

Abstract
We and others have proposed that coactivator binding inhibitors, which block the interaction of estrogen receptor and steroid receptor coactivators, may represent a potential class of new breast cancer therapeutics. The development of coactivator binding inhibitors has been limited, however, because many of the current molecules which are active in in vitro and biochemical assays are not active in cell-based assays. Our goal in this work was to prepare a coactivator binding inhibitor active in cellular models of breast cancer. To accomplish this, we used molecular dynamics simulations to convert a high-affinity stapled peptide with poor cell permeability into R4K1, a cell-penetrating stapled peptide. R4K1 displays high binding affinity for estrogen receptor α, inhibits the formation of estrogen receptor/coactivator complexes, and distributes throughout the cell with a high percentage of nuclear localization. R4K1 represses native gene transcription mediated by estrogen receptor α and inhibits proliferation of estradiol-stimulated MCF-7 cells. Using RNA-Seq, we demonstrate that almost all of the effects of R4K1 on global gene transcription are estrogen receptor-associated. This chemical probe provides a significant proof-of-concept for preparing cell-permeable stapled peptide inhibitors of the estrogen receptor/coactivator interaction.
Introduction
Approximately 75% of breast tumors are estrogen receptor (ER) positive, representing over 170,000 new cases annually.[1] Patients with these tumors usually receive endocrine therapy, such as the selective ER modulator (SERM) tamoxifen to block ER activity and/or aromatase inhibitors to block estrogen production. By 10 years after diagnosis nearly 50% of ER+ tumors treated with endocrine therapy will relapse or recur [2, 3]. Of the ER+ tumors that recur following endocrine therapy, nearly 70% retain ER expression[4], but the function of ER has changed such that it no longer responds to endocrine therapies. Thus, effective treatment for women with resistant/recurrent ER+ cancer that has metastasized represents a major obstacle in the clinic.
Because of its pathophysiological role in ER+ breast cancer, ERα is the target of many endocrine therapies, but it also regulates many physiological processes, including reproduction, proliferation, and development, among others.[5] Binding of estrogen agonists, like the native ligand 17β-estradiol (E2), to the ligand-binding domain induces folding of ERα into a conformation that allows the recruitment of coactivator proteins to activation function 2 when ER is bound to estrogen response elements, sequences of DNA within a gene promoter that bind to ER.[6] Coactivators, in turn, recruit other members of the basal transcription machinery, such as p300/CBP, and effect gene transcription. Selective estrogen receptor modulators, like 4-hydroxytamoxifen (the active metabolite of tamoxifen), oppose the action of 17β-estradiol in the breast by repressing many stimulated target genes. They do so by inducing a conformation of ERα that disfavors binding of coactivators and favors corepressors.[7, 8]
The most well-characterized coactivators are the steroid receptor coactivators (SRCs). These coactivators bind to ERα over two turns of an α-helix though an LXXLL motif, also known as a nuclear receptor box (NR-box).[9, 10] In addition to their effects at ERα, coactivators also regulate the activity of other transcription factors, including other members of the nuclear receptor superfamily. In particular, SRC3, also known as AIB1 (amplified in breast cancer 1), is upregulated in up to 60% of breast cancer cases[11] and is correlated with poor survival rates.
A mechanistic hypothesis in the breast cancer literature has been that directly blocking the ER/coactivator interaction may provide an alternative to antagonizing ER, and that this approach may be useful in treating ER+ breast cancers that have become refractory to current endocrine therapy.[12] A major limitation to testing this hypothesis has been in developing peptides and small molecules that are active in cellular models of ER+ breast cancer.[13–16] [17] With a notable exception, [18] many of the reported small molecule and peptide coactivator binding inhibitors show activity in in vitro assays of ER binding and activity but not in more advanced assays of native gene regulation or of ER+ breast cancer phenotypes.
A powerful method for blocking α-helical protein-protein interactions--like the ER/coactivator interaction--employs “stapled” peptides, which are peptides that are constrained by linking sidechains through olefin metathesis[19]. Optimized by Verdine and coworkers[19], stapled peptides have been used to inhibit many different α-helical protein-protein interactions[20–26]. Stapled peptides confer several benefits, including conformational stability, proteolytic stability, and, in some cases, cell permeability, [27] and they are being tested in the clinic for acute myeloid leukemia, peripheral T-cell lymphoma, and myelodysplastic syndrome (ClinicalTrials.gov ID: NCT02909972 and NCT02264613).
In this work, we used molecular dynamics simulations to design a cell-permeable stapled peptide, R4K1, that inhibits the ER/coactivator interaction in vitro with low nanomolar potency. R4K1 is taken up by breast cancer cells, blocks ERα-mediated gene transcription, and inhibits the proliferation of breast cancer cells. We also examine the effects of R4K1 on global gene transcription using RNA-Seq. R4K1 provides a significant proof-of-concept for preparing cell-permeable stapled peptide inhibitors of the ER/coactivator interaction.
Results
Molecular dynamics-guided design of cell-permeable “stapled” peptide coactivator binding inhibitors
Because the LXXLL motif of coactivators occurs over two turns of an α-helix, stapled peptides provide a good starting point for developing ERα/coactivator binding inhibitors. Indeed, a group at Pfizer used the LXXLL motif to design stapled peptides with nanomolar affinity for ERα.[28] Unfortunately, there are no reports of cellular activity associated with the Pfizer peptides, and we found that several stapled peptides reported by us [29] and Phillips, et al.[28] were unable to decrease expression of native genes that are under the control of ERα (Figures S1 and S2). This finding may be explained by poor cell penetration (see microscopy studies below). Guided by this hypothesis, we set out to design a high-affinity stapled peptide that would also show cell-permeability.
Chu et al. recently published a comprehensive study aimed at understanding cell penetration by stapled peptides. They found that stapled peptides with a formal charge of +5 at pH 7.5 can display high levels of cellular uptake.[30] To replicate this approach, we examined our previously reported crystal structure of ERα bound to stapled peptide SRC2-SP4 (PDB: 5DXE) to decide where to place additional charged residues. ERα contains four surface-exposed aspartate/glutamate residues near the N-terminal region of the SRC2-SP4 binding site (Figure 1A). We reasoned that this region of ERα may provide electrostatic complementarity for positively charged residues of SRC2 because three of six residues preceding the SRC2-Box2 LXXLL motif are lysines (-KEKHKILHRLL-). Replacement of -KEKHK- with -RRRRK- would generate an SRC stapled peptide with a +5 formal charge that mimics the structural motif of primary amphipathic cell penetrating peptides[31] and contains a variation of the putative nuclear localization signal sequence.[32]
Figure 1.

(A) The ligand-binding domain of estrogen receptor α (gray) contains four negatively charged residues (red) at the N-terminal region of our previously reported stapled peptide SRC2-SP4 (magenta, PDB: 5DXE). (B) Snapshot of an MD simulation showing hydrogen bond interactions between arginine residues of R4K1 (beige/magenta) and nearby acidic residues E380, D538, E542, and D545 (cyan) of estrogen receptor. C) Sequences of the nuclear receptor interacting box 2 of steroid receptor coactivator 2 (SRC2-box2) and peptides R4K1, SRC2-SP4, SRC2-wt used in this study.
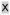 indicates the position of stapling amino acid S5. (D) MD simulations were carried out for 3 × 250 ns using estrogen receptor ligand-binding domain and either R4K1 (beige) or SRC2-SP4 (magenta). The mean percentage of simulations in which E380, D538, E542, or D545 formed a hydrogen bond with a residue from R4K1 or SRC2-SP4 is shown. Each simulation was carried out starting from distinct peptide conformations. Error bars represent the standard deviation; *, p < 0.05.
indicates the position of stapling amino acid S5. (D) MD simulations were carried out for 3 × 250 ns using estrogen receptor ligand-binding domain and either R4K1 (beige) or SRC2-SP4 (magenta). The mean percentage of simulations in which E380, D538, E542, or D545 formed a hydrogen bond with a residue from R4K1 or SRC2-SP4 is shown. Each simulation was carried out starting from distinct peptide conformations. Error bars represent the standard deviation; *, p < 0.05.
To provide evidence of electrostatic complementarity, we carried out a total of 1.5 μs of molecular dynamics simulations of ERα bound to either SRC2-SP4 (magenta in Fig. 1A/1B) or a version of SRC2-SP4 that contains four Arg residues (R4K1, beige in Fig. 1A/1B). The percentage of time in each simulation that SRC2-SP4 (magenta) or R4K1 (beige) formed at least one H-bond with negatively charged residues Glu380, Asp538, Glu542, or Asp545 is shown (Figure 1D). In the simulations, R4K1 showed a statistically significant increase in the number of H-bonds formed at three of four residues, although the effect seemed to be most pronounced at residue Glu542, part of the so-called “charge clamp.”[7] While arginine residues were incorporated to increase cell permeability, these data suggested that they may also contribute to higher binding affinity.
R4K1 is taken up by cells
We hypothesized that inclusion of the Arg4 sequence should increase cell permeability. To examine this, we carried out confocal microscopy studies of fluorescein isothiocyanate (FITC)-labeled peptides. MCF-7 cells were treated with 10 nM estradiol and 15 μM FITC-SRC-WT, FITC-SRC-SP, or FITC-R4K1 for 4, 8, and 24 hours, and confocal images were obtained. FITC-R4K1 was taken up more substantially by MCF-7 cells than either FITC-SRC-WT or FITC-SRC-SP (Fig. 2, S3 and S4).
Figure 2. MCF-7 cells show enhanced uptake of R4K1.
MCF-7 cells were treated for 24 hours with 15 μM fluorescein isothiocyanate (FITC)-labeled SRC2-WT (top), SRC2-SP (center), or R4K1 (bottom). Images from left to right include FITC channel, Hoechst stained nucleus and FITC/Hoechst overlay at 63× magnification.
At 24 hours FITC-R4K1 was fully distributed throughout the cell, with enhanced accumulation in nucleoli, similar to previously reported results (Supporting Information Figure S12). By comparing overlap of Hoechst stain and FITC-R4K1, we quantitated the percentage of nuclear volume containing FITC-R4K1 as 78 ± 2%. We also quantitated the cytoplasmic volume that contained FITC-R4K1 by comparing brightfield images to FITC images. According to this analysis, 89 ± 5.6% of the cytoplasmic area also contained R4K1. These data indicated that R4K1 was present in both nucleus and cytoplasm, so that it was available to bind either cytosolic- or nuclear-localized ER.
R4K1 binds tightly to estrogen receptor
In order to measure dissociation constants of SRC2-WT, SRC2-SP4 or R4K1 for ERα, we used a surface plasmon resonance (SPR) assay in which the ligand-binding domain of ERα was immobilized onto a CM5 chip. The Kd of R4K1 (19 nM) for ERα was 22-fold higher than that of SRC2-SP4 (420 nM) and 137-fold higher than the Kd of SRC2-WT (2600 nM) (Fig. 3). The only difference between the sequences of SRC2-SP4 and R4K1 were four additional arginines, implying that the enhanced binding affinity of R4K1 was mediated through the appended arginines, in agreement with the molecular dynamics simulations.
Figure 3. Stapled peptides bind to estrogen receptor.

A surface plasmon resonance (SPR) assay using immobilized estrogen receptor α ligand binding domain was used to determine Kd for R4K1 (beige, A), SRC2-SP2 (magenta, B), and SRC2-WT (blue, C). Data were analyzed using a steady-state fit.
Stapled Peptides inhibit ER/SRC interaction
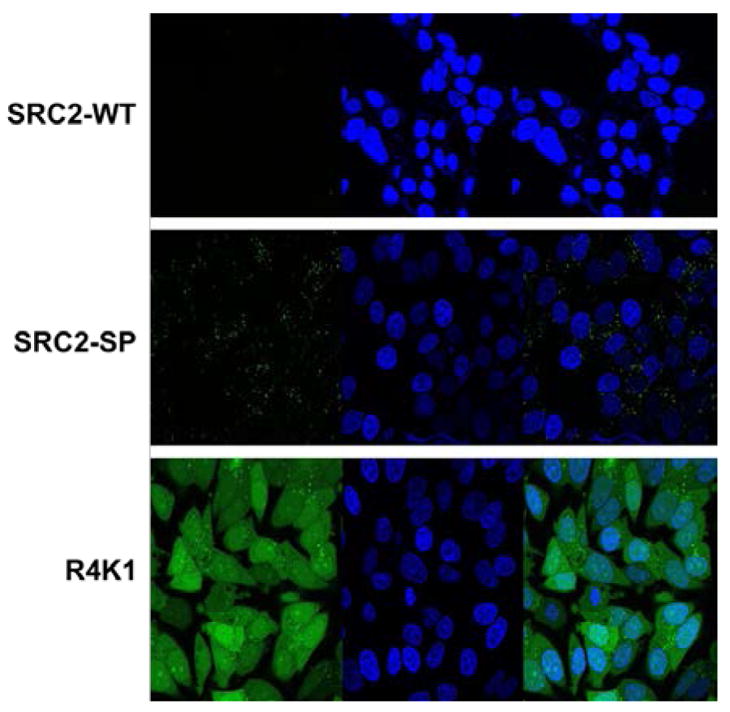
To provide evidence that the peptides would block the ER/coactivator interaction, we measured the ability of the peptides to block recruitment of a fluorescein-labeled SRC fragment to a terbium-labeled ERα ligand binding domain using time-resolved Förster resonance energy transfer (TR-FRET).[33] The IC50 value decreased from 1100 nM to 380 nM as the ILXXLL motif of SRC2-WT (blue) was replaced by the S5LXXS5L motif of SRC2-SP4 (magenta, Fig. 4). The IC50 further decreased to 5.1 nM as the Arg sequence of R4K1 (beige) was appended. These data were in good agreement with the SPR assay and implied that the stapled peptides bind at the coactivator binding region and inhibit interaction of ER with coactivator.
Figure 4.
R4K1 inhibits the ER/coactivator interaction with high potency. Interaction of estrogen receptor α ligand-binding domain, labeled with a long lifetime time-resolved fluorescence resonance energy transfer (TR-FRET) donor (terbium), and a steroid receptor coactivator fragment, labeled with TR-FRET acceptor fluorescein, was inhibited with increasing concentrations of R4K1 (beige), SRC2-SP4 (magenta), or SCR2-WT (blue). The ratio of fluorescent emissions of fluorescein acceptor and terbium donor is plotted along the y-axis (F520/F495), and the log of molar concentration of inhibitor is plotted along the x-axis. Error bars represent the standard deviation.
Quantitation of Membrane Integrity after Treatment with R4K1
Appending positively charged residues onto a peptide is a commonly used procedure for enhancing uptake of peptides by cells;[34, 35] however, some groups have shown that incorporating many positively charged residues may lead to loss of membrane integrity.[36–38] To guard against this possibility, we carried out lactate dehydrogenase release assays to determine safe concentrations to use in our cell-based experiments. In this assay, increased release of the cytoplasmic protein lactate dehydrogenase is indicative of membrane disruption and can be quantified relative to maximum lysis with sodium dodecylsulfate (SDS).
We carried out LDH release assays at one concentration (30 μM) after one-hour treatment of two ER+ breast cancer cell lines, MCF-7 and T47D, with peptides. None of the peptides showed release of LDH that was significantly different from vehicle (Supporting Information Figure S5).
To guard against the potential for bias introduced by examining the conditions above, we measured lactate dehydrogenase release in a time vs. concentration vs. response mode. Specifically, we measured release of lactate dehydrogenase at four timepoints and at five concentrations (Figure 5). There was no significant lactate dehydrogenase release at 5, 10, and 15 μM, even at 24 hours. These data suggested that 15 μM R4K1, even at long time points, would not damage the membrane and would be a safe concentration to use to examine cellular effects of R4K1.
Figure 5.

R4K1 does not cause loss of membrane integrity at efficacious concentrations. MCF-7 cells were treated with 5, 10, 15, 30 or 50 μM stapled peptide R4K1. Release of lactate dehydrogenase (LDH) was measured at 1, 2, 4, or 24 hours after treatment. %LDH release is plotted vs. time and concentration, relative to maximum lysis with sodium dodecyl sulfate.
R4K1 inhibits transcription of ER-regulated, but not NFκB-regulated, native genes
Our central hypothesis was that blockade of coactivator recruitment with R4K1 should show repression of estradiol-mediated gene expression, similar to that of selective estrogen receptor modulators, like 4-hydroxytamoxifen. We further hypothesized that R4K1 would not show estrogenic activity.
We first carried out a screening assay to determine what anti-estrogenic benefit R4K1 might have over SRC2-WT or SRC2-SP4. We treated MCF-7 cells with 10 nM estradiol and 15 μM SRC2-WT, SRC2-SP4, or R4K1 (Supporting Information Figure S6). For each of these treatment conditions, we measured transcript levels of five genes known to be stimulated by estradiol: PTGES, PR, PS2, EGR3, and IGFBP4. Treatment with estradiol showed upregulation of all genes. When cells were also treated with R4K1, all five genes showed a decrease in gene expression that was not seen with SRC2-SP4 or SRC2-WT, although only four reached statistical significance.
Based on the results of the screening assay, we compared estrogenic and anti-estrogenic activities of R4K1 to 4-hydroxytamoxifen in two different breast cancer cell lines. MCF-7 and T47D cells were treated with vehicle, 4-hydroxytamoxifen (4OHT) or R4K1 in the presence or absence of estradiol (E2) (Fig. 6). Estradiol induced expression of each of the five genes from above. This effect was reversed by co-treatment with estradiol plus 4OHT or estradiol plus R4K1, although the magnitude of the effect of R4K1 was smaller than that of 4OHT. There was no statistically significant difference in gene expression between cells treated with vehicle alone (i.e., no estradiol) and those that were treated with 4OHT or R4K1 alone. These effects were similar in a second ER+ breast cancer cell line (Fig. S7), suggesting that R4K1 can broadly inhibit ER activity and is not acting in a cell-specific manner. Taken together, these data supported our hypothesis that blocking the ER/coactivator interaction represses estrogen-stimulated gene expression.
Figure 6.

R4K1 inhibits transcription of ER-regulated native genes, but not NFκB-regulated genes. mRNA levels for ER-regulated genes PTGES (A), PR (B), PS2 (C), EGR3 (D), and IGFBP4 (E) were examined in MCF-7 cells by RT-QPCR. Cells were pretreated with R4K1 (15 μM, 24 hrs), 4OHT (1 μM, 2 hrs) or DMSO control, followed by 10 nM 17β-estradiol (E2) treatment for 2 hrs. Data were normalized to 36B4 (A–E) or GAPDH (F–H) internal controls and presented as fold change relative to DMSO vehicle. Error bars represent the standard deviation. n.s., not statistically significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
We also tested whether R4K1 had effects on genes regulated by a different transcription factor, NFκB, which is also coactivated by SRC3.[39] We measured expression of NFκB-regulated genes RelB, ICAM1, and TNFα in the presence or absence of NFκB-stimulating cytokine, TNFα. There was little, if any, difference in expression between those genes that were treated with vehicle versus those treated with R4K1 (Figure 6F–H). These data implied that R4K1 did not non-specifically repress the activity of all transcription factors.
R4K1 reverses estradiol-stimulated proliferation of MCF-7 cells
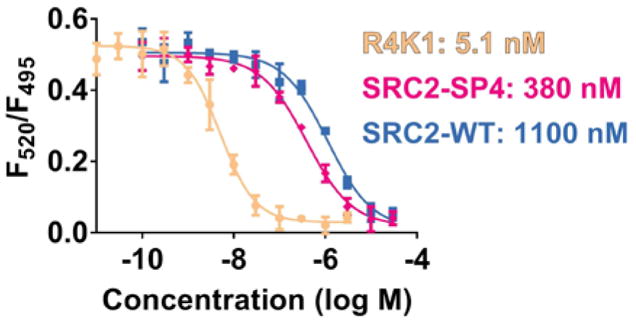
Proliferation of ER+ breast cancer cell lines is enhanced by treatment with estradiol. We treated the ER+ breast cancer cell line MCF-7 with 15 μM R4K1 in the presence or absence of 10 nM estradiol and measured proliferation of these cells (Figure 7). Estradiol stimulated proliferation of MCF-7 cells, but R4K1 alone had no effect on proliferation of MCF-7 cells. Administering R4K1 with estradiol decreased proliferation to vehicle-treated levels. These data implied that blocking the ER/coactivator complex can repress proliferation that is stimulated by estradiol.
Figure 7.
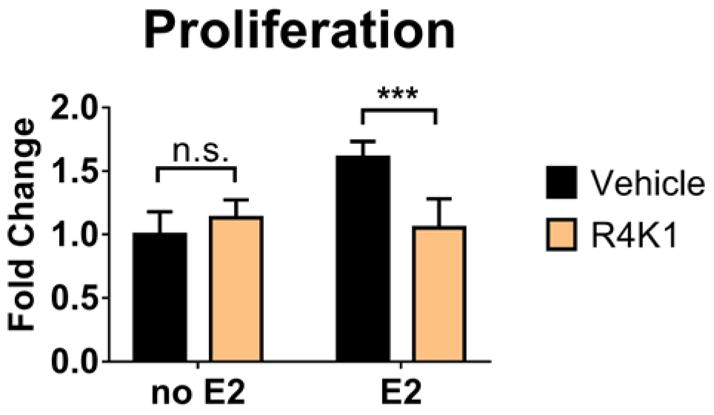
R4K1 reverses estradiol-stimulated proliferation. MCF-7 cells were treated with vehicle or 15 μM stapled peptide R4K1 in the presence or absence of 10 nM estradiol (E2). Treatment was initiated on Day 3 and cell numbers were measured 24 hours later. Fold change was determined relative to vehicle control for three independent experiments. Error bars represent the standard deviation. n.s., not statistically significant; ***, p<0.001.
R4K1 has little, if any, estrogen receptor-degrading activity
Tamoxifen is a selective estrogen receptor modulator (SERM). It induces a conformation of ER that recruits a distinct set of coactivators and corepressors, so that it has both agonist and antagonist properties, depending on context and tissue type. Another class of ER ligands are referred to as selective estrogen receptor degraders (SERDs), which include the breast cancer drug fulvestrant. SERDs cause degradation of ER, but SERMs do not.
We were curious whether the mechanism-of-action of R4K1 would more closely resemble that of SERMs or SERDs. We measured protein levels of ER in the presence or absence of R4K1 and with or without estradiol in both MCF-7 and T47D cells (Figure 8). R4K1 showed no significant difference from vehicle-treated cells in the presence or absence of estradiol. There was a statistically significant decrease in ER levels in vehicle-treated T47D cells in the absence of estradiol, but this effect disappeared in the presence of estradiol. These data implied that, if R4K1 has SERD activity, it is only modest at best.
Figure 8.
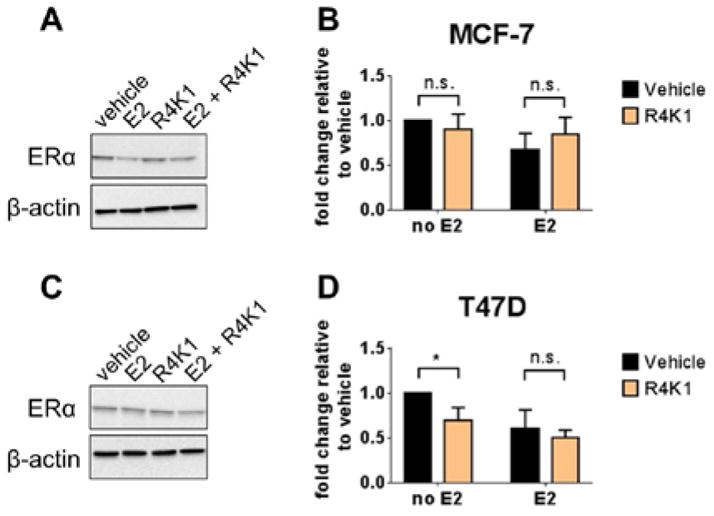
R4K1 has little, if any, estrogen receptor-degrading activity. MCF-7 (top) or T47D (bottom) cells were pretreated with R4K1 (15 μM, 24 hrs), 4OHT (1 μM, 2 hrs) or DMSO control, followed by 10 nM 17β-estradiol (E2) treatment for 2 hrs. Western blot was performed for ERα. β-actin was used as loading control. Vehicle-treated sample value was used as one arbitrary unit. Error bars represent the standard deviation from three independent experiments. n.s., not statistically significant; *, p <0.05.
R4K1’s effects on global gene expression
To more fully understand a ligand’s effects on transcription mediated by ER, it is necessary to look at global gene expression, rather than individual genes. We used RNA-Seq to analyze the transcriptome of MCF-7 cells to compare the global effects of R4K1 with those of 4-hydroxytamoxifen.
MCF-7 cells were treated under six different conditions, shown as columns in the heatmap of Figure 9: 1) 10 nM 17β-estradiol (E2), 2) 1 μM 4-hydroxytamoxifen (4OHT), 3) 10 nM E2 + 1 μM 4OHT, 4) 15 μM R4K1, 5) 10 nM E2 + 15 μM R4K1 and vehicle alone. Across the five experimental conditions, 1,041 transcripts were expressed at levels that were at least 2-fold different from vehicle control (FDR<0.05, Figure 9A). These genes clustered into nine sets, depicted by rows in the heatmap of Figure 9A: 1) genes up-regulated similarly by E2, 4OHT, and to a lesser extent by R4K1; 2 and 3) genes down- or up-regulated by E2 that were fully reversed by 4OHT and partially by R4K1; 4) genes down-regulated similarly by E2 and 4OHT, and, to a lesser extent, by R4K1; 5) genes up-regulated by E2 that were reversed by 4OHT and not R4K1; 6) genes up-regulated by 4OHT that were not affected by E2 or R4K1; 7) genes repressed by R4K1 that were reversed by E2; 8) genes repressed by 4OHT that were reversed by E2; and 9) genes up-regulated by R4K1 that were reversed by E2.
Figure 9.

(A) RNA-Seq heatmap for 1,041 mRNA transcripts differentially expressed in MCF7 cells treated with 10 nM estradiol (E2), 1 μM 4-hydroxytamoxifen (4OHT), 10 nM E2 + 1 μM 4OHT, 15 μM R4K1, and 10 nM E2 + 15 μM R4K1. Data are normalized to vehicle treatment. Blue bars represent transcripts that are repressed relative to vehicle, and red bars represent transcripts that are stimulated relative to vehicle. Genes were grouped into 9 clusters using the k-means algorithm embedded within Gene Cluster 3.0 (B) Box-and-whiskers plot for cluster 2, mRNA transcripts repressed by E2 (green) that are reversed by co-treatment with either 4OHT (purple) or R4K1 (beige). (C) Box-and-whiskers plot for cluster 3, mRNA transcripts stimulated by E2 that are repressed by co-treatment with either 4OHT or R4K1. (D) Box-and-whiskers plot for cluster 7, mRNA transcripts repressed by R4K1 that are reversed by co-treatment with E2. (E) Box-and-whiskers plot for cluster 9, mRNA transcripts stimulated by R4K1 that are repressed by co-treatment with E2. In panels B, C, D, and E, the box represents the first through third quartiles, and the vertical “whiskers” represent the range. n.s., not statistically significant; ****, p<0.0001.
Of particular relevance were clusters 2 and 3, which contained E2-regulated genes that were reversed by R4K1 (Fig. 9B/C). Cluster 2 contained 226 E2-stimulated genes that were reversed fully by 4OHT and partially by R4K1. Cluster 3 contained 87 E2-repressed genes that were reversed fully by 4OHT and partially by R4K1. These data indicated that R4K1 reversed E2-regulated genes, but not to the same extent as 4OHT. R4K1 also showed gene regulation that was distinctly different from 4OHT but was reversed by E2, as seen in clusters 7 and 9 (Fig. 9D/E), suggesting that these genes may be related to ER activity.
DISCUSSION
R4K1 is among the first stapled peptides that block the action of ER in cellular models of breast cancer. There have been several other peptides and small molecules that block the ER/coactivator interaction in vitro, but, generally, the extant studies are limited in nature. Much of the characterization for these molecules has been restricted to in vitro studies (see [17, 40] for reviews of the literature), and of those molecules that have cellular characterization, there are several common features: many are active in reporter gene and mammalian two-hybrid assays, but whether they can repress the activity of native genes or breast cancer phenotypes regulated by ER is unknown. [41] [42] There are a few exceptions, including peptides synthesized by Brunsveld and coworkers, [13] as well as Li and coworkers[43], although even these most advanced examples have been characterized using only one native gene.
The most well-characterized molecule for inhibiting the ER/coregulator interaction comes from Raj et al. who recently described ERX-11, a small molecule that is active in several different models of ER+ breast cancer, including a tumor xenograft model.[18] ERX-11 is an oligoamide that was designed to bind to ER at the coregulator-binding region, but even after careful experimentation and design, the precise binding site and mode of action is not fully understood for ERX-11, demonstrating the difficult nature of designing inhibitors of this protein-protein interaction.
To address the lack of cell-permeable, well-characterized ER/coactivator binding inhibitors, we redesigned a cell-impenetrant stapled peptide so that it would show cell permeability and activity in cell-based models of ER function. The computationally informed placement of arginine residues led to an increase in binding affinity as a result of enhanced hydrogen bonding to negatively charged residues. This finding is in agreement with with strategies that have been previously used to prepare high affinity peptides for ER via proline-primed helices, isoaspartic-acid cyclized peptides, and lysine-to-arginine substituted peptides. In this work, we have been guided by the principle of linking a thorough understanding of the molecular basis of ER/stapled peptide binding with in vitro and cellular studies. The product of this work, R4K1, represents a significant proof-of-principle molecule for the future design of cell-permeable stapled peptides to inhibit the ER/coactivator interaction.
Our studies suggest that R4K1 is taken up by cells, and that relatively long incubation times may be required for R4K1 to distribute throughout the cell so that it can have its effects at the nucleus. These studies also indicate that, for this molecule, the arginine sequence that we have used is necessary for cell penetration, as poor cell penetration was seen with both SRC2-WT and SRC2-SP. Mechanistically, R4K1 acts more similarly to selective estrogen receptor modulators (SERMs) rather than selective estrogen receptor degraders (SERDs) in that it does not cause ER degradation. Based on our understanding of R4K1 binding, R4K1 does not expose hydrophobic residues, nor does it cause exposure of hydrophobic ER residues. Exposure of hydrophobic residues could lead to degradation, so that this finding is in keeping with our understanding of the mechanism of action of blocking coactivator binding.
Most importantly, our studies suggest that essentially all gene regulatory actions of R4K1 are ER-associated. First, RNA-seq data suggest that R4K1 acts similarly to 4OHT on both up- and down-regulated genes (Clusters 1–5), albeit with lower magnitude. Given our mechanism of blocking coactivator recruitment, understanding how R4K1 reverses E2-stimulated genes is straightforward, but the mechanism by which R4K1 reverses E2-repressed genes is unclear. One explanation is that coregulators may directly have dual activating and repressive functions. Some coregulators (e.g., PELP-1 [44], RIP140 [45]) are known to have different activities at different transcription factors, but the extent to which they repress or stimulate gene expression at a single transcription factor is not completely understood, so that an improved version of R4K1 could be used to shed light on this problem. Second, there are essentially no R4K1-specific effects that are not reversed by E2 (Clusters 7 and 9). If R4K1 were non-selective for ER and able to affect other transcription factors, we might expect to see up- or down-regulation of gene expression that is not reversed by E2. This is further supported by the lack of R4K1 effect on NFκB target genes. The mechanism by which R4K1 regulates genes in the presence of unliganded ER is unclear. One possible explanation for these activities could be that R4K1 shows a preference for binding to folded, liganded ER, but that, in the absence of folded, liganded ER, R4K1 may bind to a subset of transcription factors and block coactivator recruitment, which could also help to explain how R4K1 reverses E2-repressed genes. This explanation is similar to the “squelching” hypothesis in the coactivator literature, wherein binding of a limited pool of coactivators at one transcription factor may lead to repression of genes regulated by other transcription factors. [46] [47] Overall, the work here could lay the groundwork for providing tools to probe incompletely understood mechanisms of coregulators, including the dual-function and squelching mechanisms.
In conclusion, we have described a cell-permeable stapled peptide, R4K1, that modulates the activity of estrogen receptor in breast cancer cell lines. These studies are informed by a detailed molecular understanding of inhibiting the estrogen receptor/coactivator interaction. R4K1 provides a proof-of-concept that cell-permeable stapled peptides may be used to inhibit the estrogen receptor/coactivator interaction and that this disruption may prove advantageous in models of ER+ breast cancer. While R4K1 is a promising proof-of-principle probe, these studies also suggest that future cell-permeable stapled peptides need to show higher efficacy, which could come from increased uptake and/or higher affinity for estrogen receptor.
Experimental Methods
General Considerations
Peptide synthesis was accomplished using a literature procedure.[48] The TR-FRET assay protocol was carried out as previously described.[29, 49] Molecular dynamics simulations were performed using XSEDE resources [50] as previously described [29] with some exceptions that are fully explained in the SI. Unless otherwise noted, cell culture experiments were carried out in the presence of charcoal-dextran-stripped 5% fetal bovine serum (FBS). Lower concentrations of FBS resulted in low estrogen-responsiveness (data not shown).
Surface Plasmon Resonance (SPR)
The SPR assay protocol was based on previously reported conditions, [28] with the following changes: SPR analysis was performed on a BiacoreT200; ERα ligand binding domain construct contained amino acids 299–554, including N-terminal 6His-tag; final ERα surface density was ~9500 RU; stapled peptide solutions at a series of increasing concentrations were applied to flow cells at a 30 μL/min flow rate using a contact time of 60 s and a dissociation time of 120 s; KD values were determined by fitting reference subtracted data to a steady-state affinity equation embedded in the Biacore T200 evaluation software 3.0; and kinetic fittings were done using the two state reaction binding equation embedded in the Biacore T200 evaluation software 3.0 (Figure S8).
Confocal Microscopy
Breast cancer cells were cultured as previously described. [51] For microscopy studies, cells were incubated with 15 μM FITC-conjugated stapled peptides for 4, 8, or 24 hr. Hoechst dye was used for nuclear staining, at a concentration of 4 mg/mL for 30 minutes (Life Technologies). Images were taken with a Zeiss confocal LSM 710 microscope. The percentage of stapled peptide in the nucleus was determined by FITC and Hoechst co-localization, and the percentage cytoplasmic stapled peptide relied on FITC and brightfield overlap. Corrected total cell fluorescence was evaluated using Image J software with the SEM for each treatment group.
Lactate Dehydrogenase Assay
Cytotoxicity was measured according to the manufacturer’s instructions (CytoTox 96, Promega). Absorbance was read at 490 nm on a BioTek Synergy HT plate reader. A maximum release LDH reagent (provided with the kit) was used as a positive control. All samples were evaluated as a percentage of LDH released relative to maximum.
RNA and RT-qPCR
Total RNA was isolated and RT-qPCR was performed as previously described. [52] 36B4 or GAPDH were used as internal controls, and fold change was calculated using the ΔΔCt method. qPCR primers are listed in Table S3.
Western Blot
Whole cell extracts were prepared using radioimmunoprecipitation assay buffer (RIPA) buffer; proteins were denatured and separated by SDS-PAGE using a 5–12% gradient gel (Invitrogen) and then transferred to nitrocellulose membranes. The membranes were blocked for 1 hr in 5% non-fat dry milk. Membranes were incubated overnight at 4 °C with appropriate primary antibody (ERα [Cell Signaling #8644] or β-actin [Sigma #A5441]. The next day, membranes were washed and incubated in horseradish peroxidase conjugated secondary antibodies. The signal was visualized using Chemi-doc XRS (Bio-Rad laboratories) following incubation with the Pierce Supersignal West Pico Chemiluminescent Substrate.
Proliferation Assay
Cell counts were determined using an imaging cytometer (Celigo) on the brightfield channel following 24 hr of treatment. Fold change was calculated relative to vehicle control.
RNA-seq Experimental Design and Data Analysis
RNA isolated for qPCR was provided to the Genomics Core Facility, RRC at UIC, for RNA-Seq analysis. Libraries were prepared from two biological replicates per condition. Sequencing libraries were prepared using magnetic beads similar to described previously using barcoded adapters (NextFlex, Bioo Scientific). [53] RNA was sequenced on an Illumina HiSeq 2000 according to the manufacturer’s instructions. RNA-Seq results were trimmed and aligned to the hg19 assembly using ELAND allowing up to 2 mismatches. Differential gene expression was determined using edgeR as a component of the HOMER software suite.[54] Detailed instructions for analysis can be found at http://homer.ucsd.edu/homer/. Genes were considered differentially regulated if fold change >2 and p-value <0.05 compared to vehicle treatments. Heatmaps were generated using CLUSTER and visualized using JavaTreeView software [55] Box-and-whiskers plots were prepared using Graphpad Prism 7.03. A paired t-test was used to calculate statistical significance. All data are publicly available through GEO (accession # TBD).
Supplementary Material
Acknowledgments
We would like to thank Kathryn Carlson and John Katzenellenbogen for providing plasmids for biochemical studies and Irida Kastrati for helpful discussions. This work was funded by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust (to JMF and TWM) and by the University of Illinois Cancer Center (to TWM). TES was funded by training grant T32AT007533, Office of The Director, National Institutes Of Health (OD) and National Center For Complementary & Integrative Health (NCCIH). This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges at the Pittsburgh Supercomputing Center through allocation TG-MCB170062, which is supported by National Science Foundation grant number ACI-1548562.
References
- 1.Fan C, et al. Concordance among Gene-Expression–Based Predictors for Breast Cancer. New England Journal of Medicine. 2006;355(6):560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 2.Yu KD, et al. Hazard of Breast Cancer-Specific Mortality among Women with Estrogen Receptor-Positive Breast Cancer after Five Years from Diagnosis: Implication for Extended Endocrine Therapy. The Journal of Clinical Endocrinology & Metabolism. 2012;97(12):E2201–E2209. doi: 10.1210/jc.2012-2423. [DOI] [PubMed] [Google Scholar]
- 3.Jatoi I, et al. Breast Cancer Adjuvant Therapy: Time to Consider Its Time-Dependent Effects. Journal of Clinical Oncology. 2011;29(17):2301–2304. doi: 10.1200/JCO.2010.32.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoehn JL, Plotka ED, Dickson KB. Comparison of estrogen receptor levels in primary and regional metastatic carcinoma of the breast. Annals of Surgery. 1979;190(1):69–71. doi: 10.1097/00000658-197907000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heldring N, et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiological Reviews. 2007;87(3):905. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 6.Johnson AB, O’Malley BW. Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol. 2012;348(2):430–9. doi: 10.1016/j.mce.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiau AK, et al. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95(7):927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 8.Watson PJ, Fairall L, Schwabe JWR. Nuclear hormone receptor co-repressors: Structure and function. Molecular and Cellular Endocrinology. 2012;348–135(2–3):440–449. doi: 10.1016/j.mce.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heery DM, Kalkhoven E. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387(6634):733. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 10.Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. The Journal of Peptide Research. 2004;63(3):207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- 11.Anzick SL, et al. AIB1, a Steroid Receptor Coactivator Amplified in Breast and Ovarian Cancer. Science. 1997;277(5328):965. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 12.Norris JD, et al. Peptide Antagonists of the Human Estrogen Receptor. Science. 1999;285(5428):744–746. doi: 10.1126/science.285.5428.744. [DOI] [PubMed] [Google Scholar]
- 13.Carraz M, et al. Perturbation of Estrogen Receptor α Localization with Synthetic Nona-Arginine LXXLL-Peptide Coactivator Binding Inhibitors. Chemistry & Biology. 2009;16(7):702–711. doi: 10.1016/j.chembiol.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Galande AK, et al. Potent Inhibitors of LXXLL-Based Protein–Protein Interactions. ChemBioChem. 2005;6(11):1991–1998. doi: 10.1002/cbic.200500083. [DOI] [PubMed] [Google Scholar]
- 15.Nagakubo T, et al. Development of Cell-Penetrating R7 Fragment-Conjugated Helical Peptides as Inhibitors of Estrogen Receptor-Mediated Transcription. Bioconjugate Chemistry. 2014;25(11):1921–1924. doi: 10.1021/bc500480e. [DOI] [PubMed] [Google Scholar]
- 16.Jiang Y, et al. Switching substitution groups on the in-tether chiral centre influences backbone peptides’ permeability and target binding affinity. Organic & Biomolecular Chemistry. 2017;15(3):541–544. doi: 10.1039/c6ob02289h. [DOI] [PubMed] [Google Scholar]
- 17.Moore TW, Mayne CG, Katzenellenbogen JA. Minireview: Not Picking Pockets: Nuclear Receptor Alternate-Site Modulators (NRAMs) Molecular Endocrinology. 2010;24(4):683–695. doi: 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raj GV, et al. Estrogen receptor coregulator binding modulators (ERXs) effectively target estrogen receptor positive human breast cancers. eLife. 2017;6:e26857. doi: 10.7554/eLife.26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schafmeister CE, Po J, Verdine GL. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. Journal of the American Chemical Society. 2000;122(24):5891–5892. [Google Scholar]
- 20.Walensky LD, et al. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science. 2004;305(5689):1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wachter F, et al. Mechanistic Validation of a Clinical Lead Stapled Peptide that Reactivates p53 by Dual HDM2 and HDMX Targeting. Oncogene. 2017;36(15):2184–2190. doi: 10.1038/onc.2016.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitra S, et al. Stapled peptide inhibitors of RAB25 target context-specific phenotypes in cancer. Nature Communications. 2017;8:660. doi: 10.1038/s41467-017-00888-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cromm PM, et al. Protease-Resistant and Cell-Permeable Double-Stapled Peptides Targeting the Rab8a GTPase. ACS Chemical Biology. 2016;11(8):2375–2382. doi: 10.1021/acschembio.6b00386. [DOI] [PubMed] [Google Scholar]
- 24.Rennie YK, et al. A TPX2 Proteomimetic Has Enhanced Affinity for Aurora-A Due to Hydrocarbon Stapling of a Helix. ACS Chemical Biology. 2016;11(12):3383–3390. doi: 10.1021/acschembio.6b00727. [DOI] [PubMed] [Google Scholar]
- 25.Thomas JC, et al. Inhibition of Ral GTPases Using a Stapled Peptide Approach. Journal of Biological Chemistry. 2016;291(35):18310–18325. doi: 10.1074/jbc.M116.720243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards AL, et al. Challenges in Targeting a Basic Helix–Loop–Helix Transcription Factor with Hydrocarbon-Stapled Peptides. ACS Chemical Biology. 2016;11(11):3146–3153. doi: 10.1021/acschembio.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cromm PM, Spiegel J, Grossmann TN. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chemical Biology. 2015;10(6):1362–1375. doi: 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- 28.Phillips C, et al. Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc. 2011;133(25):9696–9. doi: 10.1021/ja202946k. [DOI] [PubMed] [Google Scholar]
- 29.Speltz TE, et al. Stapled Peptides with γ-Methylated Hydrocarbon Chains for the Estrogen Receptor/Coactivator Interaction. Angewandte Chemie International Edition. 2016;55(13):4252–4255. doi: 10.1002/anie.201510557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu Q, et al. Towards understanding cell penetration by stapled peptides. MedChemComm. 2015;6(1):111–119. [Google Scholar]
- 31.Ziegler A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Advanced Drug Delivery Reviews. 2008;60(4):580–597. doi: 10.1016/j.addr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Boulikas T. Putative nuclear localization signals (NLS) in protein transcription factors. Journal of Cellular Biochemistry. 1994;55(1):32–58. doi: 10.1002/jcb.240550106. [DOI] [PubMed] [Google Scholar]
- 33.Gunther JR, et al. A Set of Time-Resolved Fluorescence Resonance Energy Transfer Assays for the Discovery of Inhibitors of Estrogen Receptor-Coactivator Binding. Journal of Biomolecular Screening. 2009;14(2):181–193. doi: 10.1177/1087057108329349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang H, et al. Helical Poly(arginine) Mimics with Superior Cell-Penetrating and Molecular Transporting Properties. Chemical science (Royal Society of Chemistry. 2010;4(10):3839–3844. doi: 10.1039/C3SC51328A. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt N, et al. Arginine-rich cell-penetrating peptides. FEBS Letters. 2010;584(9):1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 36.Bechinger B. Structure and Functions of Channel-Forming Peptides: Magainins, Cecropins, Melittin and Alamethicin. The Journal of Membrane Biology. 1997;156(3):197–211. doi: 10.1007/s002329900201. [DOI] [PubMed] [Google Scholar]
- 37.Herce HD, et al. Arginine-Rich Peptides Destabilize the Plasma Membrane, Consistent with a Pore Formation Translocation Mechanism of Cell-Penetrating Peptides. Biophysical Journal. 2009;97(7):1917–1925. doi: 10.1016/j.bpj.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li YC, et al. A versatile platform to analyze low affinity and transient protein-protein interactions in living cells in real time. Cell reports. 2014;9(5):1946–1958. doi: 10.1016/j.celrep.2014.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu RC, et al. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator activity by IκB kinase. Molecular and cellular biology. 2002;22(10):3549–3561. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caboni L, Lloyd DG. Beyond the Ligand-Binding Pocket: Targeting Alternate Sites in Nuclear Receptors. Medicinal Research Reviews. 2013;33(5):1081–1118. doi: 10.1002/med.21275. [DOI] [PubMed] [Google Scholar]
- 41.Weiser PT, et al. 4, 4′-Unsymmetrically substituted 3, 3′-biphenyl alpha helical proteomimetics as potential coactivator binding inhibitors. Bioorganic & medicinal chemistry. 2014;22(2):917–926. doi: 10.1016/j.bmc.2013.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gunther JR, et al. Amphipathic benzenes are designed inhibitors of the estrogen receptor α/steroid receptor coactivator interaction. ACS chemical biology. 2008;3(5):282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie M, et al. Structural Basis of Inhibition of ERα-Coactivator Interaction by High-affinity N-terminus Isoaspartic Acid Tethered Helical Peptides. Journal of Medicinal Chemistry. 2017 doi: 10.1021/acs.jmedchem.7b00732. [DOI] [PubMed] [Google Scholar]
- 44.Choi YB, Ko JK, Shin J. The transcriptional corepressor, PELP1, recruits HDAC2 and masks histones using two separate domains. Journal of Biological Chemistry. 2004;279(49):50930–50941. doi: 10.1074/jbc.M406831200. [DOI] [PubMed] [Google Scholar]
- 45.Wei LN, et al. Receptor-interacting protein 140 directly recruits histone deacetylases for gene silencing. Journal of Biological Chemistry. 2000;275(52):40782–40787. doi: 10.1074/jbc.M004821200. [DOI] [PubMed] [Google Scholar]
- 46.Min G, et al. Inhibitory cross-talk between estrogen receptor (ER) and constitutively activated androstane receptor (CAR) CAR inhibits ER-mediated signaling pathway by squelching p160 coactivators. Journal of Biological Chemistry. 2002;277(37):34626–34633. doi: 10.1074/jbc.M205239200. [DOI] [PubMed] [Google Scholar]
- 47.Guertin MJ, et al. Transient Estrogen Receptor Binding and p300 Redistribution Support a Squelching Mechanism for Estradiol-Repressed Genes. Molecular Endocrinology. 2014;28(9):1522–1533. doi: 10.1210/me.2014-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YW, Grossmann TN, Verdine GL. Synthesis of all-hydrocarbon stapled [alpha]-helical peptides by ring-closing olefin metathesis. Nat Protocols. 2011;6(6):761–771. doi: 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- 49.Gunther JR, et al. A set of time-resolved fluorescence resonance energy transfer assays for the discovery of inhibitors of estrogen receptor-coactivator binding. Journal of biomolecular screening. 2009;14(2):181–193. doi: 10.1177/1087057108329349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Towns J, et al. XSEDE: accelerating scientific discovery. Computing in Science & Engineering. 2014;16(5):62–74. [Google Scholar]
- 51.Kastrati I, Canestrari E, Frasor J. PHLDA1 expression is controlled by an estrogen receptor-NFκB-miR-181 regulatory loop and is essential for formation of ER+ mammospheres. Oncogene. 2015;34(18):2309–2316. doi: 10.1038/onc.2014.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frasor J, et al. Positive cross-talk between estrogen receptor and NF-κB in breast cancer. Cancer research. 2009;69(23):8918–8925. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garber M, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Molecular cell. 2012;47(5):810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.