

Abstract
The TATA box binding transcription factor TFIID of S. cerevisiae was used as a ligand for affinity chromatography. Polypeptides that bind specifically to yeast TFIID (TFIID-associated proteins, DAPs) were purified from human HeLa (heDAPs) and calf thymus (ctDAPs) whole cell extracts. Both heDAP and ctDAP fractions altered the binding of TFIID to the TATA element, and substituted for the TFIIA transcription activity in a reconstituted in vitro system. The heDAP fraction also behaved like TFIIA in its ability to form a promoter-TFIID-TFIIA complex and to recruit TFIIB to such a complex. The interaction of DAPs with TFIID can confer heat-resistance (47°C) on recombinant yeast or human TFIID. SDS-PAGE analysis revealed that three polypeptides from HeLa extracts specifically bound to yTFIID columns (heDAP35, heDAP21, and heDAP12). These data suggest that a multi-subunit transcription factor with the properties of TFIIA can bind to TFIID in the absence of DNA.
Initiation of transcription by RNA polymerase II (pol II) is a highly regulated process which requires a number of auxiliary transcription factors. Most sequence-specific DNA-binding factors are required for the optimal transcription only of particular genes, while the general transcription factors are necessary for the transcription of all pol Il-transcribed genes (reviewed in Mitchell et al., 1989). Separation and purification of the general transcription factors have revealed that at least five different fractions are required in addition to RNA pol II in cell-free systems to produce a basal level of transcription: TFIIA, TFIIB, TFIID, TFIIE, and TFIIF (or RAP 30/74; reviewed in Mermelstein et al., 1989; Mitchell et al., 1989; Saltzman and Weinmann, 1989). A polypeptide contained in the TFIID fraction binds to the TATA element located upstream of most pol Il-transcribed genes (Davison et al., 1983; Reinberg et al., 1987; Butatowski et al., 1988; Cavallini et al., 1988; VanDyke et al., 1988; VanDyke et al., 1989). At least one general transcription factor has been shown to facilitate the binding of TFIID to the promoter (Samuels et al., 1982; Davison et al., 1983; Egly et al., 1984; Fire et al., 1984; Samuels and Sharp, 1986; Reinberg et al., 1987; Buratowski et al., 1988; Hahn et al., 1989; Maldonado et al., 1990). This activity, known as TFIIA (Reinberg et al., 1987; Maldonado et al., 1990), AB (Fire et al., 1984; Samuels and Sharp, 1986), or STF (Davison et al., 1983; Egly et al., 1984), has been purified to varying extents. TFIIA retards the mobility of cloned TFIID in gel mobility shift assays and extends the TFIID footprint at the promoter (Buratowski et al., 1989; Hahn et al., 1989; and Maldonado et al., 1990). The other transcription factors and pol II are believed to be recruited onto the template after TFIID and TFIIA to form a functional preinitiation complex (Davison et al., 1983; Fire et al., 1984; Reinberg et al., 1987; Reinberg and Roeder, 1987; VanDyke et al., 1988; Horikoshi et al., 1988; Buratowski et al., 1989; VanDyke et al., 1989; Conaway et al., 1990; Maldonado et al., 1990). Such an initiation complex must be stabilized by multiple protein-protein interactions, and some of these interactions have been detected by protein-affinity chromatography (see, for example, Sopta et al., 1985). Additional interactions between TFIID or TFHB and particular regulatory factors have also been detected (Stringer et al., 1990; Lin et al., 1991; Horikoshi et al., 1991; Lee et al., 1991).
General initiation factors derived from yeast and mammalian sources are to some extent interchangeable. Thus, the cloned TATA-binding TFIID polypeptide (Buratowski et al., 1988; Cavallini et al., 1988) and TFIIA (Hahn et al., 1989) derived from S. cerevisiae can replace their human counterparts in basal transcription reactions containing mammalian RNA polymerase II and general initiation factors. Since protein-affinity chromatography can precisely identify intermolecular contacts between the general initiation factors, we have employed the recombinant yeast TFIID (yTFIID) polypeptide as an immobilized ligand to detect mammalian polypeptides that bind to TFIID in the absence of DNA. The eluate from a yTFIID column was analyzed for its effect on the binding of TFIID to the adenovirus major late promoter using gel mobility shift and DNase I footprinting assays, for its role in transcription using a reconstituted in vitro system, and for its protein content using SDS-PAGE. Our data identify three mammalian proteins (TFIID-associated proteins or DAPs) that specifically bind to yTFIID and indicate that they probably comprise transcription factor TFIIA.
Materials and methods
Purification of recombinant yeast and human TFIIDs
Yeast TFIID was produced in E. coli using a phage T7 promoter expression system (Studier and Moffatt, 1986). A BamH I DNA fragment containing the yTFIID coding sequence was purified from plasmid pASY2D (Schmidt et al., 1989) and subcloned into the BamH I site of plasmid pAR3038 (Studier and Moffatt, 1986). E coli (BL21[DE3])-transformed cells were grown in 11 L of LB medium to an OD550 of 1, induced with 0.5 mM isopropyl-thiogalacto-pyranoside (IPTG) for 2 hours, harvested, and lysed in 400 ml buffer containing 25 mM Tris-HCl, pH7.8, 10 mM EDTA, 2% sucrose, 1 mM dithiotreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.25 mg/ml lysozyme for 15 minutes at 0°C. KC1 was then added to a final concentration of 0.4 M. The lysate was clarified by centrifugation in a 50.2Ti rotor (Beckman) for 90 minutes at 45000 rpm, and proteins were precipitated using (NH4)2S04 (60% saturation). The pellet was resuspended in 50 ml ACB (10 mM Hepes, pH 7.9,0.2 mM EDTA, 20% glycerol and 1 mM DTT) and dialyzed against 2.5 L ACB containing 0.1 M NaCl. This extract was loaded onto a 400 ml DEAE-cellulose (Whatman DE52) column at a flow rate of 300 ml per hour. Flow-through fractions (25 ml/fraction) containing yTFIID were pooled (total of 225 ml) and loaded onto a 25 ml heparin-agarose column (BRL) at a flow rate of 50 ml per hour. The column was washed with 75 ml ACB containing 0.1 M NaCl and eluted with a 300 ml linear salt gradient (0.2 to 0.8 M NaCl) in the same buffer. Fractions (25 ml) were analyzed by SDS-PAGE and the elution peak of yTFIID was at 0.55 M NaCl. Fractions containing yTFIID were pooled (see Fig. 6D) and used for affinity chromatography and DNA-binding assays. Eleven liters of cell culture allowed the purification of 10 mg of yTFIID.
Figure 6.
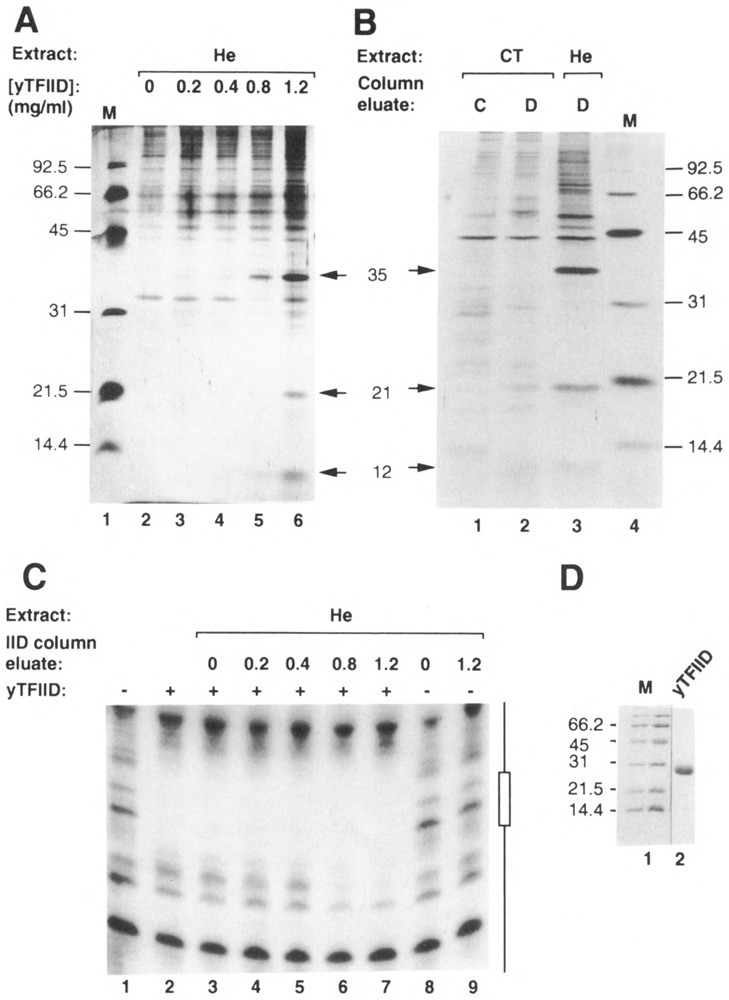
Affinity chromatography using yTFIID as a ligand. A. A volume of 400 μl of HeLa WCE (He) was chromatographed through 20 μl affinity columns containing different concentrations of immobilized yTFIID (0, 0.2, 0.4, 0.8, and 1.2 mgof yTFIID/ml of wet Affi-gel 10; lanes 2–6). Columns were washed with 200 μl loading buffer and eluted with loading buffer containing 0.5 M NaCl (Sopta et al., 1985). High salt eluates were analyzed using SDS-PAGE, followed by silver staining. Positions of DAP35, 21, and 12 (arrows), as well as molecular weight markers (M; lane 1), are indicated. B. Calf thymus WCE (CT) was chromatographed through control (C; lane 1) and yTFIID (D; lane 2) columns (0 and 2.5 mg of ligand/ml wet Affi-gel 10 respectively) and analyzed as described in A. A HeLa (He) eluate from a similar yTFIID column was included for comparison (lane 3). Positions of heDAP35, 21, and 12 (arrows), as well as positions of molecular weight standards (M; lane 4), are indicated. C. Eluates from yTFIID columns loaded with HeLa whole cell extract (He; see A) were analyzed using DNase I footprinting both in the absence (−) and presence (+) of yTFIID (20 ng). Lane 1: no added proteins; lane 2: yTFIID alone; lanes 3–7: yTFIID and eluates (1.2 μl) from 0, 0.2, 0.4, 0.8, or 1.2 mg/ml-yTFIID columns; lanes 8 and 9: eluates from the 0 and 1.2 mg/ml-yTFIID columns alone. The probe was the non-coding strand of the Ad2 ML promoter (positions −50 to +33) labeled at its 3′ end. The position of the TATA box is indicated as an open box. D. The yTFIID used as a ligand for affinity chromatography was analyzed by SDS-PAGE and stained with Coomassie blue. Positions of molecular weight markers are indicated (M).
Recombinant human TFIID was purified as previously described (Peterson et al., 1990). Active fractions eluting from the heparin-sepharose column were used in in vitro transcription assays.
Protein affinity chromatography
Whole cell extracts (WCEs) were prepared from 10g of either HeLa cells of calf thymus. Cells or tissue pieces were homogenized in 20 ml buffer A (10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KC1, 0.5 mM DTT, and 0.5 mM PMSF). A 15 ml volume of buffer B (50 mM Hepes, pH 7.9, 0.6 mM EDTA, 1.5 mM MgCl2, 0.5 mM DTT, 0.5 mM PMSF, 1.26 M NaCl, and 75% [vol:vol] glycerol) was added and the extract was homogenized again, and stirred on ice for 30 minutes. The calf thymus extract was passed through cheesecloth. Extracts were then centrifuged in a 70Ti rotor (Beckman) for 3 hours at 37000 rpm. The supernatant was dialyzed against 4L ACB containing 0.1 M NaCl. The precipitate formed during dialysis was spun down, and this whole cell extract was stored in aliquots at −70°C.
A detailed procedure for protein-affinity chromatography was described previously (Sopta et al., 1985) and essentially followed here. For micro-affinity chromatography, a 400 μl volume of whole cell extract was chromatographed through 20 μl affinity columns containing different concentrations of immobilized yTFIID (0, 0.2, 0.4, 0.8, and 1.2 mg of yTFIID/ml of wet Affi-gel 10). Columns were washed with 10-column volumes of loading buffer and eluted with 3-column volumes of loading buffer containing 0.5 M NaCl. These high-salt eluates were analyzed by SDS-PAGE followed by silver staining. Affinity chromatography (Fig. 6B) was performed using 1.5 ml columns containing 0 or 2.5 mgof TFIID/ml of wet Affi-gel l0. Columns were washed and eluted as described above. The HeLa and calf thymus eluates were used in DNA-binding and in vitro transcription assays.
Protein/DNA-binding assays
Protein/DNA-binding reactions for both gel mobility and DNase I footprinting assays were performed as described by Horikoshi et al. (1989). For gel mobility shift assays shown in Figure 1, approximately 0.1 ng of a 5′-end-labeled (Maniatis et al., 1982; Buratowski et al., 1989) double-stranded oligonucleotide containing either the wild-type Ad2 MLP TATA element (5′-TTTCTGAAGGGGGGCTATAAAAGGGGGT GGGGG-3′) or a variant containing mutations (TAGAGAA instead of TATAAAA) was used as probe. Incubations were at 25°C for 30 minutes. Electrophoresis was performed under the conditions described by Horikoshi et al. (1989). The mobility Shift assay shown in Figure 2 was performed using the Ad2 MLP (−50 to +33) which was 3′-end-labeled according to Buratowski et al. (1989). About 0.05–0.1 ng of probe was used per reaction. Incubations of proteins and DNA, as well as electrophoresis conditions, were according to Flores et al. (1991). The reactions contained 50 ng of yTFIID and 30 ng of recombinant TFIIB (rTFIIB) where indicated. The highly purified TFIIA fraction and the rTFIIB were kind gifts from D Reinberg, O. Flores, I. Ha, and P. Cortes.
Figure 1.

Gel mobility shift assays using yTFIID and DAPs. HeLa (He) and calf thymus (CT) eluates both from control (C) and yTFIID (D) columns were tested for their ability to bind DNA either in the presence (+) or absence (−) of yTFIID (25 ng). Lanes 1 and 14: no added proteins; lanes 2, 7, and 15: yTFIID alone; lanes 3–5: 0.5, 1.5, and 4.5 μl of He D eluate in the presence of yTFIID; lane 6: 4.5 μl of He C eluate in the presence of yTFIID; lanes 8–10: 0.5, 1.5, and 4.5 μl of CT D eluate in the presence of yTFIID; lane 11: 4.5 μl of CT C eluate in the presence of yTFIID; lane 12: 4.5 μl of He D eluate alone; lane 13: 4.5 μl of CT D eluate alone; lane 16: 4.5 μl of He D eluate in the presence of yTFIID; and lane 17: 4.5 μl of CT D eluate in the presence of yTFIID. Probes were either wild-type (TATAAAA; lanes 1-13) or mutated (TAGAGAA; lanes 14–17) oligonucleotides derived from the Ad2 ML promoter (from position −44 to −16). The positions of the yTFIID-specific complex and the complex of lower mobility are indicated by arrows.
Figure 2.

Human DAPs but not calf thymus DAPs can form TFIIA-TFIID- and TFIIATFIID-TFIIB-type complexes. Proteins and the DNA probe (Ad2 MLP from position −50 to +33) were mixed together and incubated for 30 minutes at 28°C. Fifty ng of yTFIID were used per lane, and 30 ng of recombinant human TFIIB where indicated. Lane 1: free probe; lane 2: yTFIID plus 4 μl yTFIID-column eluate from HeLa cell extracts (He); lane 3: yTFIID plus 1 μl TFIIA fraction; lane 4: yTFIID plus 2 μl TFIIA fraction; lane 5: yTFIID plus 4 μl yTFIID-column eluate from calf thymus extracts (CT); lane 6: yTFIID plus 4 μl yTFIID-column eluate from HeLa cell extracts plus TFIIB; lane 7: yTFIID plus 1 μl TFIIA fraction plus TFIIB; lane 8: yTFIID plus 2 μl TFIIA fraction plus TFIIB; and lane 9: yTFIID plus 4 μl yTFIID-column eluate from calf thymus extract plus TFIIB. The positions of the complexes are indicated.
For DNase I footprinting, the DNase I reactions on DNA-protein complexes were done at 25°C for 2 minutes and stopped using in vitro transcription stop buffer (Burton et al., 1986). Probes used for DNase I footprinting were made using the Ad2 ML promoter (positions −50 to +33) labeled either on the coding or non-coding strand as previously described (Buratowski et al., 1989). Reactions were extracted with phenol-chloroform, precipitated with ethanol, and analyzed on 10% urea-polyacrylamide sequencing gels.
In vitro transcription assays
Nuclear extract was prepared from HeLa cells using the method of Shapiro et al. (1988). A volume of 1.5 ml of nuclear extract (about 15 mg protein) was chromatographed through a 1 ml-Pll (Whatman) column in conditions described by Reinberg and Roeder (1987). Flow-through (0.1 M KC1) and step elution (0.5 and 1 M KC1) fractions were designated TFIIA, TFIIB/E/F, and TFIID.
In vitro transcription reactions and product analyses were performed as previously described (Burton et al., 1986). Heat treatment of nuclear extract or recombinant TFIID was for 20 minutes at 47°C in the presence of 1 mg/ml of BSA. Each reaction using the reconstituted system contained fraction TFIIB/E/F (2 μl; 3.6 μg protein), the TFIID fraction (2 μl; 1.8 μg protein), and purified calf thymus RNA pol II (a gift from Dr. S. McCracken; 0.1 ng; Sopta et al., 1985) in the presence of either fraction TFIIA (1 μl; 7.5 Hg protein) or various column eluates (as described in the legend of Figure 4). The DNA template (0.5 μg/reaction) was plasmid pML (C2AT)A-50 linearized with restriction enzyme EcoR I (Sawadogo and Roeder, 1985; Sumimoto et al., 1990).
Figure 4.

In vitro transcription using a reconstituted system and DAPs. HeLa nuclear extract was loaded onto a P11 column, and fractions were generated by step-elution (flow-through 0.1 M = TFIIA fraction; 0.5 M = TFIIB/E/F fraction; and 1 M = TFIID fraction. Transcription reactions were performed using a mixture containing fraction TFIIB/E/F (2 μl; 1.8 mg/ml protein), purified RNA pol II (0.1 μl), and the fraction TFIID (2 μl; 0.9 mg/ml protein) (lanes 1–9) in the absence (lanes 2) or in the presence (lane 3) of fraction TFIIA (1 μl; 7.5 mg/ml protein), HeLa control-column eluate (+He C [2.5 μl]; lane 4), HeLa yTFIID column eluate (He D [2.5 and 5 μl]; lanes 5 and 6 respectively), calf thymus control-column eluate (CT C [2.5 μl]; lane 7), and calf thymus yTFIID-column eluate (CT D [2.5 and 5 μl]; lanes 8 and 9 respectively). The DNA template was plasmid pML(C2AT)Δ-50 linearized with restriction enzyme EcoR I. The formation of runoff transcripts (indicated by an arrow) was monitored on a denaturing-polyacrylamide gel. Positions of DNA molecular weight markers (M; lane 1) are indicated.
Results
Mammalian DAPs affect the binding of yTFIID to the TATA element
In order to identify and isolate polypeptides that bind to immobilized yTFIID, whole cell extract prepared from HeLa cells or calf thymus was loaded onto 1.5 ml-columns containing purified yTFIID coupled to Affigel 10 at a concentration of 2.5 mg of protein/ml of packed resin (yTFIID column) or onto control columns of Affigel 10 without coupled proteins. The columns were washed with loading buffer and eluted with buffer containing 0.5 M NaCl. Proteins found in the salt eluates of yTFIID-columns that specifically bound to TFIID were called DAPs (yTFIID-associated proteins). These salt eluates were first analyzed for their effect on the binding of yTFIID to the TATA element. Gel mobility shift assays were performed using a 5′-end-labeled double-stranded oligonucleotide containing the Ad2 MLP TATA element as a probe. As shown in Figure 1 (lanes 2 and 7), yTFIID bound to the DNA and formed a small amount of a TFIID-DNA complex. This complex was specific because it was not formed using an oligonucleotide mutated in the TATA box (TAGAGAA instead of TATAAAA) (Fig. 1, lane 15). Eluates from control columns had no effect on the TFIID mobility shift (Fig. 1, lanes 6 and 11). Addition of increasing amounts of yTFIID-column eluates from HeLa and calf thymus extracts (lanes 3–5 and 8–10) increased the yield of the yTFIID-DNA complex, presumably because they increased either the stability of the complex or its rate of formation. The HeLa DAPs (heDAPs) also allowed the formation of an additional complex with lower mobility (Fig. 1, lanes 3–5). This complex also was specific because it could not be formed using an oligonucleotide mutated in the TATA box (Fig. 1, lanes 16 and 17). Both complexes were TFIID-dependent since neither the HeLa nor the calf thymus DAPs (ctDAPs) bound the probe in the absence of yTFIID (Fig. 1, lanes 12 and 13). These data indicated that we had isolated polypeptides which bind to TFIID in the absence of DNA and alter the interaction of TFIID with the TATA element.
Human DAPs are indistinguishable from partially purified TFIIA in gel mobility shift assays
Because it has been shown previously that TFIIA affects the binding of TFIID to promoter elements (see, for example, Maldonado et al., 1990), we wished to determine whether the mobility shift complexes formed using yTFIID and the DAPs resembled TFIID-TFIIA complexes. Consequently, we performed mobility shift assays in which partially purified TFIIA (a gift of P. Cortes, O. Flores, and D Reinberg) was used as a standard. We observed that the mobility of the complex produced by mixing TFIIA and yTFIID with a Ad2 MLP probe (Fig. 2, lanes 3 and 4) was the same as that of the low mobility complex produced by mixing heDAPs and yTFIID (lane 2). A similar complex could not be seen with ctDAPs and yTFIID (lane 5). We next tested whether this TFIIDTFIIA-type complex could recruit TFIIB as described previously (Buratowski et al., 1989; Maldonado et al., 1990; Ha et al., 1991). We found that partially purified TFIIA and the heDAP fraction both cooperated with yTFIID to recruit recombinant human TFIIB (Fig. 2, lanes 6, 7, and 8) and produced DNA-protein complexes with indistinguishable mobilities. The ctDAP fraction could not produce the TFIIDTFIIATFIIB-type complex (Fig. 2, lane 9). These data suggest that a component of DAPs from HeLa extracts, which is not present in DAPs from calf thymus extract, is essential for producing a TFIIDTFIIA-type complex which is able to recruit TFIIB (see below).
The mammalian DAPs alter the TFIID footprint on the TATA element
We next examined how DAPs affected the TFIID footprint on the TATA element of the Ad2 ML promoter. DNase I footprinting was performed on both strands using 5′end-labeled restriction fragments. Footprinting reactions were done in the presence of yTFIID alone, yTFIID with either HeLa or calf thymus DAPs, and yTFIID with the eluates of HeLa or calf thymus control columns (Fig. 3). Both the calf thymus and HeLa DAPs produced the same qualitative changes in the yTFIID footprint on each strand of the DNA (Fig. 3, lanes 3–6 and 11–14). These changes are shown schematically at the bottom of Figure 3. They were not observed when eluates from control columns were used (Fig. 3, lanes 15 and 16). Of particular interest was the upstream extension from −38 to −41 of the footprint produced by yTFIID on the non-coding strand, which we observed here and which has been shown previously to be characteristic of mammalian and yeast TFIIA activity (Buratowski et al., 1989; Hahn et al., 1989).
Figure 3.

DNase I footprinting using yTFIID and DAPs. HeLa (He) and calf thymus (CT) eluates from both control (C) and yTFIID (D) columns were used in DNase I footprinting experiments in the presence of yTFIID (25 ng). Lanes 1 and 9: no added proteins; lanes 2 and 10: yTFIID alone; lanes 3 and 11: 2 μl of He D eluate in the presence of yTFIID; lanes 4 and 12: 4 μl of He D eluate in the presence of yTFIID; lanes 5 and 13: 2 μl of CT D eluate in the presence of yTFIID; lanes 6 and 14: 4 μl of CT D eluate in the presence of yTFIID; lanes 7 and 15: 4 μl of He C eluate in the presence of yTFIID; and lanes 8 and 16: 4 μl of CT C eluate in the presence of yTFIID Footprinting analyses were performed on both the coding (lanes 1-8) and the non-coding (lanes 9-16) strands of the Ad2 ML promoter (position −50 to +33). DNA coordinates, as well as the position of the TATA box (open boxes), are indicated; they were deduced from G + A sequencing reactions run on a separate gel. A schematic representation of the DNA probes and protection patterns is shown at the bottom. Arrows indicate sites hypersensitive to DNase I digestion.
The mammalian DAPs can substitute for TFIIA transcription factor activity
Since heDAPs behaved like TFIIA in DNA binding assays, the ability of the DAPs to replace the TFIIA fraction in an in vitro transcription assay was examined. Nuclear extract prepared from HeLa cells was chromatographed on a phosphocellulose column (Reinberg and Roeder, 1987). Flow-through (0.1 M KC1) and step elution (0.5 and 1.0 M KC1) fractions were pooled and designated as TFIIA, TFIIB/E/F, and TFIID fractions for use in in vitro transcription experiments. The template DNA was the G-less cassette plasmid pML (C2AT)Δ-50, which lacks the upstream regulatory sequence of the Ad2 MLP. We observed that purified calf thymus RNA polymerase II as well as the TFIIB/E/F and TFIID fractions were all essential for accurate initiation of transcription (data not shown). The TFIIA fraction stimulated transcription (Fig. 4, lanes 2 and 3), as previously described by some investigators (Samuels et al., 1982; Egly et al., 1984; Samuels and Sharp, 1986; Buratowski et al., 1988), but it was not essential, presumably because our TFIID fraction contained TFIIG (Sumimoto et al., 1990), or because other fractions used in the experiment were still contaminated with some TFIIA activity. Addition of either HeLa or calf thymus DAPs similarly and reproducibly stimulated transcription (Fig. 4, lanes 5, 6, 8, and 9), while eluates from control columns had no effect (lanes 4 and 7). From these data we concluded that the TFIIA fraction contains a general transcription factor that stimulates transcription in this partially purified system, and that similar activities in HeLa and calf thymus whole cell extracts bound selectively to our yTFIID columns.
Mammalian DAPs can confer heat-resistance to recombinant TFIID, but not to TFIID in a nuclear extract
We also analyzed the effect of DAPs on other properties of TFIID. It has been reported previously that the treatment of a HeLa nuclear extract at 47°C for 15 minutes destroys its transcription activity as measured using run-off assays (Nakajima et al., 1988; and Fig. 5A, lanes 1 and 2). This effect is the result of the in-activation of the TFIID activity in the extract (Nakajima et al., 1988), since addition of recombinant yeast or human TFIID to such a heat-inactivated extract fully restores its transcription activity (Fig. 5A, lanes 8 and 9). Both recombinant yeast and human TFIID can also be inactivated by a treatment at 47°C (Fig. 5B, lanes 2 and 8; and 5C, lanes 2 and 7). This heatlability can be reversed for both recombinant TFIIDs by the addition of DAPs directly to the TFIID prior to heat-treatment (Fig. 5B, lanes 3–5 and 9–11); neither the addition of eluates from control columns (Fig. 5B, lanes 6 and 12) nor the addition of BSA (present at 1 mg/ml in each heat-treated reaction) produced a similar effect. The observation that the TFIID activity of a nuclear extract is heat-sensitive implies that DAPs cannot protect TFIID from heat inactivation in the extract. Consistent with this, when DAPs were added to a nuclear extract prior to heat-treatment, no heat-resistance was conferred (Fig. 5A, lanes 3–6). In addition, when recombinant TFIIDs were added to heat-treated nuclear extract, incubated for 20 minutes, and then heated again in the presence or absence of DAPs, no heat-resistance was conferred on hTFIID (Fig. 5C, lanes 2–5), and only partial resistance on yTFIID (Fig. 5C, lanes 7–10). These data further indicate that DAPs cannot protect human TFIID from heat-inactivation in a nuclear extract.
Figure 5.

DAPs can confer heat-resistance to recombinant TFIIDs, but not to endogenous nor exogenous human TFIID present in a nuclear extract. A. Nuclear extract from HeLa cells was used in in vitro transcription assays either before (lane 1) or after (lanes 2–9) treatment for 20 minutes at 47°C. Increasing amounts of a yTFIID-column eluate (D eluate or DAPs; 0.5 [IX], 1 [2X], 2 [4X], and 4 μl [8X] in lanes 3-6 respectively), as well as a control-columneluate (C eluate; 2 μl [4X]; lane 7) were added to the nuclear extract prior to a heat-treatment. Recombinant TFIID (rTFIID), either human (h; lane 8) or yeast (y; lane 9), was added to the reaction after heat-treatment. Reactions were performed for 60 minutes at 30°C, using a template containing the Ad2 ML promoter. B. Recombinant TFIID (rTFIID), either human (h; lanes 1–6) or yeast (y; lanes 7–12), was added to the reaction (lanes 1 and 7) or heated at 47°C for 20 minutes in the absence (lanes 2 and 8) or in the presence of either increasing amounts of yTFIID-column eluate (0.5 [IX], 1 [2X], and 2 μl [4X]; lanes 3 and 9, 4 and 10, and 5 and 11 respectively) or control-columneluate (2 μl [4X]; lanes 6 and 12). Each reaction contained previously heat-treated nuclear extract. C. Recombinant TFIID (rTFIID), either human (h) or yeast (y), was added to heat-treated nuclear extract, incubated for 20 minutes at 30°C, and either used in transcription reactions (lanes 1 and 6) or heated again in the absence (lanes 2 and 7) or in the presence of increasing amounts of yTFIID-column eluates (1 [2X], 2 [4X] and 4 μl [8X]; lanes 3 and 8, lanes 4 and 9, and lanes 5 and 10 respectively). For heat-treatment, each reaction contained approximately 1 mg/ml BSA. The template and conditions were the same as in Figure 4, except that 2 μl of nuclear extract (15 mg/ml protein), 5 ng of ryTFIID, and 2ng of rhTFIID were used where indicated.
Three polypeptides specifically bind to a yTFIID-affinity column
In order to identify the human proteins that bind specifically to yTFIID, whole cell extract prepared from HeLa cells was loaded onto micro-columns containing purified yTFIID (see Fig. 6D) coupled to Affi-gel 10 at concentrations of 0, 0.2, 0.4, 0.8, and 1.2 mg of protein/ml of packed resin. As for the preparation of DAP fractions described above, the columns were washed with loading buffer and eluted with buffer containing 0.5 M NaCl. These column eluates were analyzed by SDS-PAGE followed by silver staining. Three polypeptides with apparent molecular weights of 35,000, 21,000, and 12,000 bound to the two columns having the highest concentrations (0.8 and 1.2 mg/ml) of yTFIID (Fig. 6A, lanes 5 and 6). These polypeptides were named heDAP35, heDAP21, and heDAP12. When whole cell extract prepared from calf thymus was chromatographed in the same conditions through yTFIID and control columns, bovine polypeptides with apparent molecular weights of 21,000 (ctDAP21) and 12,000 (ctDAP12) bound selectively to the TFIID column (Fig. 6B, compare lanes 2 and 3). When the eluates analyzed using SDS-PAGE (Fig. 6A) were analyzed by DNase I footprinting, the upstream extension of the footprint was seen only with the eluates of columns containing 0.8 and I.2 mg/ml yTFIID (Fig. 6C). Therefore, the eresence of this TFIIA footprint correlated precisely with the appearance of the specific TFIID-binding proteins heDAP35/21/12.
Discussion
We have described the use of affinity chromatography to purify, in a single step, polypeptides in human and bovine whole cell extracts that specifically bind to the yeast TATA box protein TFIID. These polypeptides, which we called DAPs for yTFIID-associated proteins, stimulated the formation of yTFIID-DNA complexes on the Ad2 ML promoter and extended the DNase I footprint produced by yTFIID on the Ad2 ML promoter from −38 to −41. DAPs purified from HeLa extracts allowed the formation of an additional complex (a DNATFIID-TFIIA-type complex) with lower gel mobility than a yTFIID-DNA complex. They also allowed binding of TFIIB to form a DNATFIID-TFIIATFIIB-type complex. DAPs also stimulated transcription in vitro in reactions containing TFIIB/E/F and TFIID fractions, as well as purified RNA pol II. These data indicate that DAP fractions contain a transcription factor which can modulate transcription by interacting with TFIID. Since all the properties we have described for the DAP fraction are also properties of TFIIA, we conclude that TFIIA can bind directly to TFIID in the absence of DNA.
There was a major qualitative difference between ctDAP and heDAP fractions in gel mobility shift assays. CtDAPs did not allow the formation of a stable low mobility complex (a DNATFIID-TFIIA-type complex) or allow the recruitment of TFIIB. Because the only apparent difference in polypeptide content between heDAPs and ctDAPs is a 35 kDa protein in the heDAP fraction, this heDAP35 polypeptide is likely to be an important component of TFIIA activity. DAP21 and DAP12, the two TFIID-binding polypeptides found in both ctDAPs and heDAPs, are similar in size to the polypeptides previously observed in a TFIIA fraction highly purified from calf thymus (Samuels and Sharp, 1986). This calf thymus TFIIA behaved like our calf thymus DAPs in that it stimulated transcription in a reconstituted system, but did not behave like HeLa TFIIA in gel mobility shift assays (Buratowski et al., 1989). Therefore, we think heDAP35, heDAP21, and heDAP12 likely compose human HeLa cell TFIIA. Of course, we cannot presendy exclude the possibility that the three HeLa cell DAPs are derived by proteolysis from a single precursor protein. Neither DAPs nor TFIIA purified by Samuels and Sharp (1986) resemble TFIIA fractions described by Egly et al. (1984) or Usuda et al. (1991).
Do DAPs contain a component which is absolutely essential for in vitro transcription? In the course of this work we compared the basal transcription activity of nuclear extracts that have been passed one to four times over yTFIID and control columns. This was done on several occasions, and at no time did we find any significant reduction in the transcription activity of an extract passed over the yTFIID column. This observation would suggest that DAPs do not contain a required general initiation factor. The requirement for TFIIA as a general initiation factor has been controversial. Perhaps an explanation can be found in the observation by Sumimoto et al. (1990) that TFIIG can replace TFIIA as a required pol II general initiation factor.
DAPs may serve functions other than that of a required basal transcription factor. One possibility is an involvement in transcriptional activation as a co-activator; the existence of such co-activator factors has been proposed recently and co-activator factors have been found in fractions containing TFIID-binding proteins (Dynlacht et al., 1991; Meisterernst et al., 1991). Another attractive possibility would be the involvement of DAPs along with TFIID in transcription by RNA pol III (see Lobo et al., 1991). Alternatively, DAPs may be involved in positive or negative regulation of transcription in special biological conditions. For example, DAPs could stabilize TFIID in heat-shock or other stress conditions.
We have also shown that DAPs can confer heat-resistance on recombinant TFIID. Our data indicate that neither the endogenous DAPs nor exogenously added DAPs can make the TFIID in a nuclear extract resistant to heat-inactivation. One possible explanation for this observation is suggested by the report of Dynlacht et al. (1991), which shows that TFIID (or TBP, TATA box-binding protein) is bound very tightly to several polypeptides (TAFs or TBP-associated factors) in an extract from Drosophila. Human TFIID may interact more tightly with putative human TAFs than it does with DAPs, and such human TAFs could prevent DAPs from making TFIID resistant to heat-inactivation. Our DAP fraction may not contain human TAFs because we used yTFIID, rather than hTFIID, for affinity chromatography. Indeed, we have observed that affinity columns containing recombinant hTFIID as a ligand bound additional HeLa extract polypeptides that were not bound by yTFIID columns (data not shown).
In conclusion, we have described here the identification and the isolation of three mammalian polypeptides which specifically bind to yTFIID in the absence of DNA. They have the properties of TFIIA and they have the ability to preserve TFIID from heat-inactivation.
Acknowledgments
We particularly thank Susan McCracken, Dan Fitzpatrick, and Ray Truant for help and advice. This work was supported by grants to C. J. Ingles and J. Greenblatt from the Medical Research Council of Canada and the National Cancer Institute of Canada. B. Coulombe was awarded a fellowship from the Medical Research Council of Canada. J. Greenblatt is an International Research Scholar of the Howard Hughes Medical Institute.
The costs of publishing this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Buratowski S., Hahn S., Sharp P. A., and Guarente L. (1988), Nature 334, 37–42. [DOI] [PubMed] [Google Scholar]
- Buratowski S., Hahn S., Guarente L., and Sharp P. A. (1989), Cell 56, 549–561. [DOI] [PubMed] [Google Scholar]
- Burton Z. F., Ortolan L. G., and Greenblatt J. (1986), EMBO J 5, 2923–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallini B., Huet J., Plassat J.-L., Sentenac A., Egly J.-M., and Chambon P. (1988), Nature 334, 77–80. [DOI] [PubMed] [Google Scholar]
- Conaway J. W., Reines D., and Conaway R. C. (1990), J Biol Chem 265, 7552–7558. [PMC free article] [PubMed] [Google Scholar]
- Davison B. L., Egly J.-M., Mulvihill E. R., and Chambon P. (1983), Nature 301, 680–686. [DOI] [PubMed] [Google Scholar]
- Dynlacht B. D., Hoey T., and Tjian R. (1991), Cell 66, 563–576. [DOI] [PubMed] [Google Scholar]
- Egly J.-M., Miyamoto N. G., Moncollin V., and Chambon P. (1984), EMBO J 3, 2363–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A., Samuels M., and Sharp P. A. (1984), J Biol Chem 259, 2509–2516. [PubMed] [Google Scholar]
- Flores O., Lu H., Killeen M., Greenblatt J., Burton Z. F., and Reinberg D. (1991), Proc Natl Acad Sci USA 88, 9999–10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha I., Lane W. S., and Reinberg D. (1991), Nature 352, 689–695. [DOI] [PubMed] [Google Scholar]
- Hahn S., Buratowski S., Sharp P. A., and Guarente L. (1989), EMBO J 8, 3379–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi M., Hai T., Lin Y.-S., Green M. R., and Roeder R. G. (1988), Cell 54, 1033–1042. [DOI] [PubMed] [Google Scholar]
- Horikoshi M., Wang C. K., Fujii H., Cromlish J. A., Weil P. A., and Roeder R. G. (1989), Proc Natl Acad Sci USA 86, 4843–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi N., Maguire K., Kralli A., Maldonado E., Reinberg D., and Weinmann R. (1991), Proc Natl Acad Sci USA 88, 5124–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W S., Kao C. C., Bryant G. O., Liu X., and Berk A. J. (1991), Cell 67, 365–376. [DOI] [PubMed] [Google Scholar]
- Lin Y.-S., Ha I., Maldonado E., Reinberg D., and Green M. R. (1991), Nature 353, 569–571. [DOI] [PubMed] [Google Scholar]
- Lobo S. M., Lister J., Sullivan M. L., and Hernandez N. (1991), Genes Dev 5, 1477–1489. [DOI] [PubMed] [Google Scholar]
- Maldonado E., Ha I., Cortes P., Weis L., and Reinberg D. (1990), Mol Cell Biol 10, 6335–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T., Fritsch E. F., and Sambrook J. (1982), Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Meisterernst M., Roy A. L., Lieu H. M., and Roeder R. G. (1991), Cell 66, 981–993. [DOI] [PubMed] [Google Scholar]
- Mermelstein F. H., Flores O., and Reinberg D. (1989), Biochim Biophys Acta 1009, 1–10. [DOI] [PubMed] [Google Scholar]
- Mitchell P. J. and Tijan R. (1989), Science 245,371–378. [DOI] [PubMed] [Google Scholar]
- Nakajima N., Horikoshi M., and Roeder R. G. (1988), Mol Cell Biol 8, 4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson M. G., Tanese N., Pugh B. F., and Tjian R. (1990), Science 248, 1625–1630. [DOI] [PubMed] [Google Scholar]
- Reinberg D., Horikoshi M., and Roeder R. G. (1987), J Biol Chem 262, 3322–3330. [PubMed] [Google Scholar]
- Reinberg D. and Roeder R. G. (1987), J Biol Chem 262, 3310–3321. [PubMed] [Google Scholar]
- Saltzman A. G. and Weinmann R. (1989), FASEB J 3, 1723–1733. [DOI] [PubMed] [Google Scholar]
- Samuels M., Fire A., and Sharp P. A. (1982), J Biol Chem 257, 14419–14427. [PubMed] [Google Scholar]
- Samuels M. and Sharp P. A. (1986), J Biol Chem 261, 2003–2013. [PubMed] [Google Scholar]
- Sawadogo M. and Roeder R. G. (1985), Proc Natl Acad Sci USA 82, 4394–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. C., Kao C., Pei R., and Berk A. (1989), Proc Natl Acad Sci USA 86, 7785–7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro D. J., Sharp P. A., Wahli W. W., and Keller M. J. (1988), DNA 7, 47–55. [DOI] [PubMed] [Google Scholar]
- Sopta M., Carthew R. W., and Greenblatt J. (1985), J Biol Chem 260, 10353–10360. [PubMed] [Google Scholar]
- Studier W. and Moffatt B. A. (1986), J Mol Biol 189, 113–123. [DOI] [PubMed] [Google Scholar]
- Stringer K. F., Ingles C. J., and Greenblatt J. (1990), Nature 345, 783–786. [DOI] [PubMed] [Google Scholar]
- Sumimoto H., Ohkuma Y., Yamamoto T., Horikoshi M., and Roeder R. G. (1990), Proc Natl Acad Sci USA 87, 9158–9162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuda Y., Kubota A., Berk A. J., and Handa H. (1991), EMBO J 10, 2305–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke M. W., Roeder R. G., and Sawadogo M. (1988), Science 241, 1335–1338. [DOI] [PubMed] [Google Scholar]
- Van Dyke M. W., Sawadogo M., and Roeder R. G. (1989), Mol Cell Biol 9, 342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]