

Abstract
Patients with primary serine biosynthetic defects manifest with intellectual disability, microcephaly, ichthyosis, seizures and peripheral neuropathy. The underlying pathogenesis of peripheral neuropathy in these patients has not been elucidated, but could be related to a decrease in the availability of certain classical sphingolipids, or to an increase in atypical sphingolipids. Here, we show that patients with primary serine deficiency have a statistically significant elevation in specific atypical sphingolipids, namely deoxydihydroceramides of 18-22 carbons in length. We also show that patients with aberrant plasma serine and alanine levels secondary to mitochondrial disorders also display peripheral neuropathy along with similar elevations atypical sphingolipids. We hypothesize that the etiology of peripheral neuropathy in patients with primary mitochondrial disorders is related to this elevation of deoxysphingolipids, in turn caused by increased availability of alanine and decreased availability of serine. These findings could have important therapeutic implications for the management of these patients.
Keywords: Serine Biosynthesis Defect, Serine deficiency, Deoxysphingolipids, Deoxydihydroceramide
1. Introduction
Serine is a non-essential amino acid that can be synthesized de novo from 3-phosphoglycerate, a glycolytic and gluconeogenic intermediate. Among the major roles of serine in the cell are to participate in the early biosynthetic steps of sphingolipid metabolism. Herein, we describe the presence of pathological sphingolipid biosynthesis in primary and secondary causes of serine deficiency.
In the first biosynthetic step of serine biosynthesis, the NAD+-dependent enzyme 3-phosphoglycerate dehydrogenase (PHGDH) converts 3-phosphoglycerate into 3-phosphohydroxypyruvate, which in turn is converted to 3-phosphoserine by phosphoserine aminotransferase (PSAT). Then the phosphoserine phosphatase (PSPH) removes the phosphate group from phosphoserine to form L-serine (Figure 1). Primary defects in all three enzymatic reactions of serine biosynthesis have been described. The most severely affected patients present with Neu-Laxova syndrome, a fetal or neonatal lethal condition characterized by severe growth retardation, arthrogryposis, microcephaly, ichthyosis, and dysmorphic facial features including ectropion and eclabion [1,2]. Patients with milder forms of the disease present with acquired microcephaly, developmental delay and intellectual disability of variable severity, ichthyosis, and progressive peripheral neuropathy.
Figure 1. Biosynthesis of serine, sphingolipids and 1-deoxy-sphingolipids.
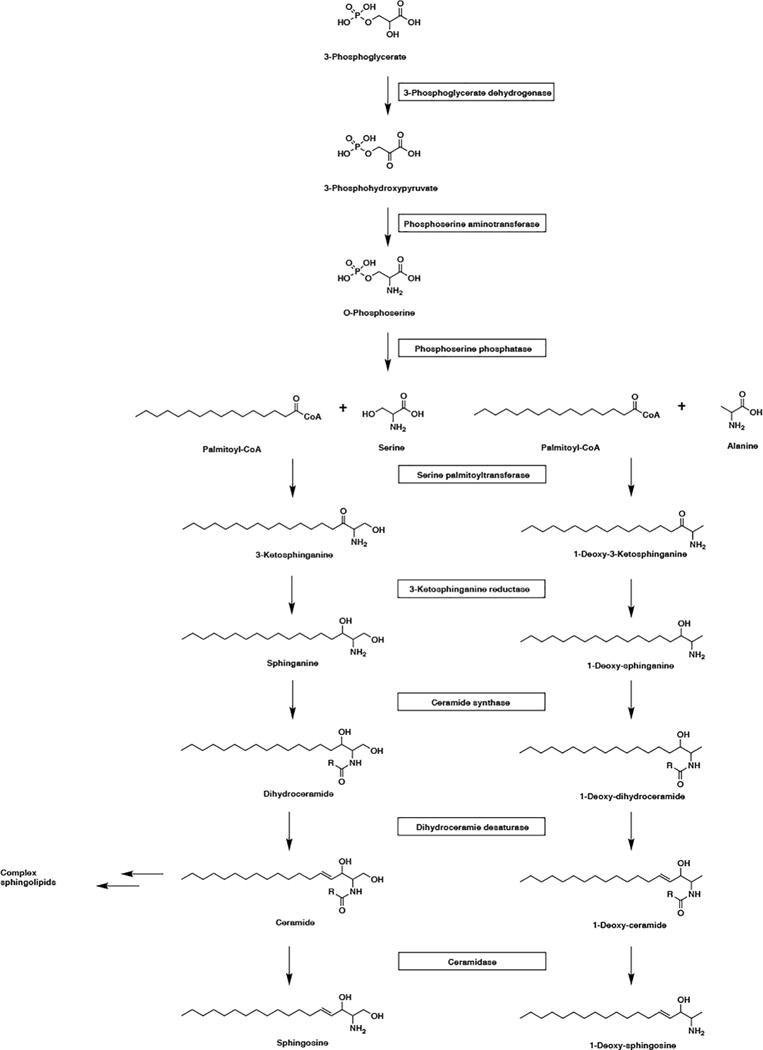
The NAD+-dependent enzyme 3-phosphoglycerate dehydrogenase converts 3-phosphoglycerate into 3-phosphohydroxypyruvate, which in turn is converted to 3-phosphoserine by phosphoserine aminotransferase. The phosphoserine phosphatase (PSPH) dephosphorylates phosphoserine to form L-serine. In classical sphingolipid de novo synthesis, serine palmitoyl transferase (SPT) condenses serine and palmitoyl CoA to synthesize 3-ketosphinganine. Next, 3-keto-dihydrosphingosine is reduced to form dihydrosphingosine by 3-ketosphinganine reductase. Dihydrosphingosine is acylated by ceramide synthase to form dihydroceramide, which is desaturated to give ceramide. Deacylation of ceramide by ceramidase generates sphingosine. The ceramide can be also converted into complex sphingolipids such as sphingomyelin and glycosphingolipids. Atypical sphingolipids are synthesized when SPT uses a non-canonical substrate such as alanine in the first step of sphingolipid synthesis. Following same sequence as classical sphingolipid de novo synthesis, 1-deoxy-ceramide is produced and converted into 1-deoxy-sphingosine. Due to lack of 1-hydroxyl group, no analogs of complex sphingolipids can be synthesized from 1-deoxy-ceramide.
The pathogenesis of the aforementioned neurological manifestations is not fully understood. One possibility is limited serine substrate for the rate-limiting enzyme of sphingolipid biosynthesis, serine palmitoyltransferase (SPT) leading to decreased total sphingolipids. Indeed, Phgdh knockout mice show decreased brain levels of serine-derived lipids, such as phosphatidylserine (PS), sphingomyelin, and ganglioside GD3 [3,4]. Moreover, Glinton et al. recently described reduced levels of various phospholipid species including glycerophosphoethanolamine, glycerophosphocholine, and sphingomyelin in individuals with untreated primary serine deficiency [5]. They found that these deficiencies normalized with serine and glycine supplementation.
A second possibility is that elevation of atypical sphingolipids, in addition to deficiency of classical sphingolipids, contributes to the neurological manifestations of serine synthetic disorders. Atypical sphingolipids are synthesized when SPT uses a non-canonical substrate–such as alanine or glycine–instead of serine in the key step of sphingolipid synthesis (Figure 1). SPT is an enzymatic complex composed of subunits encoded by the ubiquitously expressed SPTLC1 and SPTLC2, and in some tissues–but not the brain–also SPTLC3. Mutations in STPLC1 and SPTLC2 lead to hereditary sensory and autonomic neuropathy types IA and IC (HSAN1A and HSAN1C), respectively [6,7].
In support of this mechanism, it has been shown that the neuropathy in SPTLC1 mutant mice is caused by accumulation of neurotoxic 1-deoxysphingolipids, and that supplementation with oral L-serine reduces this toxic accumulation [8]. Moreover, the deprivation of exogenous L-serine in mouse embryonic fibroblasts lacking PHGDH leads to the accumulation of cytotoxic 1-deoxysphingolipids, the most abundant of which is 1-deoxydihydroceramide (DoxDHCer) [9]. This accumulation was normalized after the addition of L-serine to the medium.
Variable availability of substrates for SPT, including serine and alanine, is not limited only to primary disorders of serine biosynthesis, and can be seen in primary mitochondrial disease (PMD). This is particularly relevant because peripheral neuropathy is an important feature in many affected patients [10]. In addition to elevation of alanine, a common amino acid finding in PMD caused by increased transamination from pyruvate [11], we have observed persistently low plasma serine levels in a cohort of individuals with primary mitochondrial disease with peripheral neuropathy. We considered that this low serine and elevated alanine could lead to aberrant SPT substrate utilization, and production of atypical sphingolipids, with resultant peripheral neuropathic disturbances.
In this study we compare serine, sphingolipids, and 1-deoxysphingolipids in plasma from patients with primary serine deficiency (PSD) and PMD with peripheral neuropathy to control subjects. We show, for the first time, that patients with both primary and secondary forms of serine biosynthetic abnormalities exhibit elevation of atypical sphingolipids in the form of DoxDHCer. We also show a highly significant correlation between the plasma alanine/serine ratio and levels of atypical sphingolipids in PMD. These atypical sphingolipids likely contribute to the pathogenesis of neuropathy in the setting of functional serine deficiency.
2. Materials and methods
2.1 Patients
Two patients with primary serine synthetic defects were evaluated at the National Institutes of Health Clinical Center after enrollment under protocol #76-HG-0238, “Diagnosis and Treatment of Patients with Inborn Errors of Metabolism or Other Genetic Disorders,” approved by the National Human Genome Research Institute (NHGRI) Institutional Review Board (IRB). Patients or their guardians gave written, informed consent. PSD patient 1, enrolled in the NIH Undiagnosed Diseases Program [12–14], had PSAT deficiency, while PSD patient 2 (previously reported [15]) has PSPH deficiency. Plasma samples were obtained when the patients were not receiving serine or glycine supplementation.
Plasma samples from individuals with molecularly confirmed PMD and from normal controls (individuals without a known inborn error of metabolism or other genetic condition) were obtained from the Kennedy Krieger Biochemical Genetics Laboratory under IRB study NA_00069372, Metabolic Analysis of Archived Biofluids (PI. H. Vernon). All samples were drawn at 4-5 hours fasting.
We identified plasma samples from 6 individuals with molecularly confirmed primary mitochondrial disease (3 with MERRF, 1 with autosomal dominant POLG related mtDNA deletion disorder, 1 with LHON, and 1 with MELAS), and for 4 of these individuals we were able to obtain two independently drawn plasma samples from different dates (Table 1). The 3 MERRF patients and the POLG patient had significant sensory neuropathy of their lower extremities diagnosed by physical exam and/or consistent nerve conduction studies. The patients with LHON and MELAS had no features of peripheral neuropathy based on history or physical exam, though nerve conductions studies were not performed.
Table 1.
Deoxydihydroceramides in patients with PSDs and controls.
| Patient | ||
|---|---|---|
| DoxDHCer(m18:0) | DoxDHCer(m20:0) | DoxDHCer(m22:0) |
| 0.37962 | 0.32331 | 0.64632 |
| 0.81978 | 0.72654 | 1.23255 |
| Controls | ||
| DoxDHCer(m18:0) | DoxDHCer(m20:0) | DoxDHCer(m22:0) |
| 0.12822 | 0.19015 | 0.40674 |
| 0.10687 | 0.18399 | 0.40536 |
| 0.06363 | 0.09508 | 0.20939 |
| 0.04315 | 0.11518 | 0.30621 |
| 0.06606 | 0.15612 | 0.41883 |
| 0.09674 | 0.16519 | 0.36892 |
| 0.00338 | 0.07192 | 0.23018 |
| 0.09916 | 0.18439 | 0.51814 |
| 0.14335 | 0.15616 | 0.31697 |
| 0.08866 | 0.15725 | 0.34316 |
| 0.08177 | 0.18487 | 0.41326 |
| 0.26016 | 0.29861 | 0.47123 |
Ages of controls ranged from 12-76 years (mean 35.7 years +/− 24 SD), with 4 males and 7 females. Ages of patients ranged from 12-72 years (mean 36.5 years +/− 25 SD), with 2 males and 4 females. Patients’ specific mutations and characteristics can be found in supplementary table 1.
2.2 Plasma amino acid analysis
Amino acids were quantified via ion exchange chromatography. Plasma was de-proteinized, centrifuged, and the supernatant used for further analysis. Quantification of amino acids was conducted via ion-exchange liquid chromatography with ninhydrin detection on a Biochrom 30 Amino Acid Analyzer.
2.3 Sphingolipid/ceramide analysis
For the 12 controls and the patients with PSD, lipids were extracted with protein precipitation with methanol in the presence of internal standards. The d31-SM(16:0) was used as internal standard for SM. Cer(17:0) was the internal standard for Cer, DHCer, DoxCer, and DoxDHCer; PS(28:0) were internal standards for PS; d5-GluC(18:0) was internal standard for GluC; d3-LC(16:0) was internal standard for LC; C17 sphingosine-1-phosphate as internal standard for sphingosine, sphinganine, S1P, 1-deoxysphingosine, and 1-deoxysphinganine. The lipids were separated by column-switching high-performance liquid chromatography (HPLC) and monitored by multiple-reaction monitoring (MRM) detection on an Applied Biosystems Sciex 4000QTRAP tandem mass spectrometer (MS/MS) equipped with an electrospray ion source. A quality control (QC) sample was prepared from pooled study plasma samples and injected every five study samples to monitor the LC-MS/MS assay performance.
The results of the QC samples provided the basis for accepting or rejecting the data. Only the lipid species in QC samples with coefficient of variation of less than 20% are reported. Since at this stage this was a pilot project screening for potential biomarkers, no absolute quantification method was developed for these lipids, but we used instead a relative quantification method. The DoxDHCer(m18:0), DoxDHCer(m20:0), and DoxDHCer(m22:0) data are expressed as peak area ratios of these deoxydihydroceramides to their internal standard (Cer(17:0)). Unpaired Student’s t-test was performed with Prism version 6.0c (Graphpad Software Inc, La Jolla, CA). Statistical significance was assigned to a two-tailed p-value <0.05.
For the patients with PMD and peripheral neuropathy, ceramides and sphingoid bases were extracted from plasma according to the method described by Bligh and Dyer [16]. The lower phase, containing both ceramides and sphingoid bases, was dried and the residue was dissolved in sodium hydroxide in methanol. Samples were either deacylated for analysis of ceramides or not for analysis of sphingoid bases. Next, internal standards were added to each sample (d7-sphingosine, d7-sphinganine and d3-deoxysphinganine) and samples were dried and dissolved in butanol and water. Samples were analyzed by LC-MS/MS. Levels of ceramides and sphingoid bases were calculated using calibration lines, according to the internal standard ratio method. Sphingoid bases were separated by RP-UPLC using an Acquity I-Class UPLC with BEH C18 column, 2.1 × 50 mm with 1.7-μm particle size (Waters Corporation, Milford, MA, USA) and detected by electron spray ionization in positive ionization mode (ESI+) using a MS/MS-instrument (Xevo TQ MS, Waters Corporation) in multiple reaction monitoring (MRM) mode. Levels of the biomarkers were calculated using calibration curves within the appropriate concentration range, according to the internal standard ratio method.
It is worth noting that the ceramides and sphingoids in PMD and PSD patients were detected using different sample preparation and extraction methodologies. In the PSD patients, the ceramide and spingoid base species were measured intact, thus allowing for measurement of individual ceramide species. In the PMD patients, the DoxDHCer(m18:0) and Cer(d18:1) species of different acyl chain lengths were deacylated to Dox-Sphinganine(m18:0) and Sphingosine(d18:1) respectively, and measured as a sum total of the bases (DoxSO(m18:1) was measured in the non-de-acylated sample). However, here we have shown the validity of both methodologies in detecting relevant variant ceramides in these patient groups.
3. Results
3.1 Sphingolipids in PSD
The values of DoxDHCer (m18:0), (m20:0) and (m22:0) were significantly elevated compared to controls (Figure 2). For DoxDHCer(m18:0), the mean peak ratio for controls was 0.0984 ± 0.018 vs 0.5997 ± 0.220 for patients (P-value <0.0001); for DoxDHCer(m20:0), the control mean was 0.1632 ± 0.016 vs 0.5249 ± 0.202 in patients (P-value 0.0004); for DoxDHCer(m22:0), the control mean was 0.3674 ± 0.026 vs 0.9394 ± 0.293 for affected patients (P-value 0.0003) (figure 3). The specific peak area ratios of the different DoxDHCer are provided in table 1.
Figure 2. Deoxysphingolipids in patients with PSDs.

DoxDHCer species as measured by peak area ratio compared to internal standard were significantly elevated in individuals with PSD (n=2) compared to controls (n=12). a) DoxDHCer (m18:0), b) DoxDHCer (m20:0) c) DoxDHCer (m22:0). DoxDhCer, deoxydihydroceramide.
Figure 3. Deoxysphingolipids in patients with PMDs.
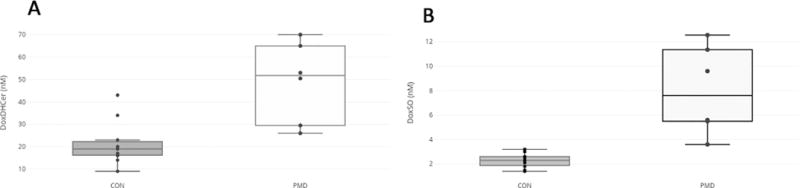
Total DoxDHCer (a) and Total DoxSO (b) were significantly elevated in individuals with PMD (n=6) compared to controls (n=11). DoxDhCer, deoxydihydroceramide; DoxSO, deoxysphingoids.
3.2 Plasma amino acids in PMDs
Plasma amino acids were compared between the PMD cohort and the control cohort (supplementary table 1). For patients having two independent samples measured, the results were averaged prior to cohort comparisons. The results from the LHON patient were removed from the comparison analysis, since the serine level was a significant outlier for the PMD the population. Full plasma amino acid profiles can be found in Supplementary table 2.
The mean plasma serine level was significantly different between the PMD and control cohort (71.5 umol/L +/−22 SD vs 106.9 umol/L +/−18 SD, respectively), with a P-value of 0.017. Alanine was significantly different between the PMD and control cohort (444.2 umol/L +/− 55 SD vs. 361.2 umol/L +/− 59 SD, respectively), with a P-value of 0.025. Serine/Alanine ratios were significantly increased in the PMD population compared to controls (6.12 +/− 2.8 and for the control cohort was 3.41 +/− 0.5, respectively), with a P-value of 0.038).
3.3 Sphingolipids in PMDs
We compared DoxDHCer(m18:0), deoxysphingosine (DoxSO)(m18:1), and the ratios of these metabolite classes to ceramide(d18:1) (DoxDHCer(m18:0)/Cer(d18:1), DoxSO(m18:1)/Cer(d18:1)) between PMD patients and controls (Table 2). DoxDHCer(m18:0) was significantly elevated in PMD patients compared to controls (49.7nM+/−17 SD vs. 21.2nM+/−10 SD, (P-value 0.00043)) (Figure 3). DoxSO(m18:1) was also significantly elevated in PMD patients compared to controls (8.1uM+/−4 SD vs. 2.3nM+/−0.6 SD (P-value 0.0011)) (Figure 3). Both ratios were also elevated in PMD patients compared to controls (DoxDHCer(m18:0)/Cer(d18:1): 0.012+/−004 vs. 0.003+/− 0.002, P-value 0.00001, and DoxSO(m18:1)/Cer(d18:1): 0.002+/−0.001vs/ 0.00035+/− 0.0001 (P-value 0.00092)).
Table 2.
Deoxysphingolipids in patients with PMDs and controls.
| PMD Patients | ||||
|---|---|---|---|---|
| DoxDHCer (nM) | DoxSO (nM) | DoxDHCer/Cer | DoxSO/Cer | |
| Patient 1, Sample 1 | 68 | 9.1 | 0.0122 | 0.0016 |
| Patient 1, Sample 2 | 72 | 13.6 | 0.0136 | 0.0026 |
| Patient 2 | 65 | 9.6 | 0.0133 | 0.002 |
| Patient 3, Sample 1 | 40 | 4.4 | 0.01 | 0.0011 |
| Patient 3, Sample 2 | 61 | 6.6 | 0.0178 | 0.0019 |
| Patient 4, Sample 1 | 56 | 4.4 | 0.0157 | 0.0012 |
| Patient 4, Sample 2 | 50 | 2.8 | 0.0144 | 0.0008 |
| Patient 5, Sample 1 | 28 | 11.4 | 0.0085 | 0.0035 |
| Patient 5, Sample 2 | 31 | 13.7 | 0.0094 | 0.0042 |
| Patient 6 | 26 | 5.6 | 0.0065 | 0.0014 |
| Controls | ||||
| DoxDHCer (nM) | DoxSO (nM) | DoxDHCer/Cer | DoxSO/Cer | |
| 14 | 2.5 | 0.002 | 0.0004 | |
| 17 | 2.1 | 0.0019 | 0.0002 | |
| 19 | 3.2 | 0.0033 | 0.0006 | |
| 16 | 3 | 0.0021 | 0.0004 | |
| 9 | 1.5 | 0.0011 | 0.0002 | |
| 43 | 1.8 | 0.0083 | 0.0003 | |
| 34 | 2.6 | 0.0048 | 0.0004 | |
| 20 | 1.4 | 0.0043 | 0.0003 | |
| 19 | 2.6 | 0.0029 | 0.0004 | |
| 19 | 2.1 | 0.0027 | 0.0003 | |
| 23 | 2.3 | 0.0038 | 0.0004 | |
When comparing the plasma serine/alanine ratio to the plasma DoxDHCer (m.18:0) in the PMD patients, there is a strong correlation with DoxDHCer (m.18:0) (R2 = 0.74, figure 4). When comparing the plasma serine/alanine ratio with DoxSO, there is not a significant direct correlation between the two values (R2 = 0.29); however, there is a clear clustering of PMD patients versus controls (Figure 4).
Figure 4. Correlation between plasma amino acids and deoxysphingolipids in patients with PMDs.
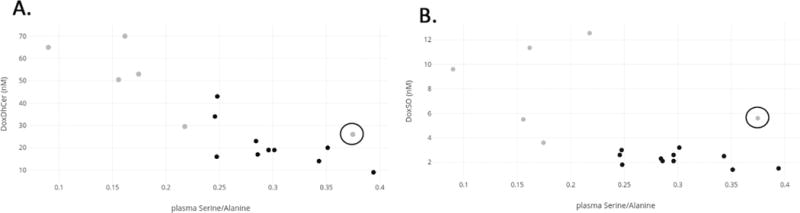
a) Strong correlation between plasma serine/alanine ratio and DoxDHCer in PMD patients (light gray) vs controls (dark gray) (R2 = 0.74). b) No significant direct correlation between plasma serine/alanine ratio and DoxSO (R2 = 0.29), although there is clear clustering of PMD patients (light gray) versus controls (dark gray). DoxDhCer, deoxydihydroceramide; DoxSO, deoxysphingoids. The PMD data points differentiated by a circle around represent the patient with LHON plus (patient 6) who does not manifest peripheral neuropathy and who has an above average plasma serine level of 142 uM.
4. Discussion
Dysregulation in both sphingolipid biosynthesis and breakdown have been implicated in the etiology of various peripheral neuropathies. This includes the biosynthetic sphingolipid defects of SPT in HSAN1 leading to accumulation of deoxysphingolipids. More recently, defects in sphingosine 1-phosphate lyase (SPL), which catalyzes the final step of sphingolipid catabolism, have been shown to underlie an autosomal recessive form of Charcot-Marie-Tooth neuropathy [17]. This enzymatic defect leads to elevations in sphingosine 1-phosphate and in the sphingosine/sphinganine ratio.
We have shown, for the first time, that abnormal deoxysphingolipids are elevated in the plasma of individuals with both primary and secondary causes of abnormal serine availability. We hypothesize that this is due to a mechanism involving alternative sphingolipid metabolism similar to that seen in the SPTLC abnormalities, HSAN1A and HSAN1C. Specifically, that mechanism consists of alternative amino acid substrate utilization (alanine vs. serine) by serine palmitoyltransferase in sphingolipid synthesis. In the situations presented here, however, this alternative substrate utilization is due to substrate availability rather than abnormal enzyme function.
For PSD patients, abnormal deoxysphingolipids levels are probably due to straightforward serine availability for SPT, whereas the etiology of these abnormal deoxysphingolipids in PMD may be more complex. The strong association between the serine/alanine ratio and DoxDHCer identified in this study suggests that availability of both amino acid substrates is relevant. There are several possible factors contributing to the abnormal amino acid availability for SPT and the consequent production of DoxSO in PMD. Firstly, elevated alanine, resulting from increased transamination from elevated pyruvate, is commonly seen in PMD [11]. The potential reasons for decreased serine availability are less straightforward. One possible explanation for serine deficiency in PMD could be a functional deficiency of the NAD+-dependent first enzyme of serine biosynthesis, 3-phosphoglycerate dehydrogenase, due to deficiency of NAD+ [18,19]. Another possible explanation could be potential shunting of serine away from sphingolipid metabolism, and towards stress response pathways of glutathione synthesis and one-carbon metabolism; these pathways are upregulated in multiple models of primary mitochondrial disease including the Deletor mouse model of mtDNA deletions [20,21].
It is not yet clear if all PMD patients are at risk for functional serine deficiency, or if this occurs in only a subset of PMD patients. Of the cases presented here, most PMD patients had abnormally low plasma serine, with some in the extreme low range seen in PSD. Retrospective studies of PMD cohorts suggest that peripheral neuropathy is common, and potentially undiagnosed [10]. While further functional studies are necessary to determine the precise cause of the serine deficiency in individuals with mitochondrial disease, the presence of these abnormal sphingolipids has the potential to serve not only as a biomarker for mitochondrial disease, but also potentially as a biomarker for those with PMD who are at risk for peripheral neuropathy. Further studies may also reveal whether serine supplementation could lead to prevention of peripheral neuropathy in PMD or improvement in clinical status of those affected by peripheral neuropathy, as it has been shown to do in PSD and HSAN1.
Supplementary Material
Highlights.
Atypical sphingolipids are elevated primary serine deficiency disorders
Atypical sphingolipids are elevated in some cases of primary mitochondrial disease
This may be due to aberrant amino acid substrates for serine palmitoyltransferase
Acknowledgments
The authors wish to thank the patients and their families for their cooperation and support. This study is supported in part by the National Institutes of Health Intramural Research Program of the National Human Genome Research Institute and the Common Fund, Office of the Director; the NIH grants 5RO1-N5021328-030 and RO1OD010944-05.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
none
References
- 1.Shaheen R, Rahbeeni Z, Alhashem A, Faqeih E, Zhao Q, Xiong Y, Almoisheer A, Al-Qattan SM, Almadani HA, Al-Onazi N, Al-Baqawi BS, Saleh MA, Alkuraya FS. Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am J Hum Genet. 2014;94:898–904. doi: 10.1016/j.ajhg.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acuna-Hidalgo R, Schanze D, Kariminejad A, Nordgren A, Kariminejad MH, Conner P, Grigelioniene G, Nilsson D, Nordenskjöld M, Wedell A, Freyer C, Wredenberg A, Wieczorek D, Gillessen-Kaesbach G, Kayserili H, Elcioglu N, Ghaderi-Sohi S, Goodarzi P, Setayesh H, van de Vorst M, Steehouwer M, Pfundt R, Krabichler B, Curry C, MacKenzie MG, Boycott KM, Gilissen C, Janecke AR, Hoischen A, Zenker M. Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am J Hum Genet. 2014;95:285–293. doi: 10.1016/j.ajhg.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida K, Furuya S, Osuka S, Mitoma J, Shinoda Y, Watanabe M, Azuma N, Tanaka H, Hashikawa T, Itohara S, Hirabayashi Y. Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J Biol Chem. 2004;279:3573–3577. doi: 10.1074/jbc.C300507200. [DOI] [PubMed] [Google Scholar]
- 4.Hirabayashi Y, Furuya S. Roles of l-serine and sphingolipid synthesis in brain development and neuronal survival. Prog Lipid Res. 2008;47:188–203. doi: 10.1016/j.plipres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Glinton KE, Benke PJ, Lines MA, Geraghty MT, Chakraborty P, Al-Dirbashi OY, Jiang Y, Kennedy AD, Grotewiel MS, Sutton VR, Elsea SH, El-Hattab AW. Disturbed phospholipid metabolism in serine biosynthesis defects revealed by metabolomic profiling. Mol Genet Metab. 2018;123:309–316. doi: 10.1016/j.ymgme.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. 2001;27:309–312. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 7.Rotthier A, Auer-Grumbach M, Janssens K, Baets J, Penno A, Almeida-Souza L, Van Hoof K, Jacobs A, De Vriendt E, Schlotter-Weigel B, Löscher W, Vondráček P, Seeman P, De Jonghe P, Van Dijck P, Jordanova A, Hornemann T, Timmerman V. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am J Hum Genet. 2010;87:513–522. doi: 10.1016/j.ajhg.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garofalo K, Penno A, Schmidt BP, Lee HJ, Frosch MP, von Eckardstein A, Brown RH, Hornemann T, Eichler FS. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest. 2011;121:4735–4745. doi: 10.1172/JCI57549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esaki K, Sayano T, Sonoda C, Akagi T, Suzuki T, Ogawa T, Okamoto M, Yoshikawa T, Hirabayashi Y, Furuya S. L-Serine Deficiency Elicits Intracellular Accumulation of Cytotoxic Deoxysphingolipids and Lipid Body Formation. J Biol Chem. 2015;290:14595–14609. doi: 10.1074/jbc.M114.603860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luigetti M, Sauchelli D, Primiano G, Cuccagna C, Bernardo D, Lo Monaco M, Servidei S. Peripheral neuropathy is a common manifestation of mitochondrial diseases: a single-centre experience. Eur J Neurol. 2016;23:1020–1027. doi: 10.1111/ene.12954. [DOI] [PubMed] [Google Scholar]
- 11.Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Gahl WA, Tifft CJ. The NIH Undiagnosed Diseases Program: lessons learned. JAMA. 2011;305:1904–1905. doi: 10.1001/jama.2011.613. [DOI] [PubMed] [Google Scholar]
- 13.Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DMD, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med Off J Am Coll Med Genet. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, Adams DR, Tifft CJ. UDN, The NIH Undiagnosed Diseases Program and Network: Applications to modern medicine. Mol Genet Metab. 2016;117:393–400. doi: 10.1016/j.ymgme.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byers HM, Bennett RL, Malouf EA, Weiss MD, Feng J, Scott CR, Jayadev S. Novel Report of Phosphoserine Phosphatase Deficiency in an Adult with Myeloneuropathy and Limb Contractures. JIMD Rep. 2016;30:103–108. doi: 10.1007/8904_2015_510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 17.Atkinson D, Nikodinovic Glumac J, Asselbergh B, Ermanoska B, Blocquel D, Steiner R, Estrada-Cuzcano A, Peeters K, Ooms T, De Vriendt E, Yang X-L, Hornemann T, Milic Rasic V, Jordanova A. Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy. Neurology. 2017;88:533–542. doi: 10.1212/WNL.0000000000003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, Goessling W, Regev A, Carr SA, Clish CB, Mootha VK. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. ELife. 2016;5 doi: 10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikkanen J, Forsström S, Euro L, Paetau I, Kohnz RA, Wang L, Chilov D, Viinamäki J, Roivainen A, Marjamäki P, Liljenbäck H, Ahola S, Buzkova J, Terzioglu M, Khan NA, Pirnes-Karhu S, Paetau A, Lönnqvist T, Sajantila A, Isohanni P, Tyynismaa H, Nomura DK, Battersby BJ, Velagapudi V, Carroll CJ, Suomalainen A. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016;23:635–648. doi: 10.1016/j.cmet.2016.01.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.