

Abstract
At least four regulatory cis-acting DNA sequences, CCAAAAGTGG (element A), TTATTTTTA (element B), TATTTATT (element C), and TATTACCTTTAT (element S), were identified in cardiac myosin light chain-2 (MLC2) proximal promoter as target sites for sequence-specific binding of nuclear proteins. For muscle-specific transcription, the proximal promoter (−53 to +1) consisting only of elements B and C is required. Addition of element A to this promoter results in a musclespecific up-regulation, whereas the addition of element S exerts a negative effect on transcription. The negative and positive regulatory effects of elements S and A respectively were demonstrated by site-specific mutations of the promoter following transient transfection of cardiac muscle cells in culture. Elements S and A interact separately with distinct nuclear protein factor present in both muscle and non-muscle cells, even though their regulatory activities are restricted to muscle cells. Among the multiple complexes resulting from the interaction of nuclear proteins and elements S and A DNAs, one requires both S and A sequences together for binding. Element B, which exerts a muscle-specific positive effect on transcription, binds to a nuclear protein present in cardiac muscle, but not in non-muscle cells. DNA-protein binding assays and mutational analysis of the MLC2 promoter suggest that the contribution of the functionally opposed cis-elements depends upon an interplay between the positively and negatively acting DNA-binding proteins via protein-protein interactions to mediate opposite regulatory effects on gene transcription.
The tissue- and developmental-specific expression of muscle contractile proteins, which accounts for muscle cell differentiation during myogenesis, provides a highly useful system to investigate the mechanisms involved in the control of gene transcription. It is known that expression of muscle-specific genes is controlled primarily at the level of transcriptional initiation, which is regulated through interactions between cis-acting DNA elements and trans-acting protein factors (Schwartz and Roth-blum, 1981; Whalen et al., 1981; Bains et al., 1984; Lomper et al., 1984; Wieczorek et al., 1985; Hayward and Schwartz, 1986; Hayward et al., 1988). It is believed that a concerted activity of both positive and negative regulatory elements ensures the tissue-specific transcription of muscle genes (Chow and Schwartz, 1990). A number of gene promoter sequences and enhancer elements potentially involved in muscle-specific transcription have been described (Jaynes et al., 1986; Minty and Kedes, 1986; Miwa and Kedes, 1987; Bouvagnet et al., 1987; Sternderg et al., 1988; Gustafson and Kedes, 1989; Braun et al., 1989; Chow and Schwartz, 1990; Shen et al., 1991). Among these elements a 10 bp sequence CC(A/T)6GG (CArG box or C/BAR), originally identified in the promoter of muscle α-actin gene (Grichnik et al., 1986; Minty and Kedes, 1986; Muscat et al., 1988; Mohun et al., 1989); an A/T-rich hnhancer requence, MEF (ATAAAAATAA), found in the mouse skeletal muscle creatine kinase gene (Buskin and Hauschka, 1989); a BSe-element (GTGTCAGTCA; Yu and Nadal-Ginard, 1989) in rat skeletal myosin heavy chain (MHC) gene; and a skeletal muscle-specific distal promoter element (M-CAT) in cardiac troponin-T gene (Mar and Ordahal, 1990) appear to be involved in the regulation of transcription of the respective genes. To date, however, no single regulatory element or factor has been identified which would suggest a common regulatory mechanism for muscle-tissue specificity.
In recent years there has been an explosion of studies that define and elucidate the role of DNA-binding proteins in activation of gene transcription, but relatively little is known about proteins that mediate repression of transcription. For cardiac MLC-2 gene, the proximal promoter consisting of about 60 bp upstream to the initiation site is sufficient for muscle-specific transcription (Braun et al., 1989, and see below). We have recently described a sequence (CSS) involved in repression of transcription of cardiac MLC-2 gene in skeletal muscle (Shen et al., 1991) and an upstream element required for fos-dependent inhibition of transcription of MLC-2 gene (Goswami et al., 1992). Another element located at −64 in the same gene was reported earlier to exert a negative effect on MLC-2 transcription in HeLa cells (Braun et al., 1989). In order to delineate further the regulatory elements in MLC-2 promoter and understand their role in control of transcriptions, we undertook a fine mapping of the proximal promoter of the gene by functional analysis of a variety of deletion mutants of MLC-2 promoter. The results indicate that elements denoted as A and B (see below), located at −65 and −46 respectively, have muscle-specific positive regulatory activity, whereas element S exerts a negative effect on transcription. Among the proteins that recognize element B, MBF1 is present in cardiac muscle cells, but not in fibroblasts. On the other hand, the element S binding protein SF1 is present in both myoblasts and fibroblasts, even though the latter cells do not support transcription of the cardiac MLC-2 gene. Element A, whose binding properties have been described in a recent communication (Qasba et al., 1992), is a muscle-specific activator of transcription and is recognized by nuclear proteins (ABFs) common to both muscle and non-muscle cells. Thus, the muscle-specific positive regulation of transcription appears to be mediated primarily by element B and its binding proteins, whereas the negative role is afforded by element S. Gel mobility shift competition assay and mutational analysis of elements S and A suggest that close interplay, presumably involving intra-protein interactions, must occur between the negative and positive protein factors which mediate the opposite regulatory consequences in MLC-2 gene transcription.
Materials and methods
Primary cultures of chicken cells
Chicken embryos (13 days old) were used to prepare primary cell cultures as described earlier by Shen et al. (1991). Fibroblast cells were removed from the cardiac cells by three 60-minute cycles of differential attachment. Primary cultures of the chicken brain were prepared as described by Borden et al. (1984), and fibroblast cell cultures were according to Chow and Schwartz (1989).
Preparation of nuclear extracts
The nuclear extract was prepared by a modification of the method of Dignam et al. (1983) as described earlier (Shen et al., 1991). The extract usually contained 4 to 6 mg protein/ml and remained stable in liquid nitrogen for several months.
DNase-I footprinting
The footprinting was performed essentially according to Jones et al. (1985) and as described earlier (Shen et al., 1991). Radiolabeled DNA fragment (corresponding to the nucleotide sequence −120 to +38 of the chicken cardiac MLC-2 promoter) was obtained from the 1.3 kb promoter region of pLC106CAT plasmid DNA by treatment with restriction enzymes Hind III and Hinf I, followed by the end-labeling of the fragment with Klenow DNA polymerase in the presence of [α-32P]dCTP. DNase-I digestion of end-labeled DNA was performed with 100-fold freshly diluted Dnase I for 60 seconds. The reaction mixture was subjected to phenol-chloroform extraction, and the DNA was analyzed on an 8% polyacrylamide sequencing gel.
Construction of 5′-deleted and site-directed MLC-2 mutants
All of the deletion mutants were derived from pLC106CAT, which contains 1.3 kb of the chicken MLC-2 promoter region in pSVOCAT (Zarraga et al., 1986). Following the digestion of the plasmid DNA with restriction enzyme Xho I, which has a unique site at −241, the DNA was digested by Bal 31 exonuclease. The reaction was terminated at different time points, and DNAs were digested by Sma I, which has a unique restriction site at −1285. The ends were filled with Klenow fragment of DNA polymer-ase, self-ligated, and transformed into E. coli strain JM109. The resultant mutants were characterized by restriction enzyme analysis and subsequent DNA sequencing.
Site-directed mutants corresponding to elements S and A sites were obtained by polymerase chain reaction (PCR) using two sets of synthetic oligonucleotides. The first set of oligos contained primer A, corresponding to the Hind III site located downstream from the S site; and primer B, containing a Kpn I restriction site in place of the S and A oligonucleotide. The second set of oligos contains primer C, which is complementary to primer B; and primer D, which corresponds to the Nde I site located upstream from the S and A site. One of the PCR reaction products has the original Nde I restriction site at the 5′ end and Kpn I restriction site in place of S or A oligonucleotide at the 3′ end. The second reaction product has the Kpn I site in place of S or A sequence at the 5′ end and the original Hind III site at its 3′ end. These two PCR products were treated with Kpn I and ligated to produce site-specific mutants in S or A sequence.
Transfection of primary culture cells and analysis of the promoter activity
Cells were transfected by calcium phosphate precipitation method (Gorman et al., 1982). The cells were plated at a density of 2 × 106 per plate and re-fed with fresh medium for 4 hours prior to the addition of different DNAs. Calcium phosphate-DNA precipitate was added at a concentration of 20 μg per plate, except in the case of control pSV2CAT DNA, which was added at 4 μg per plate. After 4 hours the culture medium was replaced with fresh medium, and cells were harvested after 48 hours to prepare cell extract. The CAT gene expression was assayed by thin-layer chromatography (Gorman et al., 1982). The relative CAT activity of different mutants was calculated by dividing the CAT activity of the mutant by that of RSVβGal. Normalization of CAT activity was also done, when needed, by co-transfection with plasmid RSVpGal harboring β-galactoside gene according to Borras et al. (1988).
Southwestern hybridization assay
This assay was performed according to Jones et al. (1985) and White et al. (1985). Nuclear protein (25 μg per well) was fractionated on 10% SDS polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane at 4°C overnight. The membrane was incubated twice in 6 M guanidine hydrochloride for 5 minutes each and washed successively with a twofold dilution of the 6 M guanidine hydrochloride in buffer A (20 mM HEPES pH 7.9, 3 mM MgCl2, 40 mM KC1), followed by two washes with buffer A alone for 5 minutes each. The membrane was then blocked with 5% nonfat dry milk and 250 ng/ml salmon-sperm DNA in buffer A and washed with buffer A containing 0.25% nonfat milk. Hybridization was performed by incubating the membrane in buffer A containing 0.25% nonfat milk and 106 cpm/ ml radio-labeled oligonucleotide for 10 hours at 4°C in a rocking platform. Filters were then washed three times with buffer A and two times with 1.5X buffer A for 5 minutes each. The membrane was then dried and exposed to X-ray film.
Gel mobility shift assay
Double-stranded DNA fragments obtained by renaturation of chemically synthesized single-stranded oligonucleotides were radio-labeled at the 5′ end by polynucleotide kinase in presence of γ-32P-ATP and used for the binding assay. DNA fragment (4,000 cpm), 2 μg of poly (dl-dC), and 6 to 10 μg of nuclear protein extract were incubated in 10 mM HEPES (pH 7.9), 50 mM KC1, 5 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, 12.5% glycerol at room temperature for 30 minutes and separated on an 8% polyacrylamide gel with circulating Tris-borate buffer. The gel was then dried under vacuum and exposed to X-ray film for autoradiography.
Results
DNase footprinting assay of MLC-2 proximal promoter
The promoter of the chicken cardiac MLC-2 gene contains two A/T-rich sequence elements, TATTTAT (element C) and TTATTTTTA (element B), located at −24 and −46 respectively; a CArG-like sequence, CCAAAAGTGG (element A), located at −64 (Minty and Kedes, 1986); and a BSe-like sequence, GTGTCTCGCA (element D2), located at −177 (Yu and Nadal-Ginard, 1989; see Fig. 1). These nucleotide sequences are conserved during evolution and have perfect sequence homology with the rat MLC-2 gene promoter (Handerson et al., 1989). To examine whether these sequences are the target sites for binding of nuclear proteins, we performed DNase-I footprinting using the nuclear extract from embryonic chicken cardiac muscle cells. The radiolabeled DNA fragment corresponding to nucleotide sequence −120 to +38 of the MLC-2 promoter produced four defined, protected regions in the footprinting assay (Fig. 2). Three of these protected regions (elements C, B, and A) correspond to previously identified protected sequences (Braun et al., 1989), but a new 12-base protected region with nucleotide sequence TAT TACCTTTAT, designated S, was also identified. The element S sequence is also conserved in the rat MLC-2 gene promoter, suggesting it might be functionally important.
Figure 1.

Partial nucleotide sequence of chicken cardiac MLC-2 gene promoter. The transcription initiation site is marked as +1. The cis-acting DNA elements C, B, A, and S in the proximal region and D in the distal region are underlined. D1 is a nuclear protein binding site (data not shown). D2 is a BSe-like element (see text). Nucleotide sequence of synthetic oligonucleotides containing SA, S, A, B, and C elements respectively are indicated; ΔS indicates the nucleotide sequence of the mutated S oligonucleotide.
Figure 2.
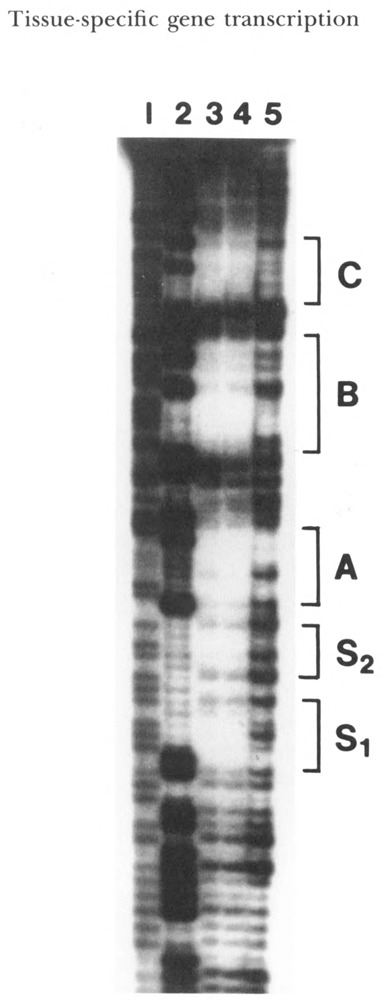
DNase footprinting analysis of the cardiac MLC-2 gene proximal promoter. DNA fragment corresponding to −120 to +38 was end-labeled and incubated with nuclear extracts prepared from chicken cardiac tissue and primary cardiac cells. Following the partial digestion of labeled fragment with DNase I, the DNA was analyzed by sequencing gel. Lanes 1 and 2: T/C and G ladder sequences of the DNA sequencing reaction; lane 3: DNA sequence protected by the nuclear protein prepared from cardiac tissue; lane 4: DNA sequence protected by nuclear protein prepared from primary cardiac cells; lane 5: the negative control lacking the nuclear protein extract. Five protected regions are marked C, B, A, S1, and S2.
Analysis of proximal MLC-2 promoter function by sequential 5′-deletions
In order to understand the putative role of these elements, we constructed a series of recombinant molecules containing sequentially 5′-deleted MLC-2 promoter fused to the CAT gene (Fig. 3). The transcriptional activity of these DNAs was tested following their transfection into primary cultures of chicken embryonic heart, fibroblast, and brain cells (see Materials and Methods). The results, summarized in Figures 3 and 4, demonstrate that multiple nucleotide sequence elements exist in the MLC-2 promoter that are involved in the positive and negative regulation of its gene expression. The deletion plasmid pLCΔ15CAT, which contains a 15 bp segment upstream from the transcription initiation site (+1) and does not include any of the putative regulatory cis-elements (S, A, B, and C), was transcriptionally inactive in both muscle and non-muscle cells, fibroblasts, and brain cells. Recombinant pLCΔ31CAT, which contains a 31 bp segment upstream from the start site and includes only element C, exhibited a low but consistent level of activity (5 % of the level of the positive control, pSV2CAT) in cardiac muscle cells. A very similar level of activity was observed with this construct in primary cultures of chicken brain and HeLa cells (data not shown), whereas the level of expression in fibroblasts was less than 0.2% compared with cardiac muscle cells. Addition of elements B in pLCΔ53CAT and of elements B and A in pLCΔ69CAT resulted in a 4- and 9-fold increase respectively in CAT expression in muscle cells, but not in brain cells, indicating that these two elements are involved in the positive regulation in muscle cells. The same constructs exhibited a decrease in activity in brain cells (data not shown). To avoid possible errors in reproducibility in CAT expression, we took several precautions (see Materials and Methods). Repeated experiments with multiple plasmid preparations provided identical results. When quantitation was desired, a recombinant expression vector harboring β-galactosidase gene was co-transfected, and the CAT activity was normalized (Borras et al., 1988).
Figure 3.
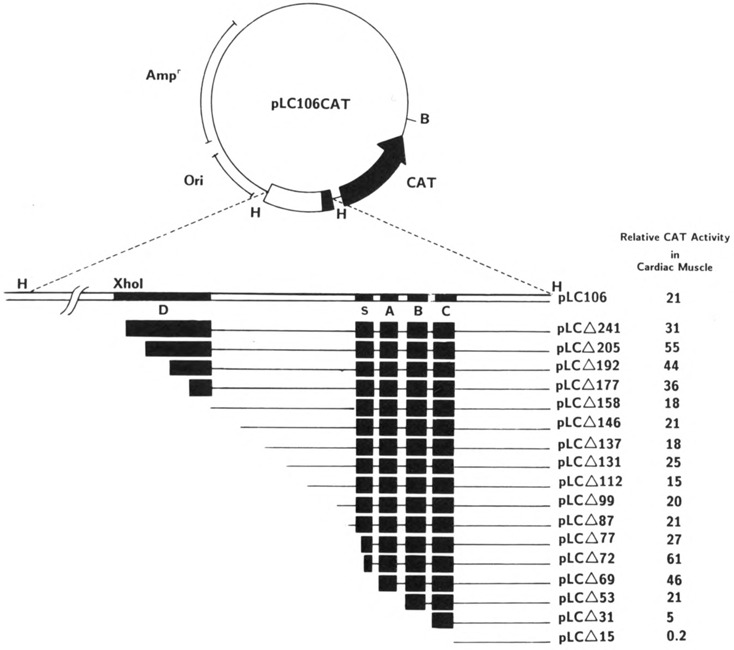
Construction of sequential 5′-deletion mutants of chicken cardiac MLC-2 promoter. The linearized plasmid pLC106CAT was digested by Bal 31, as described in Materials and Methods. The end points of deletion mutants are indicated by the number of nucleotide bases in the 5′ flanking region of the promoter. The relative CAT activity of the mutant represents the percentage of CAT conversion relative to pSV2CAT under identical conditions.
Figure 4.
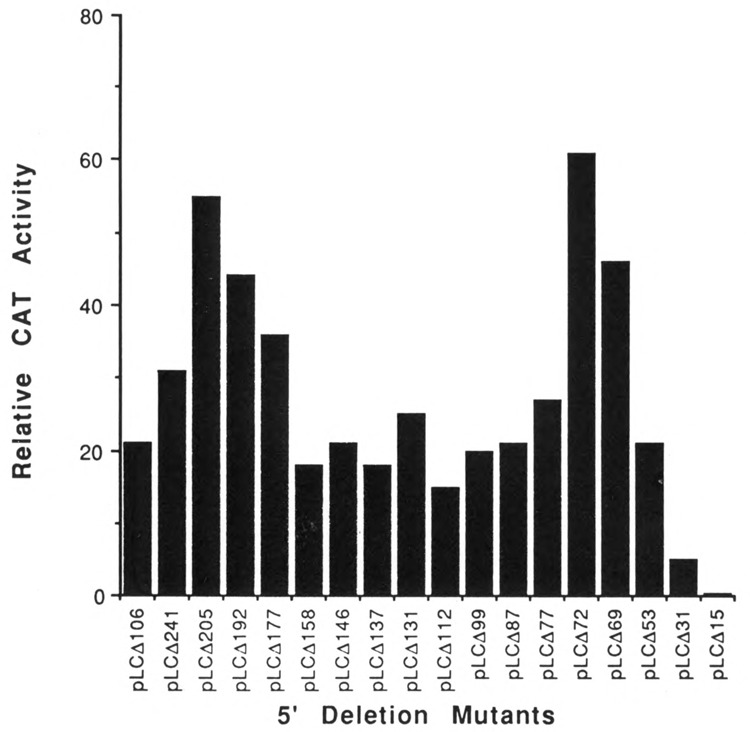
Promoter activity of the sequentially 5′-deleted mutants of MLC-2 gene in primary cardiac muscle cells. The plasmid pLC106CAT and the deletion mutants described (Fig. 3) were used to transfect primary cultures of 13-day-old chicken cardiac embryonic muscle cells. CAT activity was measured as described in Materials and Methods. The bars represent the mean value of three experiments.
Element A core sequence has a striking similarity to the evolutionary conserved sequence CC(A/T)6 GG (CArG box or C/BAR), which has been implicated in muscle-specific up-regulation of several muscle genes (Minty and Kedes, 1986; Muscat et al., 1988; Mohun et al., 1989). Interestingly, pLCΔ72CAT, which contains only three additional nucleotides in the 5′ direction of the element A sequence, increased the promoter activity by 33% when compared to the level of pLCΔ69CAT, suggesting that the functional element A is extended by three additional nucleotides, even though the protein-binding boundary was restricted to 10 nucleotides (−68 to −59; see Fig. 2). The nucleotide sequence further upstream to element A, on the other hand, has a negative effect on transcription, since both plasmids pLCΔ77CAT and pLCΔ87CAT exhibited a reduction in promoter activity relative to pLCΔ72CAT. The negative effect of S sequence was also confirmed by introducing site-specific mutation in element S and the resultant mutant, pLCΔ87SCAT, was used to compare the expression of CAT gene with wild-type pLCΔ87CAT. Figure 5 shows that deletion or substitution of element S sequence alone abolished the negative effect of S, and an increase in CAT activity with increase in DNA concentrations was observed. These experiments thus established unambiguously the negative role of element S sequence. Previous studies using breast muscle cells for transfection (Braun et al., 1989) did not identify the negative role of elementS in the proximal promoter, although an element at −64 down-regulated MLC-2 transcription in HeLa cells. A second set of positive and negative elements was located in the distal region between −158 and −241, which contains a BSe-like element at −177 (Yu and Nadal-Ginard, 1989) and another protein-binding site at −192 (DI). The functional analysis of these sequences is in progress.
Figure 5.
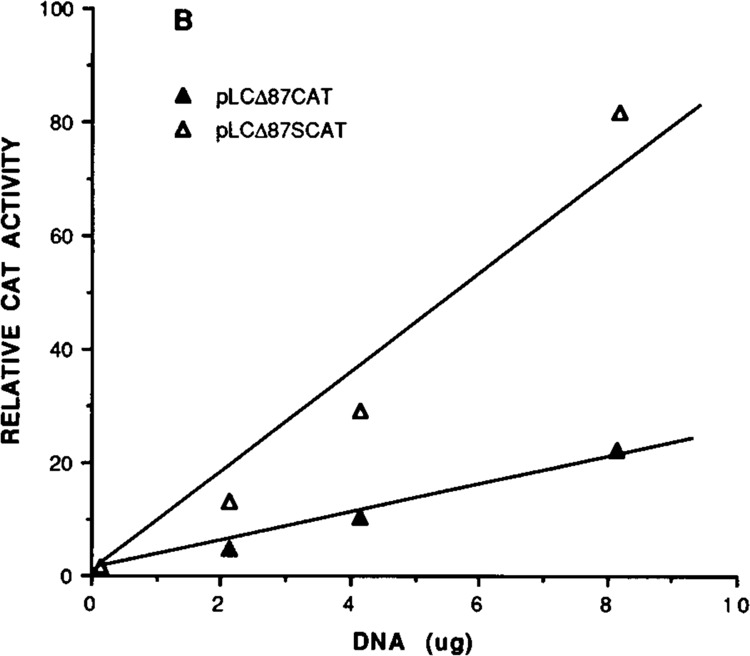
Negative regulation of transcription by S element demonstrated by site-directed mutational analysis. Relative CAT activity with different amount of DNA of plasmids pLCΔ87CAT and pLCΔ87SCAT (with site-mutated S sequence) was obtained by dividing the activity of the mutant with the CAT activity of pSV2CAT used as positive control (see Materials and Methods).
Consistent with the physiological expression of the cardiac MLC-2 gene, the MLC-2 promoter containing the 1.3 kb 5′ flanking region in pLC106CAT was found to be transcriptionally inactive in fibroblasts (data not shown). It is believed that at least one embryonic form of MLCs (LC23) is transiently expressed in chicken embryonic brain (Uetsuki et al., 1990). In the case of cardiac MLC-2, element C alone produced a low level of expression in brain cells, whereas the addition of 5′ sequences that include B and A (pLCΔ69CAT) caused a reduction in CAT activity—unlike muscle cells, where elements B and A exerted a positive effect. Element S (pLCΔ77CAT), which has a negative effect in muscle cells, exerted a stimulatory effect in brain cells. These results suggest that the positive regulation of elements A and B and the negative effect of element S are restricted to muscle cells.
Tissue-specific interaction of DNA elements with nuclear proteins
In an attempt to identify the cell-specific factors involved in the regulation of MLC-2 gene expression, oligonucleotides containing elements S, A, B, and C (see Fig. 1) were used separately to interact with nuclear proteins in extracts prepared from the cardiac muscle, fibroblast, and brain cells in the gel mobility shift assay. Binding properties of nuclear proteins to element A sequence have been described previously (Qasba et al., 1992). As shown in Figure 6A and B, the binding of proteins to element B and C sequences resulted in the formation of multiple complexes, and the recognition of these elements by nuclear proteins from the three tissues was consistent with their roles in transcription. For example, among the element B binding proteins, at least one complex, MBF1, was muscle-specific, as it was not produced with the fibroblast and brain nuclear extracts (Fig. 6A). Non-labeled B-DNA competed effectively for MBF1, but S and A elements containing oligonucleotides did not. The fibroblast and brain extracts also produced binding complexes which were different from those of cardiac extracts. The proteins that bind with element C-containing DNA produced at least two major complexes, CBF1 and CBF2 (Fig. 6B). CBF2 appears to be ubiquitous, as cardiac as well as fibroblast and brain cells extracted produced the complex, whereas CBF1 was present in cardiac muscle and brain cells, but not in fibroblasts. Element B, which has a muscle-specific function, bound to a protein (MBF1) present in muscle only, whereas element C, which serves as a basal promoter in cardiac muscle and brain cells, but not in fibroblasts, bound to the protein CBF1 present in muscle and brain tissues.
Figure 6.
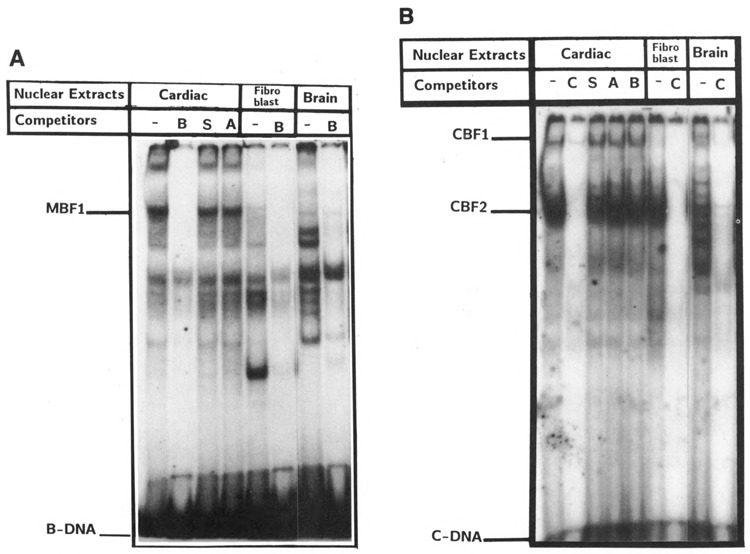
Nuclear proteins from cardiac, fibroblast, and brain cells interact with elements B and C. The radiolabeled oligonucleotides −36 to −55 (A) and −12 to −31 (B) were incubated with cardiac, fibroblast, and brain cell nuclear proteins (10 ng) without competitor (−) or with 100-fold excess unlabeled oligonucleotides containing A, B, C, and S sequences respectively (see Figure 1 for the nucleotide sequence of oligonucleotides). The muscle-specific protein/ DNA complex is marked as MBF-1 in A, and element C-specific protein/DNA complexes are marked as CBF-1 and CBF-2 in B.
Binding of nuclear protein(s) to negative element S
To investigate the interaction of protein factors with element S, chemically synthesized oligonucleotides containing elements S and A separately or together (SA) were used in a gel mobility shift assay. As shown in Figure 7, the DNA probe containing S element alone produced two closely migrating protein complexes (SBF1), both of which were displaced by the excess of non-labeled S DNA, but not with mutated S-(S′) or A-containing DNAs. We also performed a Southwestern analysis with S-DNA as probe, using nuclear extracts fractionated on poly-acrylamide gel, and found that the S sequence was recognized by a single protein of approximately 50 kD (Fig. 8). It is possible that a single DNA-binding protein produces multiple complexes in which other protein(s) participates through protein-protein interaction. Alternatively, the denaturation/renaturation process in the Southwestern blotting assay could destroy the interaction for one of the polypeptides. Extracts from fibroblasts which do not support MLC-2 gene transcription also showed the S-binding protein, but at a relatively low level. A complex formation similar to cardiac muscle was also observed with extract from brain cells (data not shown).
Figure 7.
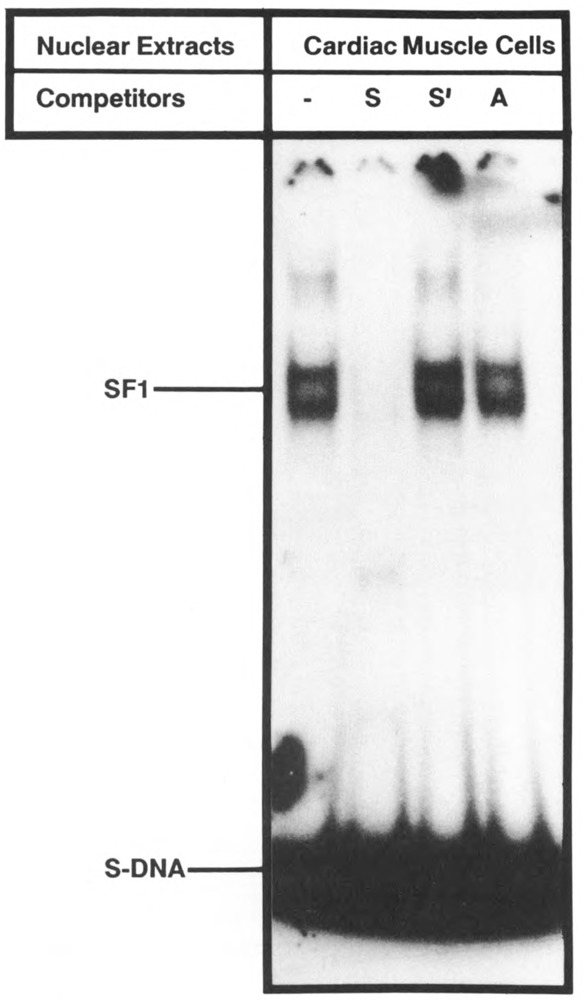
Sequence-specific binding of nuclear protein with element S. Chemically synthesized end-labeled oligonucleotide S DNA was incubated with cardiac nuclear extract, and the complex was analyzed by gel mobility shift assay. Lane 1: without competition; lanes 2 and 3: on competition with 100-fold excess of unlabeled DNA containing element S, mutated element S, and element A separately (see Figure 1 for nucleotide sequence).
Figure 8.
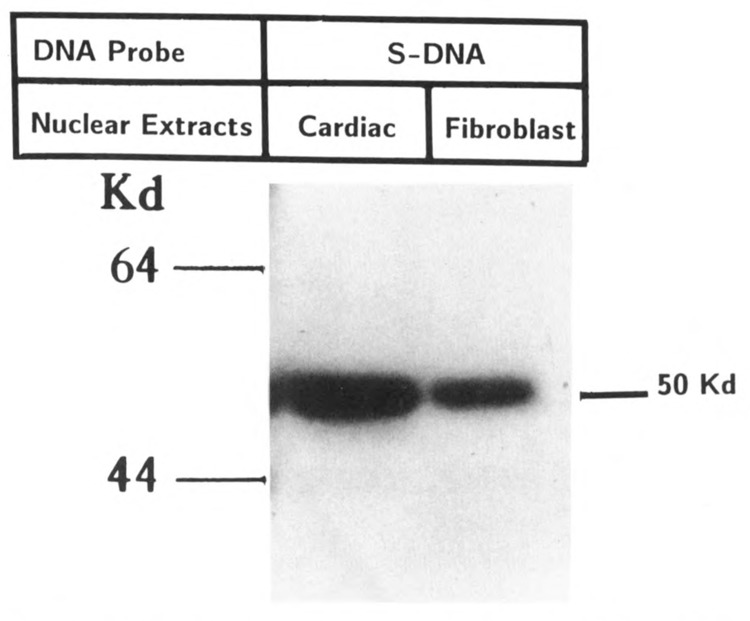
Southwestern analysis of nuclear proteins from cardiac and fibroblast cells with oligonucleotide S. Nuclear protein from cardiac cells was separated by 10% polyacrylamide gel electrophoresis and analyzed by Southwestern blotting, using end-labeled 20 bp oligonucleotide (−87 to −68) containing element S sequence (see Materials and Methods). This oligonucleotide binds to a protein of approximately 50 kDa present in both cardiac and fibroblast cells. The estimate of molecular size was based on migration of three size markers (the low size marker is not shown).
Elements A and S are separated by only three nucleotides, yet oligonucleotide A did not compete successfully for the protein-DNA complexes formed by element S (see Fig. 7A). To examine the pattern of DNA complexes when elements A and S are present together on the same DNA, a 42 bp synthetic oligonucleotide SA was used as a probe in a gel shift assay. Multiple complexes were obtained with fragment SA (Fig. 9). Complex I was displaced by fragment SA competition, as well as by S or A separately, whereas complex II (which consists of two closely related complexes) was displaced by SA competition, but not as effectively as was observed with either S or A alone. Proteins in complexes III and IV were possibly generated by the displacement of complex I by A and S competition respectively, as they were generated only upon the addition of S and A DNAs respectively. A similar pattern of SA-DNA binding was observed when nuclear extracts from brain cells and fibroblasts were used (data not shown). Thus, despite the spatial proximity, both S and A sequences bound to protein factors separately, but at least one complex was formed in which both sequences were used simultaneously. Since A and S sequences are involved in positive and negative transcription respectively, it would be of interest to explore whether SA-specific protein controls, in some way, the S-and A-specific regulatory functions.
Figure 9.
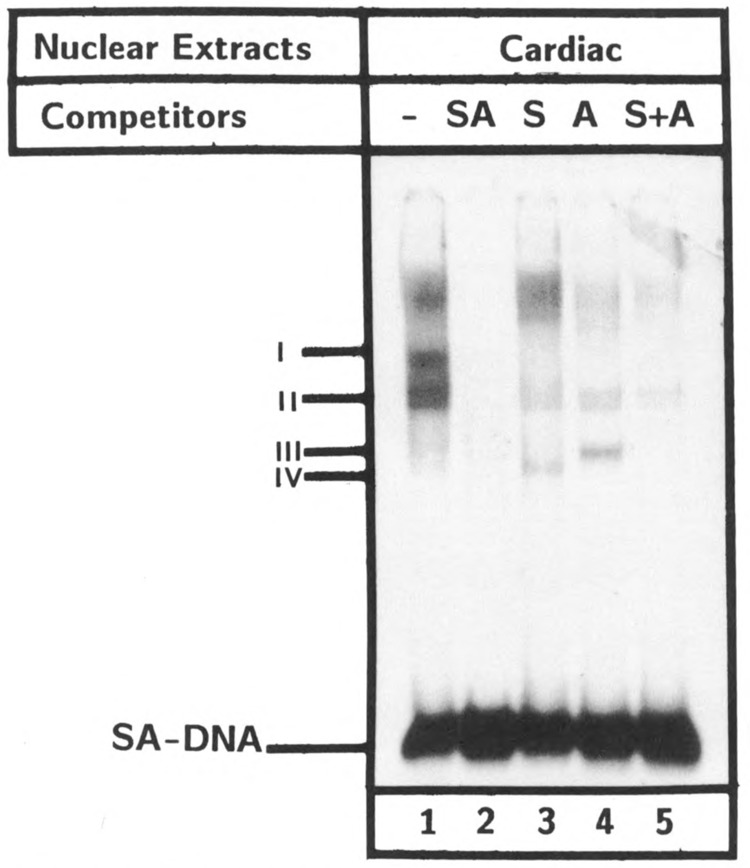
Multiple nuclear proteins interact with SA domain in vitro: The end-labeled DNA probe containing both elements A and S (−88 to −49) was incubated with cardiac cell nuclear extracts (10 μg) and analyzed by gel shift analysis without competition (−) and with 100-fold excess unlabeled oligonucleotides SA, S, A, and S +A. Protein complexes are indicated as I, II, III, and IV.
Interplay between proteins binding to elements S and A
We next examined whether the nuclear protein(s) that binds to element S regulates the expression of MLC-2 gene by a mechanism independent of element A requirement. Site-specific mutations were created in element S (pLCΔ87CAT), element A (pLCΔ87ACAT), and elements A and S (pLCΔ87SACAT) using the parent pLCΔ87CAT DNA, as described in Materials and Methods. These expression vectors were used in a transient transfection assay using chicken embryonic cardiac cells, as above. Results summarized in Figure 10 indicate that mutation in element S alone (pLCΔ87SCAT) caused an increase in CAT expression as expected, when compared to the expression of pLCΔ87CAT, confirming the negative transcriptional role of element S. The mutation of element A alone in pLCΔ87ACAT decreased CAT gene expression, confirming the positive role of element A. However, when both elements A and S were mutated, the expression of the resultant plasmid pLCΔ87SACAT still remained low, although one would expect that it would reach a level intermediate to those of pLCΔ87SCAT and pLCΔ87ACAT, should the S binding protein act independently of element A binding protein. To ascertain reproducibility, multiple transfections were done normalizing the CAT activity, as previously. We interpret these results to suggest that nuclear transcription factors binding to elements S and A regulate the expression of MLC-2 gene through interaction with each other. Taken together, these observations point to the complexity of S and A DNA-protein interactions and suggest that a close interplay between the positive and negative transcription factors associated with elements A and S is involved in the regulated expression of MLC-2 gene.
Figure 10.
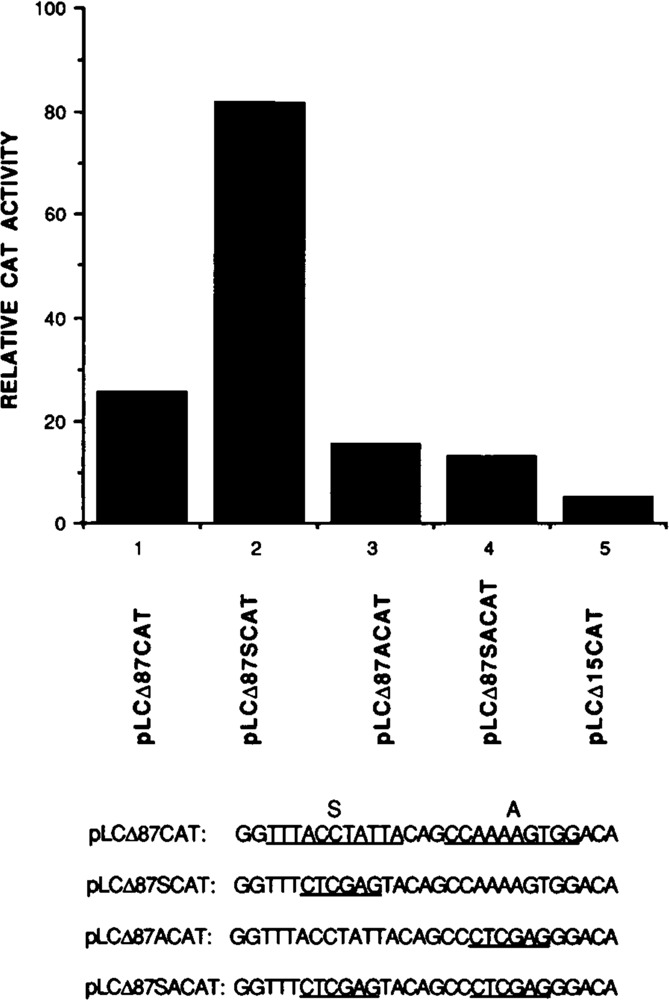
The negative effect of element S depends on the participation of element A. The relative CAT activity of different mutant promoters was compared with the wild-type promoter in a transient transfection assay in chicken cardiac cells. pLCΔ87CAT: control; pLCΔ87SCAT: mutated S sequence promoter; pLCΔ87ACAT: mutated A promoter; pLCΔ87SACAT: mutated A and S sequence; pLCΔ15CAT: deletion mutant containing 15 bp upstream to +1.
Discussion
A previous study (Braun et al., 1989) using cardiac MLC-2 promoter and breast muscle cell cultures detected two functional sites, designated distal (DPE) and proximal promoter element (PPE), corresponding to elements B and C identified in this study. Here, we have not only confirmed the role of these sequences, but have undertaken a detailed analysis of the entire promoter, using appropriate muscle and non-muscle cells in cultures to understand the mechanism underlying the tissue-specific transcription. We have identified two additional sequence elements, S and A, as well as their binding proteins, and have delineated further the role of elements B and C. The A/T-rich element C is analogous to the TATA box of the early genes of SV40 promoter (TATTTAT) but differs from the TATA box of the cardiac α-actin promoter (TATAAA). It serves as a basal promoter in breast muscle cells, as described previously (Braun et al., 1989), and in cardiac muscle cells, as observed in this study. Based on SI mapping of RNA isolated from COS cells transiently transfected with cardiac MLC 2/CAT recombinant, we have previously proposed that element B serves as the TATA promoter (Zarraga et al., 1986). In muscle cells, however, the same sequence has a muscle-specific activation role. Thus, it appears that element B can function both as the basal promoter and as an activator, depending on the cell type. The potential of A/T-rich sequences in muscle genes to function both as promoter and enhancer has previously been described (Horlick et al., 1990).
Consistent with the transcriptional roles of elements B and C is the evidence concerning their DNA-binding proteins. MBF1, which binds to element B, is found in cardiac muscle cells but not in brain and fibroblast, whereas element C-specific factor, CBF1, is present in both muscle and brain cells. Other factors which recognize elements B, but do not exhibit tissue specificity, are also found in the same extracts. Interestingly, element B sequence shares a remarkable similarity to the mouse skeletal muscle creatine kinase (MCK) gene enhancer sequence (Buskin and Hauschka, 1989; Gossett et al., 1989), which binds to the myotube-specific nuclear protein, MEF-2. Both sequences activate their respective promoters. Whether the two binding proteins are functionally interchangeable remains to be seen. Another regulatory element upstream to elements B in MLC-2 gene is a CArG-like motif, element A, which is an essential component of the sarcomeric actin gene promoters in a wide variety of vertebrates (Hayward et al., 1988; Gustafson and Kedes, 1989; Mohun et al., 1989). Multiple nuclear factors, CBF1, CBF2, CBF3, and the ubiquitous serum responsive factor SRF, bind to CArG box and mediate the up-regulation of the human cardiac α-actin gene (Gustafson and Kedes, 1989). The results presented here show that element A plays a positive role in the MLC-2 transcription in cardiac cells, but not in fibroblast or brain cells, and therefore suggest that the mechanism by which cardiac MLC-2 gene is up-regulated through element A and its binding factors may be distinct from that of actin genes. Since the factors that bind to element A are present in both muscle and non-muscle cells and differ from the CArG binding proteins (Qasba et al., 1992), it appears that the element A mediated enhancement of muscle-specific transcription may occur via the participation of proteins associated with element B, the muscle-specific activator. Together the two cis-acting elements A and B account for approximately a 30-fold increase in the transcriptional activity in primary cardiac cells relative to the activity in brain cells. Element C alone is sufficient for the basal level transcription, but elements A and B appear to contribute significantly to muscle-specific up-regulation of transcription. It was previously suggested that chicken cardiac MLC-2 promoter is active in both muscle and non-muscle cells in a transient transfection assay, even though the physiological expression of cardiac muscle is restricted to cardiac muscle only (Braun et al., 1989). Those studies used the same cardiac MLC-2 gene promoter as we did, but with an addition of 0.8 kb segment of the first intron; they showed that MLC-2 promoter was active in both breast muscle and fibroblast cells. Our evidence, however, indicates that the construct containing element B, without the first intron, is inactive in fibroblasts (as well as in skeletal muscle cells). We have recently identified (Shen et al., 1991 and unpublished data) a potent activator element which resides in the first intron of the gene and exerts a strong positive effect, thus making it difficult to interpret the results in terms of tissue specificity.
Located immediately upstream to element A and separated by only 3 bp is the negative element S. The results clearly demonstrate the existence of the sequence as a separate protein-binding domain responsible for pronounced transcriptional repression in muscle cells. The gel mobility shift assay suggests that the element S and element A binding proteins form multi-protein complexes requiring that elements A and S both be present. Recent evidence suggests that binding of nuclear proteins to negative elements can repress transcription by a variety of mechanisms (Levine and Manley, 1989). These include direct repression, wherein the negative protein blocks the activity of the basal promoter by interfering with the function-but not binding-of the activator, or by competing for the DNA-binding sites. The proximity of S and A sites, with the two motifs most likely on the same side of the DNA helix, could conceivably result in directly blocking the interaction or one or more proteins at the activator site A. The binding of one sequence element must interfere with the binding activity of the other, and the binding of the protein(s) to both S and A together may influence, in some way, the binding at individual sites to form the positive and negative transcriptional complexes. We speculate that a close interplay, presumably involving intra-protein interactions, must occur between the factors involved, which should affect the opposite regulatory paths in transcription. The presence of discrete repressing and activating domains in MLC-2 promoter is intriguing, but the relative contribution of these two functionally opposing sequences may depend upon a variety of parameters, including the nature of the protein factors involved, their interactions with other proteins, the conformational states, and possibly the selective and combinative utilization of proteins in functional complex formation. To elucidate the precise mechanism, further analysis of the DNA-protein interactions with purified nuclear proteins is necessary.
Acknowledgment
This work was supported in part by an NIH grant 1R01HL43159.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Bains W., Ponte P., Blau H., and Kedes L. (1984), Mol Cell Biol 4, 1449–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borras T., Peterson C. A., and Piatgorsky J. (1988), Dev Biol 127, 209–219. [DOI] [PubMed] [Google Scholar]
- Borden L. A., Czajkowski C. M., Chan C. Y., and Farb D. H. (1984), Science 226, 857–859. [DOI] [PubMed] [Google Scholar]
- Bouvagnet P., Strehler E. E., Whit G. E., Strehler-Page M. A., Nadal-Ginard B., and Mahdavi V. (1987), Mol Cell Biol 7, 4377–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Tannich E., Buschhausen-Denker G., and Arnold H. H. (1989) Mol Cell Biol 9, 2513–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskin J. N. and Hauschka S. D. (1989), Mol Cell Biol 9, 2627–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow K.-L. and Schwartz R. J. (1990), Mol Cell Biol 10, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam J. D., Lebovitz R. M., and Roeder R. G. (1983), Nucl Acids Res 11, 1474–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman C. M., Moffat L. F., and Howard B. H. (1982), Mol Cell Biol 2, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett L. A., Kelvin D. J., Sternberg E. A., and Olson E. N. (1989), Mol Cell Biol 9, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S., Zarraga A. M., Morgenstern D., Martin M. E., and Siddiqui M. A. Q. (1992), Cell Mol Biol 38, 49–58. [PubMed] [Google Scholar]
- Grichnik J. M., Bergsman D. J., and Schwartz R. J. (1986), Nucl Acids Res 14, 1683–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T. A. and Kedes L. (1989), Mol Cell Biol 9, 3269–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handerson S. A., Spencer M., Sen A., Kumar C., Siddiqui M. A. Q., and Chien K. R. (1989), J Jiol Chem 262, 18142–18148. [PubMed] [Google Scholar]
- Hayward L. J. and Schwartz R. J. (1986), J Cell Biol 102, 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward L. J., Zhu Y. Y, and Schwartz R. J. (1988), J Cell Biol 106, 2077–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlick R. A., Hobson G., Patterson J. H., Michell M. T., and Benfield P. A. (1990), Mol Cell Biol 10, 4826–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaynes J. B., Johnson J. E., Buskin J. N., Gartside C. L., and Hauschka S. D. (1986), Mol Cell Biol 8, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. A., Yamamoto K. K., and Tjian R. (1985), Cell 42, 559–572. [DOI] [PubMed] [Google Scholar]
- Levine M. and Manley J. L. (1989), Cell 59, 405–408. [DOI] [PubMed] [Google Scholar]
- Lomper A.-M., Nadal-Ginard B., and Mahdavi V. (1984), J Biol Chem 259, 6437–6446. [PubMed] [Google Scholar]
- Mar J. H. and Ordahl C. P. (1990), Mol Cell Biol 10, 4271–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minty A. and Kedes L. (1986), Mol Cell Biol 6, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa T. and Kedes L. (1987), Mol Cell Biol 7, 2803–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohun T. J., Taylor M. V., Garrett N., and Gurdon J. B. (1989), EMBO J 8, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat G. E. O., Gustafson T. A., and Kedes L. (1988), Mol Cell Biol 8, 4120–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qasba P., Lin E., Kumar A., and Siddiqui M. A. Q. (1992), Mol Cell Biol 12, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R. J. and Rothblum K. N. (1981), Biochemistry 20, 4122–4129. [DOI] [PubMed] [Google Scholar]
- Shen R., Goswami S. K., Mascareno E., Kumar A., and Siddiqui M. A. Q. (1991), Mol Cell Biol 11, 1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternderg E. A., Spizz G., Vizard D., Weil T., and Olson E. N. (1988), Mol Cell Biol 8, 2896–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetsuki T., Nabeshima Y., Fujisawa-Sehara A., and Nabeshima Y. (1990), Mol Cell Biol 10, 2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen R. G., Bulter-Browne S. M. G. S., Schwartz K., Bouveret P., and Pinset-Harstrom I. (1981), Nature 270, 725–727. [DOI] [PubMed] [Google Scholar]
- White B. A., Preston G. M., Lufkin T. C., and Bancroft C. (1985), Mol Cell Biol 5, 2967–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek D. F., Periasamy M., Butler-Browne G. S., Whalen R. G., and Nadal-Ginard B. (1985), J Cell Biol 101, 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.-T. and Nadal-Ginard B. (1989), Mol Cell Biol 9, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarraga A. M., Danishefsky K., Deshpande A., Nicholson D., Mendola C., and Siddiqui M. A. Q. (1986), J Biol Chem 261, 13852–13860. [PubMed] [Google Scholar]