

Abstract
Central tolerance checkpoints are critical for the elimination of autoreactive B cells and the prevention of autoimmunity. When autoreactive B cells encounter their antigen at the immature B cell stage, BCR crosslinking induces receptor editing, followed by apoptosis if edited cells remain autoreactive. While the transcription factor Foxo1 is known to promote receptor editing, the role of the related factor Foxo3 in central B cell tolerance is poorly understood. We find that BCR-stimulated immature B cells from Foxo3-deficient mice demonstrate reduced apoptosis compared to wild type cells. Despite this, Foxo3−/− mice do not develop increased autoantibodies. This suggests that the increased survival of Foxo3−/− immature B cells allows additional rounds of receptor editing, resulting in more cells “redeeming” themselves by becoming non-autoreactive. Indeed, increased Igλ usage and increased RS recombination among Igλ-expressing cells were observed in Foxo3−/− mice, indicative of increased receptor editing. We also observed that deletion of high affinity autoreactive cells was intact in the absence of Foxo3 in the anti-hen egg lysozyme (HEL)/mHEL model. However, Foxo3 levels in B cells from Systemic Lupus Erythematosus (SLE) patients were inversely correlated with disease activity and reduced in patients with elevated anti-dsDNA antibodies. While this is likely due in part to increased B cell activation in these SLE patients, it is also possible that low affinity B cells that remain autoreactive after editing may survive inappropriately in the absence of Foxo3 and become activated to secrete autoantibodies in the context of other SLE-associated defects.
Introduction
The development of a diverse B cell repertoire is crucial for normal humoral immune responses. However, this diversity comes at a price, as many of the B cells generated in the bone marrow express B cell receptors (BCRs) that recognize self-antigens. Failure of tolerance checkpoints that eliminate or inactivate these autoreactive B cells can lead to autoimmune diseases such as Systemic Lupus Erythematosus (SLE), in which autoantibodies are produced and form immune complexes that induce inflammation and tissue damage. At the immature B cell stage of development, the BCR is first fully assembled and tested for functionality. A basal or tonic signal through an unligated, innocuous (non-autoreactive) BCR is necessary for continued cell survival and maturation (1–3). This is mediated by PI3K signaling (2, 4). Disruption of this tonic signal, inhibition of the PI3K pathway, or strong engagement of the BCR by self-antigen result in receptor editing, in which B cells continue light chain rearrangements in an attempt to change their specificity. Cells remaining autoreactive after a few rounds of editing are eliminated by clonal deletion (2–6).
Foxo transcription factors are downstream targets of PI3K that have pro-apoptotic and anti-mitogenic effects in numerous cell types (7, 8). Two Foxo family members, Foxo1 and Foxo3, have each been shown to play unique roles at several stages of B cell development (9–14). Upon activation of mature B cells via the BCR, PI3K signaling is activated and downregulates Foxo function at two levels: 1) by reducing their expression at the mRNA level (10, 14) and 2) by inducing their phosphorylation by Akt and their subsequent exclusion from the nucleus (7, 9). In contrast, BCR crosslinking blocks activation of PI3K in immature B cells (2), resulting in nuclear localization of both Foxo1 and Foxo3 (11, 15). The activation of Foxo family transcription factors in antigen-engaged immature B cells suggests that they might play a role in central B cell tolerance. Indeed, Foxo1 is known to promote Rag expression in immature B cells and thus receptor editing, while the role of Foxo3 in these processes is poorly understood (11–14). We previously demonstrated that while Foxo3−/− mice have reduced numbers of pre B cells (for unknown reasons), they have normal numbers of immature B cells (14). We hypothesized that this relative increase from the pre B to the immature B stage could be indicative of increased immature B cell survival in the absence of Foxo3 due to a role for Foxo3 in immature B cell apoptosis.
Here we show that Foxo3 plays a unique role in promoting apoptosis of BCR-stimulated immature B cells. Our results suggest that receptor editing is unimpaired and in fact enhanced in Foxo3−/− mice, as measured by both Igλ expression and RS recombination. This is likely a result of a longer editing window due to reduced apoptosis, as germline Igλ expression was not significantly elevated in Foxo3−/− pre B cells. These results support a model in which Foxo1 and Foxo3 promote receptor editing and apoptosis, respectively, in immature B cells expressing a non-functional or autoreactive BCR. While Foxo3−/− mice do not develop autoantibodies, reduced expression of Foxo3 mRNA was observed in B cells from SLE patients with anti-dsDNA antibodies and high disease activity compared to patients without these characteristics. While this is likely due in part to increased B cell activation in these SLE patients, it is also possible that low affinity B cells that remain autoreactive after editing may survive inappropriately in the absence of Foxo3 and become activated to secrete autoantibodies in the context of other SLE associated defects.
Materials and methods
Mice
Animal studies were approved by the UT Southwestern Institutional Animal Care and Use Committee. Foxo3−/− mice (16) and wild type controls were on the FVB background. MD4 (anti-HEL Ig) (JAX 002595) (17) and KLK (mHEL) (JAX 002598) (18) mice on the C57BL/6 background were obtained from JAX Labs and crossed to Foxo3−/− mice which had been backcrossed three generations onto the C57BL/6 background. Foxo3−/− mice and littermate controls backcrossed to C57BL/6 (B6) for 4 or 5 generations were also used in some experiments. Experimental and control mice were age and gender matched and littermate controls were used whenever possible.
Flow cytometric analysis
Apoptosis was measured by staining with annexin V FITC (BD Biosciences). To assay Igλ usage, splenocytes were depleted of red blood cells and stained with anti-CD21-FITC, anti-CD23-PE, anti-B220-PerCP and anti-Igλ1,2,3-biotin (BD Biosciences or Tonbo Biosciences). The biotinylated antibody was detected with streptavidin- allophycocyanin (APC) (BD Biosciences). Bone marrow cells were depleted of red blood cells and stained with anti-B220 FITC, anti-Igλ1,2,3-biotin, anti-B220-PerCP, and anti-CD93/AA4.1-APC (BD Biosciences or Tonbo Biosciences). The biotinylated antibody was detected with streptavidin-PE (BD Biosciences). T3 cells were identified by staining splenocytes with anti-B220 FITC, anti-CD23-PE, anti IgM-PerCP, and anti-AA4.1 APC (BD Biosciences or Tonbo Biosciences). In studies with the MD4 anti-HEL Ig transgene system, splenocytes were depleted of red blood cells and stained with combinations of anti-IgMb-FITC, anti-IgMa-PE, anti-B220 PerCP, anti-CD19 APC (antibodies from BD Biosciences or Tonbo Biosciences), and hen egg lysozyme (Sigma) labeled with Alexa 488 using an Alexa Fluro 488 Microscale Protein Labeling Kit (Molecular Probes) according to the manufacturer’s instructions.
Samples were run on a FACS Calibur (Becton Dickinson). Data were analyzed with CellQuest (BD Biosciences) or Flowjo (Treestar).
Cell purification
Pre and immature B cells:
Bone marrow was first depleted of red blood cells and B cells were then purified with anti-B220 magnetic beads using the IMag system (BD Pharmingen). B cells were subsequently stained with anti-IgM-PE, anti-B220-PerCP or anti-B220-FITC, and anti-CD93/AA4.1-biotin or anti-CD93/AA4.1-APC (BD Biosciences or Tonbo Biosciences). The biotinylated antibody was detected with streptavidin-APC (Caltag or BD Biosciences). Immature (B220+, IgM+, AA4.1+) and pre (B220+, IgM-, AA4.1+) B cells were sorted on a FACS Aria (Becton Dickinson) or a MoFlo (Cytomation) cell sorter. Samples were kept at 4° at all times prior to and during the sort and until stimulation to avoid activating the BCR with the sorting antibodies (11). Purified cells were either harvested immediately or stimulated with 10 μg/ml goat anti-mouse IgM F(ab’)2 (Jackson ImmunoResearch Laboratories) for the indicated times at 106 cells/ml in RPMI + 10% FBS. Alternatively, bone marrow B lineage cells were expanded by culturing for 4–6 days in 10 ng/ml IL-7 (R & D Systems) at 2 × 106 cells/ml and pre and immature B cells sorted as above.
Splenic B cell subsets:
Total splenic B cells were purified from red blood cell-depleted splenocytes by either negative selection with anti-CD43 magnetic beads (Miltenyi) or by positive selection with anti-B220 beads (BD Biosciences) according to the manufacturer’s instructions. To purify Igλ+ and Igλ− (and thus Igκ+) cells, Igλ+ cells were purified from splenocytes using anti-Igλ1,2,3-biotin and streptavidin magnetic beads (BD Biosciences). B220+ cells were then purified from the Igλ-depleted cells by positive selection with magnetic beads (BD Biosciences). To purify Igκ+ and Igκ− (and thus Igλ+) cells, Igκ+ cells were purified from splenocytes using anti-Igκ-biotin and streptavidin magnetic beads (BD biosciences), and B220+ cells were purified from the Igκ-depleted cells by positive selection with magnetic beads (BD Biosciences). To purify marginal zone (B220+CD21+CD23-) and transitional (B220+CD21-CD23-) B cells, splenocytes were depleted of CD23+ cells using anti-CD23 magnetic beads (Miltenyi Biotech) according to the manufacturer’s instructions, stained with anti-CD21 FITC, anti-CD23 PE, and anti-B220 PerCP or APC (BD Biosciences or Tonbo Biosciences), and sorted on a FACS Aria (Becton Dickinson) or a MoFlo (Cytomation) cell sorter.
Real-time PCR
Total RNA was prepared using the RNeasy kit (Qiagen). cDNA was generated with a cDNA Archive Kit (Applied Biosystems). Real-time PCR was performed using a Biorad CFX96 Real-Time System using TaqMan reagents for mouse Bcl2l11 (Bim) and the internal control GAPDH (Applied Biosystems). Data were normalized to GAPDH using the delta comparative threshold cycle method.
Western Blots
Total cell lysates were generated by boiling cells in 2× Laemmli sample buffer. Samples were run on a 4–15% gradient SDS-page gel (Biorad) and transferred to nitrocellulose (GE Healthcare Amersham). Blots were blocked in 5% milk and probed with a 1:1000 dilution of anti-Bim (Cell Signaling Technology clone C34C5) or anti-β-actin (Cell Signaling Technology clone D6A8) rabbit monoclonal antibodies. Blots were washed three times in TBST, probed with a 1:2500 dilution of goat-anti-rabbit HRP (Biorad), washed three times in TBST and developed with Clarity ECL substrate (Biorad). Bands were detected using a Chemidoc System (Biorad) and quantified with Image Lab Software (Biorad).
Germline Igλ expression analysis
Total RNA was prepared from pre B cells using the RNeasy kit (Qiagen). cDNA was generated with a cDNA Archive Kit (Applied Biosystems). Serial dilutions of cDNA were subjected to PCR for germline transcripts for Igλ1, Igλ2, and Igλ3, as well as β-actin as a loading control, as described in (19, 20) using the following primers: Igλ1: Stlambda1for2 5’-CTTGAGAATAAAATGCATGCAAGG-3, Clambda1R3 5’-TGATGGCGAAGACTTGGGCTGG-3’; Igλ2: Stlambda2for 5’-GAGAACAGGACCAGGTGCTG-3’, Clambda2rev 5’-ACACGGTGAGAGTGGGAGTG-3’; Igλ3: Stlambda3for 5’-CCCAGGTGCTTGCCCCAC-3’, Clambda3R3 5’-TGTTTTCCTGGAGCTCCTCAGG-3’; β-actin: betaActfor 5’-CTTTTCCAGCCTTCCTTCTT-3’, betaActrev 5’-ACGCAGCTCAGTAACAGTCC-3’. PCR reactions were run on an agarose gel and band intensities were quantified using VisionWorksLS software (UVP).
RS PCR
Genomic DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer’s instructions. Serial dilutions of DNA were subjected to PCR to detect RS recombination with the following primers as described in (21–23): Vk degenerate Primer, 5’GGCTGCAGSTTCAGTGGCAGTGGRTCWGGRAC 3’(S = G or C, R = A or G, W = T or A) and RS-101, 5′ ACATGGAAGTTTTCCCGGGAGAATATG 3′. Primers from the Ets1 gene (5’ AAAGCTGACCCATGTGCTCT 3’ and 5’ TGCAGTGGATTCAGGCATTA 3’) were used as a loading control. PCR reactions were run on an agarose gel and band intensities were quantified using VisionWorksLS software (UVP).
AutoAg array
Autoantibody reactivity against a panel of 124 autoantigens were measured using an autoantigen microarray platform developed by the University of Texas Southwestern Medical Center (https://microarray.swmed.edu/products/category/protein-array/) (24). Briefly, serum samples were pretreated with DNAse-I and then diluted 1:50 in PBST buffer for autoantibody profiling. The autoantigen array bearing 124 autoantigens and 4 control proteins were printed in duplicates onto Nitrocellulose film slides (Grace Bio-Labs). The diluted serum samples were incubated with the autoantigen arrays, and autoantibodies were detected with cy3-labeled anti-mouse IgG and cy5-labeled anti-mouse IgM using a Genepix 4200A scanner (Molecular Device) with laser wavelength of 532 nm and 635 nm. The resulting images were analyzed using Genepix Pro 6.0 software (Molecular Devices). The median of the signal intensity for each spot were calculated and subtracted the local background around the spot, and data obtained from duplicate spots were averaged. The background subtracted signal intensity of each antigen was normalized to the normalization factors generated based on the overall background signal of each array. Finally, the net fluorescence intensity (NFI) for each antigen was calculated by subtracting a PBS control which was included for each experiment as negative control. Signal-to-noise ratio (SNR) was used as a quantitative measurement of the true signal above background noise. SNR values equal to or greater than 3 were considered significantly higher than background, and therefore true signals. The NFI of each autoantibody was used to generate heatmaps using Cluster and Treeview software (http://rana.bl.gov/EisenSoftware.htm). Each row in the heatmap represents an autoantibody and each column represents a sample. Red color represents the signal intensity higher than the mean value of the raw and green color means signal intensity is lower than the mean value of the raw. Grey or black color represents the signal is close or equal to the mean value of the raw.
ELISAs
Total splenic B cells, marginal zone B cells, transitional B cells, or Igκ-depleted B cells were cultured at 106 cells/ml in complete RPMI media with or without 5 mg/ml LPS (Sigma). Supernatants were harvested after 5 days of culture.
Anti-dsDNA and/or anti-ssDNA ELISAs were performed on serial dilutions of serum (1:100, 1:400, 1:1600) or culture supernatant (neat, 1:5, 1:25, 1:125) as described in (25). Anti-IgM, IgG, anti-Igκ, or anti-Igλ alkaline phosphatase-conjugated detection antibodies (Southern Biotech) were used as indicated in the figure legends.
Analysis of SLE patient data
Publically available gene expression data from GSE10325 (26) was analyzed for correlation of Foxo3 (204131_s_at) expression in B, T, and myeloid cells with SLEDAI (as reported in (26)) and for the level of Foxo3 (204131_s_at) expression in B, T, and myeloid cells in patients with versus without anti-dsDNA antibodies (as defined in (26)). Publically available gene expression data from GSE49454 (27) was analyzed for the level of Foxo3 (ILMN_1844692) expression in patient samples with <50U versus >60U anti-dsDNA antibodies. Statistical analysis was performed using Graph Pad.
Results
We previously demonstrated that while Foxo3−/− mice have reduced numbers of pre B cells (for unknown reasons), they have normal numbers of immature B cells (14). We confirmed this in an additional cohort of mice (Figure 1a, Supplemental Figure 1a). Given the known pro-apoptotic role of Foxo3 in other cell types (7, 8), we hypothesized that this relative recovery of the immature B cell population in Foxo3−/− mice may be due to a reduction in apoptosis during central tolerance mechanisms. Indeed, BCR-stimulated primary immature B cells from Foxo3−/− mice showed increased survival compared to their wild type counterparts (Figure 1b,c). The pro-apoptotic BH3 family member Bim is a known Foxo3 target in other cell types (7) and has been shown to promote immature B cell apoptosis (28, 29). However, although Bim mRNA expression was decreased in BCR-stimulated Foxo3−/− immature B cells in some experiments, this was not consistent (Figure 1d), nor was there a significant difference in Bim protein expression (Figure 1e,f). Thus, Foxo3 promotes apoptosis in BCR-stimulated immature B cells via mechanisms other than control of Bim expression.
Figure 1: Foxo3 promotes apoptosis in BCR-stimulated immature B cells.
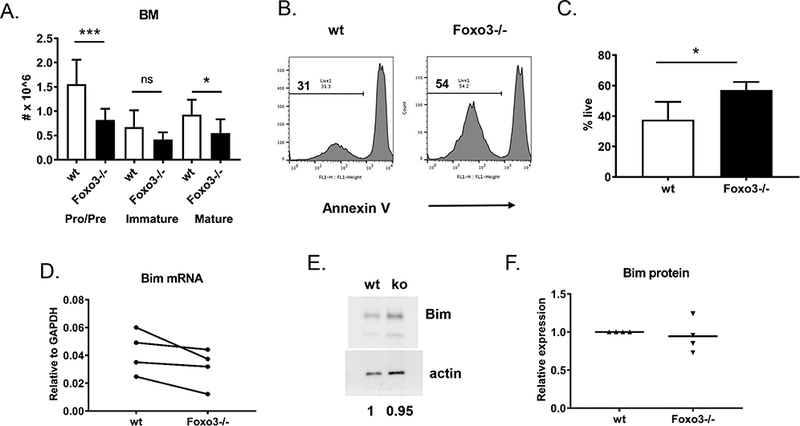
A) Bone marrow cells from mice on the FVB background were stained with antibodies against B220, IgM, AA4.1, and Igλ1,2,3. The total number of pro/pre (B220loIgM-), immature (B220loIgM+), and mature (B220hiIgM+) cells is indicated. Data are presented as mean +/− SD, n = 11–12 (A). *** p < 0.001, * p < 0.05 by Student’s t-test. Representative FACS plots are shown in Supplemental Figure 1. B, C) Bone marrow immature B cells (B220+, IgM+, AA4.1+) were sorted (see Supplemental Figure 1d) from pools of 5 mice and incubated for 18 h in the presence of 10 μg/ml anti-IgM F(ab’)2 fragments. Cells were stained with annexin V and analyzed by flow cytometry. B) Representative annexin V staining is shown, gated on all cells. C) The percentage of live cells (defined by live FSC vs. SSC gate AND annexin negative) among all cells is indicated. Data are presented as mean +/− SD, n = 4 experiments. *p < 0.05 by Student’s t-test. D, E, F) FACS-sorted immature B cells (B220+, IgM+, AA4.1+) from pools of 4–5 mice on the FVB background were stimulated for 18–24 hrs with 10 ug/ml anti-IgM F(ab)’2 fragments. D) RNA and cDNA were prepared and analyzed for expression of Bcl2l11, which encodes Bim, by real-time PCR. Expression levels were normalized to GAPDH. Symbols connected by lines represent individual experiments. E. F) Protein extracts were subjected to Western blot for Bim, with actin as a loading control. A representative blot is shown in (E). Results were quantified (F) by normalizing Bim levels to actin, then setting the wild type to 1 in each experiment.
In addition to undergoing apoptosis, immature B cells can undergo receptor editing in response to antigen encounter. This has been shown to be mediated by Foxo1, while the role of Foxo3 in this process is controversial (11, 12). To determine whether Foxo3−/− mice exhibit signs of altered receptor editing, we first assessed their usage of Igλ. Rearrangement of the kappa light chain locus occurs first, and production of a functional kappa light chain inhibits rearrangement of the lambda locus. Thus, the lambda locus will usually only undergo recombination if an in-frame kappa light chain cannot be produced or if receptor editing occurs due to an autoreactive kappa light chain (30). We thus examined bone marrow and splenic B cell subpopulations for Igλ expression by flow cytometry. We found that a significantly larger proportion of immature B cells in the bone marrow and spleen of Foxo3−/− expressed Igλ than was the case in wild type mice (Figure 2, Supplemental Figures 1a, 1b, 2a, 2b). The frequency of Igλ+ cells in more mature B cell populations – follicular and marginal zone B cells in the spleen, and mature recirculating cells in the bone marrow – was more similar between wild type and Foxo3−/− mice, however, although still slightly increased in some animals in the absence of Foxo3 (Figure 2).
Figure 2: Igλ usage in Foxo3−/− bone marrow and splenic B cells.
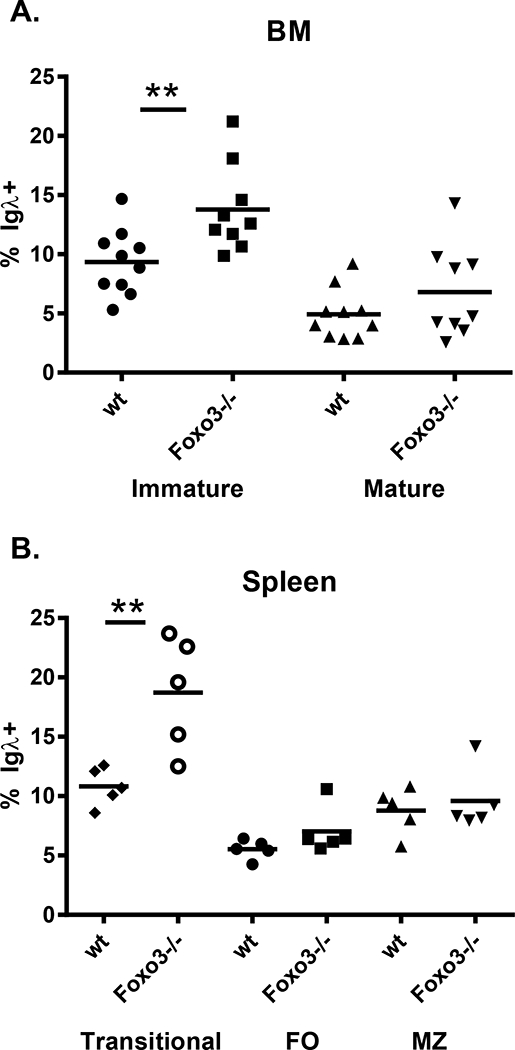
A) Bone marrow cells from mice on the FVB background were stained with antibodies against B220, IgM, AA4.1, and Igλ1,2,3. The frequency of Igλ+ cells among immature (B220lo IgM+ AA4.1+) and mature recirculating (B220hi IgM+ AA4.1-) bone marrow cells is indicated. Each symbol represents a mouse, and the bar is the mean. ** p < 0.01, by Student’s t test. Representative FACS plots are shown in Supplemental Figure 1. B) Splenocytes from mice on the FVB background were stained with antibodies against B220, CD21, CD23, and Igλ1,2,3. The frequency of Igλ+ cells among transitional (B220+CD21-CD23-), marginal zone (B220+CD21hiCD23-/lo), and follicular (B220+CD21+CD23+) B cells is indicated. Each symbol represents a mouse, and the bar is the mean. ** p < 0.01, by Student’s t test. Consistent with our previous observations (14), the total number of cells did not differ between wild type and Foxo3−/− mice in any of these splenic B cell subsets. This data and representative FACS plots are shown in Supplemental Figure 2.
To determine whether the increased Igλ usage could result in part from enhanced accessibililty of the Igλ locus, we examined expression of Igλ germline transcripts in pre B cells from wild type and Foxo3−/− mice (Figure 3). No significant difference was observed. This suggests that receptor editing not only occurs independently of Foxo3, but appears to be increased in its absence. As a more direct measure of receptor editing, we performed a PCR assay for Vκ-RS recombination, which occurs when B cells have exhaustively rearranged an Igκ allele (21, 22). While RS recombination levels in total, Igλ− (and thus Igκ+) and Igκ+ splenic B cells were normal in Foxo3−/− mice, both Igλ+ and Igκ− (and thus Igλ+) B cells from Foxo3−/− mice demonstrated increased Vκ-RS recombination (Figure 4). This suggests that developing autoreactive Foxo3−/− B cells are more likely to edit their Igκ alleles to exhaustion prior to utilizing Igλ due to a longer editing window.
Figure 3: Normal expression of germline IgL transcripts in Foxo3−/− pre B cells.
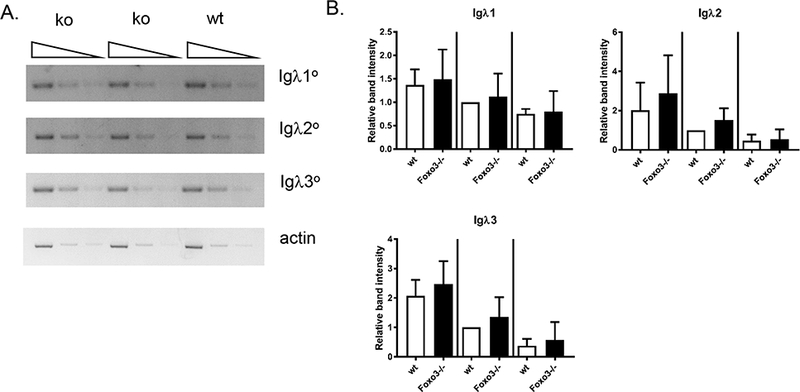
(A) Serial dilutions of cDNA from wild type and Foxo3−/− bone marrow pre B cells (on the FVB background) underwent PCR to detect Igλ1, Igλ2, and Igλ3 germline transcripts. β-actin was used as a loading control. Shown is one of 5 experiments which are quantified in (B). (B) Band intensities were quantified using VisionWorksLS software (UVP). Igλ values were normalized to the loading control, and the normalized Igλ value for the middle dilution of the wild type sample set to 1 in each experiment. Data are shown as mean +/− SD, n = 5 wild type and 6 Foxo3−/−.
Figure 4: Increased RS recombination in Foxo3−/− Igλ+ cells.
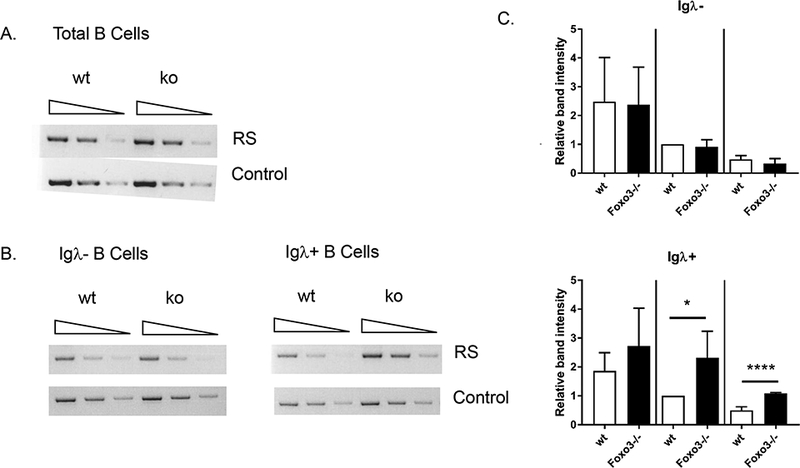
Serial dilutions of genomic DNA from wild type and Foxo3−/− splenic B cells (on the FVB background) underwent PCR to detect RS recombination as an indication of the amount of receptor editing. Primers for the Ets1 gene were used as a loading control. Results are representative of 3 independent experiments for total B cells (A), Igλ− (and thus Igκ+) B cells (B, left), and Igλ+ B cells (B, right) and 1 experiment for Igκ+ and Igκ− (and thus Ig λ+) cells. C) Quantification of RS PCR experiments. Band intensities were quantified using VisionWorksLS software (UVP). RS values were normalized to the loading control, and the normalized RS value for the middle dilution of the wild type sample set to 1 in each experiment. Data are shown as mean +/− SD, n = 4. * p < 0.05, **** p < 0.0001 by Student’s t-test.
Reduced apoptosis of immature B cells in the absence of Foxo3 may result in the inappropriate release of autoreactive B cells that remain autoreactive even after editing. We thus sought to determine whether Foxo3−/− mice demonstrate signs of humoral autoimmunity. We first asked whether Foxo3−/− mice accumulated anti-DNA antibodies with age. There was no difference between anti-ssDNA or anti-dsDNA IgM levels between wild type and Foxo3−/− mice at 9–11 months of age (Figure 5a). To obtain a broader view of potential autoreactivity, we analyzed serum from aged wild type and Foxo3−/− mice using an autoantigen array that allows the simultaneous interrogation of autoreactivity against approximately 100 self-antigens commonly targeted in autoimmune disease (24). We did not observe an increase in either IgM or IgG autoantibodies in Foxo3−/− mice using this approach (Figure 5b, Supplemental Tables 1, 2).
Figure 5: Foxo3−/− mice do not demonstrate increased serum autoantibodies.
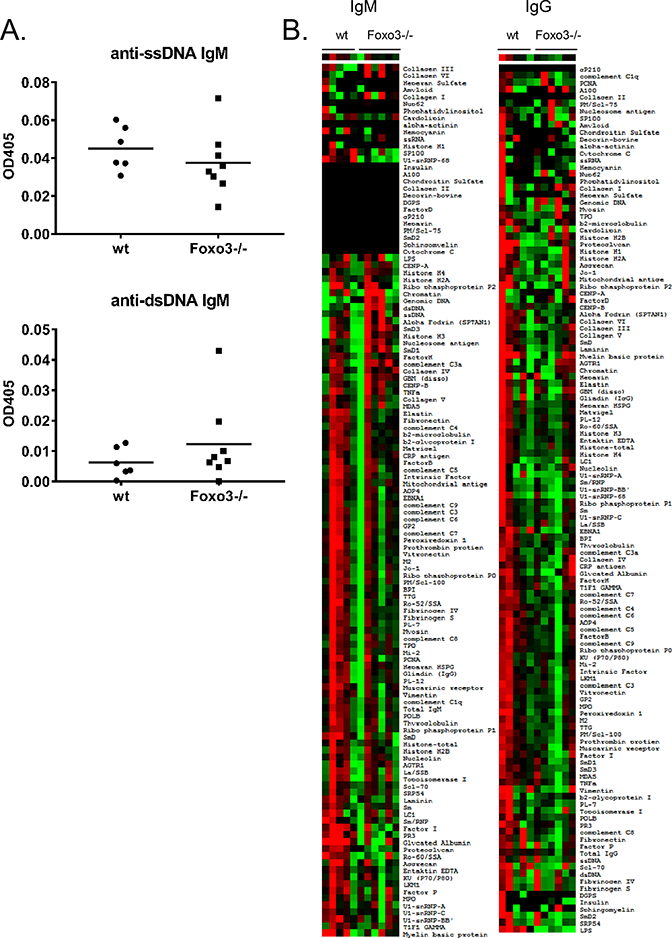
A) anti-ssDNA (left) and anti-dsDNA (right) IgM was measured in serum from 9–11 month old Foxo3−/− mice on the FVB background. Results from a 1:100 dilution of serum are shown. Each symbol represents a mouse, the bar is the mean. B) Serum from 12–16 month old female wt (n = 5) and Foxo3−/− (N = 6) mice on the FVB background was hybridized to an autoantigen array containing more than 100 commonly targeted autoantigens and IgM (left) and IgG (right) detected as described in Materials and Methods. Red indicates reactivity greater than the mean for each row, green is less than the mean for each row, and gray or black is close to the mean for each row. There was no significant increase in reactivity of Foxo3−/− mice to any of the antigens. The complete data sets plotted in the heat map in (B) are provided in Supplemental Tables 1 and 2.
To test whether autoreactive B cells may be present at increased frequencies in Foxo3−/− mice but not activated in vivo, we purified B cells from wild type and Foxo3−/− mice by negative selection with anti-CD43 magnetic beads, stimulated them with LPS to induce differentiation and antibody secretion, and measured anti-dsDNA antibodies by ELISA. There was no difference in the level of anti-dsDNA Igκ between genotypes, and anti-dsDNA Igλ was not detected (Figure 6a). This was not due to reduced response of Foxo3−/− B cells to LPS (14). We also purified B cells with anti-B220 magnetic beads in case a CD43+ B cell subset was enriched for autoreactivity in Foxo3−/− mice. Still, there was no increase, and in fact a slight decrease, in the secretion of anti-dsDNA antibodies in response to LPS by Foxo3−/− B220+ cells (Figure 6b). We also did not observe enhanced production of anti-dsDNA antibodies by marginal zone, transitional, or Igκ-depleted B cells (thus enriched for Igλ+ cells) from Foxo3−/− mice (Figure 6c-e). We considered the possibility that there was an increase in autoreactive cells that were not activated by LPS due to their anergic state, and thus enumerated T3 cells (B220+ AA4.1+ CD23+ IgMlo), which are known to be enriched in anergic, autoreactive B cells (31, 32). However, there were normal numbers of these cells in young Foxo3−/− mice (wild type 1.36 +/− 0.72 × 106, Foxo3−/− 1.13 +/− 0.75 × 106, n = 8), (Supplemental Figure 2e) and this population was in fact reduced, rather than increased, in aged Foxo3−/− mice (wild type 8.32 +/− 1.75 × 105, Foxo3−/− 1.93 +/− 0.95 × 105, n = 16). Taken together, these results suggest that the increased window of receptor editing afforded by reduced apoptosis of Foxo3−/− bone marrow immature B cells allows for cells to redeem themselves away from autoreactivity.
Figure 6: Foxo3−/− B cells do not secrete increased autoantibodies.
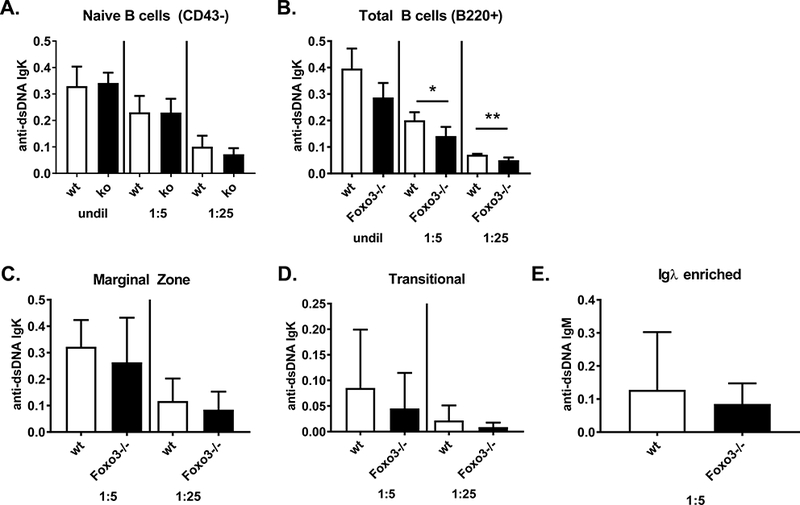
A-D) B cell subpopulations were isolated from wild type and Foxo3−/− mice on the FVB background and stimulated with 5 ug/ml LPS for 5 days. anti-dsDNA Igκ and Igλ antibodies were measured by ELISA in the indicated dilutions of culture supernatant. Results are shown for Igκ as mean +/− SD, n = 4–6. *p < 0.05, **p < 0.01. Igλ anti-DNA antibodies were not detected. Subpopulations analyzed are as follows: (A) naïve B cells purified by negative selection with anti-CD43 beads; (B) total B cells purified by positive selection with anti-B220 beads; (C) marginal zone (B220+CD21hiCD23-/lo) and (D) transitional (B220+CD21-CD23-) B cells isolated as described in Materials and Methods. (E) Igκ-depleted B cells (and thus Igλ+) were isolated from spleens of wild type and Foxo3−/− mice on the FVB background and stimulated with 5 ug/ml LPS for 5 days. Anti-dsDNA IgM (E) and IgG (not detected and thus not shown) antibodies were measured by ELISA in the indicated dilutions of culture supernatants. Results are presented as mean +/− SD, n = 7.
To determine whether autoreactive Foxo3−/− cells that should be deleted survive in the periphery in situations where receptor editing does not efficiently eliminate autoreactivity, we used the anti-HEL Ig (MD4) x mHEL (KLK) transgenic system. The anti-HEL Ig transgene, which confers high affinity reactivity with the antigen HEL, is not in the endogenous Ig locus and thus is not edited in the presence of self-antigen (17, 18, 33). Instead, exposure to mHEL (membrane bound HEL) results in deletion of HEL-reactive cells (17, 18, 33). We generated anti-HEL Ig x mHEL x Foxo3−/− mice and asked whether anti-HEL B cells were present in the periphery in greater numbers than in anti-HEL Ig x mHEL mice. Surprisingly, Foxo3−/− anti-HEL Ig B cells were deleted normally in the presence of mHEL (Figure 7). This suggests that the high affinity BCR/Ag interaction in the anti-HEL/mHEL system provides a strong enough signal to overcome a requirement for Foxo3 in BCR-induced apoptosis.
Figure 7: anti-HEL Foxo3−/− B cells are deleted in response to mHEL.
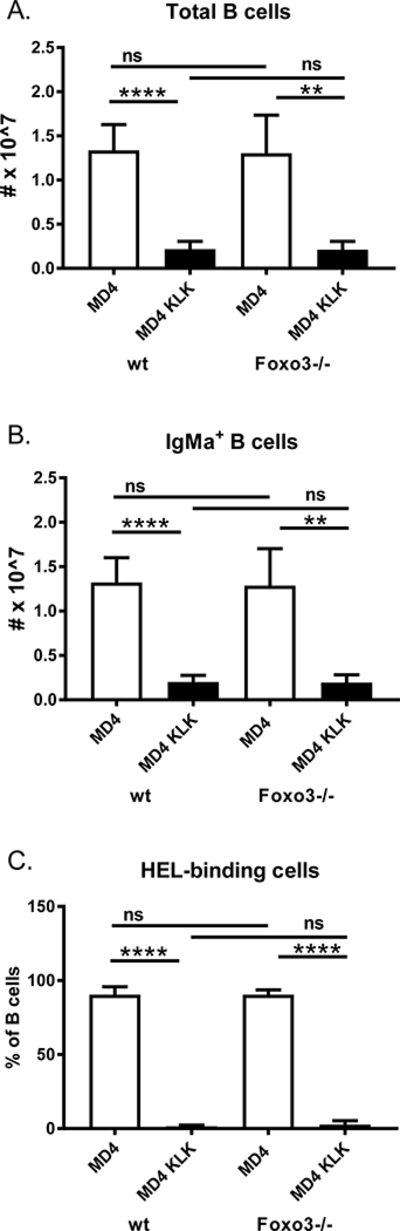
Splenic B cells from anti-HEL Ig transgenic mice (MD4) with or without a mHEL transgene (KLK) on a wild type (wt) or Foxo3−/− background were analyzed by flow cytometry with antibodies against IgMa, IgMb, B220, CD19, and Alexa 488-labeled HEL. A) The total number of B220+CD19+ cells. B) The total number of IgMa+B220+CD19+ cells. C) The frequency of HEL-binding cells among B220+CD19+ cells. Data represent mean +/− SD, n = 3–5. ** p < 0.01, **** p < 0.0001 by Student’s t-test.
Reduced Foxo3 levels have been observed in B cells from polygenic mouse models of lupus (34). We thus asked whether alterations in Foxo3 expression were observed in B cells from SLE patients by analyzing previously published (26) gene expression profiling data. While Foxo3 levels did not differ overall between healthy controls and SLE patients (not shown), SLE patients with high anti-dsDNA levels (>60 IU) had lower levels of Foxo3 mRNA expression in B cells, but not T or myeloid cells, compared to patients with normal anti-dsDNA levels (< 50 IU) (Figure 8). Furthermore, Foxo3 levels in CD19+ B cells, but not CD4+ T cells or CD33+ myeloid cells, correlated inversely with disease activity as measured by SLEDAI (Figure 8). We also analyzed another publically available study in which whole blood gene expression analysis was performed on SLE patients (27). We divided these patients into groups based on anti-DNA titers (<50 IU and >60 IU) using the same criteria as in the first study (26) and found that whole blood Foxo3 mRNA levels were also reduced in patients with high anti-DNA titers (Figure 8).
Figure 8: Foxo3 levels in SLE B cells:
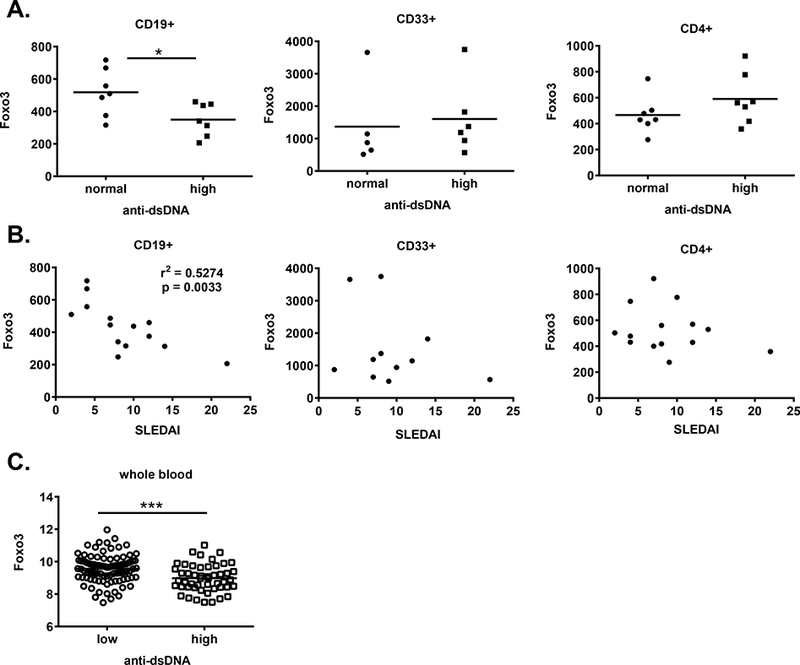
A, B) Analysis of data from (26) (GSE10325). Foxo3 (204131_s_at, Affymetrix platform) levels in sorted CD19+ B cells, but not CD33+ myeloid cells or CD4+ T cells, from adult SLE patients were (A) reduced in patients with high dsDNA levels (*p < 0.05 by Student’s t-test) and (B) inversely correlated with SLEDAI (p = 0.0033) based on dsDNA and SLEDAI levels reported in (26). Each symbol represents an individual patient, the bar is the mean. C) Data from GSE49454 (27) was analyzed for the level of Foxo3 (ILMN_1844692) expression in patient samples with <50U versus >60U anti-dsDNA antibodies. *** p = 0.001 by Student’s t-test.
Discussion
Elimination of autoreactive B cells during development is critical to limit the frequency of potentially pathogenic B cells in the periphery and prevent autoimmune disease. This central tolerance checkpoint involves receptor editing, followed by apoptosis of cells that remain autoreactive after several rounds of editing. Here, we define the role of the transcription factor Foxo3 in these processes.
Upon antigen encounter or inhibition of the tonic BCR signal, immature B cells express Rag and undergo receptor editing. If this fails to produce a functional, innocuous receptor, the cells die by apoptosis. These processes are inhibited by PI3K (2, 6, 15, 35) and promoted by Foxo family members (11, 12). Upregulation of Rag in BCR-stimulated immature B cells is mediated primarily by Foxo1. Although overexpression of constitutively active Foxo3 promotes BCR-induced Rag expression (12), shRNA targeting of Foxo1 inhibits this process almost completely (11). Knockdown of Foxo3 has little effect on Rag expression (11), and Foxo3−/− pre-B cells express normal Rag levels (14). Increased Igλ usage and RS recombination in Igλ+ B cells from Foxo3−/− mice suggests that receptor editing is not only unimpaired in the absence of Foxo3, but actually enhanced. Apoptosis is reduced in anti-IgM stimulated Foxo3−/− immature B cells, however, indicating a unique role for Foxo3 in this process. This likely explains our observation that immature B cell numbers are normal in Foxo3−/− mice despite a reduced number of pre B cells (14).
Together these studies support the following model for Foxo regulation and function in immature B cells. Pre-existing Foxo protein is largely phosphorylated and sequestered in the cytoplasm, where it may be degraded (11, 15). Disruption of tonic BCR signaling or engagement of the BCR with self-antigen leads to the loss of Akt activity (2). Foxo proteins, no longer phosphorylated by Akt, enter the nucleus (11, 15). Foxo1 then activates Rag transcription, initiating light chain receptor editing (11). If receptor editing is successful, tonic signaling by the newly generated BCR restores Akt activity, drawing the Foxo proteins out of the nucleus and averting cell death. If editing is unsuccessful, however, and receptors remain non-functional or autoreactive, then Foxo3 promotes the death of these cells via apoptosis.
Although Foxo3 is known to control the expression of Bim in other cell types (7), and Bim is thought to contribute to the deletion of autoreactive immature B cells (28, 29), we did not observe a consistent change in Bim expression in Foxo3−/− immature B cells. This suggests that Foxo3 promotes immature B cell apoptosis via Bim-independent mechanisms, an idea supported by two recent studies. Foxo3 binding sites in the Bim locus were shown not to be required for Bim to mediate apoptosis in response to cytokine withdrawal (36), and mice lacking Bim specifically in the B lineage did not have an increase in the bone marrow immature B cell pool (37). Foxo3 can also promote the expression of another pro-apoptotic BH3 family member PUMA (38), which could possibly contribute to immature B cell apoptosis. In addition, Foxo3 has been reported to inhibit NFkB (39, 40). NFkB activity is enhanced in cells undergoing receptor editing (41), and its loss results in a cell intrinsic reduction of immature B cell numbers (42). It is thus possible that the balance of Foxo3 and NFκB activity plays a role in determining whether receptor editing or apoptosis occurs in immature B cells, with NFκB favoring editing and Foxo3 favoring apoptosis.
Despite impaired BCR-induced apoptosis in Foxo3−/− immature B cells, Foxo3−/− mice do not develop autoantibodies. Delayed apoptosis in the absence of Foxo3 may extend the window for Foxo1 to successfully edit autoreactive receptors via its promotion of Rag expression. The increase in the frequency of Igλ expressing B cells and the increase in RS recombination in these cells in Foxo3−/− mice is consistent with this idea. This scenario suggests a system of checks and balances to maintain B cell tolerance; when Foxo3-mediated deletion is reduced, Foxo1-mediated editing is increased. Since the block in immature B cell apoptosis is not complete in Foxo3−/− mice, some autoreactive B cells may still be eliminated by deletion in vivo. We show that this is indeed the case for high affinity B cells in the anti-HEL Ig x mHEL model. In addition, the increased frequency of Igλ-expressing B cells drops off between the transitional and mature B cell stage in the spleens of Foxo3−/− mice, suggesting that some Igλ+ cells that remain autoreactive and escape the bone marrow may be eliminated in the periphery. Finally, we did not observe enhanced secretion of autoantibodies by Foxo3−/− B cells in vitro, nor did we observe an increase in the T3 population, known to be enriched in autoreactive, anergic B cells, in Foxo3−/− mice. Taken together, these data suggest that loss of Foxo3 does not breach B cell tolerance despite impairing immature B cell apoptosis.
These studies were performed in whole body Foxo3−/− mice. It is likely that the effects we observe on immature B cell survival and receptor editing are B cell intrinsic, as defects in BCR-induced apoptosis are observed in sort-purified immature B cells and we believe that the increase in receptor editing results from increased survival. However, we cannot rule out a role for Foxo3 in other cell types in our results. Foxo3 is known to limit inflammatory cytokine production in myeloid cells (43–45). In T cells, it can promote pathogenic Th1 differentiation (46), limit the maintenance of CD4 and CD8 T cell memory (47–50), and contribute to the development of Tregs (51). While these parameters are unlikely to affect central B cell tolerance checkpoints, alterations in T or myeloid cell function in the absence of Foxo3 may alter the ability of autoreactive B cells to survive or become activated in the periphery. Future studies using mice with B cell specific deletion of Foxo3 will shed light on this issue. It is also important to consider that the majority of these studies were performed with Foxo3−/− mice on the FVB background. It is possible that other genetic backgrounds may reveal a more dramatic effect of Foxo3-deficiency on B cell tolerance. We have begun to address this issue by backcrossing the FVB.Foxo3−/− mice to the C57BL/6 background. Preliminary analysis of these mice after 4–5 generations of backcrossing suggests that the increased frequency of Igλ-expressing B cells, particularly among immature and transitional cells, is retained (Supplementary Figure 1c, 2d).
Foxo3 levels are reduced in B cells from murine lupus models (34). Similarly, analysis of publically available gene expression profiling data indicate that Foxo3 levels are reduced in B cells from SLE patients with high levels of anti-dsDNA antibodies. There are several models, not mutually exclusive, that reconcile our data with these observations. A B cell intrinsic defect upstream of Foxo3 that also affects Foxo1 levels could result a more severe loss of central B cell tolerance in SLE patients than in Foxo3−/− mice due to an impairment of both editing and apoptosis. Indeed, central tolerance defects have been observed in SLE patients (52, 53). Furthermore, SLE is a complex disease which results from a combination of increased autoantigen availability, loss of adaptive immune tolerance, and innate immune system hyperactivity (54). It is thus possible that preexisting low levels of Foxo3 in a subset of patients preferentially promote the survival of low affinity autoreactive B cells that remain after extensive editing. These cells could then become activated to secrete autoantibodies in the context of additional SLE associated defects, which would likely not have been present in the Foxo3−/− mice. Finally, low levels of Foxo3 in B cells may be a consequence of increased peripheral B cell activation (55) or a more inflammatory environment in patients who have elevated anti-DNA antibodies or are undergoing disease flares, rather than a cause of autoantibody production. Consistent with this idea, reduced expression of PTEN, an inhibitor of the PI3K pathway, has been observed in some SLE patients’ B cells in the periphery and correlates with disease activity (56). Ongoing studies to address the effect of Foxo3-deficiency in murine lupus models will address this issue.
Supplementary Material
Acknowledgements
We thank Arturo Menchaca, Lyndsay Joson, Hansaa Abbasi, Elizabeth Curry, Julia McLouth, Ian Matthews, and Angela Mobley for excellent technical assistance.
This work was supported by NIH grants AI049248, AI005284, and AR067625. A.B.S. is a Southwestern Medical Foundation Scholar in Biomedical Research and holds the Peggy Chavellier Professorship for Arthritis Research and Treatment. E.B. was supported UT Southwestern Summer Medical Student Research Program.
Abbreviations:
- APC
allophycocyanin
- HEL
hen egg lysozyme
- SLE
systemic lupus erythematosus
- SLEDAI
systemic lupus erythematosus disease activity index
References
- 1.Tze LE, Schram BR, Lam KP, Hogquist KA, Hippen KL, Liu J, Shinton SA, Otipoby KL, Rodine PR, Vegoe AL, Kraus M, Hardy RR, Schlissel MS, Rajewsky K and Behrens TW. 2005. Basal immunoglobulin signaling actively maintains developmental stage in immature B cells. PLoS Biol. 3: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verkoczy L, Duong B, Skog P, Ait-Azzouzene D, Puri K, Vela JL and Nemazee D. 2007. Basal B cell receptor-directed phosphatidylinositol 3-kinase signaling turns off RAGs and promotes B cell-positive selection. J Immunol. 178: 6332–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schram BR, Tze LE, Ramsey LB, Liu J, Najera L, Vegoe AL, Hardy RR, Hippen KL, Farrar MA and Behrens TW. 2008. B cell receptor basal signaling regulates antigen-induced Ig light chain rearrangements. J Immunol. 180: 4728–4741. [DOI] [PubMed] [Google Scholar]
- 4.Llorian M, Stamataki Z, Hill S, Turner M and Martensson IL. 2007. The PI3K p110delta is required for down-regulation of RAG expression in immature B cells. J Immunol. 178: 1981–1985. [DOI] [PubMed] [Google Scholar]
- 5.Melamed D, Benschop RJ, Cambier JC and Nemazee D. 1998. Developmental regulation of B lymphocyte immune tolerance compartmentalizes clonal selection from receptor selection. Cell. 92: 173–182. [DOI] [PubMed] [Google Scholar]
- 6.Cheng S, Hsia CY, Feng B, Liou ML, Fang X, Pandolfi PP and Liou HC. 2009. BCR-mediated apoptosis associated with negative selection of immature B cells is selectively dependent on Pten. Cell Res. 19: 196–207. [DOI] [PubMed] [Google Scholar]
- 7.Tran H, Brunet A, Griffith EC and Greenberg ME. 2003. The many forks in FOXO’s road. Sci STKE. 2003: RE5. [DOI] [PubMed] [Google Scholar]
- 8.Hedrick SM 2009. The cunning little vixen: Foxo and the cycle of life and death. Nat Immunol. 10: 1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yusuf I, Zhu X, Kharas MG, Chen J and Fruman DA. 2004. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood. 104: 784–787. [DOI] [PubMed] [Google Scholar]
- 10.Hinman RM, Bushanam JN, Nichols WA and Satterthwaite AB. 2007. B cell receptor signaling down-regulates forkhead box transcription factor class O 1 mRNA expression via phosphatidylinositol 3-kinase and Bruton’s tyrosine kinase. J Immunol. 178: 740–747. [DOI] [PubMed] [Google Scholar]
- 11.Amin RH and Schlissel MS. 2008. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol. 9: 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herzog S, Hug E, Meixlsperger S, Paik JH, DePinho RA, Reth M and Jumaa H. 2008. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol. 9: 623–631. [DOI] [PubMed] [Google Scholar]
- 13.Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA and Rickert RC. 2008. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol. 9: 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinman RM, Nichols WA, Diaz TM, Gallardo TD, Castrillon DH and Satterthwaite AB. 2009. Foxo3−/− mice demonstrate reduced numbers of pre-B and recirculating B cells but normal splenic B cell sub-population distribution. Int Immunol. 21: 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandramohan V, Jeay S, Pianetti S and Sonenshein GE. 2004. Reciprocal control of Forkhead box O 3a and c-Myc via the phosphatidylinositol 3-kinase pathway coordinately regulates p27Kip1 levels. J Immunol. 172: 5522–5527. [DOI] [PubMed] [Google Scholar]
- 16.Castrillon DH, Miao L, Kollipara R, Horner JW and DePinho RA. 2003. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 301: 215–218. [DOI] [PubMed] [Google Scholar]
- 17.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K and et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 334: 676–682. [DOI] [PubMed] [Google Scholar]
- 18.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A and Goodnow CC. 1991. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 353: 765–769. [DOI] [PubMed] [Google Scholar]
- 19.Engel H, Rolink A and Weiss S. 1999. B cells are programmed to activate kappa and lambda for rearrangement at consecutive developmental stages. Eur J Immunol. 29: 2167–2176. [DOI] [PubMed] [Google Scholar]
- 20.Kersseboom R, Ta VB, Zijlstra AJ, Middendorp S, Jumaa H, van Loo PF and Hendriks RW. 2006. Bruton’s tyrosine kinase and SLP-65 regulate pre-B cell differentiation and the induction of Ig light chain gene rearrangement. J Immunol. 176: 4543–4552. [DOI] [PubMed] [Google Scholar]
- 21.Retter MW and Nemazee D. 1998. Receptor editing occurs frequently during normal B cell development. J Exp Med. 188: 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vela JL, Ait-Azzouzene D, Duong BH, Ota T and Nemazee D. 2008. Rearrangement of mouse immunoglobulin kappa deleting element recombining sequence promotes immune tolerance and lambda B cell production. Immunity. 28: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlissel MS and Baltimore D. 1989. Activation of immunoglobulin kappa gene rearrangement correlates with induction of germline kappa gene transcription. Cell. 58: 1001–1007. [DOI] [PubMed] [Google Scholar]
- 24.Li QZ, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, Mohan C, Wakeland EK and Olsen NJ. 2007. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol. 147: 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whyburn LR, Halcomb KE, Contreras CM, Lowell CA, Witte ON and Satterthwaite AB. 2003. Reduced dosage of Bruton’s tyrosine kinase uncouples B cell hyperresponsiveness from autoimmunity in lyn−/− mice. J Immunol. 171: 1850–1858. [DOI] [PubMed] [Google Scholar]
- 26.Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, Li QZ, Lian Y, Wu T, Reimold AM, Olsen NJ, Karp DR, Chowdhury FZ, Farrar JD, Satterthwaite AB, Mohan C, Lipsky PE, Wakeland EK and Davis LS. 2013. SLE Peripheral Blood B Cell, T Cell and Myeloid Cell Transcriptomes Display Unique Profiles and Each Subset Contributes to the Interferon Signature. PLoS One. 8: e67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiche L, Jourde-Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, Anguiano E, Quinn C, Burtey S, Berland Y, Kaplanski G, Harle JR, Pascual V and Chaussabel D. 2014. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 66: 1583–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM and Strasser A. 2003. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J Exp Med. 198: 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granato A, Hayashi EA, Baptista BJ, Bellio M and Nobrega A. 2014. IL-4 regulates Bim expression and promotes B cell maturation in synergy with BAFF conferring resistance to cell death at negative selection checkpoints. J Immunol. 192: 5761–5775. [DOI] [PubMed] [Google Scholar]
- 30.Tiegs SL, Russell DM and Nemazee D. 1993. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 177: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ and Cambier JC. 2006. Identification of anergic B cells within a wild-type repertoire. Immunity. 25: 953–962. [DOI] [PubMed] [Google Scholar]
- 32.Teague BN, Pan Y, Mudd PA, Nakken B, Zhang Q, Szodoray P, Kim-Howard X, Wilson PC and Farris AD. 2007. Cutting edge: Transitional T3 B cells do not give rise to mature B cells, have undergone selection, and are reduced in murine lupus. J Immunol. 178: 7511–7515. [DOI] [PubMed] [Google Scholar]
- 33.Fang W, Weintraub BC, Dunlap B, Garside P, Pape KA, Jenkins MK, Goodnow CC, Mueller DL and Behrens TW. 1998. Self-reactive B lymphocytes overexpressing Bcl-xL escape negative selection and are tolerized by clonal anergy and receptor editing. Immunity. 9: 35–45. [DOI] [PubMed] [Google Scholar]
- 34.Nakou M, Bertsias G, Stagakis I, Centola M, Tassiulas I, Hatziapostolou M, Kritikos I, Goulielmos G, Boumpas DT and Iliopoulos D. 2010. Gene network analysis of bone marrow mononuclear cells reveals activation of multiple kinase pathways in human systemic lupus erythematosus. PLoS One. 5: e13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerji L, Glassford J, Lea NC, Thomas NS, Klaus GG and Lam EW. 2001. BCR signals target p27(Kip1) and cyclin D2 via the PI3-K signalling pathway to mediate cell cycle arrest and apoptosis of WEHI 231 B cells. Oncogene. 20: 7352–7367. [DOI] [PubMed] [Google Scholar]
- 36.Herold MJ, Rohrbeck L, Lang MJ, Grumont R, Gerondakis S, Tai L, Bouillet P, Kaufmann T and Strasser A. 2013. Foxo-mediated Bim transcription is dispensable for the apoptosis of hematopoietic cells that is mediated by this BH3-only protein. EMBO Rep. 14: 992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu R, King A, Bouillet P, Tarlinton DM, Strasser A and Heierhorst J. 2018. Proapoptotic BIM Impacts B Lymphoid Homeostasis by Limiting the Survival of Mature B Cells in a Cell-Autonomous Manner. Front Immunol. 9: 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A and Mak TW. 2006. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 203: 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin L, Hron JD and Peng SL. 2004. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 21: 203–213. [DOI] [PubMed] [Google Scholar]
- 40.Thompson MG, Larson M, Vidrine A, Barrios K, Navarro F, Meyers K, Simms P, Prajapati K, Chitsike L, Hellman LM, Baker BM and Watkins SK. 2015. FOXO3-NF-kappaB RelA Protein Complexes Reduce Proinflammatory Cell Signaling and Function. J Immunol. 195: 5637–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cadera EJ, Wan F, Amin RH, Nolla H, Lenardo MJ and Schlissel MS. 2009. NF-kappaB activity marks cells engaged in receptor editing. J Exp Med. 206: 1803–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Claudio E, Saret S, Wang H and Siebenlist U. 2009. Cell-autonomous role for NF-kappa B in immature bone marrow B cells. J Immunol. 182: 3406–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dejean AS, Beisner DR, Ch’en IL, Kerdiles YM, Babour A, Arden KC, Castrillon DH, DePinho RA and Hedrick SM. 2009. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat Immunol. 10: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luron L, Saliba D, Blazek K, Lanfrancotti A and Udalova IA. 2012. FOXO3 as a new IKK-epsilon-controlled check-point of regulation of IFN-beta expression. Eur J Immunol. 42: 1030–1037. [DOI] [PubMed] [Google Scholar]
- 45.Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, Raman A, Rust AG, Bergthaler A, Aitchison JD and Aderem A. 2012. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature. 490: 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stienne C, Michieletto MF, Benamar M, Carrie N, Bernard I, Nguyen XH, Lippi Y, Duguet F, Liblau RS, Hedrick SM, Saoudi A and Dejean AS. 2016. Foxo3 Transcription Factor Drives Pathogenic T Helper 1 Differentiation by Inducing the Expression of Eomes. Immunity. 45: 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riou C, Yassine-Diab B, Van grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, Campos-Gonzalez R, Balderas RS, Kelvin D, Sekaly RP and Haddad EK. 2007. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med. 204: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tzelepis F, Joseph J, Haddad EK, Maclean S, Dudani R, Agenes F, Peng SL, Sekaly RP and Sad S. 2013. Intrinsic role of FoxO3a in the development of CD8+ T cell memory. J Immunol. 190: 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Grevenynghe J, Procopio FA, He Z, Chomont N, Riou C, Zhang Y, Gimmig S, Boucher G, Wilkinson P, Shi Y, Yassine-Diab B, Said EA, Trautmann L, El Far M, Balderas RS, Boulassel MR, Routy JP, Haddad EK and Sekaly RP. 2008. Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med. 14: 266–274. [DOI] [PubMed] [Google Scholar]
- 50.Sullivan JA, Kim EH, Plisch EH, Peng SL and Suresh M. 2012. FOXO3 regulates CD8 T cell memory by T cell-intrinsic mechanisms. PLoS Pathog. 8: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch’en IL, Stockmann C, Katayama CD and Hedrick SM. 2010. Foxo transcription factors control regulatory T cell development and function. Immunity. 33: 890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V and Nussenzweig MC. 2005. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 201: 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panigrahi AK, Goodman NG, Eisenberg RA, Rickels MR, Naji A and Luning Prak ET. 2008. RS rearrangement frequency as a marker of receptor editing in lupus and type 1 diabetes. J Exp Med. 205: 2985–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z and Davidson A. 2012. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 18: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, Schieferl S, Hom J, Jenks S, Feldman RJ, Mehr R, Wei C, Lee FE, Cheung WC, Rosenberg AF and Sanz I. 2015. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol. 16: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, Chen H, Zhao LD, Zeng XF, Zhang FC, Tang FL, He W, Cao XT, Zhang X and Lipsky PE. 2014. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med. 6: 246ra299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.