

Abstract
Recessive lethal mutations in the β subunit of eIF-2 that restore HIS4 expression in the absence of an AUG start codon were isolated from diploid Saccharomyces cerevisiae strains. DNA sequence analysis of these alleles and of eIF-2β suppressor alleles isolated from haploid strains, identified point mutations that altered one of six amino acids that map to a Cys-X2-Cys-X19-Cys-X2-Cys “zinc finger” motif and immediately adjacent residues. Five of the affected amino acids are identical in the human and yeast eIF-2β protein. Together with earlier studies (Donahue et al., 1988), these point mutations implicate the zinc finger domain of eIF-2β in start-site selection during the scanning process. We have supplemented the mutations obtained by genetic selection with an additional set of constructed mutations in this region. Our studies indicate that the cysteine residues and the intervening amino acids of this motif are essential for eIF-2β function in translation in itiation in vivo. However, the effects observed in cells containing a copy of eIF-2β with a deletion of this motif suggest that this mutated form is still able to associate with other components of the initiation complex, imparting defects on translation initiation. Thus, this motif may be re quired only for later events that lead to initiator codon recognition. Alterations in defined positions, as found in our suppressor alleles, could lead to recognition of non-AUG codons.
A number of DNA binding proteins contain a sequence motif commonly referred to as a “zinc finger,” in which a zinc atom is coordinated to either two pairs of cysteines (C2-C2 motif), as in the steroid hormone receptors (reviewed in Schwabe and Rhodes, 1991), or two cysteines and two histidines (C2-H2 motif), as found in the transcription factor TFIIIA (Berg, 1990; Lee et al., 1989). The structures in solution of both sequence motifs, derived from two-dimensional nuclear magnetic resonance spectroscopy, indicate that these two types of sequences can assume distinct folding patterns which have in common the role of the zinc atom in stabilization of a rigid conformation of a domain in which an α helical region exposes basically charged residues on the surface for nucleic acid contact. For these proteins it has been shown that the zinc fingers represent domains that directly interact with DNA in a zinc-dependent manner.
Proteins that are implicated in RNA binding also contain zinc finger motifs, such as the gene 32 protein of phage T4 (Gauss et al., 1987), aminoacyl tRNA synthetases (reviewed in Berg, 1986), the retroviral gag proteins (Summers et al., 1990), and the β subunit of the eukaryotic translation initiation factor eIF-2 (Donahue et al., 1988). Nevertheless, it remains unclear whether these sequence motifs represent structural domains of these proteins that bind zinc and function directly in binding to RNA.
Biochemical studies have shown that eIF-2 is composed of three protein subunits—α, β, and γ—and that eIF-2 functions during one of the earliest steps in translation initiation, binding the initiator tRNA in a GTP-dependent fashion (for review see Maitra et al., 1982; Moldave, 1985). This ternary complex then binds the 40S ribosomal subunit, which in turn binds the 5′ end of the mRNA and scans the leader sequence for the first AUG codon in which protein synthesis begins. At the time of 80S complex formation, GTP is hydrolyzed, and eIF-2 is ejected from the 80S complex.
We have previously reported the isolation of the Saccharomyces cerevisiae SUI3 gene encoding eIF-2β (Donahue et al., 1988). The β sub-unit of eIF-2 contains a putative zinc finger motif with the sequence Cys-X2-Cys-X19-Cys-X2-Cys. SUI3 was identified genetically as an extragenic suppressor that restored HIS4 expression in the absence of an AUG start codon. Three independent suppressor mutations in yeast eIF-2β were shown to have altered amino acid residues that resided either within the region flanked by the two pairs of cysteines in the zinc finger motif, or slightly 3′ to the carboxyl cysteine pair. Moreover, these suppressor mutations altered amino acid positions that are either identical or conserved in the human eIF-2β subunit (Donahue et al., 1988; Pathak et al., 1988; also see Fig. 1). Amino terminal sequence analysis of the His4 protein produced when no AUG start codon is present at HIS4 indicated that the SUI3 suppressor gene confers the ability to the ribosome to initiate translation at a UUG codon present in the early HIS4 coding region (Donahue et al., 1988). Recently, we have extended this analysis to show that a suppressor mutation in eIF-2β allows the initiator Met-tRNA to base-pair incorrectly with the UUG codon (Yoon and Donahue, 1991). Taken together, these studies suggested that in addition to its initiator tRNA binding activity, eIF-2 is also involved in the start site selection process.
Figure 1.
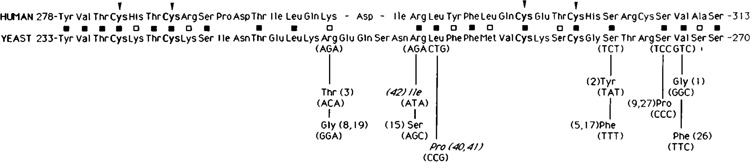
Suppressor mutations in eIF-2β. Amino acid residues of the C2-C2 motif of yeast and human proteins are depicted with closed squares representing identical amino acids and open squares representing conserved substitutions. Alterations found in the suppressor proteins are shown below the amino acid sequent, with the allele numbers in parentheses. Recessive lethal mutations are in italics.
Since suppressor mutations in yeast eIF-2β map to a zinc finger motif, and since this motif is highly conserved in the human eIF-2β protein, it seemed reasonable to assume that this motif represents an important structural domain that functions during the start-site selection process. However, a number of attempts have been unsuccessful in detecting either zinc binding to eIF-2β or a requirement for zinc for eIF-2 function. Mass spectroscopy analysis of partially purified human eIF-2β was not able to detect zinc (Pathak et al., 1988). In vitro GTP-dependent initiator Met-tRNA binding to the yeast eIF-2 complex occurs normally in preparations extensively dialysed against zinc-free buffers (A. M. Cigan and T. F. Donahue, unpublished observations). In addition, we have been unable to detect the binding of 65Zn to filter immobilized yeast eIF-2β and higher concentrations of zinc in growth medium have no effect on the strength of SUI3 suppression events at his4 UUG as measured either on SD-histidine plates or by assays of his4 UUG -lacZ expression (G M. Thompson, B. Castilho-Valavicius, and T. F. Donahue, unpublished observations).
Thus, to gain further insight into the importance of the Cys-X2-Cys-X19-Cys-X2-Cys motif in yeast eIF-2β for the start site selection process, we have characterized eleven additional dominant SUI3 suppressor alleles. Three of these alleles were identified from diploid yeast cells and represent a new class of SUI3 suppressor mutations which confers a recessive lethal phenotype in haploid yeast strains. In these strains, the start site selection process may be altered in favor of non-AUG initiation events; therefore, a wild-type copy of eIF-2β must be present to promote levels of AUG initiation events that are compatible with cell viability. Each of these 11 alleles contains a mutation that alters one of 6 amino acids that map to the carboxyl half of the putative zinc finger motif of eIF-2β. Five of the amino acid residues which are mutated represent amino acids that are identical at the same relative position in the human eIF-2β motif. Hence, evidence from both mutations (in a total of 14 independently isolated SUI3 suppressor genes) and sequence conservation clearly define specific regions of this motif as being functionally relevant to the start site selection process.
To extend this analysis, we also constructed mutations, in either a SUI3 wild-type or suppressor allele, to alter those features that are predicted to be important for the structure and function of a zinc finger. All constructed mutations that deleted this motif, altered the X2 spacing, or mutated the cysteine residues led to inviable yeast cells and a non-suppressor phenotype, implicating the cysteine residues in maintaining the structural features of this region. The deletion of this motif in one of two copies of eIF-2β, however, imparted properties to the cell that are consistent with this mutated eIF-2β protein still associating with components of the initiation complex, but conferring a defect in translation initiation.
Materials and methods
Yeast strains, media, and genetic methods
The yeast strains used in this analysis and their complete genotypes are listed in Table 1. All strains are related to TD28, an ascospore derivative of S. cerevisiae S288C (MATα). Standard genetic techniques and media used for these studies have been described (Rose et al., 1988).
Table 1.
Yeast strains
| Strain | Genotype |
|---|---|
| TD28 | MATa, ura3-52, ino1-13 |
| 45-3 B | MATα, his4-401, ura3-52, leu2-3,-112 |
| JAJ15 | MATa/α, his4 ACC-300/his4-401, ino1-13/INO1, leu2-3,-112/LEU2, ura3-52/ura3-52::his4 ACC-lacZ (Ura+) |
| JAJ18 and JAJ19 | MATa/α, his4 CTG-302/his4-401, ino1-13/INO1, leu2-3,-112/LEU2, ura3-52/ura3-52::his4 CTG-lacZ (Ura+) |
| JAJ20 and JAJ21 | MATa/α, his4 AUU-303/his4-401, ino1-13/INO1, leu2-3,-112/LEU2, ura3-52/ura3-52::his4 AUU-lacZ (Ura+) |
| JAJ25 | MATa/α, his4 ACG-301/his4-401, ino1-13/INO1, leu2-3,-112/LEU2, ura3-52/ura3-52::his4 ACG-lacZ (Ura+) |
| JAJ26 and JAJ27 | MATa/α, his4 GTG-305/his4-401, ino1-13/INO1, leu2-3,-112/LEU2, ura3-52/ura3-52::his4 GTG-lacZ (Ura+) |
| BCV59 | MATa/MATα, ura3-52/ura3-52, leu2-3,-112/leu2-3,-112, his4 UUG-306/his4 UUG-306, SUI3/sui3::URA3 |
| B76-3B | MATα, his4-401, ura3-52::his4 UUG-lacZ [Ura+], leu2-3,-112 |
| TD28-1 | MATa, ura3-52, leu2-3,-112, trp1-Δ63::GCN4-lacZ |
Diploid yeast strains used for reversion analysis were generated by crossing haploid his4 initiator codon mutant strains with haploid strains that contained a deletion at HIS4 and the identical initiator codon mutation at the his4-lacZ fusion integrated at the ura3-52 locus as part of a YIp5 vector as previously described (Donahue et al., 1988). The construction of haploid strains containing initiator codon mutations at HIS4 and the corresponding his4-lacZ fusion construct has also been described (Donahue et al., 1988).
Isolation and characterization of SUI3 suppressor strains
Spontaneous revertants of the His–, haploid initiator codon mutant strains, were selected by demanding growth in the absence of histidine and tested for His4-β-galactosidase expression on SD complete plates containing the X-gal indicator (5-bromo-4-chloro-3indolyl-β-d-galactoside) as previously described (Donahue et al., 1988). The identification and genetic characterization of the haploid SUI3 suppressor strains used in this analysis have been previously reported (Castilho-Valavicius et al., 1990).
The diploid suppressor strains were derived from the identical selection scheme as His+ colonies that were blue on X-gal indicator plates. These revertants were then induced to undergo meiosis and subjected to tetrad analysis. Diploid His+, blue revertants that segregated only 2 viable: 2 inviable meiotic products—each viable product having a His− phenotype and appearing white on X-gal indicator plates - represented candidates containing a suppressor mutation that conferred a lethal phenotype to haploid yeast strains.
Diploid revertants containing a recessive lethal suppressor mutation were then reverted to Ura− by selection on plates containing 5-fluoroorotic acid (5-FOA). This resulted in loss of the his4-lacZ fusion, which is part of the Ura3+, YIp5 vector integrated at the ura3-52 locus in these strains. These strains were then transformed with plasmid pBE30. Plasmid pBE30 contains the intact SUI3 wild-type gene (Donahue et al., 1988) as part of a 1.8kb Hind III DNA fragment in the Ura3+, centromere containing vector YCp50. The SUI3 + gene on this centromere vector was then tested for its ability to rescue the recessive lethal phenotype conferred by the suppressor mutations. Transformed, diploid suppressor strains were sporulated and subjected to tetrad analysis. Those strains which now yielded 3 viable : 1 inviable and/or 4 viable : 0 inviable meiotic products represented candidates containing a suppressor mutation in the SUI3 gene that was lethal in a haploid strain. Transformed, diploid suppressor strains that yielded only 2 viable : 2 in-viable meiotic products represented candidates containing a recessive lethal suppressor mutation in some gene other than SUI3, as the presence of the wild-type SUI3 + gene on the centromere containing vector was incapable of rescuing the lethal phenotype of the haploid meiotic products.
The SUI3 suppressor alleles that were determined to confer a recessive lethal phenotype were isolated from the corresponding Ura− diploid strains by the integration-excision method (Roeder and Fink, 1980), using plasmid p494, which contains a 2.0kb Hind III fragment contiguous to the chromosomal SUI3 gene, inserted at the Hind III site of YIp5, as previously described (Donahue et al., 1988). SUI3 suppressor alleles obtained from the haploid selection scheme were isolated by the same procedure from Ura− derivatives obtained by reversion on 5-FOA plates. Plasmids were isolated in E. coli, and the DNA sequence of the distal SUI3 region was determined by the chain termination method (Sanger et al., 1977), using the primer 5′-TACGTCCGGTTCTGTTGA-3′, which is complementary to a sequence located 106-89 nucleotides 5′ to the region of the zinc finger motif in the SUI3 coding region (Donahue et al., 1988).
Construction and characterization of yeast strains containing site-directed mutations at SUI3
The wild-type SUI3 gene and the SUI3-2 suppressor allele (Donahue et al., 1988) were sub-cloned as a 1.8kb Hind III DNA fragment into the Hind III site of the bacteriophage vector M13mpl0 and used as a target for mutagenesis by the two-primer method (Zoller and Smith, 1982). Oligonucleotides 5′TTGGAGTATGTCA-CTCTCGAGGGTTCTACCAGATCC-3′ and 5′-TTGGAGTATGTCACTCTCGAGGGTTATACCA-GATCC-3′ were used to construct a complete deletion of the zinc finger region in the SUI3 + (SUI3-104, Fig. 2) and SUI3-2 (SUI3-105, Fig. 2) genes, respectively. Each construct deletes amino acid positions 237 to 261 (Fig. 1) and changes the most proximal and distal cysteine residues of the finger region (Cys236 and Cys262) to leucine and aspartic acid, respectively (Fig. 2). Two different oligonucleotides were required for each construct, as the SUI3-2 suppressor mutation (TAT, Tyr) is located at amino acid position 264 (Fig. 1) and differs from the wild-type sequence (TCXSer). Oligonucleotides 5′-CTTTAT-GGTCCACAAAAGTCACGGTTCTACC-3′ and 5′-CTTTATGGTCCACAAAAGTCACGGTTATACC-3′ were used to mutate the most distal cysteine pair (Cys259 and Cys262 to histidine in the SUI3 + (SUI3-102) and SUI3-2 (SUI3-108) genes, respectively (Fig. 2). The remaining constructs were limited to the SUI3-2 suppressor gene. One construct, SUI3-101, mutated the most proximal cysteine residue (Cys236) to histidine using the oligonucleotide 5′TATGTCACTCACAAAACTTGT-3′ (Fig. 2). Two constructs, SUI3-103 and SUI3-107, mutated Cys239 to proline and serine, respectively (Fig. 2), using the corresponding oligos 5′-TGTAAAACTCCAAAGAGTATT-3′ and 5′-TGTAAAACTTCAAAGAGTATT-3′. SUI3-106 was constructed to contain a precise deletion of the two amino acids which intervene the most proximal cysteine pair (Fig. 2), using the oligonucleotide 5′-TATGTCACTTGTTGTAAGAGTATT-3′.
Figure 2.
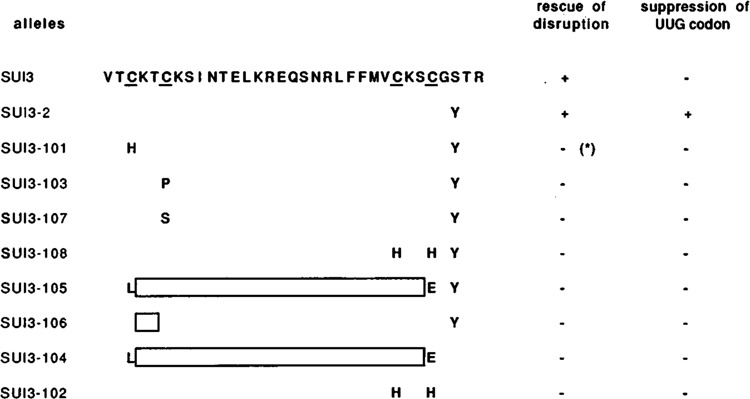
Functional analysis of synthetic alleles of SUI3. Mutant forms of eIF-2β were assayed for function by their ability to rescue the lethality of a strain containing a disruption of the chromosomal copy of the SUI3 gene, and by their ability to initiate translation at a UUG codon at the HIS4 locus. Identical results were obtained when the synthetic SUI3 alleles were present in high copy plasmids (data not shown) except that the cysteine to histidine alteration (allele sui3-101), designated by * allowed germination and growth of a microcolony that could not be tested for its His phenotype.
The presence of each site-directed mutation at either SUI3 or SUI3-2 was confirmed by DNA sequencing as described above. Each approximately 1.8kb Hind III DNA fragment was then isolated and subcloned into the Hind III site of either the LEU2, CEN4 vector pSB32 or the highcopy, LEU2 vector YEp351 (Hill et al., 1986). These plasmids were then used to transform the diploid yeast strain BCV59 to Leu2+ and were tested for their ability to functionally substitute for the wild-type gene in yeast. BCV59 is a diploid yeast strain that contains two chromosomal copies of SUI3, one of which is nonfunctional due to a gene disruption constructed by replacing an internal Xba I fragment in the SUI3 coding sequences with a Hind III fragment containing the URA3 gene, as described by Donahue et al. (1988). Upon sporulation and tetrad analysis, this strain yields 2 viable : 2 in-viable meiotic products. The inviability observed with two of the meiotic products is a result of these haploid segregants containing the disrupted allele of SUI3, which is an essential gene (Donahue et al., 1988). However, this inviability can be rescued when the wild-type SUI3 gene is present on either of the two Leu2+ vectors, pSB32 or YEp351. Hence, strains harboring the mutated SUI3 genes on these plasmids were subjected to tetrad analysis to determine the ability of the mutant eIF-2β proteins to rescue the lethality associated with the gene disruption in BCV59. The BCV59 strain also contains an initiator codon mutation, UUG, at both chromosomal copies of HIS4. Therefore, simply by testing these transformants for growth on SD-His plates, we could determine whether any of the constructed mutations either conferred a suppressor function to the SUI3 wild-type gene, or altered the ability of the SUI3-2 allele to act as a dominant suppressor.
Two other assays were employed for an indication of in vivo function of these mutated forms of SUI3. One assay measured the ability of these mutated SUI3 genes to restore expression to a his4-lacZ initiator codon mutant strain as a more sensitive indication of suppressor function. Mutated SUI3 genes as part of plasmid pSB32 were used to transform yeast strain B76-3B to Leu2+. This strain contains an in-frame his4-locZ construct with a UUG initiator codon mutation. The second assay measured the ability of these mutated forms of SUI3 to alter GCN4 expression, which is regulated at the level of translation initiation (Hinnebusch, 1988). The mutated SUI3 genes as part of plasmid pSB32 were used to transform yeast strain TD28-1 to Leu2+. This strain contains an in-frame GCN4-locZ construct that is integrated at the trpl locus. The ability of the mutated genes to either suppress initiator codon mutations or alter GCN4 expression was assayed by measuring β-galactosidase activity in cell extracts, as previously described (Castilho-Valavicius et al., 1990), from cells grown to an OD600 of 1.2 in minimal SD liquid medium lacking leucine (plasmid selection), and supplemented with histidine, uracil, and inositol. The protein concentration of each extract was determined by the dye-binding method of Bradford (Bradford, 1976). Standard protein curves were performed with bovine serum albumin. The specific activity of β-galactosidase in cell extracts was determined from three independent experiments.
Results
Isolation and characterization of recessive lethal alleles of SUI3
Mutations that implicated a zinc finger motif in eIF-2β in specifying initiator codon recognition were previously identified as revertants of His– haploid strains containing a mutated initiator codon at both the HIS4 locus and a HIS4-lacZ fusion reporter gene, by their ability to grow in the absence of histidine and to form blue colonies on X-gal indicator plates (Donahue et al., 1988). As part of this genetic analysis, spontaneous His+ revertants that formed blue colonies on X-gal plates were also isolated from six diploid strains that contained the HIS4 initiator codon mutated to either ACC, CUG, AUU, GUG, or ACG, and the identical initiator codon mutation at a HIS4-lacZ construction which was integrated at the ura3-52 locus (Table 1). As shown in Table 2, twenty-two of these diploid revertants have been subjected to tetrad analysis, which identified two classes of suppressor mutations based on spore germination and the segregation pattern of the His+ phenotype in the haploid ascospores. Class I diploid revertants (Table 2), gave rise predominantly to four or three viable meiotic products, some of which were His+. For most of the Class I revertants, the segregation patterns of the His+ phenotype were those expected for a suppressor mutation that is unlinked to HIS4 (2+:2−; 1+:3−; 0+:4−). In contrast, Class II diploid revertants yielded predominantly 2 viable : 2 inviable meiotic products, and all viable meiotic products were His− (Table 2). This segregation pattern indicated the presence of a dominant suppressor mutation in the diploid parent strain, which confers a recessive lethal phenotype when present as the sole allelic copy in a haploid meiotic product.
Table 2.
Tetrad analysis of His+ blue revertants from diploid initiator mutant strains.
| Revenants | Segregation of His phenotype in tetrads | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 spores | 3 spores | 2 spores | ||||||||||
| 4+:0− | 3+:1− | 2+:2− | 1+:3− | 0+:4− | 3+:0− | 2+:1− | 1+:2− | 0+:3− | 2+:0− | 1+:1− | 0+:2− | |
| Class 1 | ||||||||||||
| JAJ15R12 | 0 | 0 | 0 | 8 | 1 | |||||||
| JAJ15R26 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 4 | 0 | 0 | 0 | 1 |
| JAJ15R44 | 0 | 0 | 2 | 2 | 0 | 0 | 3 | 1 | 1 | |||
| JAJ15R45 | 0 | 0 | 0 | 3 | 0 | 0 | 1 | 2 | 0 | |||
| JAJ19R10 | 0 | 0 | 1 | 6 | 0 | 0 | 0 | 1 | 1 | |||
| JAJ20R7 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 3 | 0 | |||
| JAJ21R3 | 0 | 0 | 4 | 8 | 1 | 0 | 1 | 0 | 0 | |||
| JAJ26R3 | 0 | 0 | 4 | 6 | 2 | 0 | 0 | 5 | 1 | |||
| Class II | ||||||||||||
| JAJ15R5 | 0 | 0 | 8 | |||||||||
| JAJ15R16 | 0 | 0 | 4 | |||||||||
| JAJ15R17 | 0 | 0 | 9 | |||||||||
| JAJ18R1 | 0 | 0 | 6 | |||||||||
| JAJ18R2 | 0 | 0 | 0 | 1 | 0 | 0 | 5 | |||||
| JAJ18R7 | 0 | 0 | 3 | |||||||||
| JA]19R2 | 0 | 0 | 7 | |||||||||
| JAJ19R3 | 0 | 0 | 8 | |||||||||
| JAJ20R2 | 0 | 0 | 0 | 4 | 0 | 0 | 10 | |||||
| JAJ20R6 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 7 |
| JAJ20R8 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 12 | ||||
| JAJ25R1 | 0 | 0 | 2 | 0 | 0 | 0 | 8 | |||||
| JAJ27R1 | 0 | 0 | 0 | 1 | 0 | 0 | 3 | |||||
| JAJ27R3 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 6 | ||||
In light of the facts that SUI3, which encodes eIF-2β, is an essential gene, and that the SUI3 mutations which were identified from the haploid selection are dominant suppressors in diploid strains (Donahue et al., 1988), we reasoned that some of the dominant suppressor mutations obtained in the diploid selection could represent unusual, recessive lethal allelic forms of SUI3. These unique alleles of SUI3 would represent different suppressor mutations not obtained in our haploid selection; therefore, the isolation and characterization of these SUI3 mutations might provide new insight into regions of the eIF-2β protein which function in start-site selection. In order to determine which of the Class II revertants might contain a SUI3 suppressor allele, we tested the ability of a wild-type copy of SUI3 to rescue the recessive lethal phenotype associated with Class II suppressor mutations. Seven Class II strains were chosen for this analysis, representing suppressor mutants isolated by independent reversion events. The diploid strains were initially reverted to Ura3− by selection on plates containing 5-fluoro-orotic acid (5-FOA) to remove the Ura3+, YIp5 vector that was originally used to insert his4-lacZ at the ura3-52 locus (Table 1). The Ura− derivatives were then transformed with a Ura3+, YCp50 plasmid containing the wild-type SUI3 gene. These transformants were then subjected to tetrad analysis (Table 3). Three diploid suppressor strains—BCV45, BCV49, and BCV55, derived respectively from JAJ27R3, JAJ25R1, and JAJ20R8—were now able to produce either four or three viable meiotic products. Those meiotic products that were His+ were always Ura+, demonstrating that in order to observe the His+, suppressor phenotype in a haploid meiotic product, the wild-type SUI3 gene must be present as part of the Ura3+, YCp50 plasmid. These results indicated that the wild-type SUI3 gene was able to rescue the lethal effect of these suppressor mutations; thus, each was a recessive lethal form of SUI3. It is important to note that these SUI3 suppressor alleles conferred lethality not only to germinating haploid meiotic products but also to vegetatively growing cells. His+, Ura+ ascospores obtained from BCV45, BCV49, and BCV55 (Table 3), when plated on 5-FOA, do not give rise to Ura3−, His+ colonies (data not shown). Therefore, the SUI3 suppressor allele as the sole copy of SUI3 is incompatible with maintenance of cell viability.
Table 3.
Rescue of lethal suppressors by wild-type SUI3.
| Suppressor strain derivatives | Segregation of His phenotype in tetrads | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 spores | 3 spores | 2 spores | ||||||||||
| 4+:0− | 3+:1− | 2+:2− | 1+:3− | 0+:4− | 3+:0− | 2+:1− | 1+:2− | 0+:3− | 2+:0− | 1+:1− | 0+:2− | |
| BCV37 (JAJ15R5) | 0 | 0 | 3 | 1 | 0 | 1 | 15 | |||||
| BCV45 (JAJ27R3) | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 3 | 1 | 0 | 1 | 13 |
| BCV49 (JAJ25R1) | 0 | 0 | 14 | 1 | 3 | 0 | 5 | 7 | 2 | 1 | 2 | 8 |
| BCV51 (JAJ19R2) | 0 | 0 | 0 | 1 | 0 | 0 | 13 | |||||
| BCVS3 (JAJ15R17) | 0 | 0 | 14 | |||||||||
| BCV55 (JAJ20R8) | 0 | 0 | 2 | 7 | 3 | 0 | 0 | 13 | 5 | 0 | 1 | 6 |
| BCVS7 (JAJ18R7) | 0 | 0 | 1 | 0 | 0 | 0 | 16 | |||||
In contrast, tetrad analysis of the other diploid strains—BCV37, BCV51, BCV53, and BCV57, derived respectively from JAJ15R5, JAJ19R2, JAJ15R17, and JAJ18R7—produced no 4-spore tetrads and no or relatively few viable 3-spore products. Instead, these strains produced predominantly 2 viable : 2 inviable meiotic products, despite having been transformed with the wild-type SUI3 gene (Table 3). The majority of the viable spores are His−. Mating type tests indicated that the few His+ meiotic products observed were diploid cells. These results indicated that these diploid revertants contained dominant suppressor mutations in genes other than SUI3 that also conferred a recessive lethal phenotype. These suppressor mutants were excluded from further characterizations.
Each recessive lethal allele of SUI3 identified in BCV45 (SUI3-40), BCV49 (SUI3-41), and BCV55 (SUI3-42) was isolated by the integration-excision method, and its nucleotide sequence, corresponding to the carboxyl end of the SUI3 coding region, was determined. Each allele contained a single base change altering amino acids that are identical at the same relative position in the C2-C2 motif shared between the wild-type yeast and human eIF-2β proteins (Fig. 1). The mutations in the sui3-40 and sui3-41 alleles were identical, both changing the leucine residue at position 254 in the yeast sequence to a proline. The mutation in the sui3-42 allele changed the arginine at position 253 to isoleucine. Thus, the analysis of three recessive lethal SUI3 suppressor alleles identified two amino acids in the “finger” region of the C2-C2 motif of eIF-2β that can be mutated to alter the start-site selection process in yeast.
Characterization of additional viable suppressor mutations in elF 2β
In our initial genetic characterization of suppressors of initiator codon mutations, a large group of dominant suppressors selected from haploid strains was identified, and thirty-two isolates were shown in genetic crosses to be allelic to SUI3 (Castilho-Valavicius et al., 1990). The amino acid changes of three of these alleles, SUI3-1, SUI3-2, and SUI3-3, have been characterized previously (Donahue et al., 1988) and are shown in Figure 1. To define other amino acid residues in eIF-2β which can be mutated to alter the start-site selection process, we have cloned and sequenced eight additional SUI3 suppressor alleles chosen from the above group. Because each of these SUI3 alleles was derived from different parent strains, each represents an independent mutational event.
As shown in Figure 1, five alleles contained point mutations that altered one of three amino acids located slightly 3′ to the carboxyl cysteine pair of the C2-C2 motif. Mutations in the SUI3-9 and SUI3-27 alleles define a new position (Ser267), which can be mutated (from TCC to CCCPro) to confer suppression properties to eIF-2β. The SUI3-5 and SUI3-17 alleles and the SUI3-26 allele define amino acid positions 264 and 268, respectively, as being important for start-site selection, confirming results reported previously for SUI3-2 and SUI3-1 alleles (Donahue et al., 1988). The amino acid changes are different for the various alleles, however, as shown in Figure 1.
The remaining three SUI3 suppressor alleles mapped to amino acid positions in the “finger” region of the C2-C2 motif (Figure 1). The SUI3-8 and the SUI3-19 alleles and the SUI3-15 allele redefined amino acid positions 248 and 253, respectively, as being important for start-site selection; each amino acid position was found to be mutated in the SUI3-3 allele (Donahue et al., 1988), and in the recessive lethal suppressor allele SUI3-42, respectively. However, the amino acid changes are different, as shown in Figure 1.
Functional analysis of in vitro constructed mutations in elF-2β
The repeated isolation of mutations in the same set of residues indicated that we had nearly reached saturation with respect to eIF-2β mutations that lead to a suppressor phenotype, at least within the limits of our selection scheme for spontaneous revertants. The mutations described above specifically identify the carboxyl half of the C2-C2 motif—and only the C2-C2 motif—as being important for start site selection. All substitutions involved residues that are identical or conserved between the yeast and human proteins, with the introduction of side chains that are predicted to impart marked effects either on the native structure or chemistry of this region.
To gain further insight into the amino acid sequences in the C2-C2 region that are important for start-site selection, we constructed a series of mutations, specifically altering features that are believed to be essential for the structural integrity or function of zinc finger motifs. Mutations were constructed by site-directed mutagenesis in the dominant suppressor allele SUI3-2, which was identified in the reversion analysis of haploid yeast strains (Castilho-Valavicius et al., 1990) and shown to contain a mutation that alters an amino acid residing 3′ to the carboxyl cysteine pair (Donahue et al., 1988; Fig. 1). The site-directed, mutated forms of the SUI3-2 gene were cloned into a single copy CEN4 vector and a high copy YEp351 vector, both of which contain the selectable marker Leu2+. Each plasmid was then used to transform the diploid strain BCV59 to Leu2+. The BCV59 diploid strain has one of the two chromosomal SUI3 alleles disrupted by the URA3 + gene (Table 1). This strain gives rise to 2 viable : 2 inviable meiotic products, as one functional copy of SUI3 is essential for the viability of yeast strains (Donahue et al., 1988). Therefore, tetrad analysis of these transformants enabled us to assess whether any of the mutated forms of SUI3-2 were fully functional in yeast by their ability to rescue ascospores that contained the SUI3:URA3 + gene disruption. The BCV59 strain also contains an initiator codon mutation, UUG, atHIS4. Hence, with these transformants we can also assess whether the mutations constructed at SUI3-2 have any effect on the ability of the dominant suppressor mutation in SUI3-2 to alter the start site selection process as his4.
The results of this analysis are shown in Figure 2. The SUI3-2 alleles constructed to contain an amino acid substitution in either of the two proximal cysteine residues—SUI3-101 (Cys236 to His), SUI3-103 (Cys239 to Pro), SUI3-101 (Cys239 to Ser)—or an amino acid substitution in both of the distal cysteine residues—SUI3-108 (Cys259-X2-Cys262 to His-X2-His)—were incapable of rescuing haploid SUI3 gene disruption products from lethality. In addition, none of these constructed alleles could confer a His+ suppressor phenotype to the BCV59 strain (Fig. 2). In contrast, the SUI3 + gene or the SUI3-2 suppressor allele, when part of either the single copy or high copy plasmid, was capable of rescuing the lethality caused by the gene disruption at SUI3, and the SUI3-2 allele conferred a His+ suppressor phenotype to BCV59 (Fig. 2). These results demonstrate not only that mutation of these cysteines is incompatible with normal eIF-2β function in yeast, but that these cysteines are needed for the SUI3-2 allele to mediate initiation at a non-AUG codon in his4. Figure 2 also shows that deletion of the two amino acids that separate the proximal cysteine pair (SUI3-106) is incompatible with normal function and suppression; the same results were observed when the entire C2-C2 motif was deleted (SUI3-105). Thus mutations which alter the cysteine residues or their spacing abolish the ability of eIF-2β to function in yeast, as might indeed be anticipated if this region constituted a structural domain defined by the Cys residues.
A deletion of the C2-C2 motif (SUI3-104) and an amino acid substitution in both of the distal cysteine residues— SUI3-102 (Cys259-X2-Cys262 to His-X2-His)—were also constructed in the wild-type SUI3 gene. Both of these mutations were incompatible with normal SUI3 function (Fig. 2). Furthermore, neither of these constructed mutations could confer a His+ phenotype (Fig. 2). Our data suggest that the structural integrity of this region must be maintained for suppressor activity as well as for normal elF-2β function.
Deletion of the C2-C2 region in elF-2β alters GCN4 expression in yeast
As a more sensitive assay for suppression, we also determined whether any of our site-directed mutations at SUI3, when present in the haploid yeast strain B76-3B (Table 1), could increase the expression of a his4-lacZ construct that contained a UUG initiator codon mutation. Each of the SUI3 synthetic alleles, as well as the wild-type SUI3 gene and the SUI3-2 suppressor allele contained on the CEN plasmid, was used to transform B76-3B. The level of his4-lacZ expression was determined by measuring the specific activity of His4-β-galactosidase in cell extracts derived from each transformant. As shown in Figure 3, the presence of the SUI3-2 suppressor allele on a low copy plasmid resulted in a 6.8-fold increase in His4-p-galactosidase expression when compared to the activity measured from transformants that contained the wild-type SUI3 gene. In contrast, the His4-P-galactosidase activity measured from strains that contained the site-directed SUI3 constructs on a plasmid showed no significant increase over the wild-type control strain with two exceptions: strains containing the SUI3-104 and the SUI3-105 alleles showed an approximately twofold increase in His4-β-galactosidase expression (Fig. 3). This effect cannot be attributed to the SUI3-2 suppressor mutation (used in obtaining SUI3-105), because the effect is also seen with SUI3-104, which was constructed in the wild-type SUI3 gene. Instead, this effect appears to be a result of deleting the C2-C2 motif.
Figure 3.
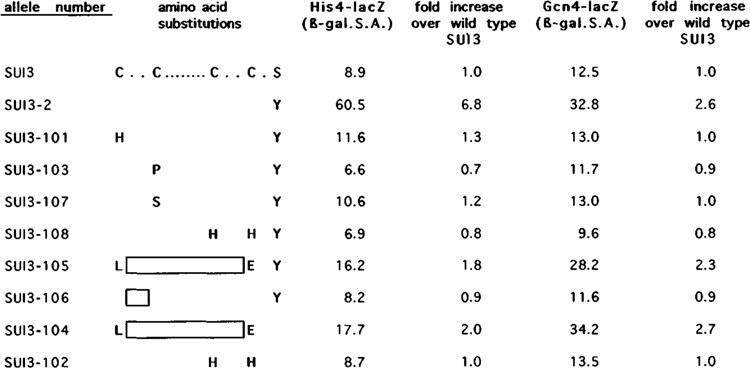
Quantitation of HIS4 and GCN4 expression. The ability of the synthetic constructs present on centromeric plasmids to initiate protein synthesis at a non-AUG codon was measured from a chromosomal in-frame lacZ fusion to HIS4, in which the normal initiator codon was altered to UUG. GCN4 expression was measured from an isogenic strain containing a lacZ fusion to GCN4, inserted in the chromosome.
One possible interpretation of these studies is that deletion of the C2-C2 motif confers suppression properties to eIF-2β, but the efficiency of suppression is too low to produce a His+ phenotype (Fig. 2) and is only detected by the more sensitive β-galactosidase assay (Fig. 3). However, an alternative interpretation is that the twofold increase in His4-β-galactosidase activity reflects an increase in transcription, rather than translational suppression. This alternative interpretation merits consideration, inasmuch as the expression of the HIS4 gene (including our HIS4lacZ fusion constructs) is regulated by the positive transcriptional activator GCN4, and GCN4 itself is under a form of translational control that is sensitive to the concentration and forms of eIF-2 (Hinnebusch, 1990; Abastado et al., 1991; Dever et al., 1992).
To differentiate between an increase in His4-β-galactosidase activity mediated at the level of His4 translation versus an increase in his4-lacZ transcription secondary to disruption of the control of GCN4 expression, we assayed the levels of GCN4 expression in yeast cells that contained the C2-C2 deletion alleles SUI3-104 and SUI3-105. These and the other constructs contained on a CEN plasmid were transformed into the haploid strain TD28-1, which carries a chromosomal GCN4-lacZ fusion. This represents the entire upstream regulatory region of GCN4 and its initial coding sequences fused in-frame with lacZ and has been used extensively for characterizing the GCN4 regulatory response (Dever et al., 1992). β-galactosidase activity was determined from cell extracts prepared from each of these transformants as an indication of GCN4 expression levels and was compared to a strain carrying a wild-type plasmid-borne copy of SUI3 or the SUI3-2 allele. As shown in Figure 3, the presence of SUI3-2 in this strain caused a 2.6-fold increase in the expression of the GCN4-lacZ fusion compared to the SUI3 wild-type control strain. Thus, as previously demonstrated, a SUI3-2 suppressor allele not only leads to suppression of an initiator codon mutation at HIS4 and HIS4-lacZ but also increases the level of GCN4 expression (Castilho-Valavicius et al., 1990; Williams et al., 1989). In contrast, all the site-directed SUI3 constructions when present in this strain did not increase GCN4-lacZ expression with two exceptions: the SUI3-104 and SUI3-105 alleles, which have the C2-C2 deletion mutation in common, resulted in a 2.7- and 2.3-fold increase in GCN4-lacZ, respectively. Significantly, this is the same magnitude of effect on GCN4-lacZ expression as that seen with the SUI3-2 allele (Fig. 3). In addition, this increase in GCN4 expression correlates with the increased level of his4-lacZ expression (twofold) that was observed with the C2-C2 deletion constructs (Fig. 3). Thus, the SUI3, C2-C2 deletion alleles probably do not increase his4-lacZ expression by suppression of the initiator codon mutation, but rather by increasing Gcn4 levels. Again, these effects cannot be attributed to the SUI3-2 suppressor mutation, as the SUI3-104 allele was constructed in the wild-type SUI3 gene. Also, these effects cannot be attributed to differences in genetic backgrounds, as strain isogenicity was maintained for the assay of the effect of each construct on his4-lacZ or GCN4-lacZ expression. Instead, these results imply that, in contrast to the other site-directed mutations in eIF-2β, elF-2β with a deleted C2-C2 motif maintains some function in yeast cells that alters the translational control of GCN4 expression.
Discussion
We have described here an extensive mutational analysis of the β subunit of yeast eIF-2, involving the C2-C2 motif located in the carboxyl end of the protein. The repeated isolation of suppressor mutations in the same set of amino acid residues seems to present a strong argument that the C2-C2 motif is implicated in conferring start-site selection properties to the ribosome. In addition, site-directed mutations that delete this motif, change the cysteine residues, or alter their spacing not only abolish suppressor activity of the SUI3-2 allele but destroy the ability of SUI3 to maintain cell viability. Since all substitutions involved residues that are identical or conserved between the C2-C2 motif of yeast and human eIF-2β, this data must also be relevant to the function of the region of the mammalian eIF-2β protein during the start site selection process.
One significant observation we have made is that deletion of the C2-C2 motif in eIF-2β disrupts the translational control of GCN4. Recent studies have mapped out how eIF-2 functions in the regulation of GCN4. This regulatory response, mediated by four small upstream reading frames (uORFs) in the leader region of its mRNA, can be simplified by considering the events that occur at uORFl and uORF4 (Hinne-busch, 1990). Under normal growth conditions ribosomes initiate at uORFl, terminate translation, and reinitiate at uORF4 (Abastado et al., 1991). After translation of uORF4, ribosomes presumably fall off the mRNA, precluding initiation at the GCN4 start codon. Under amino acid starvation conditions, the signal for the general control response, ribosomes still initiate translation at uORFl, but only 50% of the ribosomes reinitiate at uORF4. The remainder bypass uORF4 and reinitiate at the GCN4 start site (Abastado et al., 1991). The basis for this is that the signal for general control induces an eIF-2 kinase, Gcn2, to phosphorylate the a subunit of eIF-2 at the Ser51 position (Dever et al., 1992). Based on the effects of phosphorylation of Ser51 of eIF-2α by mammalian eIF-2 kinases, this would block the GDP to GTP exchange reaction and lead to accumulation of eIF-2-GDP, which is inactive. As the eIF-2-GTP bound form is required for ternary complex formation (the ability of eIF-2 to bind the initiator tRNA), the net effect is that lower levels of eIF-2 diminish the efficiency of reinitiation at uORF4, enabling ribosomes to bypass this region. The increased scanning distance from uORF4 to the GCN4 start codon would therefore allow more time for the ribosome to reacquire eIF-2-GTP and initiate at the GCN4 start site.
Based on these studies, we suggest that the increased levels of GCN4 expression we observed in strains that contained a deletion of the C2-C2 in eIF-2β (Fig. 3) is related to the mutant form of β causing reduced levels of active eIF-2 in the cell, thus leading to an increase in GCN4 expression. One possible explanation for this effect is that, despite a deletion of the C2-C2 motif, eIF-2β maintains partial activity that still enables it to associate with the α and γ subunits of eIF-2. This would lead to two populations of eIF-2 in these cells. One population is composed of the α, γ, and wild-type β subunits; the latter is encoded by the SUI3 + allele and maintains normal translation initiation events for cell viability. The other population of eIF-2 is composed of α and γ but contains the β protein encoded by the C2-C2 deletion allele. This latter complex, however, is defective in either initiator-tRNA binding or perhaps AUG codon recognition. The net effect is that the level of fully functional eIF-2 in the cell is reduced, and as a result reinitiation at uORF4 is less efficient. Consequently the ribosome bypasses the barrier presented by uORF4, allowing increased scanning time for the active eIF-2 to reassemble with the ribosome at the GCN4 start site, and in turn yielding increased levels of Gcn4. This interpretation is consistent with our observation that SUI3 suppressor mutations, the sole copy of eIF-2β in the cell, also disrupt the translational control of GCN4. These suppressor strains do maintain sufficient activity for normal “first AUG codon” recognition but may reduce the level of active eIF-2 in the cell for reinitiation at GCN4.
Another indication that the C2-C2 deletions of eIF-2β were functional in yeast derives from growth curves performed with cells harboring these alleles in the high copy plasmids (data not shown). The SUI3-2 suppressor allele in high copy number caused a twofold increase in the generation time of this strain, compared to cells carrying the vector or the wild-type SUI3 allele. The same increase in generation time was also observed in cells containing the SUI3 deletion alleles in high copy number, but this effect was not observed with the isogenic strains that carried the other synthetic mutations in elF-2β. However, the polysome profiles observed from these strains were virtually indistinguishable from each other (data not shown). Apparently, alterations in polysome profiles may not be sensitive enough to reveal defects in translation initiation with these mutants, in contrast to their effects on cell growth rate or the disruption of the control of GCN4 expression. The latter assay, we believe, is a more sensitive barometer of the “state of translation initiation” in the cell.
Unfortunately, we were unable to detect the C2-C2 deleted form of eIF-2β in these cells by Western blot. We believe that this is not a result of either the absence or the poor expression of this protein in yeast. Instead, the inability to detect this protein represents a technical limitation with our anti-Sui3 antisera, which is directed against the carboxyl end of the SUI3 coding region, including the C2-C2 motif that is deleted in these constructs (Donahue et al., 1988). However, more impprtantly, we eould detect by Western blot that all of the other site-directed mutated forms of eIF-2β, which had no effect on growth or GCN4 expression, were overproduced in these strains (data not shown). These studies therefore present a strong argument that eIF-2β in the absence of the C2-C2 motif maintains partial activity in yeast, most likely maintaining functions that allow association with the other subunits of eIF-2. We can draw an analogy to transcription factors which maintain DNA binding ability despite a deletion of an activation domain (Mitchell and Tjian, 1989).
One basic question that remains is why a deletion of the C2-C2 motif is compatible with partial eIF-2β function in yeast, but not the other mutated forms of eIF-2β that were constructed in the C2-C2 region of SUI3. The simplest interpretation is that mutations that either alter the cysteine residues or delete the amino acids that intervene in the proximal pair of cysteine residues destroy the ability of the C2-C2 motif to assume a structure that is analogous with a zinc finger domain. These mutations would result in unfolding of the domain, and consequently interfere with the ability of other regions of eIF-2β to take on a normal conformation. Hence, these forms of eIF-2β may not be able to associate with the α and γ subunits of eIF-2 or other components of the preinitiation complex and are completely non-functional in yeast, as we detected. In contrast, the deletion of the C2-C2 motif does not interfere with the ability of other portions of eIF-2β to assume its normal conformation. Instead, the carboxyl region of eIF-2β which contains a deletion of C2-C2, at least in part, mimics the normal structure of this region. That structure is where the sequences adjoining the motif are brought to close proximity, whether by disulfide bonds or coordination of a metal atom in the wild-type protein, or as accomplished by the deletion of the C2-C2 region. Thus, the deletion of C2-C2 may not interfere with the ability of the amino terminal end of eIF-2β to assume its normal conformation and to retain partial function.
A second important question to resolve is how the suppressor mutations alter the start site selection process to allow non-AUG initiation events. Specific initiation codon recognition may be associated with the C2-C2 region and immediately flanking residues, as identified by the suppressor alleles of P, involving those residues that are conserved between the human and yeast sequences. These conserved positions could either define specific contact points or contribute to making contact with other components of the initiation complex, such as tRNA or mRNA. Alterations such as those found in the haploid strains would lead to AUG and UUG recognition (Donahue et al., 1988; Yoon and Donahue, 1992), while alterations found in the diploid revertants may lead to only UUG utilization, rendering the cells non-viable in the absence of a wild-type protein. In this regard, SUI3 suppressor alleles would represent “gain-of-function” mutations, having acquired a new nucleic acid binding specificity; alternatively, SUI3 suppressor alleles could be “loss-of-function” mutations, having reduced stringency for AUG codon recognition and permitting the initiator-tRNA to base-pair incorrectly with UUG codon (Yoon and Donahue, 1992). Loss-of-function mutations might be compatible with the amino acid substitutions found in the suppressor mutants occurring in charged residues that might be predicted to make nucleic acid contact. It is also worth noting that Arg248 is highly conserved in position to positively charged residues in seven other sequences known to bind zinc, whose structures have been defined, when these regions are aligned by their carboxyl cysteine or histidine pair (Hard et al., 1990; Lee et al., 1989; Parraga et al., 1988; Omichinski et al., 1990; Frankel et al., 1987). Suppressor mutations that insert Pro in this region could also reduce C2-C2 function by altering the structure of this domain. Our previous observations that SUI3 suppressor alleles confer an in vitro defect to eIF-2 in initiator-tRNA binding activity may be the key link in determining the relationship between the effects of these mutations on the start-site selection and what this region may be binding to arrive at this goal.
In summary, although we have not yet been able to demonstrate that eIF-2β binds zinc, it is difficult to reconcile the data obtained from these mutational studies with any other interpretation than that the evolutionarily conserved C2-C2 motif of eIF-2β constitutes a structural domain analogous to a zinc finger. This study presents a striking parallel to mutational analyses of zinc finger DNA binding proteins for which mutations in similarly positioned amino acids alter the function and specificity of these proteins (Schena et al., 1989). In addition, mutation of any one of four cysteine residues that coordinate the zinc atom in such DNA binding proteins has been shown to be incompatible with protein function—the same result we have shown by site-directing mutations at the cysteine residues of the C2-C2 motif in eIF-2β.
Clearly, what are needed next are biochemical studies to verify that the structural conformation of this region exists, as we have proposed, and to determine the binding specificity of this region. The mutations we have described should be invaluable for these analyses, and such studies are currently under way.
Acknowledgments
We thank Tom Dever and Alan Hinnebusch for the GCN4-lacZ fusion strain, Julie A. Johansen for excellent technical assistance, and Heejeong Yoon for contributing Table 3 to this manuscript.
This work was supported by grant 410642/89-4 from CNPq (Brazil) awarded to Beatriz Castilho-Valavicius and by a U. S. Public Health Service grant GM32263 from the National Institutes of Health awarded to Thomas F. Donahue. Gloria M. Thompson is a pre-doctoral student supported by CNPq.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Abastado J.-P., Miller P. F., Jackson B. M., and Hinnebusch A. G. (1991), Mol Cell Biol 11, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J. (1986), Science 232, 485–487. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976), Anal Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Castilho-Valavicius B., Yoon H., and Donahue T. F. (1990), Genetics 124, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever T. F., Feng L., Wek R., Cigan A. M., Donahue T. F., and Hinnebusch A. G. (1992), Cell 68, 585–596. [DOI] [PubMed] [Google Scholar]
- Donahue T. F., Cigan A. M., Pabich E. K., and Castilho-Valavicius B. (1988), Cell 54, 621–632. [DOI] [PubMed] [Google Scholar]
- Gauss P., Krassa K. B., McPheeters D. S., Nelson M. A., and Gold L. (1987), Proc Natl Acad Sci USA 84, 8515–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A. D., Berg J. M., and Pabo C. O. (1987), Proc Natl Acad Sci USA 84, 4841–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hard T., Kellenbach E., Boelens R., Maler B. A., Dahlman K., Freedman L. P., Carlstedt-Duke J., Yamamoto K. R., Gustafson J. A., and Kaptein R. (1990), Science 249, 157–160. [DOI] [PubMed] [Google Scholar]
- Hill J. E., Myers A. M., Koerner T. J., and Tzagoloff A. (1986), Yeast 2, 163–167. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A. G. (1988), Microbiol Rev 52,248–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch A. G. (1990), Trends Biochem Sci 15, 148–152. [DOI] [PubMed] [Google Scholar]
- Lee M. S., Gippert G. P., Soman K. V., Case D. A., and Wright P. E. (1989), Science 245, 635–637. [DOI] [PubMed] [Google Scholar]
- Maitra U., Stringer E. A., and Chaudhuri A. (1982), Annu Rev Biochem 51, 869–900. [DOI] [PubMed] [Google Scholar]
- Mitchell P. J. and Tjian R. (1989), Science 245, 371–378. [DOI] [PubMed] [Google Scholar]
- Moldave K. (1985), Annu Rev Biochem 54, 1109–1149. [DOI] [PubMed] [Google Scholar]
- Omichinski J. G., Clore G. M., Appella E., Sakaguchi K., and Gronenborn A. M. (1990), Biochemistry 29, 9324–9334. [DOI] [PubMed] [Google Scholar]
- Parraga G., Horvarth S. J., Eisen A., Taylor W. E., Hood L., Young E. T., and Klevit R. F. (1988), Science 241, 1489–1492. [DOI] [PubMed] [Google Scholar]
- Pathak V. K., Nielsen P. J., Trachsel H., and Hershey J. W. B. (1988), Cell 54, 633–639. [DOI] [PubMed] [Google Scholar]
- Roeder G. S. and Fink G. R. (1980), Cell 21, 239–249. [DOI] [PubMed] [Google Scholar]
- Rose M. D., Winston F., and Hieter P. (1988), Methods in Yeast Genetics, Cold Spring Harbor Laboratories Press, Cold Spring Harbor, NY. [Google Scholar]
- Sanger F., Nicklen S., and Coulson A. R. (1977), Proc Natl Acad Sci USA 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M., Freedman L. P., Yamamoto K. R. (1989), Genes Dev 3, 1590. [DOI] [PubMed] [Google Scholar]
- Schwabe J. W. R. and Rhodes D. (1991), Trends Biochem Sci 16, 291–296. [DOI] [PubMed] [Google Scholar]
- Summers M. F., South T. L., Kim B., and Hare D. R. (1990), Biochemistry 29, 329–340. [DOI] [PubMed] [Google Scholar]
- Williams N. P., Hinnebusch A. G., and Donahue T. F. (1989), Proc Natl Acad Sci USA 86, 7515–7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H. and Donahue T. F. (1992), Mol Cell Biol 12, 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller M. J. and Smith M. (1984), DNA 3, 479–488. [DOI] [PubMed] [Google Scholar]