

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a serious blood disorder characterized by dysregulated complement activation on blood cells. Eculizumab, the current standard therapy and a humanized anti-C5 mAb relieves anemia and thrombosis symptoms of PNH patients by preventing complement-dependent intravascular hemolysis. However, up to 20% of PNH patients on long term Eculizumab treatment still suffer from significant anemia and are transfusion-dependent due to extravascular hemolysis of C3-opsonized PNH erythrocytes. Here we show that function-blocking anti-properdin (P) mAbs dose-dependently inhibited autologous complement-mediated hemolysis induced by factor H dysfunction. Furthermore, anti-human P mAbs potently and dose-dependently inhibited acidified serum-induced hemolysis of PNH erythrocytes (Ham’s test). In contrast to erythrocytes rescued by anti-C5 mAb, non-lysed PNH erythrocytes rescued by anti-P mAb incurred no activated C3 fragments deposition on their surface. These results suggested that anti-P mAbs may prevent extravascular as well as intravascular hemolysis of PNH erythrocytes. To test the in vivo efficacy of anti-human P mAbs in preventing extravascular hemolysis, we generated a P “humanized” mouse by transgenic expression of human P in P knockout mice (hP-Tg/P−/−). In a murine extravascular hemolysis model, complement-susceptible erythrocytes were completely eliminated within 3 days in control mAb-treated hP-Tg/P−/− mice, whereas such cells were protected and persisted in hP-Tg/P−/− mice treated with an anti-human P mAb. Collectively, these data suggest that anti-P mAbs can inhibit both intravascular and extravascular hemolysis mediated by complement and may offer improved efficacy over Eculizumab, the current standard therapy for PNH.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired blood disorder caused by somatic mutations in hematopoietic stem cells of the gene encoding phosphatidylinositol glycan class A (PIGA), a key enzyme involved in the biosynthesis of glycosylphosphatidylinositol anchor of membrane proteins (1, 2). Among the proteins affected in PNH are two membrane complement regulators, decay accelerating factor (DAF, CD55) and CD59 (1). Due to the lack of these proteins on them, affected PNH blood cells are highly susceptible to complement-mediated attack and injury, leading to debilitating symptoms in the patient that include hemolytic anemia, platelet activation, thrombotic attack and stroke(1, 3). Until about a decade ago when a humanized anti-C5 mAb (Eculizumab) was approved and became the standard therapy, PNH was largely untreatable and carried a significant risk of mortality and morbidity(4). Despite the remarkable success of Eculizumab in the clinic, however, up to 20% of PNH patients on the drug remain transfusion-dependent (5), suggesting that Eculizumab is not completely effective in all patients(6).
The major reason for the lack of complete efficacy of Eculizumab in some PNH patients relates to its mode of action and the mechanism of PNH pathogenesis. The absence of DAF, a C3 convertase inhibitor, and CD59, a membrane attack complex (MAC) inhibitor, renders PNH erythrocytes susceptible to both C3 activation and injury by MAC(1). While Eculizumab can effectively block MAC-mediated intravascular hemolysis (IVH), it does not inhibit C3 activation on PNH erythrocytes(1). Indeed, C3 fragment (C3b/iC3b/C3d)-opsonized PNH erythrocytes were detected abundantly in the circulation of patients after Eculizumab treatment(7–9). It is believed that C3 fragment-opsonized erythrocytes have a much shorter half-life due to complement receptor-mediated phagocytosis, a process commonly referred to as extravascular hemolysis (EVH)(10)Thus, while Eculizumab is effective at inhibiting IVH, it does not address the problem of EVH which becomes unmasked after Eculizumab treatment, potentially explaining its lack of complete efficacy in some patients.
In the present study, we have tested the concept of blocking properdin (P) as an alternative and more effective therapeutic strategy for PNH. P is a plasma glycoprotein and a component of the alternative pathway (AP) of complement activation (11). It is the only known positive regulator of AP and works by stabilizing the AP C3 convertase C3bBb (12, 13). Using in vitro hemolytic assays of normal and PNH erythrocytes as surrogate of IVH and a murine model of EVH, we demonstrated that anti-P mAbs can potently inhibit both IVH and EVH mediated by complement. Our findings suggest that anti-P mAbs may offer improved efficacy over Eculizumab, the current standard therapy for PNH.
Materials and Methods
Generation and screening of anti-human properdin mAbs
To generate anti-human P mAbs, properdin knock out (P−/−) mice (14) were each intraperitoneally immunized with 50 μg (in 100 μl PBS) purified human fP (CompTech Inc) emulsified with 100 μl Titermax adjuvant (Sigma). At day 14 and day 21, mice were again immunized with 50 μg purified human fP emulsified with Titermax adjuvant. One week later, the mice were examined for serum anti-fP titer. Mice with an antibody titer of 1:10,000 or higher were used for hybridoma fusion experiments. Two days prior to fusion experiment, mice were injected (i.p) again with 50 μg purified human fP without any adjuvant (in 100 μl PBS). Spleen was harvested and isolated single cells were fused with P3-X63-Ag8.653 myeloma cells (ATCC). Positive clones were selected by human P binding and inhibitory activity in AP complement activation assay. Single clones were grown in culture flasks in Hypoxanthine-Thymidine medium (Sigma-Aldrich) containing Hybridoma-SFM (Life Technologies) and supplemented with FBS (HyClone), gentamicin (Life Technologies), and penicillin/streptomycin (Life Technologies). mAbs were purified from serum-free culture supernatant by Protein G affinity chromatography using the AKTA FPLC system (GE life Sciences). To screen for hP-binding mAbs, Maxisorp plates (Nunc, Roskilde, Denmark) were coated overnight at 4°C with 50 μl human properdin (2 μg/ml). Wells were washed three times in washing buffer (PBS/0.05% Tween 20) and then treated with 200 μl 1% BSA/PBS for 1 h at room temperature (RT). The plates were then incubated with 1:5 diluted hybridoma supernatant (50 μl/well) in 1% BSA/PBS for 1h RT. Wells were washed three times and incubated for 1h at RT with 50 ml HRP-anti-mouse IgG (Sigma-Aldrich; 1:4000) in 1% BSA/PBS. After washing three times wells were incubated at RT for 5 min with 100 μl tetramethylbenzidine peroxidase substrate (BD Pharmingen, San Jose, CA). Reaction was stopped with 50 μl 2 N H2SO4, and OD of the samples was measured at 450 nm.
Production of anti-human C5 mAb
An anti-human C5 mAb (QDC5) bearing same VH/VL sequences as described in Thomas et al (15) and human IgG4 Fc region with S229P mutation (16) was expressed in CHO cells using the pFuse hIgG4/pFuse ClIg hK vectors (InvivoGen, San Diego, CA) and purified by Protein G affinity chromatography using the AKTA FPLC system.
Hemolytic assays
Hemolytic assays of normal human erythrocytes were performed using previously published methods (17, 18). Normal human erythrocytes (Valley Biomedical, Winchester, VA) at 5x106 cells/reaction were added to a solution (100μl final volume) composed of GVB++ buffer (Sigma) supplemented with Mg++(5mM)-EGTA(10mM), 50% normal human serum (NHS) in the presence or absence of 30μM recombinant human fH19-20 (19) and anti-human CD55(cl BRIC216) and CD59 (MEM-43)antibodies (AbD Serotec, final concentration: 7.5 or 10 μg/ml as specified in figures). In assays with anti-properdin antibody (final concentration: 5-20μg/ml as specified in figures), undiluted NHS was incubated with anti-properdin antibody for 30 min at 4°C before use. Reaction mixtures were centrifuged (1500 rpm for 5 min) to pellet non-lysed erythrocytes. Degree of erythrocyte lysis was measured by optical density at 405 nm of an aliquot of the recovered supernatant. Percentage lysis was calculated by normalizing the OD value to that of a completely lysed erythrocyte sample (hypotonic lysis in water, considered as 100%).
For hemolytic assays of PNH erythrocytes, blood was collected from two PNH patients who had never received Eculizumab therapy and from healthy volunteers after informed consent and with approval from the appropriate institutional committee of the Institute of Hematology, Chinese Academy of Medical Sciences and in compliance with the Declaration of Helsinki. Normal human sera (from ABO-compatible healthy donors) were diluted to 50% with GVB++ buffer (0.15mM CaCl2 and 0.5mM MgCl2, from Sigma) and acidified to pH 6.4 (acidified normal human serum: aNHS) and used for assays with fresh PNH red blood cells (5x106 cells per assay in 100 μl final volume). Samples were incubated at 37°C for 30 min, and reaction was stopped by adding 200 μl of cold 20mM EDTA in PBS. In antibody blocking experiments, anti-properdin or anti C5 antibodies were incubated with aNHS for 30 min in 4°C before adding to the PNH red blood cell reaction mixture. Lysis reaction processing and percentage lysis calculation were the same as described above. Non-lysed PNH erythrocytes from these assays were stained with FITC-labelled goat anti-human C3 antibody (MP Biomedicals, Cat# 0855167) (1:100) and analyzed by FACScalibur.
LPS AP assay
Micro titer plates were coated with lipopolysaccharide (Salmonella typhosa LPS, Sigma) (2μg/well) in PBS (phosphate buffered saline) overnight at 4°C. After washing the plated wells with PBST (phosphate buffered saline and 0.05% tween) for 3 times, wells were treated with a blocking buffer (1% BSA in PBS) for 1hr at RT. Normal human serum (NHS) diluted to 50% with GVB++ buffer (Sigma) supplemented with Mg++(5mM)-EGTA(20mM), with or without pre-incubation with anti-P mAbs, were then added to the plated wells (50 μl/well). NHS samples diluted in the same way but containing EDTA (20mM) were used as positive controls of complete inhibition of AP complement activation. Pre-incubation of NHS with anti-human P mAbs was performed at 4°C for 1hr. AP complement activation in plated wells was allowed to proceed for 1 hr at 37°C, and reaction was stopped by addition of cold 10 mM EDTA in PBS (100 μl/well). After washing for 3 times with PBS-T, plated wells were incubated with a HRP-conjugated goat anti-human C3 polyclonal antibody (MP Biomedicals, Cat # 0855237)(1:4000 diluted in blocking buffer) for 1hr at room temperature. Wells were washed 3 times with PBS-T and developed with HRP substrate (100 ml tetramethylbenzidine peroxidasesubstrate (BD Pharmingen, San Jose, CA). After 5 min, reaction was stopped with 2N H2SO4 and plated wells were read at 450 nm in a micro plate reader.
Human properdin ELISA
Micro titer plates were coated with 50 μl of anti-human P mAb clone 8.1 (2μg/ml, generated in house) in PBS overnight at 4°C. After washing the plates with PBS-T (PBS + 0.05% Tween-20) for 3 times, 200 μl blocking buffer (1%BSA in PBS) was added to each well and the plate was incubated for 1hr at room temperature. Afterwards, human P transgenic mouse serum (diluted to 10% with PBS containing 1% BSA and 20mM EDTA) was added to the plate wells (50μl/well) and incubated for 1hr at room temperature. Plates were then washed 3 times with PBS-T and captured human P was detected using a biotinylated goat anti-human P antibody (CompTech, Cat # A239) and standard HRP-based ELISA detection system. NHS and wild-type mouse serum served as a positive and negative control, respectively, in this assay.
Generation of human properdin (hP) transgenic mice
Previous work in our laboratory has shown that human properdin (hP) can function effectively in the mouse AP complement system(14). To generate a properdin “humanized” mouse, we sub-cloned the human properdin cDNA (Nucleotide# NM_002621 (20)) into the eukaryotic expression vector pCAGGS which has been used by others for transgenic studies (21–23). The pCAGGS-hP plasmid was digested with restriction enzymes Sal I and Hind III to release the hP cDNA along with the CMV-IE enhancer, chicken beta-actin promoter and rabbit globin polyA sequence. The DNA was then injected into C57BL/6J mouse zygotes and transferred into pseudopregnant female recipient C57BL/6J mice. Pups were screened using hP-specific primers (5′-ATCAGAG-GCCTGTGACACC-3′ and 5′- CTG CCCTTGTAGCTCCTCA-3′) and tail DNA, as well as by hP ELISA of plasma samples. Human P-positive mice were then bred with P−/− mice to generate hP transgenic mice on P−/− background (P humanized mice). All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
In vivo functional assays of anti-human P and anti-mouse C5 mAbs in properdin humanized mice
To test the efficacy of the anti-hP mAb 19.1 in inhibiting AP complement activity and the efficacy of an anti-mouse C5 mAb (BB5.1) (24) in inhibiting terminal complement activity in vivo, properdin humanized mice were intraperitoneally injected with either 500 μg of 19.1/mouse or an isotype control mAb (MOPC), or 1 mg/mouse of the anti-mouse C5 mAb BB5.1. Serum was collected at different time points and LPS-based AP complement activation assay was performed as described above to check the blocking ability of mAb 19.1 in vivo. Properdin knockout mouse serum was used as controls for this assay. Effect of inhibition was expressed as % of AP complement activity prior to mAb 19.1 injection in the same mice. To assess the efficacy of anti-mouse C5 mAb in inhibiting terminal complement activity, mouse serum was diluted to 10% with GVB++ buffer, mixed 1:1 with similarly diluted 10% C5-deficient human serum and incubated with antibody-sensitized chicken RBC (Rockland biologicals, Cat#R401-0050) (2.5x 106 cells/reaction, final volume 50 μl) at 37°C for 30 min. Antibody-sensitized chicken RBCs were prepared by incubating the cells with a rabbit anti-chicken RBC antibody (150μg/ml, Rockland biologicals, Cat #103-41390) on ice for 1 hr(24) . Percentage of lysis was calculated by normalizing cell-free hemoglobin OD value to that of a completely lysed RBC sample through hypotonic lysis in water.
Extravascular hemolysis assay
Extravascular hemolysis test was performed as previously described(25). Briefly, DAF−/−/Crry−/−/C3−/− mouse erythrocytes (from 150 μl blood for each recipient mouse) were labeled ex vivo with carboxyfluorescein diacetate succinimidyl ester (CFSE) (Molecular Probes) in 1 mL PBS containing 5 μM CFSE. The labeling reaction was carried out at room temperature for 5 minutes, and then cells were washed several times in PBS. CFSE-labeled DAF−/−/Crry−/−/C3−/− erythrocytes were transfused intravenously (via tail vein or retro-orbital route) into hP-transgenic mice pretreated 6 hr before erythrocyte transfusion with 2 mg/mouse of either anti-hP mAb clone 19.1 or an isotype control mAb (MOPC), or 1mg/mouse of the anti-mouse C5 mAb clone BB5.1 (26). Blood was collected from tail vein at 5 min and various subsequent time points as indicated, and the percentage of labeled erythrocytes present was calculated after FACS and normalized to values at 5 minutes in each mouse.
Detection of activated C3 fragment deposited on transfused erythrocytes
For detection of activated C3 fragment deposition in vivo on transfused DAF−/−/Crry−/−/C3−/−mouse erythrocytes, cells were labelled with CellTrace™ Far Red(Cat# C34564 Thermo Fisher Scientific) according to manufacturer’s instructions, and injected intravenously (from 150 μl blood for each recipient mouse, retro-orbital route) into hP-transgenic mice pretreated with either 2mg of anti-hP mAb 19.1 or an isotype control mAb (MOPC). Blood was collected from tail vein at 5 min and 30 min, and erythrocytes were stained with a FITC-conjugated mouse anti-mouse C3/C3b/iC3b mAb (Cedelane Labs, clone 10C7/Cat#CL7631F, used at 5μg/ml). Transfused cells were gated (CellTrace™ Far Red) and analyzed for activated C3 deposition (FITC) by FACScalibur.
Western blot
Proteins were resolved on 4-12% gradient gel under non-reducing conditions and transferred to polyvinylidene difluoride membranes. Properdin was detected with mouse anti-human properdin 19.1 (2 μg/ml) for 1 h, followed by HRP-conjugated rabbit anti-mouse IgG (1:4000 dilution; Sigma)and the ECL chemiluminescent detection system (Amersham Pharmacia, Uppsala, Sweden)
Results
Generation and characterization of function-blocking mouse anti-human P mAbs
We generated mouse anti-human P mAbs by immunizing P knockout mice (P−/−) with recombinant human P protein, followed by standard hybridoma fusion and screening procedures (27). By ELISA-based binding assays, we identified a number of hybridoma clones that secreted mAbs displaying high P-binding property (Fig 1a). Four of these mAbs were shown to have strong function-blocking activities in a P-dependent alternative pathway (AP) complement activation assay using 10% normal human serum (NHS) (Fig 1b). From these clones, we chose mAb 19.1 for subsequent hemolytic experiments. In a Western blot of proteins separated by non-reducing SDS-PAGE gel, mAb 19.1 recognized purified P as well as plasma P in their native form (Fig 1c), and showed dose-dependent inhibition of AP complement activity in 50% NHS, achieving complete AP inhibition at 5 μg/ml (0.03 μM) (Fig 1d). We then tested the inhibitory activity of mAb 19.1 in an induced autologous hemolysis assay. Normal human red blood cells (RBC) are resistant to autologous complement lysis due to the presence of regulators on the cell surface and in the plasma. However, when the activity of such complement regulators is blocked experimentally, autologous complement-mediated hemolysis could occur (19). Fig 1e shows that combined inhibition of DAF on human RBC by a neutralizing mAb and factor H (FH) by a dominant-negative FH fragment (SCR19-20) rendered normal human RBCs susceptible to AP complement lysis in 50% NHS. In this induced hemolytic assay, we found mAb 19.1 to be completely effective at inhibiting human RBC lysis (Fig 1e). Further tests showed that mAb 19.1 also caused concentration-dependent inhibition of normal human RBC lysis when DAF, CD59 and FH were blocked (Fig 1f).
Figure 1. Generation and characterization of anti-human properdin antibodies.
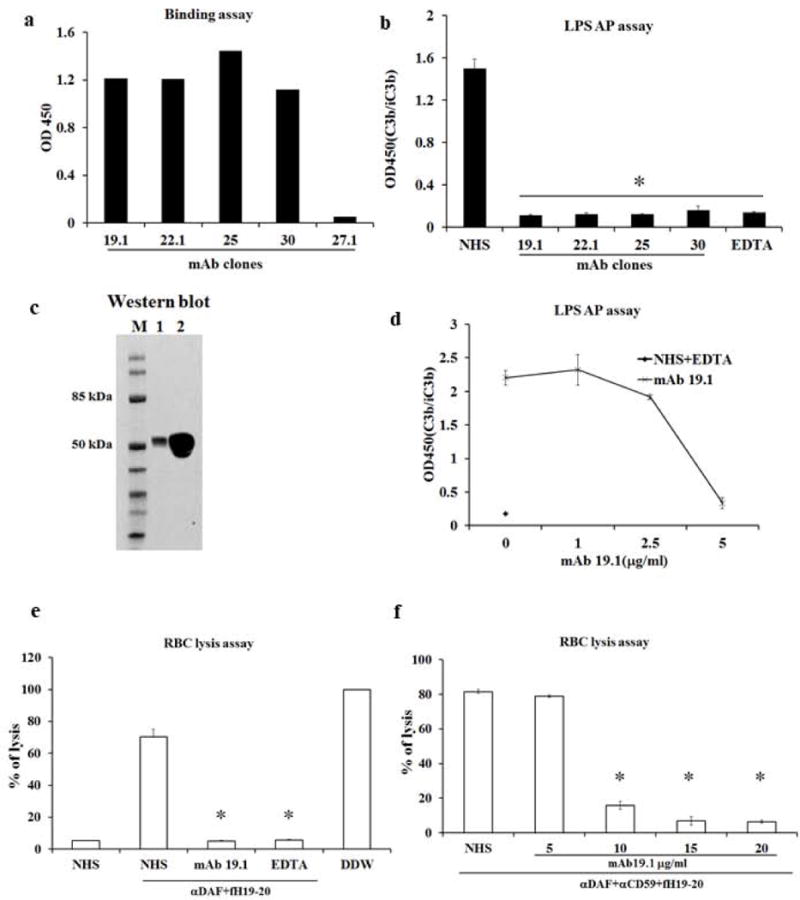
a. Screening of anti-human P mAbs by ELISA. Culture supernatant from 4 hybridoma clones, mAb 19.1, 22.1, 25 and 30, showed strong binding to plate-immobilized hP, where supernatant from a negative mAb clone 27.1 showed no binding. b. Inhibition of LPS-induced AP complement activation by affinity purified mAb 19.1, 22.1, 25 and 30. All 4 clones of mAbs effectively inhibited AP complement activation when added to 10% normal human serum (NHS) at a final concentration of 1μg/ml. A sample with EDTA added (EDTA) served as a positive control for inhibition. A sample with no mAb added (NHS) served as the baseline AP complement activation. OD450 value represents ELISA reading of relative C3 fragments deposition (i.e. activated and plate-bound C3). c. Western blot confirming that mAb clone 19.1 specifically recognizes purified properdin and properdin in NHS. Lane 1: 20 ng of purified human properdin; Lane 2: 1μl of NHS separated on non-reducing SDS-PAGE. mAb 19.1 was used at 2 μg/ml in Western blotting. d. Further functional characterization of mAb clone 19.1 showing it concentration-dependently inhibiting LPS-induced AP complement activity in 50% NHS. NHS was diluted 1:1 (i.e. to 50%) in EGTA-Mg+2 supplemented GVB++ buffer and then pretreated with or without mAb (1, 2.5 and 5μg/ml) for 1hr at 4°C before adding to LPS-coated plates. It completely blocked AP complement activity at 5μg/ml in this assay. EDTA serum (NHS+EDTA) was used as a positive control for complement inhibition. e, f. Human erythrocytes were lysed by 50% NHS in the presence of human factor H SCR19-20 and anti-DAF antibody (7.5 μg/ml, panel e) or anti-DAF and anti-CD59 antibodies (10 μg/ml for both, panel f). Lysis was prevented by EDTA or mAb 19.1 (5 μg/ml for panel e and as indicated in panel f). Assays were performed in triplicates and percent lysis was normalized to hypotonic lysis (100%) with distilled water (DDW). All data are representative of at least three independent experiments and values are expressed as mean (SD) of at least 3 replicate assays per data point. * p<0.001 compared with sample without mAb 19.1 treatment (NHS). One-way ANOVA.
Anti-P mAb prevents acidified serum-induced lysis and activated C3 fragments opsonization of PNH erythrocytes
We next tested the efficacy of mAb 19.1 in preventing acidified serum-induced lysis of PNH RBCs (Ham’s test). RBCs from two PNH patients were used for this experiment. FACS analysis showed that the patient’s RBCs contained more than 50% PNH clones (DAF-negative) (Fig 2a). As shown in Fig 2b, significant lysis of PNH RBCs occurred in 50% acidified human serum, and this lysis was inhibited by QDC5, a recombinant anti-C5 mAb with the same VH/VL sequences as Eculizumab. Notably, the inhibitory effect of the anti-C5 mAb reached a plateau at 30-40 μg/ml (0.2-0.26 μM) and further increase in antibody concentration up to 50 μg/ml (0.33 μM) did not completely eliminate residual hemolysis (Fig 2b). In contrast, the anti-P mAb 19.1 caused near complete inhibition of PNH RBC lysis at 5 μg/ml (0.03 μM) (Fig 2b). As expected, when lysis-resistant PNH RBCs rescued by anti-C5 or anti-P mAb treatment were analyzed by FACS for activated C3 fragments deposition, we found that those from anti-C5 mAb treatment incurred activated C3 fragments opsonization whereas those subjected to anti-P treatment showed no such deposition on their surface (Fig 2c). Together, these results suggested that blocking P at the C3 activation step was more effective at inhibiting PNH RBC lysis than blocking C5 at the terminal step, and that anti-P mAb had a further advantage over anti-C5 mAb in that it could also prevent activated C3 fragments opsonization of PNH RBCs.
Figure 2. Anti-human propedin mAb 19.1 effectively blocks lysis of PNH red blood cells in acidified human serum.
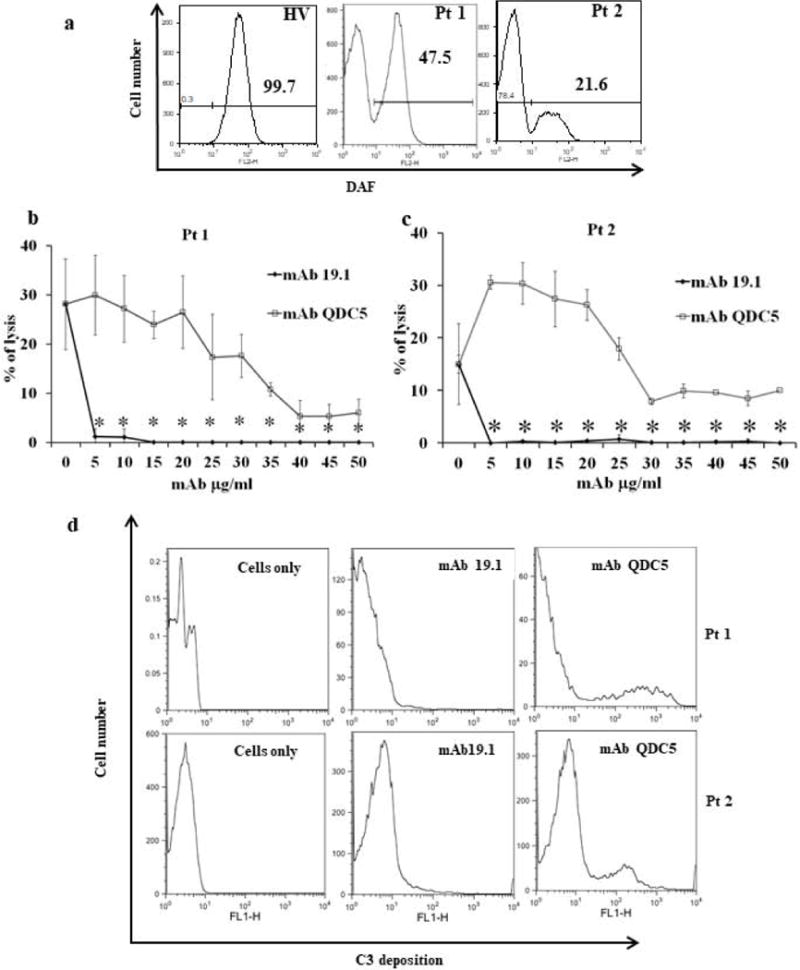
a: FACS analysis of DAF expression on RBCs of a healthy volunteer (HV) and two PNH patients (Pt1, Pt2) showing both patients having >50% of PNH cells. b: Inhibition of acidified human serum-induced lysis of PNH patient’s RBCs by anti-P (19.1) and anti-C5 (QDC5) mAbs. With RBCs from both patients, anti-P mAb 19.1 effectively inhibited lysis at 5 μg/ml (0.03 μM) concentration, whereas anti-C5 mAb (QDC5) concentration-dependently inhibited lysis but it reached a plateau effect around 30-40 μg/ml (0.2-0.26 μM) and failed to completely prevent lysis. Two independent experiments of duplicate assays were performed with similar results. Data from the two experiments were combined and presented as mean (SD). Acidified normal human serum was used at 50%. c: FACS analysis of activated C3 fragments deposition on RBCs from assays in Panel b. In each patient, a sub-population of cells with very high C3 fragments staining was present in anti-C5 mAb-treated but not mAb 19.1-treated sample. A sample of non-treated and non-stained cells from each patient was used as a negative control for FACS analysis. Results shown are representative of two independent analyses. * p< 0.001 Two-way ANOVA.
Anti-P mAb prevents complement-mediated extravascular hemolysis in a humanized mouse model
To test the effectiveness of anti-human P mAb in preventing extravascular hemolysis in vivo, we used a murine model whereby RBCs from DAF, Crry and C3 triple knockout mice, when adoptively transferred into wild-type mouse recipients, were rapidly eliminated from the circulation by an AP complement-dependent extravascular hemolysis mechanism (28). With the use of C3- and C5-deficient mice as recipients, this mouse model was previously established to involve an extravascular rather than an intravascular hemolysis mechanism (26). Because none of our anti-human P mAbs cross reacted with mouse P, we generated a P humanized mouse by firstly creating a human P transgenic mouse (hP-Tg), followed by crossing with the P knockout (P−/−) mouse (14). We constructed a human P transgene plasmid consisting of the human P cDNA under the regulation of chicken β-actin promoter/Cytomegalovirus immediate early (CMV-IE) enhancer and the rabbit β-globin polyadenylation signal sequence (Fig 3a). Injection of the transgenic construct into the pronuclei of fertilized eggs of C57BL6 mice resulted in 5 human P transgenic mouse founders (Fig 3b). ELISA analysis confirmed the presence of human P in the sera of all 5 founder lines (Fig 3c). We then crossed hP-Tg founder line #15 with a P−/− mouse and generated hP-Tg/P−/− mice. Fig 3d shows that, as previously described (14), serum from a P−/− mouse had no LPS-dependent AP complement activity, whereas sera from hP-Tg/P−/− mice displayed AP complement activity similar to that of a WT mouse, suggesting that transgenically expressed human P was fully functional in mice.
Figure 3. Generation and characterization of human properdin transgenic mice.
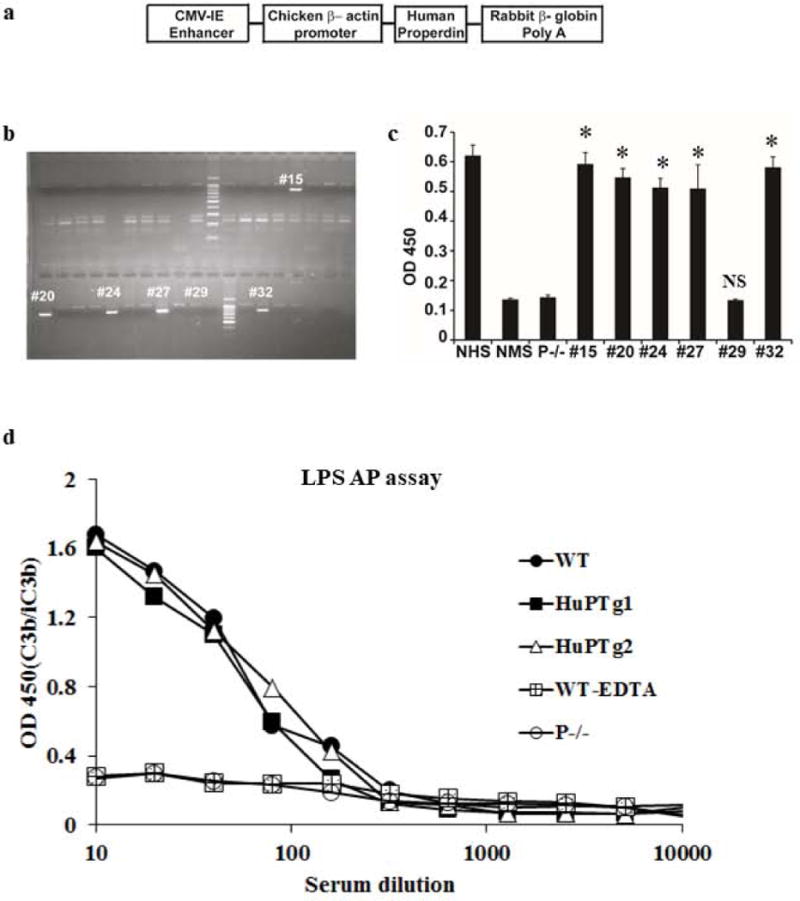
a. Human properdin cDNA construct used for transgenic mouse generation. The cDNA cassette is composed of the chicken β-actin promoter with CVM-IE enhancer, human P cDNA and the rabbit β -globin polyA tail for stable expression of the cDNA in eukaryotic cells. b. Genotyping of hP transgenic mice by PCR using hP-specific primers and tail DNA. Of 40 mice screened, five transgene-positive founders were identified by the presence of a PCR fragment of approximate 800 bp (#15, #20, #24, #27 and #32). c. Human P protein was detected by ELISA in the sera of the 5 transgene-positive mice but not in a transgene-negative mouse (#29). Normal mouse serum (NMS, i.e. serum from wild-type mouse) and serum from P−/− mouse were also negative for hP whereas normal human serum (NHS) was positive for hP. * p<0.0001, NS: non-significant. One-way ANOVA comparing with NMS or P−/− mouse serum. d. Transgenically expressed hP restored AP complement activity to P−/− mice. In contrast to P−/− mouse serum which showed no LPS-dependent AP complement activity, sera from two P−/− mice with hP transgene expression (HuPTg1 and HuPTg2) showed similar AP complement activity to that of a WT mouse. Data in panel c and d are representative of 2-3 independent experiments. Mean (SD) of triplicate assays (c) or average of duplicate assays (d) are shown.
We next used the hP-Tg/P−/− mice to test AP complement-blocking function of the anti-human P mAb 19.1 in vivo. We treated 3 hP-Tg/P−/− mice with mAb 19.1 (0.5 mg/mouse) and assessed AP complement activity in their sera at various time intervals. Fig 4a shows that injection of mAb 19.1 completely inhibited human P function in hP-Tg/P−/− mice for up to 48 hours. Finally, when complement-susceptible RBCs from DAF−/−Crry−/−C3−/− knockout mice were transfused into hP-Tg/P−/− mice treated with an isotype control mAb (MOPC), they were efficiently removed within 5 days (Fig 4c). However, such RBCs persisted in hP-Tg/P−/− mice treated with anti-P mAb 19.1 (Fig 4c). In contrast to the effectiveness of anti-P mAb, an anti-C5 mAb did not prevent the elimination of transfused DAF−/−Crry−/−C3−/− RBCs from hP-Tg/P−/− mice (Fig 4c) despite its demonstrated activity to completely inhibit lytic complement activity in hP-Tg/P−/− mice (Fig 4b). FACS analysis showed significantly higher amounts of activated C3 fragment deposition on RBCs transfused into control mAb-treated mice than those into anti-P mAb-treated mice (Fig 4d). Collectively, these data demonstrated that anti-P mAb, but not anti-C5 mAb, was capable of preventing complement-mediated extravascular hemolysis.
Figure 4. Therapeutic efficacy of anti-human P mAb 19.1 in a murine model of extravascular hemolysis.
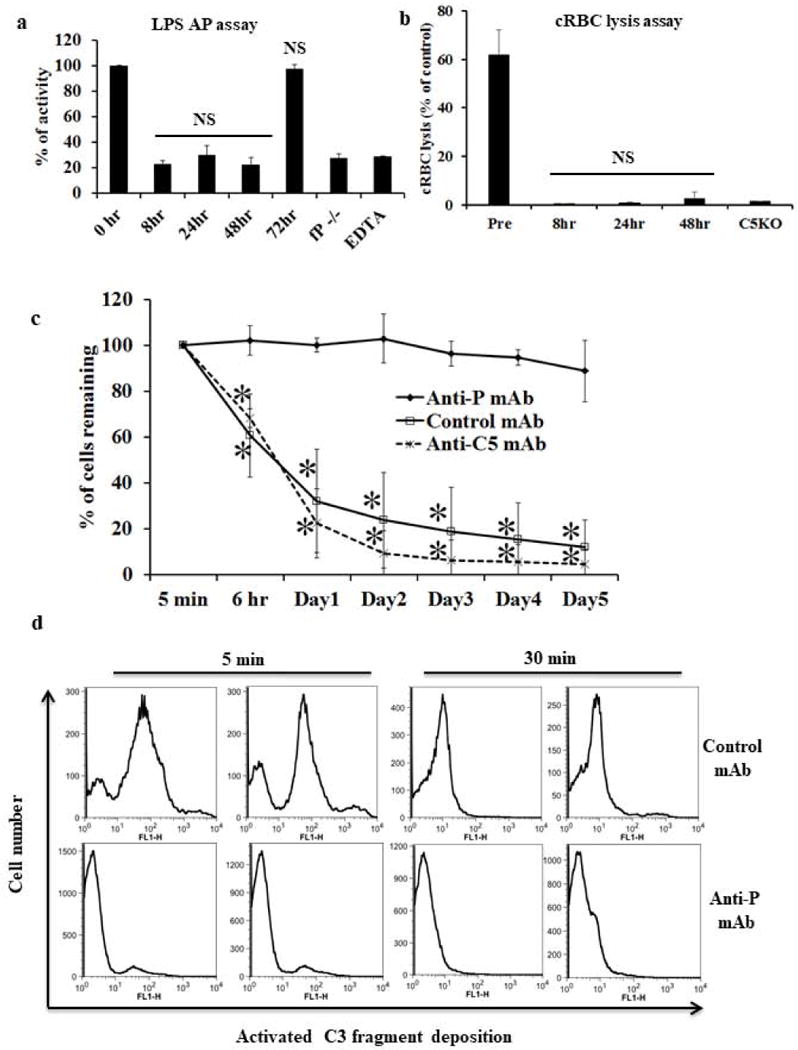
a: Pharmacodynamics of mAb 19.1 in hP transgenic mice. Each mouse was treated with 0.5 mg mAb 19.1 (n=3 mice). Serum samples were collected before and at various time points after mAb treatment and assessed for LPS-dependent AP complement activity. At this dosage of mAb 19.1, AP complement activity was suppressed to background (P−/−) level for 2 days. EDTA: time 0 sample with EDTA (20 mM) added. NS, non-significant comparing 8, 24 and 48 hr samples with fP−/− or EDTA-treated serum, or comparing 72 hr sample with 0 hr sample. One-way ANOVA. b: Pharmacodynamics of anti-mouse C5 mAb (BB5.1) in hP transgenic mice. Each mouse was treated with 1mg of anti-mouse C5 mAb (n=3). Serum samples were collected before and at various time points after mAb treatment and assessed for lytic activity using antibody-sensitized chicken RBCs. C5 knockout (KO) mouse serum was used as a control for C5 inhibition. Percentage of chicken RBC (cRBC) lysis was normalized to a sample completely lysed by hypotonic shock in double distilled water. * p<0.0001, NS: non-significant compared with C5KO serum. One-way ANOVA. c. Effect of anti-hP mAb 19.1 on the survival of transfused CFSC-labeled DAF−/−/Crry−/−/C3−/− mouse erythrocytes in hP- transgenic mice. Recipient mice were treated with anti-hP mAb 19.1 (n=4) or an isotype control mAb (n=4) or anti-mouse C5 mAb (n=3) 6 hours prior to red blood cell transfusion and blood samples were taken at 5 min, 6 hrs and then daily for 5 days. The percentage of CFSC-labeled red blood cells was measured by FACS and normalized to that determined at 5 min (100%). Transfused DAF−/−/Crry−/−/C3−/− mouse red blood cells were rapidly eliminated in control mAb- or anti-C5 mAb-treated hP transgenic mice but such an outcome was prevented by mAb 19.1 treatment. * p< 0.001, Two-way ANOVA. d. FACS analysis of activated C3 fragment deposition on DAF−/−/Crry−/−/C3−/− RBCs 5 and 30 min after their transfusion into control mAb- or mAb 19.1-treated hP-Tg/P−/− mice (representative data from two recipient mice are shown). At both time points, C3 fragment deposition was significantly higher on RBCs transfused into control mAb-treated than mAb 19.1-treated hP-Tg/P−/− mice. The reason for the marked reduction in C3 fragment deposition on RBCs in control mAb-treated hP-Tg/P−/− mice between 5 and 30 min is unknown, but could be caused by C3 fragment degradation to C3d or shedding from the cell surface or rapid removal of the C3-opsonized cells. Data in a-c are presented as mean (SD) with indicated n numbers.
Discussion
In this study, we have generated function-blocking anti-human P mAbs and used them to demonstrate the critical role of P in AP complement-mediated intravascular and extravascular hemolysis. P promotes AP complement activation and is the only known positive regulator of the complement system. Although it has not been considered to be an indispensable component of the AP, recent studies of P−/− mice have revealed a surprisingly critical role of P in several settings of AP complement activation. For example, LPS-induced AP complement activation was completely abrogated in P−/− mouse serum (14), and in several murine models of AP complement-driven diseases, P deficiency significantly attenuated disease severity (27, 29–31). These findings raised the possibility that P may represent an attractive therapeutic target.
Our data in this study has confirmed that P plays a critical role in human AP complement activation (Fig 1) and suggested that anti-P mAbs may be effective in treating human PNH disease. Eculizumab, the current standard therapy for PNH, has clinical limitations due to its inability to prevent extravascular hemolysis mediated by phagocytosis of activated C3 fragment-opsonized PNH erythrocytes. Additionally, in the setting of strong complement activation leading to deposition of high density C3b on the activating surface, an anti-C5 inhibitor such as Eculizumab may be outcompeted by the C5 convertase assembled on the cell surface for C5 binding, making residual C5 cleavage and hemolysis unavoidable (32, 33). Indeed, in an acidified serum-induced hemolytic assay of PNH erythrocytes, we observed that an Eculizumab-like recombinant anti-C5 mAb failed to completely inhibit hemolysis when used at a saturation concentration of 50 μg/ml (0.33 μM) (Fig 2), a finding that is consistent with previous data on Eculizumab activity (32, 33). In striking contrast, an anti-P mAb achieved complete hemolysis inhibition at 5-15 μg/ml (0.03-0.1 μM) (Fig 2). A similar level of efficacy of anti-P mAbs was observed in preventing autologous lysis of normal human red blood cells when DAF or DAF/CD59 and factor H functions were blocked (Fig 1).
The differential potency of anti-P and anti-C5 mAbs in preventing autologous complement-mediated hemolysis is likely related to quantitative as well as mechanistic differences between the two target proteins. The plasma concentration of P has been reported to be in the range of 5-15 μg/ml (0.09-0.27 μM based on monomeric form) whereas that of C5 is approximately 80 μg/ml (0.42 μM) (34). In addition, P works by stabilizing the AP C3 convertase and thus plays a facilitating, and in some cases, critical role in enabling AP complement activation to occur (12, 13). Accordingly, blocking P function would be expected to be more effective as by preventing AP complement activation from occurring it blocks a key initiating point in the cascade and precludes further AP complement amplification and terminal complement activation. On the other hand, given the evidence showing that P plays a less critical role in classical pathway complement activation (14), it may be expected that anti-P mAbs are less effective than anti-C5 mAbs in settings where hemolytic episodes in PNH patients are triggered and mediated by autoantibodies and the classical pathway. Additionally, it remains to be tested clinically whether anti-P mAb therapy would be sufficient to prevent hemolysis in PNH patients in settings where strong AP complement activation is triggered by infections or other inducers leading to severe bystander hemolysis.
As demonstrated in this study, another advantage of anti-P mAb over Eculizumab and other C5-based therapeutic approaches is that it prevents extravascular as well as intravascular hemolysis. It has been shown that, while PNH red blood cells in patients treated with Eculizumab were protected from intravascular lysis and could be detected in the circulation, they were heavily opsonized with activated C3 fragments and were presumably susceptible to complement receptor-mediated extravascular hemolysis(7–9). This mechanism could explain why PNH red blood cells in Eculizumab-treated patients still have abnormally shorter half-life than normal cells and thus the clinical outcome of incomplete response to Eculizumab treatment. We showed here that PNH cells rescued by anti-P mAb did not incur activated C3 fragments deposition whereas those rescued by anti-C5 had significant C3 fragments deposition. Furthermore, using a P humanized mouse model of extravascular hemolysis, we provided direct evidence that blocking P systemically by anti-P mAb, but not by an anti-C5 mAb, prevented complement-mediated extravascular hemolysis (Fig 3, Fig 4).
There are a number of ongoing studies evaluating the efficacy and safety of second generation complement inhibitors including those that target C3 or factor D (35, 36), two other key components of the alternative pathway. While these approaches are expected to be as effective as blocking properdin in preventing intravascular and extravascular hemolysis in PNH, anti-P mAb may be considered a relatively safer approach given that P is not universally required in microbial surface-triggered AP complement activation (14), whereas blocking C3 and factor D would eliminate total and AP complement activity, respectively. This notwithstanding, anti-P mAb therapy may also compromise host defense and increase certain infection risks. Reports of properdin-deficient individuals have described susceptibility to severe but generally non-recurrent Neisseria meningitides infection and to recurrent otitis media and respiratory tract infections in children and younger individuals (37, 38). Thus, as with the use of Eculizumab, anti-P mAb therapy should be preceded by vaccination against Neisseria and accompanied by prophylactic antibiotic therapy if necessary, especially in younger patients.
In summary, we have provided proof of concept data in this study to support the use of anti-P mAb as an improved therapeutic strategy for human PNH. Potential advantages of anti-P mAbs over Eculizumab, the current standard therapy for PNH, include more potent activity in preventing intravascular hemolysis to allow elimination of residual lysis and optimization of delivery frequency, dosage and route, and its ability to prevent extravascular hemolysis by inhibiting C3 activation. Given the evidence that P is not universally required for AP complement activation on microbial surfaces, a therapeutic strategy based on anti-P mAbs may also carry less infection risk in treated individuals as compared with anti-C5 and other complement inhibitors.
Acknowledgments
We thank the Transgenic and Chimeric Mouse Core of the Perelman School of Medicine of University of Pennsylvania for the service it provides in transgenic mouse production.
Footnotes
This work is supported in part by National Institutes of Health grants AI-103965, AI-44970 and AI-085596.
References
- 1.Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nature reviews Disease primers. 2017;3:17028. doi: 10.1038/nrdp.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 3.Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, Gaya A, Coyle L, de Castro C, Fu CL, Maciejewski JP, Bessler M, Kroon HA, Rother RP, Hillmen P. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111:1840–1847. doi: 10.1182/blood-2007-06-094136. [DOI] [PubMed] [Google Scholar]
- 4.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nature biotechnology. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 5.Risitano AM, Notaro R, Luzzatto L, Hill A, Kelly R, Hillmen P. Paroxysmal Nocturnal Hemoglobinuria — Hemolysis before and after Eculizumab. 2010 doi: 10.1056/NEJMc1010351. [DOI] [PubMed]
- 6.Luzzatto L. Recent advances in the pathogenesis and treatment of paroxysmal nocturnal hemoglobinuria. F1000Research. 2016;5 doi: 10.12688/f1000research.7288.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, Ricci P, Alfinito F, Camera A, Gianfaldoni G, Amendola A, Boschetti C, Di Bona E, Fratellanza G, Barbano F, Rodeghiero F, Zanella A, Iori AP, Selleri C, Luzzatto L, Rotoli B. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113:4094–4100. doi: 10.1182/blood-2008-11-189944. [DOI] [PubMed] [Google Scholar]
- 8.Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, Hillmen P. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95:567–573. doi: 10.3324/haematol.2009.007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berzuini A, Montanelli F, Prati D. Hemolytic anemia after eculizumab in paroxysmal nocturnal hemoglobinuria. The New England journal of medicine. 2010;363:993–994. doi: 10.1056/NEJMc1005108. [DOI] [PubMed] [Google Scholar]
- 10.Lin Z, Schmidt CQ, Koutsogiannaki S, Ricci P, Risitano AM, Lambris JD, Ricklin D. Complement C3dg-mediated erythrophagocytosis: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2015;126:891–894. doi: 10.1182/blood-2015-02-625871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Molecular immunology. 2013;56:191–198. doi: 10.1016/j.molimm.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science. 1954;120:279–285. doi: 10.1126/science.120.3112.279. [DOI] [PubMed] [Google Scholar]
- 13.Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. The Journal of experimental medicine. 1975;142:856–863. doi: 10.1084/jem.142.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura Y, Miwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood. 2008;111:732–740. doi: 10.1182/blood-2007-05-089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas TC, Rollins SA, Rother RP, Giannoni MA, Hartman SL, Elliott EA, Nye SH, Matis LA, Squinto SP, Evans MJ. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Molecular immunology. 1996;33:1389–1401. doi: 10.1016/s0161-5890(96)00078-8. [DOI] [PubMed] [Google Scholar]
- 16.Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, Killestein J, Polman CH, Aalberse RC, Schuurman J, van de Winkel JG, Parren PW. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nature biotechnology. 2009;27:767–771. doi: 10.1038/nbt.1553. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol. 2006;177:6308–6316. doi: 10.4049/jimmunol.177.9.6308. [DOI] [PubMed] [Google Scholar]
- 18.Heinen S, Jozsi M, Hartmann A, Noris M, Remuzzi G, Skerka C, Zipfel PF. Hemolytic uremic syndrome: a factor H mutation (E1172Stop) causes defective complement control at the surface of endothelial cells. Journal of the American Society of Nephrology : JASN. 2007;18:506–514. doi: 10.1681/ASN.2006091069. [DOI] [PubMed] [Google Scholar]
- 19.Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Herbert AP, Barlow PN, Eberhardt HU, Skerka C, Zipfel PF, Hamano T, Miwa T, Tung KS, Song WC. Combination of factor H mutation and properdin deficiency causes severe C3 glomerulonephritis. Journal of the American Society of Nephrology : JASN. 2013;24:53–65. doi: 10.1681/ASN.2012060570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nolan KF, Kaluz S, Higgins JM, Goundis D, Reid KB. Characterization of the human properdin gene. The Biochemical journal. 1992;287(Pt 1):291–297. doi: 10.1042/bj2870291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang J, Yamato E, Miyazaki J. Intravenous delivery of naked plasmid DNA for in vivo cytokine expression. Biochemical and biophysical research communications. 2001;289:1088–1092. doi: 10.1006/bbrc.2001.6100. [DOI] [PubMed] [Google Scholar]
- 22.Ray PS, Martin JL, Swanson EA, Otani H, Dillmann WH, Das DK. Transgene overexpression of alphaB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 23.Kameda S, Maruyama H, Higuchi N, Iino N, Nakamura G, Miyazaki J, Gejyo F. Kidney-targeted naked DNA transfer by retrograde injection into the renal vein in mice. Biochemical and biophysical research communications. 2004;314:390–395. doi: 10.1016/j.bbrc.2003.12.107. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim DD, Miwa T, Song WC. Retrovirus-mediated over-expression of decay-accelerating factor rescues Crry-deficient erythrocytes from acute alternative pathway complement attack. J Immunol. 2006;177:5558–5566. doi: 10.4049/jimmunol.177.8.5558. [DOI] [PubMed] [Google Scholar]
- 26.Frei Y, Lambris JD, Stockinger B. Generation of a monoclonal antibody to mouse C5 application in an ELISA assay for detection of anti-C5 antibodies. Molecular and cellular probes. 1987;1:141–149. doi: 10.1016/0890-8508(87)90022-3. [DOI] [PubMed] [Google Scholar]
- 27.Miwa T, Sato S, Gullipalli D, Nangaku M, Song WC. Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol. 2013;190:3552–3559. doi: 10.4049/jimmunol.1202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molina H, Miwa T, Zhou L, Hilliard B, Mastellos D, Maldonado MA, Lambris JD, Song WC. Complement-mediated clearance of erythrocytes: mechanism and delineation of the regulatory roles of Crry and DAF. Decay-accelerating factor. Blood. 2002;100:4544–4549. doi: 10.1182/blood-2002-06-1875. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Miwa T, Ducka-Kokalari B, Redai IG, Sato S, Gullipalli D, Zangrilli JG, Haczku A, Song WC. Properdin Contributes to Allergic Airway Inflammation through Local C3a Generation. J Immunol. 2015;195:1171–1181. doi: 10.4049/jimmunol.1401819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimura Y, Zhou L, Miwa T, Song WC. Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. The Journal of clinical investigation. 2010;120:3545–3554. doi: 10.1172/JCI41782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou HF, Yan H, Stover CM, Fernandez TM, Rodriguez de Cordoba S, Song WC, Wu X, Thompson RW, Schwaeble WJ, Atkinson JP, Hourcade DE, Pham CT. Antibody directs properdin-dependent activation of the complement alternative pathway in a mouse model of abdominal aortic aneurysm. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E415–422. doi: 10.1073/pnas.1119000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harder MJ, Kuhn N, Schrezenmeier H, Hochsmann B, von Zabern I, Weinstock C, Simmet T, Ricklin D, Lambris JD, Skerra A, Anliker M, Schmidt CQ. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129:970–980. doi: 10.1182/blood-2016-08-732800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sica M, Rondelli T, Ricci P, De Angioletti M, Risitano AM, Notaro R. Eculizumab treatment: stochastic occurrence of C3 binding to individual PNH erythrocytes. Journal of hematology & oncology. 2017;10:126. doi: 10.1186/s13045-017-0496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sjoholm AG. Complement components in normal serum and plasma quantitated by electroimmunoassay. Scandinavian journal of immunology. 1975;4:25–30. doi: 10.1111/j.1365-3083.1975.tb02596.x. [DOI] [PubMed] [Google Scholar]
- 35.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nature reviews Drug discovery. 2015;14:857–877. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Risitano AM, Marotta S. Toward complement inhibition 2.0: Next generation anti-complement agents for paroxysmal nocturnal hemoglobinuria. American journal of hematology. 2018 doi: 10.1002/ajh.25016. [DOI] [PubMed] [Google Scholar]
- 37.Schejbel L, Rosenfeldt V, Marquart H, Valerius NH, Garred P. Properdin deficiency associated with recurrent otitis media and pneumonia, and identification of male carrier with Klinefelter syndrome. Clinical immunology (Orlando, Fla) 2009;131:456–462. doi: 10.1016/j.clim.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Fijen CA, van den Bogaard R, Schipper M, Mannens M, Schlesinger M, Nordin FG, Dankert J, Daha MR, Sjoholm AG, Truedsson L, Kuijper EJ. Properdin deficiency: molecular basis and disease association. Molecular immunology. 1999;36:863–867. doi: 10.1016/s0161-5890(99)00107-8. [DOI] [PubMed] [Google Scholar]