH region. Although this second dimerization domain seems critical for DNA-binding by TFE3 (Beckmann and Kadesh, 1991; Fisher et al., 1991) and for cellular transformation by Myc (Dang et al., 1989), the USF protein isolated from sea urchin lacks such a structure (Kozlowski et al., 1991).
Here we report the isolation of cDNA clones for human USF2, a new member of the USF family. Characterization of USF2 clones sheds new light on domain conservation within the USF family. In addition, we present evidence indicating that heterodimer formation may play an important role in the regulation of gene expression by USF.
Materials and methods
Library screening and DNA sequencing
To isolate new USF clones, we initially screened a human B-cell (Namalwa cell) cDNA library constructed in a Lambda ZAP vector by Strata-gene (Scheidereit et al., 1989). The 762 bp EcoR I-Sal I fragment encompassing most of the coding region of the sea urchin USF cDNA clone was used as a probe with a low stringency hybridization protocol (Kozlowski et al., 1991). This initial screen, in which clones corresponding to the 43-kDa human USF1 were distinguished by high stringency hybridization with the Stu I fragment from clone dI2 (Gregor et al., 1990), yielded a single USF2 clone designated hUSF2-C. The BamH I fragment encompassing nucleotides 33 to 501 of hUSF2-C was in turn used to screen at high stringency a second human cDNA library derived from HeLa cells. Two new independent clones (hUSF2-C1 and hUSF2-A2) were isolated in this secondary screen. These clones were apparently identical to hUSF2-C except that they were slightly shorter at the 5′ end (see Fig. 1). DNA sequencing was performed using the dideoxy termination method (Sanger et al., 1977) with double-stranded DNA templates and Sequenase (U.S. Biochemical). The sequence of the entire hUSF2-C clone was determined by subcloning various restriction fragments as well as by using several specific internal primers. The coding region was sequenced on both strands.
Figure 1.

Nucleotide sequence of human USF2 cDNA clones and deduced amino acid sequence. Shown is the entire sequence of clone hUSF2-C that was isolated from a human Namalwa cell cDNA library. Positions of the 5′ end of two other USF2 clones (hUSF2-C1 and hUSF2-A2), isolated from a HeLa cell cDNA library, are indicated (see Materials and Methods). The presumptive polyadenylation signal is underlined.
In vitro transcription/translation of USF cDNAs
Clone hUSF2-C, which lacked an initiator methionine, was subcloned in a pBS/ET expression vector which allows in-frame insertion of coding sequences behind the initiating methionine and first 10 amino acid residues of T7 gene 10 (W.M. Perry, unpublished data). This yielded clone pBS/ET-USF2. Subclone USF2a, containing amino acids 103 to 234 of USF2, was generated by inserting the Pvu I to Sma I fragment of hUSF2-C into a pBS/ET expression vector. Subclone pBS/ETUSF2b was obtained by insertion—at the Sph I site of pBS/ETUSF2—of an oligonucleotide containing termination codons in all three frames. Subclone pBS/ETUSF2c was obtained by removal from the pBS/ET-USF2 plasmid of the Pst I fragment encoding amino acids 196 to 220 of USF2.
In vitro synthesized RNAs produced by transcription of the various USF cDNA clones with T3 or T7 RNA polymerase, as appropriate, were used to program rabbit reticulocyte lysates (Promega). Translation reactions were carried out in the presence of [35S]methionine for 2 hours at 30°C. Control reactions contained either buffer only or BMV RNA. In vitro translated products were analyzed by electrophoresis on 12% polyacrylamide-SDS gels.
Mobility shift assays
The self-complementary oligonucleotide 5′ GATCTCCGGTCACGTGACCGGA 3′, which contains the consensus binding site for USF (M. N. Szentirmay and M. Sawadogo, unpublished data), was used as a probe for mobility shift assays after end-filling with [α-32P]dATP and the Klenow fragment of DNA polymerase. The 20 μl reactions contained 50 mM Tris (pH 7.9), 5 mM DTT, 100 mM KC1,4% glycerol, 1 ng probe, 300 ng poly[d(I-C], and 1–4 μl of rabbit reticulocyte lysate. After 30 minutes incubation at 30 °C, 4 μl of ficoll were added and the samples were loaded on a 6% polyacrylamide-0.25X Trisborate-EDTA gel. After electrophoresis, auto-radiography was performed using a thin sheet of plastic between the dried gel and the film in order to shield the radiation of the [35S]-methionine in the reticulocyte lysate reactions.
Polyclonal antibodies to human USF1 and USF2
A portion of human USF1 (amino acids 106–310) was overproduced in E. coli as a fusion protein containing a stretch of six histidine residues at the N-terminus (Van Dyke et al., 1992). Purification of this protein (His6-delUSF) was achieved by affinity chromatography on immobilized Ni2+ as described earlier (Van Dyke et al., 1992), followed by a final purification step on FPLC Mono S column (Pharmacia; Sawadogo et al., 1988). The human USF2 protein was expressed in E. coli as β-galactosidase fusion protein by subcloning the Sma I fragment of hUSF2-C (nucleotides 43 to 1092) into the end-filled BamH I site of pUR278 (Rüther and Müller-Hill, 1983), yielding plasmid pUR-hUSF2. The overproduced protein was purified by electrocution from 8% polyacrylamide-SDS gels. Purified USF proteins were injected subcutaneously into male New Zealand white rabbits (50 to 100 μg per injection) with boosts at 3-week intervals.
Western blots were either stained with Fast Green FCF (Sigma) or probed with a 100,000-fold dilution of the immune sera and revealed by treatment with alkaline phosphatase-conjugated goat antibodies to rabbit IgGs (Promega) as previously described (Sawadogo et al., 1988).
Results
Isolation of cDNA clones to human USF2
Characterization of purified USF preparations has indicated that at least two different poly-peptides are responsible for the USF activity present in HeLa cells (Sawadogo et al., 1988). Isolation of a cDNA clone encoding the human 43-kDa USF (USF1 gene) has been reported (Gregor et al., 1989). We found that the 44-kDa USF polypeptide was apparently not encoded by alternatively spliced messages from this USF1 gene, since extensive screening of various human cDNA libraries yielded only the previously isolated USF1 cDNAs (E. Miley and M. Sawadogo, unpublished data). To analyze the immunological relationship between the 43- and 44-kDa aplypeptides, we eaised dplyclonal latibodies against the human USF1 protein. These antibodies were found to cross-react only weakly with the 44-kDa USF. Groups of antibodies directed against various regions of the 43-kDa protein were purified from the immune serum by reacting them with a battery of β-galactosidase-USFl fusion proteins. Each of these groups of antibodies was also found to react with the 44-kDa USF polypeptide, indicating that epitopes shared by the two proteins were present throughout the 43-kDa USF sequence (M. Sirito, unpublished observation).
Taken together, these results led us to believe that the two USF proteins were most likely the products of distinct, though related genes. To isolate new members of the USF gene family, we decided to screen human cDNA libraries with a low stringency hybridization protocol. Initial attempts, using various fragments of the human USF1 gene, were unsuccessful. However, hybridization with a DNA fragment encompassing the basic and HLH region of the sea urchin USF cDNA (Kozlowski et al., 1991), allowed identification of a new human USF cDNA clone, designated hUSF2-C. The sequence of this clone is shown in Figure 1. The insert is 1.3 kb long and contains a major open reading frame encoding a protein of 234 amino acids. Examination of the 3′ end of the cDNA revealed the presence of a poly(A) tail preceded by a consensus polyadenylation signal. Since the USF2 open reading frame continued uninterrupted towards the 5′ end of the clone, and did not contain any methionine residue before amino acid 87, it is likely that this clone is missing a portion of the coding region as well as the 5′ untranslated sequences. However, attempts to isolate full-length USF2 clones from other human cDNA libraries were unsuccessful (see Fig. 1). Given that the first 33 nucleotides of our longest clone contains 85% G and C residues, it is possible that this region of the USF2 mRNA is part of a strong secondary structure that interferes with the progress of reverse transcriptase.
Conserved domains in USF proteins
Sequence comparison between human USF2 and other USF proteins pointed out characteristic features of this family of transcription factors. A high degree of sequence conservation was found in the C-terminal half of these proteins (Fig. 2). Within this region, the exact spacing between the different motifs is invariant. The extent of amino acid conservation differs in the various domains, being highest in the basic region (82% unchanged residues) and helix 1 and 2 of the HLH domain (92% and 76% unchanged residues, respectively). The greatest divergence is found in the loop of the HLH motif. This is in agreement with previous observations indicating that the amino acid sequence in this region of HLH proteins is not important for protein dimerization and specific DNA-binding (Voronova and Baltimore, 1990). Conservation of amino acid residues in the LR region of vertebrate USFs is only moderate (64% unchanged residues).
Figure 2.

Conserved domains in the C-terminal region of the various USF proteins. The amino acid sequence of the C-terminal region of USF2 was aligned with the same region in human USF1 (amino acids 157–310; Gregor et al., 1990), Xenopus laevis USF (amino acids 150–303; Kaulen et al, 1991), and sea urchin USF (amino acids 148–265; Kozlowski et al., 1991). Location of the various conserved domains is indicated above the sequences. Unchanged residues are outlined in gray.
Most surprisingly, a high degree of amino acid conservation (73% unchanged residues) was observed in a stretch of 26 amino acids located immediately upstream of the basic region. A search against the protein sequences present in the GenBank and EMBL data bases failed to reveal any significant homology between this USF-specific region and other known proteins. In the N-terminal region of USF, only a few patches of matching amino acids residues were observed (not shown).
DNA-binding by USF2 requires an intact LR region
In order to ascertain which domains of USF2 are required for specific DNA-binding, various truncated forms of the protein (see Fig. 3A) were translated in vitro, and the ability of these mutant proteins to bind DNA was assayed by electrophoretic mobility shift assay (EMSA). Note that in vitro translation of USF2 reproducibly yielded, in addition to the expected product, a small amount of a shorter polypeptide (Fig. 3B, lane 3). This particular product may result from proteolytic degradation of the full-length USF2, or from translation initiation taking place at a downstream methionine.
Figure 3.
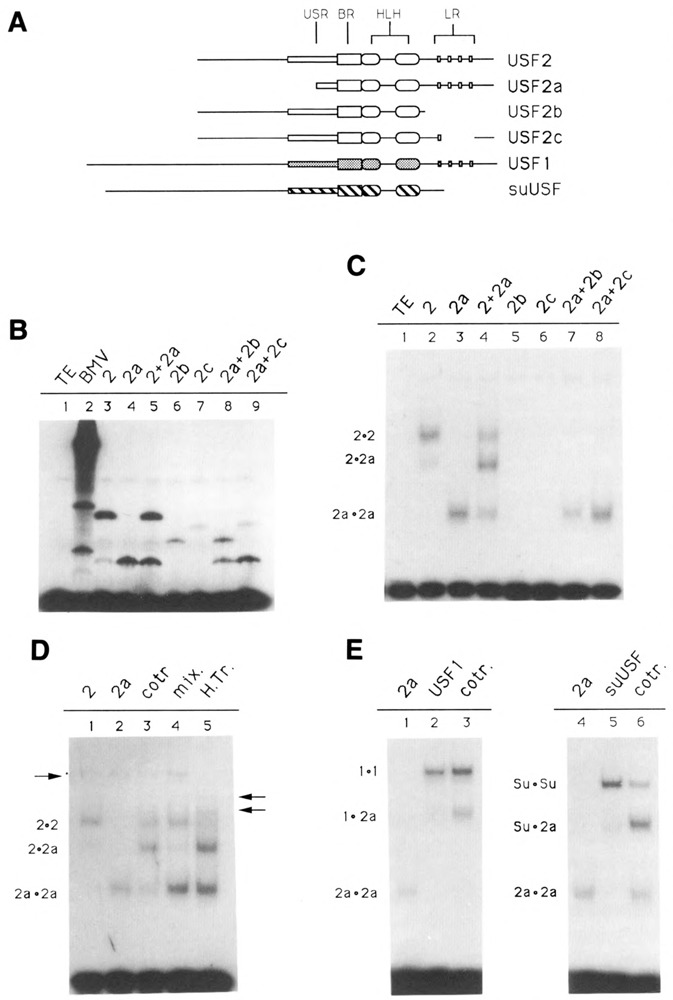
Domains of USF2 required for DNA-binding and dimer formation. A. Schematic of the various USF proteins. Shown are the USF proteins encoded by the pBS/ET-USF2, pBS/ET-USF2a, pBS/ET-USF2b, and PBS/ET-USF2c constructs (see Materials and Methods), which contain respectively amino acids 15–234, 103–234, 15–184, and 15–195/221–234 of hUSF2-C. The human USF1 (amino acids 1–310) and sea urchin USF (amino acids 1–265) were encoded by clones dI2 (Gregor et al., 1990) and 436–1 (Kozlowski et al., 1991), respectively. B. SDS gel analysis of in vitro translation products. Aliquots of reticulocyte lysates programmed with RNA derived from the various USF2 subclones, as indicated above each lane, were analyzed for the presence of translation products by SDS-PAGE. In lanes 5, 8, and 9, two different proteins were cotranslated. C. Localization of the minimum region of USF2 required for DNA-binding. The in vitro translated proteins shown in B were analyzed for DNA-binding activity by EMSA, using as a probe an oligonucleotide containing a consensus USF binding site (see Materials and Methods). The migrations of the different USF2 dimers are indicated at left. Longer exposure of the gel failed to detect any binding activity in the case of the USF2 LR mutants b and c (lanes 5 and 6). D. Formation of USF2 homodimers. USF2, or the N-terminal truncated USF2a mutant, were either translated separately (lanes 1, 2, 4, and 5) or cotranslated (lane 3), and the resulting DNA-binding species analyzed by EMSA. In lane 4, the individual proteins were simply mixed during the binding reaction. In lane 5, the two proteins were mixed, then heated for 7 minutes at 65°C prior to incubation with the oligonucleotide probe. The arrow at left indicates the migration of endogenous rabbit reticulocyte USF. The two arrows at right indicate the migration of heterodimers between rabbit USF and the human USF2 and USF2a proteins. E. Formation of USF heterodimers. The ability of USF2 to heterodimerize with human USF1 (lane 3) as well as with the sea urchin USF (lane 6) was investigated by cotranslating these proteins with the N-terminally truncated USF2a and analyzing the resulting DNA binding species by EMSA. Control reactions (lanes 1, 2, 3, and 4) were used to determine the migration of the individual homodimers, as indicated.
As expected, translation of wild-type USF2 yielded a protein capable of strong interaction with a USF binding site-containing oligonucleotide (Fig. 3C, lane 2). The specificity of this interaction was ascertained by using specific as well as nonspecific oligonucleotide competitors (not shown). Truncation of the entire N-terminal region of USF2, including most of the USF-specific region, did not alter the DNA-binding ability of the transcription factor (Fig. 3C, lane 3). This is in agreement with the results previously obtained for the USF1 protein (Gregor et al., 1990). As was also the case for USF1, cotranslation of truncated USF2 with the wild-type protein resulted in the formation of a complex of intermediate mobility, indicating that USF2 bound DNA as a dimer (Fig. 3C, lane 4). By contrast, deletion of the C-terminal region of USF2, as well as a partial deletion within the LR region, was found to abolish DNA-binding (Fig. 3C, lanes 5 and 6). This indicated that, in USF, the HLH motif alone may be insufficient for efficient dimer formation and/or DNA-binding activity. Similar results were obtained for TFE3/TFEB family members, as well as for transcription factor AP4 (Beckmann and Kadesh, 1991; Fisher et al., 1991; Hu et al., 1990).
Formation of USF homo- and heterodimers
The experiment shown in Figure 3D was designed to investigate the conditions necessary for the formation of USF2 dimers. Using in vitro translated USF2 polypeptides of two different lengths, we found that DNA complexes containing hybrid USF2 dimers could only be detected when the two proteins had been cotranslated (Fig. 3D, lane 3), but not when the individually translated proteins were mixed immediately prior to the DNA binding reaction (Fig. 3D, lane 4). This clearly indicated that the USF2 dimers were not assembling on the DNA, but rather were already present in solution prior to their interaction with the oligonucleotide probe. In addition, since little to no exchange of subunits could be observed in the mixing experiment, these USF dimers seemed to be quite stable in solution (compare Figure 3D, lanes 3 and 4). However, when the mixture of individually translated proteins was submitted to a brief heat-treatment at 65°C, dissociation of the USF dimers and exchange of subunits occurred (Fig. 3D, lane 5). Interestingly, this did not result solely in formation of the expected hybrid between the USF2 proteins, but also in the appearance of two novel USF species, as indicated by the arrows at right. Formation of these new complexes coincided with the disappearance of a specific complex attributed to endogenous rabbit USF in the reticulocyte lysate (Fig. 3D, left arrow). This result can best be explained by the formation of heterodimers between rabbit USF and the human USF2 proteins.
We next tested the possibility of heterodimer formation between the various recombinant USF proteins. Heterodimers between human USF2 and USF1 proteins formed readily upon cotranslation of the RNAs derived from the two clones (Fig. 3E, lane 3). Interestingly, heterodimer formation between human USF2 and the sea urchin USF also yielded a hybrid protein fully capable of specific DNA-binding (Fig. 3E, lane 6). Identical results were obtained using human USF1 and the sea urchin USF (not shown). Since the sea urchin protein lacks an LR, these experiments clearly indicate that interaction between two leucine repeats is not necessary for dimer formation and DNA-binding by human USF.
Expression of the USF2 gene
Southern analysis revealed thatUSF2 is encoded by a single gene. Indeed, with many restriction enzymes, unique DNA fragments were found to hybridize with the USF2 cDNA probe (Fig. 4A). Northern analysis, however, identified three different RNAs of 1.8, 2.0, and 2.6 kb (Fig. 4B). None of these corresponded to mRNAs derived from the USF1 gene which differed slightly in size and did not hybridize with the USF2 probe at high stringency (not shown). Thus it appeared that a unique USF2 gene gives rise to several different mRNAs. This heterogeneity of USF2 mRNA products may be due to differential splicing, a common feature of transcription regulatory factors.
Figure 4.
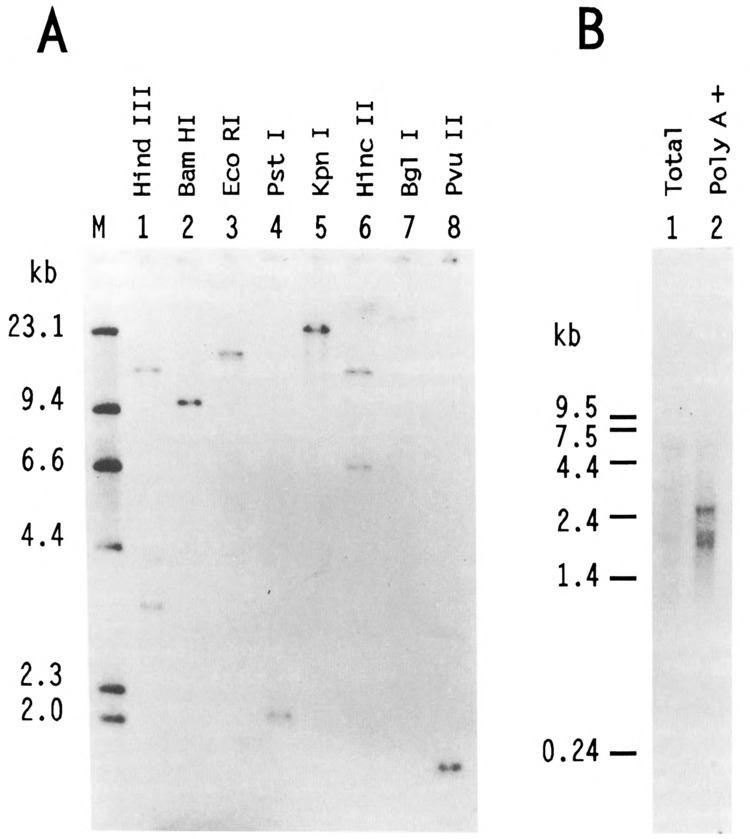
Southern and Northern analysis of human USF2. A. In each lane, 5 μg of HeLa DNA, digested by various restriction endonucleases as indicated, were resolved on a 0.8% agarose gel along with a 32P-labeled Hind III digest of λ DNA as size markers (lane M). After transfer to nitrocellulose, the blot was probed with the BamH I fragment corresponding to nucleotides 33 to 510 of hUSF2-C. Hybridization of this probe was observed with single genomic DNA fragments of 9.6 kb, 17 kb, 2 kb, 23 kb, 25 kb, and 1.7 kb after digestion with BamH I, EcoR I, Pst I, Kpn I, Bgl I, and Pvu II, respectively (lanes 2–5, and 7–8). Hybridization was also observed with 15.5- and 3.3-kb Hind III fragments (lane 1) and 13.6- and 6.5-kb Hinc II fragments (lane 6). The size of the DNA markers (in kb) is indicated at left. B. 2 μg of either total (lane 1) or poly(A)-selected HeLa RNA (lane 2) were separated on 1.2% agarose-formaldehyde gel and transferred to nylon membrane. The radioactive probe was the same as in A. The size (in kb) and migration of RNA markers run on the same gel is indicated at left.
The USF2 protein is present in purified preparations of HeLa USF
In order to investigate the immunological relationship between USF2 and the human 44-kDa USF polypeptide, we expressed a portion of USF2 in E. coli as a C-terminal fusion with β-galactosidase and raised rabbit polyclonal antibodies against this fusion protein. Western blot analysis revealed that the immune serum reacted strongly and specifically against the β-galactosidase-USF2 protein (Fig. 5, lanes 1; compare panels A and B) but showed no cross-reactivity towards bacterially expressed USF1 (lanes 2). Interestingly, when the immune serum was used to probe a purified preparation of HeLa USF (Sawadogo et al., 1988), a strong reaction was observed at the level of the 44-kDa USF protein (Fig. 5, lanes 3). A similar lack of antibody reactivity towards bacterially produced USF1, contrasting with a definite reactivity towards human USF, could also be observed by mobility shift assays (not shown). A simple explanation for these results would be that USF2 is the gene encoding the 44-kDa human USF, although final proof for this identity will await isolation of a full-length USF2 cDNA clone. Meanwhile, we can minimally conclude from these experiments that USF2 is expressed in HeLa cells, and that it copurifies with the 43-kDa USL1 through multiple chromatographic steps, presumably due to the formation of heterodimeric species.
Figure 5.
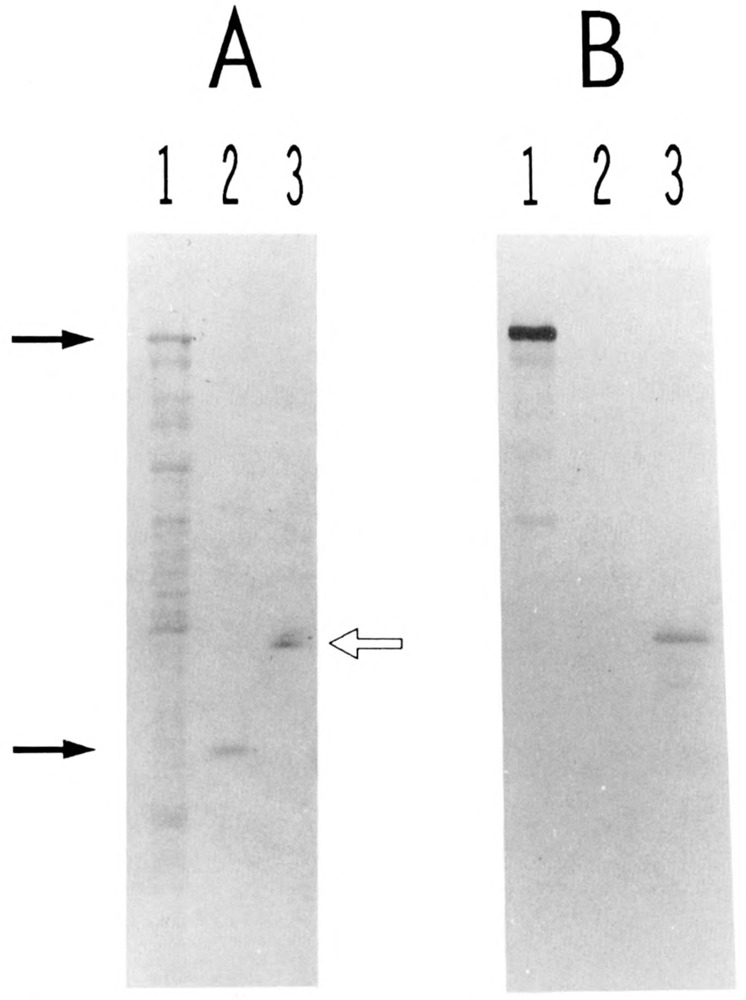
Presence of USF2 in purified preparation of HeLa USE The various protein samples were separated by SDS gel electrophoresis and transferred to nitrocellulose. One portion of the filter was stained with Fast Green (A). The rest of the filter was treated with immune rabbit serum to USF2, and specific binding of the antibodies was visualized with alkaline phosphatase-conjugated goat antibodies to rabbit IgGs (B). Lanes 1: proteins from 3 μl of an IPTG-induced culture of E. coli cells transformed with plasmid PUR-hUSF2 (see Materials and Methods). Lanes 2: 340 ng of purified His6-delUSF protein (see Materials and Methods). Lanes 3: 160 ng of purified HeLa USF (Mono S fraction). The two arrows at left indicate the respective migration of the β-galactosidase-USF2 and polyhistidine-USF1 fusion proteins. The open arrow at right indicates the migration of the 44- and 43-kDa polypeptides characteristic of purified HeLa USF preparations (Sawadogo et al., 1988).
Discussion
USF is a family of transcription factors
Biochemical characterization of USF purified to near homogeneity from HeLa nuclear extract revealed the existence of multiple forms of this transcription factor in human cells (Sawadogo et al., 1988). Using the N-terminal amino acid sequence determined for the 43-kDa HeLa USF, a first USF cDNA clone was isolated (Gregor et al., 1990). Here we used a low stringency hybridization protocol to isolate cDNA clones derived from a second human USF gene. This USF2 gene is expressed in both HeLa and Namalwa cells. Since full-length cDNA clones were not obtained, the exact relationship between the USF2-encoded proteins and the 44-kDa form of human USF could not be formally established. However, antibodies raised against bacterially expressed USF2 reacted strongly and specifically with the HeLa 44-kDa USF polypeptide. This would suggest that USF2 and the human 44-kDa USF are highly related, if not identical.
The existence of multiple family members seems to be a common feature of many eukaryotic transcription factors. In the case of Myc, some family members are ubiquitously expressed, while others display tissue-specific and developmentally regulated patterns of expression (reviewed in Zimmerman and Alt, 1990). A similar situation may exist for USF While the USF1 and USF2 proteins are both present in several cell types, expression of the sea urchin USF seems to be restricted to cells of the ectodermic lineage (Kozlowski et al., 1991). It could be that genes encoding tissue-specific USF proteins also exist in vertebrates. Since there are stretches of amino acids that are highly conserved among the various USF proteins (Fig. 2), it should now be possible to use USF-specific PCR primers to isolate potential new family members.
Involvement of the LR domain in USF DNA-binding
Leucine repeats are α-helical motifs frequently found in eukaryotic transcription factors, where they function as dimerization domains (Vinson et al., 1989). The presence of such a structure in nearly all of the USF, TFE3, Myc, and Max proteins that commonly recognize CACGTG motifs is intriguing. Indeed, a second dimerization motif, the HLH region, is also present in these proteins. In different families of transcription factors, the HLH motif is by itself sufficient for dimer formation, bringing in close proximity the contiguous basic regions of two subunits (Murre et al., 1989). However, an intact LR is essential for TFE3 dimerization (Beckmann and Kadesh, 1991; Fisher et al., 1991). In the case of USF, the function of the LR seems more ambiguous. Deletion of this region in USF2 abolished DNA-binding. On the other hand, such a deletion was only deleterious when present in the full-length USF1 protein. In an N-terminally truncated USF1, the LR was dispensable for dimerization and DNA-binding (Gregor et al., 1990). This latter result seems more consistent with the fact that the sea urchin USF, which entirely lacks an LR, nevertheless interacts with DNA as a dimer. Our experiments indicate that heterodimers between human USF and the sea urchin protein assemble efficiently and are fully capable of specific DNA-binding. Taken together, these results suggest a model in which the presence of the LR in human USF somehow modifies the secondary structure of the adjacent HLH region, thus allowing dimer formation. However, this model does not readily explain why the conformation of the HLH region in the sea urchin protein allows dimerization in the absence of an LR.
Interestingly, the DNA-binding kinetics of the sea urchin USF are quite different from those found for its mammalian counterparts (T. Lu and M. Sawadogo, unpublished observation). It could be that the presence of two potential dimerization domains in human USF allows formation of higher order complexes such as tetramers, as was observed for TFEB (Fisher et al., 1991). The existence of USF tetramers, which would contain two DNA-binding domains, would best explain our earlier observations regarding the ability of this transcription factor to directly transfer from one specific binding site to another, passing through a doubly bound intermediary complex (Sawadogo, 1988). Further experiments regarding this possibility are in progress.
Regulation of USF expression
There are many similarities between USF and Myc besides their potential interaction with common target DNA elements. One such similarity is the complex regulation imposed on the expression of these proteins. Alternatively spliced RNA messages have been observed in the case of USF1 (Gregor et al., 1990), and apparently they also exist for the USF2 gene (Fig. 4B). Additional levels of post-transcriptional regulation, such as translational control and phosphorylation state, seem to regulate the expression and activity of the sea urchin USF (M. T. Kozlowski and W. H. Klein, unpublished observations). Also, it seems quite significant that the various USF family members are capable of forming both stable homodimers and heterodimers. Indeed, these different USF polypeptides may vary in their transcription activation potential or in their interactions with additional regulators. If this is the case, one would predict that the activity of USF in a given cell will depend not only on the absolute levels of expression, but also on the exact time at which the various forms of this transcription factor are translated.
Acknowledgments
We are grateful to Claus Scheidereit for the generous gift of his Namalwa cDNA library and to W. Michael Perry for the pBS/ET plasmids. We thank Michael W. Van Dyke and Marilyn N. Szentirmay for critical review of the manuscript.
This work was supported by grants GM-38212 (M. Sawadogo) and HD-22619 (W. H. Klein) from the National Institutes of Health, and G-1195 (M. Sawadogo) and G-1210 (W. H. Klein) from the Robert A. Welch Foundation.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
Note added in proof
The C-terminal 200 amino acids of USF2 are identical to those of FIP, a protein which was recently cloned through its interaction with Fos (M. A. Blanar and W. J. Rutter [1992], Science 256, 1014–1018). Therefore USF2 and FIP are probably encoded by alternatively spliced messages from a single gene.
References
- Beckmann H., Su L. K., and Kadesh T. (1990), Genes Dev 4, 177–179. [DOI] [PubMed] [Google Scholar]
- Beckmann H. and Kadesh T. (1991), Genes Dev 5, 1057–1066. [DOI] [PubMed] [Google Scholar]
- Blackwell T. K., Kretzner L., Blackwood E. M., and Eisenman R. N. (1990), Science 250, 1149–1151. [DOI] [PubMed] [Google Scholar]
- Blackwood E. M. and Eisenman R. N. (1991), Science 252, 1211–1217. [DOI] [PubMed] [Google Scholar]
- Carr C. S. and Sharp P. A. (1990), Mol Cell Biol 10, 4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew R. W., Chodosh L. A., and Sharp P. A. (1985), Cell 43, 439–448. [DOI] [PubMed] [Google Scholar]
- Carthew R. W, Chodosh L. A., and Sharp P. A. (1987), Genes Dev 1, 973–980. [DOI] [PubMed] [Google Scholar]
- Chang L. S., Shi Y., and Shenk T. (1989), J Virol 63, 3479–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodosh L. A., Carthew R. W., Morgan G. J., Crabtree G. R., and Sharp P. A. (1987), Science 238, 684–688. [DOI] [PubMed] [Google Scholar]
- Dang C. V., McGuire M., Buckmire M., and Lee W. M. F. (1989), Nature 337, 664–666. [DOI] [PubMed] [Google Scholar]
- Fisher D. E., Carr C. S., Parent L. A., and Sharp P. A. (1991), Genes Dev 5, 2342–2352. [DOI] [PubMed] [Google Scholar]
- Greaves R. F. and O’Hare P. (1991), J Virol 65, 6705–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor P. D., Sawadogo M., and Roeder R. G. (1990), Genes Dev 4, 1730–1740. [DOI] [PubMed] [Google Scholar]
- Hu Y. F., Lusher B., Admon A., Mermod N., and Tjian R. (1990), Genes Dev 4, 1741–1752. [DOI] [PubMed] [Google Scholar]
- Kaulen H., Pognonec P., Gregor P. D., and Roeder R. G. (1991), Mol Cell Biol 11, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff E., Bister K., and Klempnauer K. H. (1991), Proc Natl Acad Sci USA 88, 4323–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski M. T., Gan L., Venuti J. M., Sawadogo M., and Klein W. H. (1991), Dev Biol 148, 625–630. [DOI] [PubMed] [Google Scholar]
- Lemaigre F. P., Courtois S. J., Lafontaine D. A., and Rousseau G. G. (1989), Eur J Biochem 181, 555–561. [DOI] [PubMed] [Google Scholar]
- Murre C., Schonleber-McCaw P., and Baltimore D. (1989), Cell 56, 777–783. [DOI] [PubMed] [Google Scholar]
- Miyamoto N. G., Moncollin V., Egly J. M., and Chambon P. (1985), EMBO J 4, 3563–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter J. J., Cheneral D., Dang C. V., Resar L. M., Mezey E., and Yang V. W. (1991), J Biol Chem 266, 15457–15463. [PubMed] [Google Scholar]
- Prendergast G. C., Lawe D., and Ziff E. B. (1991), Cell 65, 395–407. [DOI] [PubMed] [Google Scholar]
- Prendergast G. C. and Ziff E. B. (1991), Science 251, 186–189. [DOI] [PubMed] [Google Scholar]
- Reach M., Xu L. X., and Young C. S. (1991), EMBO J 10, 3439–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A. L., Meisterernst M., Pognonec P., and Roeder R. G. (1991), Nature 354, 245–248. [DOI] [PubMed] [Google Scholar]
- Rüther U. and Müller-Hill B. (1983), EMBO J 2, 1791–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., and Coulson A. R. (1977), Proc Natl Acad Sci USA 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M., Ishizawa S., Yoshida T., and Shibahara S. (1990), Eur J Biochem 188, 231–237. [DOI] [PubMed] [Google Scholar]
- Sawadogo M. (1988), J Biol Chem 263, 11994–12001. [PubMed] [Google Scholar]
- Sawadogo M. and Roeder R. G. (1985), Cell 43, 165–175. [DOI] [PubMed] [Google Scholar]
- Sawadogo M., Van Dyke M. W., Gregor P. D., and Roeder R. G. (1988), J Biol Chem 263, 11985–11993. [PubMed] [Google Scholar]
- Scheidereit C., Cromlish J. A., Gerster T., Kawakami K., Balmaceda C. G., Currie R. A., and Roeder R. G. (1988), Nature 336, 551–557. [DOI] [PubMed] [Google Scholar]
- Scotto K. W., Kaulen H., and Roeder R. G. (1989), Genes Dev 3, 651–662. [DOI] [PubMed] [Google Scholar]
- Van Dyke M. W., Sirito M., and Sawadogo M. (1992), Gene 111, 99–104. [DOI] [PubMed] [Google Scholar]
- Voronova A. and Baltimore D. (1990), Proc Natl Acad Sci USA 87, 4722–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson C. R., Sigler P. B., and McKnight S. L. (1989), Science 246, 911–916. [DOI] [PubMed] [Google Scholar]
- Zimmerman K. and Alt F. W. (1990), Crit Rev Oncog 2, 75–95. [PubMed] [Google Scholar]