

Abstract
Bone remodeling is controlled by the actions of bone-degrading osteoclasts and bone-forming osteoblasts (OBs). Aging and loss of estrogen after menopause affect bone mass and quality. Estrogen therapy, including selective estrogen receptor modulators (SERMs), can prevent bone loss and increase bone mineral density in post-menopausal women. Although investigations of the effects of estrogen on osteoclast activity are well advanced, the mechanism of action of estrogen on OBs is still unclear. The proline-rich tyrosine kinase 2 (Pyk2) is important for bone formation and female mice lacking Pyk2 (Pyk2-KO) exhibit elevated bone mass, increased bone formation rate and reduced osteoclast activity. Therefore, in the current study, we examined the role of estrogen signaling on the mechanism of action of Pyk2 in OBs. As expected, Pyk2-KO OBs showed significantly higher proliferation, matrix formation, and mineralization than WT OBs. In addition we found that Pyk2-KO OBs cultured in the presence of either 17β-estradiol (E2) or raloxifene, a SERM used for the treatment of post-menopausal osteoporosis, showed a further robust increase in alkaline phosphatase (ALP) activity and mineralization. We examined the possible mechanism of action and found that Pyk2-deletion promotes the proteasome-mediated degradation of estrogen receptor α (ERα), but not estrogen receptor β (ERβ). As a consequence, E2-signaling via ERβ was enhanced in Pyk2-KO OBs. In addition, we found that Pyk2-deletion and E2-stimulation had an additive effect on ERK phosphorylation, which is known to stimulate cell differentiation and survival. Our findings suggest that in the absence of Pyk2, estrogen exerts an osteogenic effect on OBs through altered ERα and ERβ signaling. Thus, targeting Pyk2, in combination with estrogen or raloxifene, may be a novel strategy for the prevention and/or treatment of bone loss diseases.
Keywords: Pyk2, estrogen, estrogen receptor, SERMs, osteoblast, bone formation
1. Introduction
Bone remodeling involves the coordinated actions of osteoblasts (OB) and osteoclasts, which are regulated by hormones, growth factors, transcription factors, and mechanical stimuli (Compston, 2001). Excessive resorption and/or decreased bone formation can lead to low bone mass diseases such as osteoporosis. The risk of developing osteoporosis increases with age and is common in post-menopausal women due to declining levels of the sex steroid hormone, estrogen. Evidence suggests that estrogen replacement therapy, including selective estrogen receptor modulators (SERMs), can prevent bone loss thus increasing bone mineral density in post-menopausal women (Riggs and Hartmann, 2003). Moreover, estrogen therapy decreases the risk of skeletal fractures of the hip, spine, and wrist in post-menopausal women (Compston, 2001). The osteoprotective effects of estrogen are largely the result of its pro-apoptotic actions on OCs, which reduces osteoclast numbers and bone resorption (Imai et al., 2013, Vinel et al., 2016). Soon after the onset of menopause, osteoclast numbers rapidly increase due to the declining levels of estrogen. However, in later menopause, reduced OB activity is also observed (Clarke and Khosla, 2010), which decreases the bone remodeling rate (Demontiero et al., 2012).
The actions of estrogen on target tissues are mediated by genomic and non-genomic pathways through two major estrogen receptor (ER) subtypes, ERα and ERβ (Imai et al., 2010,Krum and Brown, 2008). The genomic pathway of estrogen occurs through its binding to nuclear ERs either by direct interaction of the estrogen-ER complex with estrogen response elements (ERE) localized within transcription promoter regions or via indirect binding to EREs via other transcription factors. In contrast, the non-genomic actions of estrogen or rapid estrogen responses occur via interaction of estrogen with ERs localized at the plasma membrane or found in the cytoplasm, resulting in the activation of signal transduction pathways, including calcium flux and kinase activation within target cells (Imai et al., 2010,Ren and Wu, 2012,Kassem, 1997,Bonnelye and Aubin, 2002,Arts et al., 1997,Roforth et al., 2014,Tee et al., 2004). A novel estrogen-binding cell surface G-protein-coupled receptor (GPER1 or GPR30) has also been reported, and is known to mediate the non-genomic estrogen pathway by stimulating cyclic AMP (Ren and Wu, 2012,Heino et al., 2008,Evans et al., 2016,Hadjimarkou and Vasudevan, 2017,Ford et al., 2011).
OBs express both ERα and ERβ, but they are differentially expressed during OB differentiation (Bonnelye and Aubin, 2002,Arts et al., 1997). Although some variability in the relative expression level of these receptors is seen using different OB cell lines, ERα mRNA levels generally appear to increase early in the OB differentiation process and then plateau or decrease during mineralization (Arts et al., 1997). In contrast, ERβ mRNA levels appear to show a more constitutive expression pattern (Arts et al., 1997,Heino et al., 2008,Chen et al., 2004,Onoe et al., 1997,Wiren et al., 2002,Teplyuk et al., 2008,Noda-Seino et al., 2013,Bord et al., 2003). The ERs are also regulated by estrogen, and one study reported that ERα mRNA is increased after estradiol treatment of human primary OBs for 24 hours, but no data regarding the effect of estradiol on ERβ mRNA levels was presented (Bord et al., 2003). In addition to ERα and ERβ, OBs express GPER1 which has been shown to be abundant during the proliferation of mouse and human OBs, although it does not appear to be expressed in the MC3T3-E1 osteoblastic cell line (Teplyuk et al., 2008,Noda-Seino et al., 2013).
The proline-rich tyrosine kinase, Pyk2, belongs to the family of focal adhesion kinases. Pyk2 is linked to a variety of cellular activities including proliferation and migration (Boutahar et al., 2004,Buckbinder et al., 2007,Gil-Henn et al., 2007,Kacena et al., 2012). Pyk2 is important for the regulation of bone mass and for the function of osteoclasts and OBs (Buckbinder et al., 2007,Gil-Henn et al., 2007,Kacena et al., 2012,Bruzzaniti et al., 2005,Eleniste and Bruzzaniti, 2012,Cheng et al., 2013,Eleniste et al., 2016). Our studies and others demonstrated that female Pyk2-KO mice exhibit increased bone mass which is due to increased OB differentiation and bone formation, as well as decreased osteoclastic bone resorption (Buckbinder et al., 2007,Gil-Henn et al., 2007). In the current study, we investigated the mechanism of action of Pyk2 and estrogen in OB bone formation in vitro. Our studies suggest that the Pyk2-deletion and estrogen-stimulation have an additive effect on OB differentiation and mineralization by regulating in part the expression of the ERs in OBs.
2. Materials and Methods
2.1. Preparation of calvaria-derived OBs from WT and Pyk2-KO mice
C57BL/6 mice (WT) were obtained from Jackson Laboratories. Pyk2-KO mice (lacking the PTK2B gene) were provided by Pfizer, Groton, CT, as described previously (Buckbinder et al., 2007,Okigaki et al., 2003). Pyk2-KO mice have been back-crossed for more than 10 generations and maintained on a C57BL/6 background. All mice used in this project were handled according to the guidelines of the American Association for Laboratory Animal Science using Institutional Animal Care and Use Committee (IACUC) approved protocols (IACUC approval: DS000885R) and in according with the NIH (Guide for the Care and Use of Laboratory Animals, 1996).
For the current studies, murine calvarial cells were prepared using the previously described protocol (Kacena et al., 2012,Cheng et al., 2013). Mouse calvaria are reliable source of primary OBs which are easy to isolate, produce larger quantities of cells than bone-derived OBs. In addition, the isolation of OBs from long bones by collagenase digestion also releases osteocytes. Both long bone-derived and calvaria-derived OBs have the capacity to proliferate, express alkaline phosphatase activity and can underdo differentiation and mineralization when cultured in osteogenic media. To prepare OBs, calvaria from neonatal mice 2-3 days old were pretreated with 10 mM EDTA in PBS for 30 minutes. For each genotype, male and female 2-3 day mouse pups were combined for all preparation of calvarial OBs. Next, the calvaria were subjected to sequential collagenase digestions with 0.1% collagenase type IA (Sigma, MO, USA) from Clostridium histolyticum in serum-free α-MEM media (Hyclone, UT, USA) with 1% (v/v) Penicillin/Streptomycin for 30 minutes in each digestion at 37°C under shaking conditions (200 rpm). Cells were collected following incubation in collagenase from fractions 3–5, which consist of about 95% OBs or OB precursors (Kacena et al., 2012). Calvarial OBs were passaged twice and expanded prior to use.
To minimize any potential non-specific estrogenic effects of phenol red or serum, all cells were cultured in phenol-red free α-MEM media (Hyclone) supplemented with 2.5% FBS (Hyclone). We also compared the effects of E2 on OBs cultured in 2.5% charcoal-stripped, heat-inactivated fetal bovine serum (Valley Biomedical, BS3032CS), with similar results, and representative data are shown in the supplementary figures. Unless otherwise specified, a concentration of 100 nM E2 was used, and was determined in dose-finding studies as discussed in the results and supplementary figures. This concentration is consistent with relevant literature (Cheng et al., 2002,Taranta et al., 2002,Zhou et al., 2001). To determine if E2 was acting through ERα and/or ERβ, selective receptor agonists or antagonists were also used. The ERα-specific agonist, propyl-pyrazoletriol (PPT) and the ERβ-specific agonist, diarylpropionitrile (DPN) were purchased from Tocris. The ERβ antagonist, 4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP) was purchased from Sigma and, as reported by the manufacturer, is 36-fold more selective for ERβ over ERα. Raloxifene was purchased from Sigma.
2.2. Quantitative Real-time Polymerase Chain Reaction (QPCR)
WT or Pyk2-KO calvarial OBs, 5 × 104 cells, were cultured in 12-well plates in osteogenic media (phenol-red free α-MEM (Thermo Fisher, NY, USA) containing 50 μM ascorbic acid (AA, Sigma) and 5 mM β-glycerol phosphate (β-GP, Sigma)) in the presence or absence of 100 nM of 17β-estradiol (E2, Sigma) for 4 or 28 days. Total RNA was isolated using RNeasy® Mini Kit (Qiagen, CA, USA). The remnant genomic DNA in RNA samples were removed by digestion with DNase I enzyme (Fisher Scientific, PA, USA). Complementary DNA (cDNA) was synthesized using the Transcriptor First Strand cDNA Synthesis Kit according to the manufacturer's instructions (Roche, IN, USA). QPCR was used to quantify mRNA expression from WT and Pyk2-KO OBs. For QPCR, the SYBR® green PCR Master Mix (Applied Biosystems, Warringtons, UK) was used following the manufacturer's instructions. For each gene, a calibration curve was performed. All oligonucleotide primers were tested to ensure correct specificity and sensitivity. All samples were run in the ABI Prism® 7000 sequence detection system using STEP1 Software Solutions. The threshold cycle (Ct) for each test gene was normalized against the 18S or GAPDH housekeeping genes. The housekeeping genes were unaffected by E2 stimulation. For all graphs the ΔCt values (absolute mRNA values) are shown which were calculated using the equation ΔCt = Ct (gene of interest) – Ct (housekeeping gene). The following oligonucleotide primer sequences were used:
ERα forward primer: CTCAACCGCCCGCAGCTCAA
ERα reverse primer: GTAGGCGATGCCCGACTGGC
ERβ forward primer: ACCCTCACTGGCACGTTGCG
ERβ reverse primer: GGCTTGCGGTAGCCAAGGGG
GPER1/GPR30 forward primer: CAGTACGTGATTGCCCTCTTC
GPER1/GPR30 reverse primer: GTTGCCCACAAAGCCAATAG
c-fos forward primer: ACTTCTTGTTTCCGGC
c-fos reverse primer: AGCTTCAGGGTAGGTG
Collagen type 1 forward primer: AACCTGGTGCGAAAGGTGAA
Collagen type 1 reverse primer: AGGAGCACCAACGTTACCAA
Alkaline phosphatase forward primer: ACTGATGTGGAATACGAACTGGATGAGAAGG
Alkaline phosphatase reverse primer: CAGTCAGGTTGTTCCGATTCAATTCATACTGC
Osteocalcin forward primer: TCTCTCTGACCTCACAGATGCCAAGC
Osteocalcin reverse primer: GGACTGAGGCTCCAAGGTAGCG
Runx2 forward primer: TCCACAAGGACAGAGTCAGATTACAG
Runx2 forward primer: CAGAAGTCAGAGGTGGCAGTGTCATC
Osterix forward primer: TCTGCTTGAGGAAGAAGCTCACTATGGC
Osterix forward primer: AGGCAGTCAGACGAGCTGTGC
18S forward primer: AGTCCCTGCCCTTTGTACACA
18S reverse primer: CGATCCGAGGGCCTCACTA
GAPDH forward primer: CTTTGGCATTGTGGAAGGGC
GAPDH reverse primer: CAGGGATGATGTTCTGGGCA
2.3. Cell growth and proliferation assays
Cell proliferation was performed using the CellTiter 96® AQeous Non-Radioactive Cell Proliferation Assay (MTS) kit according to the manufacturer's instructions (Promega). Cell growth was determined by counting the number viable OBs as determined by exclusion of 0.2% (final concentration) trypan blue. WT or Pyk2-KO calvarial OBs, 5 × 104 cells, were cultured in the presence or absence of 100 nM of E2 for 1-4 days in 12-well plates. The 100 nM E2 concentration used for these studies was based on a dose-finding pilot study (shown herein) as well as relevant literature (Cheng et al., 2002,Taranta et al., 2002,Zhou et al., 2001). 100 nM E2 also produced the highest osteogenic response in primary OBs and in the pre-OB cell line, MC3T3-E1 (data not shown). OBs from each group were harvested, and the cell number per well was counted using a hemocytometer. For cell viability/cytotoxicity assessment, the Cell counting Kit-8 (Dojindo Molecular Technologies) was used as per the manufacturer's instructions.
2.4. Quantitative alkaline phosphatase (ALP) activity assay
WT or Pyk2-KO calvarial OBs were cultured in 12-well plates at 5 × 104 cells in osteogenic media containing 50 μM AA and 5 mM β-GP in the presence or absence of 100 nM of E2 or 0.1-10 nM of raloxifene (Sigma) for 4-28 days. Cells were lysed in the mRIPA buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1% NP-40, 0.25% Sodium deoxycholate) supplemented with 10 μg/mL leupeptin hydrochloride, 10 μg/mL aprotinin, 10 μg/mL pepstatin, 1 mM PMSF, 1mM sodium fluoride, and 1mM sodium orthovanadate. The lysate was sonicated for 5 minutes, and then centrifuged at maximum speed for 5 minutes and the supernatant was collected. ALP activity was assayed by adding cell lysate to ALP substrate containing 2 mg/mL p-nitrophenyl phosphate (Sigma) in 1.5 M alkaline buffer (Sigma), and then the mixture was incubated for 50 minutes at 37°C in a dark atmosphere. The enzymatic reaction was stopped by adding 20 mM NaOH to the mixture, and the absorbance at 405 nm was recorded using a spectrophotometer. ALP activity was normalized by total protein using a Pierce™ BCA protein assay kit.
2.5. Quantitative analysis of mineralization
WT or Pyk2-KO calvarial OBs, 5 × 104 cells, were cultured in 12-well plates in osteogenic media containing 50 μM AA and 5 mM β-GP in the presence of E2 (1, 10, 100 nM) or raloxifene (0.1-10 nM) for up to 28 days in 2.5 % FBS or 2.5% charcoal stripped FBS (supplementary Figure S1). Cells were washed with PBS and fixed in 4% formaldehyde/PBS for 15 min and washed twice in PBS and stored at 4°C. Prior to staining, cells were washed twice with deionized water (dH2O). Alizarin Red S (40 mM, pH 4.2, Sigma) was used to stain calcium deposits in each well for 10 minutes with agitation. The Alizarin Red S stained samples were washed 5 times with dH2O, followed by 15 minutes of PBS washing on the shaker. Next, the bound Alizarin Red S was extracted with 1% cetyl pyridinium chloride (Sigma) in 10 mM sodium phosphate (pH 7.0) for 15 minutes on the shaker at room temperature. Calcium deposition was measured by recording the absorption of extracted Alizarin Red S at 562 nm using a spectrophotometer.
2.6. Western blot analysis and co-immunoprecipitation studies
WT or Pyk2-KO calvarial OBs, 2 × 105 cells, were cultured in 6-well plates for 4 days in the presence or absence of 100 nM of E2. In addition, OBs were differentiated in osteogenic media containing 50 μM AA and 5 mM β-GP with or without 100 nM E2 for 28 days. OB cells were lysed in mRIPA buffer supplemented with protease/kinase inhibitors as previously described. The amount of protein was quantified using the Pierce™ BCA protein assay kit, and proteins were then resolved by SDS-PAGE electrophoresis. For co-immunoprecipitation studies, 5 μg primary antibody per 500 μg of cell lysate was used. Tubes were incubated at 4°C overnight with rotation. Immunoprecipitations were performed using protein G-agarose beads (Roche) for 1 hr. Immunoprecipitation of ERa was performed using a monoclonal antibody (Santa Cruz) followed by Western blotting for ubiquitin with a polyclonal antibody (Proteintech) or vice versa. Nitrocellulose membranes were incubated with the primary antibody overnight at 4°C. An anti-mouse antibody conjugated with horseradish peroxidase (HRP) or anti-rabbit HRP (Promega, WI, USA) were used as secondary antibodies. Proteins were detected using the enhanced chemiluminescence (ECL) reagent (SuperSignal™ West Pico Chemiluminescence Substrate, Sigma) according to the manufacturer's recommendations. Radiographic film was used to record the emitted signal from the membrane. As necessary, protein bands were quantified by densitometry using ImageJ software.
2.7. Statistical analyses
All data was reproduced a minimum of twice. All data were analyzed by one-way analysis of variance (ANOVA) and/or two-way ANOVA followed by post hoc multiple comparisons where appropriate. Data was analyzed by using SPSS 24 software (IBM Corporation, Armonk, NY, USA). With the exception of Western blot data, all data are shown as mean ± standard error of mean (SEM) of triplicate samples and a significant difference was determined at p ≤ 0.05.
3. Results
3.1. Pyk2-KO OBs exhibit higher basal proliferation than WT OBs
To determine if estrogen affects OB proliferation and cell growth, an equal number of calvarial-derived OBs from WT and Pyk2-KO mice were cultured in phenol-free growth medium supplemented with or without 100 nM 17β-estradiol (E2) for 4 days. The E2 concentration used was based on a dose-finding pilot study (Figure 3B and supplementary Figure S1) as well as relevant literature (Cheng et al., 2002,Taranta et al., 2002,Zhou et al., 2001). As shown in Figure 1A, Pyk2-KO OB control cultures (without estrogen) had higher OB number compared to WT OBs (p<0.05), suggesting that Pyk2-KO OBs maintain a higher level of basal proliferation than WT OBs. In cultures treated with E2 for 4 days, WT OB number was significantly increased, compared to untreated WT OBs. In contrast, E2 had no effect on the number of Pyk2-KO OBs after 1 or 4 days of culture (p<0.05).
Figure 3. Effect of Pyk2-deletion and estrogen stimulation on OB activity.
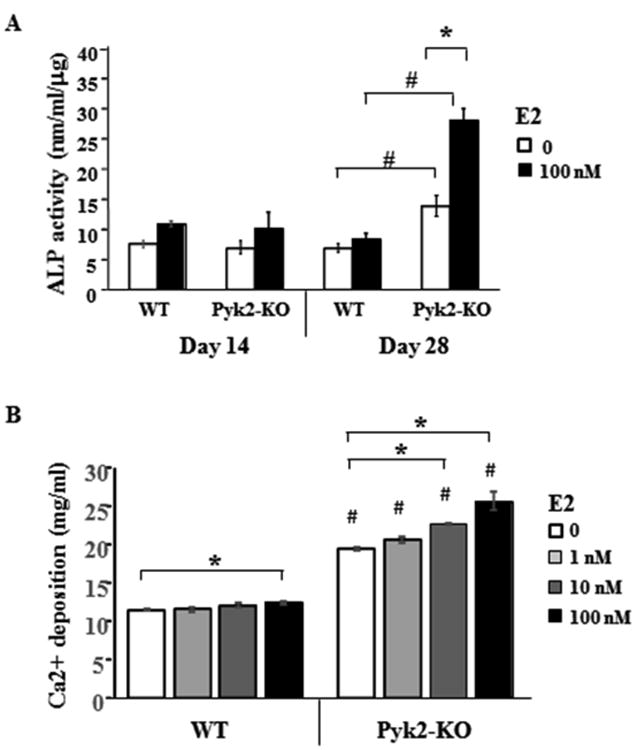
A) WT and Pyk2-KO OBs were cultured under osteogenic conditions containing ascorbic acid and β-GP, with or without 100 nM E2. ALP activity was quantified at day 14 and 28. B) WT and Pyk2-KO OBs were cultured in osteogenic media without E2 or with 1, 10 or 100 nM E2 for 28 days, and then analyzed using a quantitative mineralization assay which is based on elution of Alizarin Red S from calcium bound to the collagen matrix. All assays were performed in triplicate and error bars represent mean ± SEM. Experiments were repeated 3 or more times. Statistical significance of p<0.05 is indicated (*) within a genotype and (#) between genotypes for the matching E2-treated groups.
Figure 1. Effect of Pyk2-deletion and estrogen on OB cell number and proliferation.
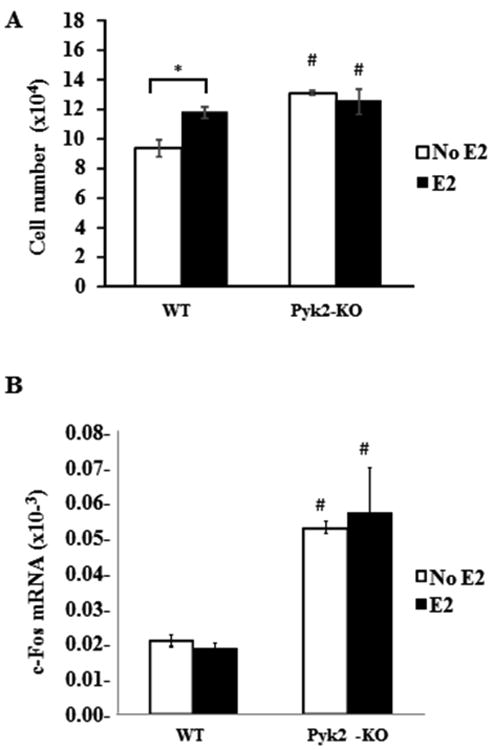
A) WT and Pyk2-KO OBs from neonatal calvaria were plated at 5×104 cells and cultured in the presence or absence of 100 nM E2. Cells were counted after 4 days. B) QPCR analysis was used to determine c-fos mRNA expression in WT and Pyk2-KO OBs cultured for 4 days in the presence or absence of 100 nM E2. 18S was used as the housekeeping control to normalize the mRNA transcript under investigation. The 18S Ct values were WT (14.26 ± 0.09) and Pyk2-KO (14.56 ± 0.21). The data are shown as mean of ΔCT ± SEM of triplicate or quadruplicate samples. Statistical significance of p<0.05 is indicated (*) for effects within a genotype and (#) for significant changes for the same treatment between WT and Pyk2-KO OBs.
Transcription factors of the AP1 complex such as c-fos are highly expressed during OB proliferation and in the initial stages of differentiation (Machwate et al., 1995,Wagner, 2002). Therefore, we examined the effects of E2 on c-fos mRNA levels in WT and Pyk2-KO OBs by QPCR (Figure 1B). These studies revealed significantly higher c-fos mRNA levels in vehicle-treated Pyk2-KO OBs, compared to WT OBs (2.5 fold, p=0.01), consistent with our findings that Pyk2-KO exhibit a higher basal proliferation rate. However, E2 stimulation of either Pyk2-KO or WT OBs did not alter c-fos mRNA expression. This latter finding differed from the E2-stimulated increase in WT OB number (Figure 1A) and may indicate that the upregulation of c-fos mRNA is a transient event.
3.2 Estrogen increases alkaline phosphatase (ALP) activity and mineralization in Pyk2-KO OBs
To investigate the effect of E2 on the expression of osteoblast markers during OB differentiation, primary calvarial OBs from WT and Pyk2-KO mice were cultured under osteogenic conditions in the presence or absence of E2 for either 4 days (early OB markers) or 28 days (mature OBs markers). QPCR analysis of collagen type 1, osterix and Runx2 (early OB differentiation) and osteocalcin (late OB differentiation) was performed. Pyk2-KO OBs exhibited significantly higher collagen type 1 mRNA expression than WTs (1.9 fold, p=0.03) (Figure 2A). Notably, E2 supplementation also significantly increased collagen type 1 mRNA expression in Pyk2-KO (p=0.04), but not WT OBs. Runx2 expression was also higher in Pyk2-KO OBs but E2 decreased Runx2 expression in Pyk2-KO OBs down to WT levels (Figure 2C). These findings are consistent with reports that Runx2 protein is upregulated in immature osteoblasts, but downregulated in mature osteoblasts (Komori, 2010). However, Osterix, which is expressed early in differentiation was not different between WT and Pyk2-KO and was also unchanged in the presence of E2 (Figure 2C). In Pyk2-KO OBs cultured for 28 days, we found increased expression of osteocalcin (OCN) mRNA compared to WT (8.75 fold, p=0.007). However, no further change in OCN mRNA levels were detected in the E2-stimulated Pyk2-KO or WT OBs (Figure 2D).
Figure 2. Effect of Pyk2-deletion and estrogen stimulation on markers of OB activity.
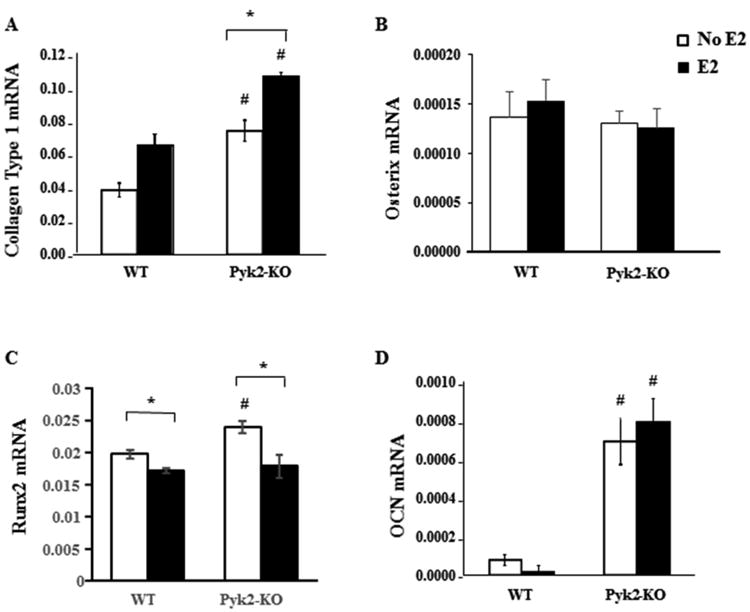
WT and Pyk2-KO OBs were cultured under osteogenic conditions containing ascorbic acid and β-glycerol phosphate (β-GP) in the presence or absence of 100 nM E2 for 4 days or 28 days. QPCR analysis was used to determine mRNA expression of collagen type 1, osterix and Runx2 (day 4) or osteocalcin (OCN; day 28). Data are absolute mRNA using 18S (A; D) or GAPDH (B; C) as the calibrator gene. The mean Ct values for the relevant housekeeping genes are: collagen (WT, 14.26 ± 0.09; Pyk2-KO, 14.56 ± 0.21); osterix (WT, 21.33 ± 0.09; Pyk2-KO, 21.65 ± 0.05); Runx2 (WT, 21.13 ± 0.07; Pyk2-KO, 20.92 ± 0.07) and OCN (WT, 14.26 ± 0.09; Pyk2-KO, 14.56 ± 0.21). The graphs shown as mean of ΔCT ± SEM of triplicate samples. Statistical significance of p<0.05 is indicated (*) within a genotype and (#) between genotypes. White and black bars represent no E2 or plus E2, respectively.
Using a quantitative biochemical ALP assay, we further examined the effects of E2 on the differentiation of OBs lacking Pyk2. WT and Pyk2-KO OBs were grown in osteogenic media supplemented without or with E2 for 14 or 28 days. No differences in ALP activity were observed between WT and Pyk2-KO OBs cultured with E2 for 14 days (Figure 3A). Similar results were obtained when cells were cultured for only 4 days (data not shown). However, in 28 day cultures, an increase in ALP activity was observed in Pyk2-KO OBs cultured without E2 (13.8±1.7 nM/mL/μg) compared to WT OBs (6.8±0.7 nM/mL/μg) (p<0.05). Moreover, E2 supplementation robustly increased ALP activity in Pyk2-KO OBs (2-fold, p=0.005) but had no effect on ALP activity in WT OBs. These results suggest that Pyk2-deletion promotes ALP catalytic activity, and that long-term culture with E2 (28 days) has an additive effect on ALP activity in OBs lacking Pyk2.
Since collagen type 1 expression and ALP activity are key steps involved in OB mineralizing activity (Burr and Allen, 2013,Datta et al., 2008,Soares et al., 2008), we examined if E2 promotes extracellular calcium deposition by Pyk2-KO OBs. For these studies, cells were cultured in osteogenic media supplemented with 100 nM E2. In addition, to determine if the E2 effects were dose-dependent, we also used 1 nM and 10 nM E2. In WT OBs, a small increase in mineralization was observed only in cells supplemented with 100 nM E2. In contrast, mineral deposition by Pyk2-KO OBs was markedly increased with both 10 nM and 100 nM E2, compared to vehicle. In addition, all Pyk2-KO OB treatment groups showed significantly higher mineralization than comparable WT OB groups (#) (p<0.05) (Figure 3B). Similar to our findings with ALP, E2 had an additive effect on calcium deposition by Pyk2-KO OBs.
3.3. Raloxifene increases ALP activity and mineral deposition in Pyk2-KO OBs
Raloxifene is an FDA-approved SERM for the treatment of post-menopausal osteoporosis (Russell, 2015,Hegde et al., 2016). Raloxifene binds ERα with approximately 3.5-fold higher affinity than ERβ, whereas estrogen has a similar binding affinity for both ERα and ERβ (Zhu et al., 2006). We investigated if raloxifene also affects the activity of WT and Pyk2-KO OBs, similar to17β-estradiol. Cells were cultured in osteogenic media plus increasing concentrations of raloxifene as indicated. After 28 days, we performed quantitative ALP activity and calcium deposition assays. Raloxifene had no effect on ALP activity in WT OBs at any of the concentrations tested, similar to our findings with E2 (Figure 3C). However, 1 nM and 10 nM raloxifene stimulated ALP activity in Pyk2-KO OBs approximately 1.7 fold and 1.4 fold, respectively, compared to the vehicle-treated Pyk2-KO OBs (p<0.05) (Figure 4A). At the higher 10 nM concentration, a decrease in mineralization was observed compared to 1 nM, which may indicate that the lower concentration of raloxifene, as used by others (Taranta et al., 2002) is better tolerated by the Pyk2-KO OBs during the long 28 day culture conditions.
Figure 4. The effect of raloxifene on ALP activity and mineralization in WT and Pyk2-KO OBs.
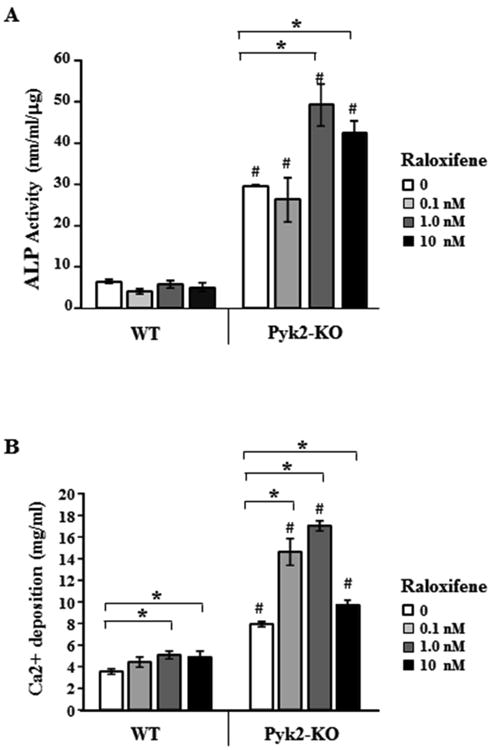
WT and Pyk2-KO OBs were cultured under osteogenic conditions with or without 0.1, 1.0 and 10 nM raloxifene for 28 days. The treatment duration was based on the most robust E2 effects, while the raloxifene concentrations were determined based on pilot studies and published literature (Lin et al., 2004,Matsumori et al., 2009). Assays were performed in triplicate and repeated 3 or more times. The error bars are mean ± SEM. A) ALP activity and B) Calcium deposition. Statistical significance of p<0.05 is indicated (*) for treatments within a genotype and (#) indicates significance between genotypes for the same raloxifene concentration.
Unlike E2 which increased the mineralization of Pyk2-KO OBs but not WT OBs (Figure 3), 1 nM and 10 nM raloxifene increased mineral deposition by both Pyk2-KO and WT OBs (Figure 4B). Nevertheless, Pyk2-KO OBs still exhibited an overall greater percentage increase in calcium deposition in the presence of 0.1, 1 and 10 nM raloxifene than comparably-treated WT OBs (e.g. 116% Pyk2-KO vs 44% WT at 1 nM). Together, these data reveal that E2 and raloxifene both exert significant osteogenic effects in Pyk2-KO OBs.
3.4. Pyk2-deletion promotes ERα protein degradation
To begin to determine the mechanism of action of Pyk2 in the E2-signaling cascade, we examined if Pyk2 regulates the expression of the estrogen receptors. We first examined if deletion of Pyk2 and/or E2 stimulation affects the mRNA expression of ERα and ERβ (Figure 5). However, QPCR analysis revealed no change in the mRNA expression of either ERα or ERβ in WT or Pyk2-KO OBs. E2 can also signal through the membrane estrogen receptor, GPER1 (also known as GPR30), which can activate ERa (Heino et al., 2008,Carmeci et al., 1997,Kang et al., 2015). Our QPCR analyses showed a higher level of GPER1 mRNA in Pyk2-KO OBs compared to WT OBs (Figure 5C). In addition, E2 increased GPER1 mRNA only in Pyk2-KO. Whether the increase in GPER1 mRNA affects protein expression and the biological activity of OBs requires further investigation.
Figure 5. Expression of ERs in WT and Pyk2-KO OBs.
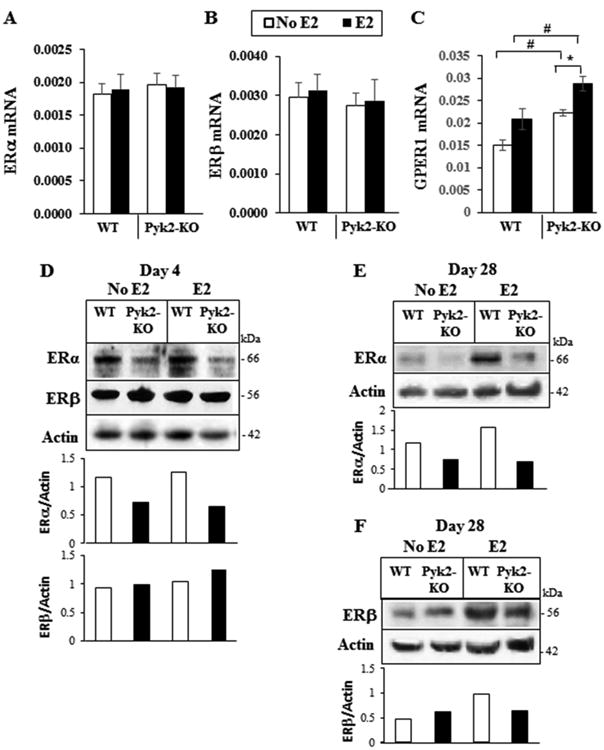
A-C) WT and Pyk2-KO OBs were cultured with or without 100 nM E2 for 4 days. The mRNA expression levels for ERα, ERβ and GPER1 (GPR30) were determined by QPCR. GAPDH was used as the housekeeping gene. The mean Ct values for the GAPDH housekeeping gene are: ERα and ERβ (WT, 14.0 ± 0.04; Pyk2-KO, 14.01 ± 0.04), and GPER1 (WT, 21.13 ± 0.07; Pyk2-KO, 20.89 ± 0.06). The data are mean ± SEM of quadruplicate samples. Statistical significance of p<0.05 is indicated as (*) for E2 treatment effects within a genotype. The (#) indicated significance between genotypes cells for the same treatment. D-F) WT and Pyk2-KO OBs were cultured for 4 days (D) or 28 days (E and F) with or without 100 nM E2. Total cell lysates from WT and Pyk2-KO OBs were resolved by SDS-PAGE and blotted using antibodies specific to ERα or ERβ as indicated (also see supplementary Figure S2). ERα and ERβ protein levels were quantified by densitometry using ImageJ software and normalized to β-actin levels.
Next, we examined possible changes in ERα and ERβ protein levels by Western blotting. Calvarial OBs were grown under osteogenic conditions for 4 days (early OBs) or 28 days (mature mineralizing OBs) in the presence or absence of E2. Western blot analysis revealed lower ERα protein levels in Pyk2-KO OBs compared to WT OBs in day 4 and day 28 cultures (Figure 5D, 5E and supplementary Figure S2). E2 stimulation did not appear to alter ERα protein levels in Pyk2-KO OBs, although a small increase was observed in WT treated with E2 for 28 days. With regards to ERβ, in 4 day cultures without E2, Pyk2-KO OBs and WT OBs exhibited similar levels of ERβ protein, while in the presence of E2, a small increase in ERβ was observed in Pyk2-KO OBs (Figure 5D). In 28 day cultures without E2, ERβ protein levels in Pyk2-KO were similar to WT OBs and although E2 had no effect on this receptor in Pyk2-KO OBs, an increase in ERβ was observed in WT OBs treated with E2 (Figure 5D and 5F).
Given that ERα was reduced in Pyk2-KO OBs cultured without E2 for 4 and 28 days, and that E2 did not appear to significantly alter ERα protein levels, we examined the possible ERα degradation mechanism. Several published studies have shown that ERα can undergo proteosome-mediated degradation (Levi-Montalcini et al., 1996,Petrel and Brueggemeier, 2003). Therefore, we examined the effect of the widely used ubiquitin-proteosome inhibitor, MG-132, on ERα protein levels (Park et al., 2009,Kretzer et al., 2010). Given that E2 did not have a major effect on ERα protein levels, for these studies, we cultured Pyk2-KO and WT OBs without E2, for 4 days and then added 20 μM MG-132 for the final 3 hours of culture. Prior to these studies, we confirmed that WT or Pyk2-KO cell viability in the presence 5 μM, 10 μM or 20 μM MG-132 were not statistically different to the controls, demonstrating it was not cytotoxic to our cells (supplementary Figure S3). After MG-132 treatment of Pyk2-KO and WT OBs, cells were then lysed and separated by centrifugation into the soluble fraction (contains cytosolic proteins) and the insoluble pellet fraction (contains proteins associated with the plasma membrane, nucleus and cytoskeleton) (Figure 6A). Western blot analysis revealed two major molecular weight forms of ERα, corresponding to approximately 66 and 54 kDa, with the lower MW form appearing mostly in the pellet fraction of WT OBs. In vehicle-treated WT OBs, total ERα was higher in the pellet fraction than the soluble fraction. However, for Pyk2-KO OBs, total ERα was similar in both fractions. Although MG-132 treatment did not affect ERα in soluble fraction of WT OBs, a decrease in ERα was observed the pellet fractions. In Pyk2-KO OBs treated with MG-132, a significant increase in ERα in the soluble fraction was observed as expected, while ERα decreased in the pellet. Although the mechanism is still unknown, this finding may indicate that MG-132 leads to the redistribution of ERα from the pellet to the soluble fraction. Alternatively, MG-132 potentially prevents the degradation of accessory proteins which may be important in ERα degradation as reported by others (Ismail and Nawaz, 2005,Nawaz et al., 1999).
Figure 6. Effect of MG-132 on the subcellular distribution of ERα.
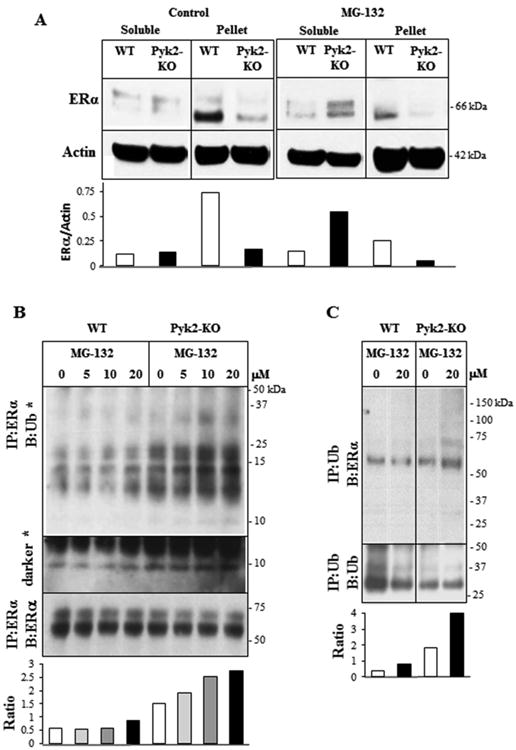
A) WT and Pyk2-KO calvarial OBs were cultured for 4 days. The proteasome inhibitor, MG-132 (20 μM), was added for the final 3 hours of culture. Cells were lysed with mRIPA buffer and then separated into the soluble and insoluble pellet fractions by centrifugation at 13,000 rpm. Western blotting and densitometry was performed to determine the ratio of ERα/β-actin. According to the manufacturer's specification sheets, the ERα antibody (sc-787) can detect multiple isoforms with the molecular weight of native ERα being 66-67 kDa. Short isoforms of 54, 48 and 36 kDa were also reported. Experiments were performed twice and representative data are shown. B) WT and Pyk2-KO OBs were cultured as above and then treated for the final 3 hours with different concentrations of MG-132 (0, 5, 10, 20 μM) as indicated. Cell lysates were subject to immunoprecipitation with a monoclonal antibody to ERα, followed by Western blotting using a polyclonal antibody to ubiquitin. (*) indicates a darker blot showing the ∼8 kDa ubiquitin band. Membranes were stripped and reblotted for ERα. The ratio of the densitometry of the IP:ERα/B:Ub blot (upper panel) to the IP:ERα/B:ERα blot (lower panel) was used to normalize for the amount of the immunoprecipitated protein (ratio graph). Please see supplementary Figure S4 for full blots. C) WT and Pyk2-KO OBs were cultured with and without E2 and then treated for 3 hours with 20 μM MG-132 for the final 3 hours. Ubiquitin was immunoprecipitated and blotted for ERα. The membrane was stripped and then reblotted for ubiquitin. The ratio of the densitometry of the IP:Ub/B:ERα blot (upper panel) to the IP:Ub/B:Ub blot (lower panel) was used to normalize for the amount of the immunoprecipitated protein (ratio graph). All major ubiquitin or ERα bands observed in blots were used for the densitometry analyses.
To further explore if ERα was being degraded via the ubiquitin proteosome pathway, we treated OBs with MG-132 as described above and then examined its association with ubiquitin (Ub) by co-immunoprecipitation followed by Western blot analysis using anti-ubiquitin (Proteintech) or anti-ERα antibodies (Figure 6B and 6C), and vice versa. MG-132 increased the levels of the ERα-Ub complex in a dose-dependent manner in Pyk2-KO OBs (Figure 6B). For WT OBs, a small increase was only observed in cells treated with 20 μM MG-132. Overall, the level of ERα-Ub complex was much higher in Pyk2-KO OBs than WTs at all MG-132 concentrations and in the vehicle control group, indicating a basal increase in ubiquitin binding to ERα in Pyk2-KO OBs. Multiple molecular weight species were also observed, with the most abundant being approximately 24 kDa. A protein species corresponding to the ubiquitin monomer (∼8 kDa) was also observed in darker blots. Similarly, when the ubiquitin antibody was used for immunoprecipitation, a higher level of the Ub-ERα complex was observed in vehicle-treated and MG132-treated Pyk2-KO OBs compared to WTs (Figure 6C). These findings suggest that MG-132 prevents the degradation of ERα via the ubiquitin proteasome pathway. However, given that Ub-ERα complex was higher in vehicle-treated Pyk2-KO OBs, other mechanisms for ERα degradation may also be involved.
3.5. ERβ signaling in Pyk2-KO OBs promotes mineralization
Based on our finding that ERα was decreased in Pyk2-KO OBs, we determined if signaling via ERβ was the predominate mechanism for the increased mineralizing activity of Pyk2-KO OB in the presence of E2. First, to determine if ERα signaling was indeed blunted in Pyk2-KO OBs, we cultured cells for 21 days under osteogenic conditions, and in the presence or absence of the ERα-specific agonist, propyl-pyrazoletriol (PPT). As expected, PPT (40 and 100 nM) had no significant effect on the mineralizing activity of Pyk2-KO OBs (Figure 7A). WT OBs also failed to show a PPT response. This result was not unexpected given that E2 had only a minor effect on WT OBs mineralization when used at 100 nM with no effect observed at 1 and 10 nM (Figure 3B). Next, we examined mineral deposition by WT and Pyk2-KO calvarial OBs cultured in the presence or absence of the ERβ-specific agonist, diarylpropionitrile (DPN). The treatment time and drug concentrations used (100 nM and 400 nM) were based on our previous experiments and relevant literature (Somjen et al., 2011,Galea et al., 2013). Although WT OBs treated with DPN showed no change in mineralization, we observed an increase in mineralization in Pyk2-KO OBs cultured with 400 nM DPN, compared to vehicle-treated Pyk2-KO OBs (Figure 7B). Lastly, WT and Pyk2-KO OBs were cultured in the presence and absence of E2 in combination with increasing concentrations of the ERβ-specific antagonist, 4-[2-Phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP). As reported by the manufacturer, PHTPP is a synthetic, highly selective antagonist of ERβ and possesses 36-fold selectivity for ERβ over ERα. As shown in Figure 7C, PHTPP (20, 50 and 100 nM) had no effect on WT OBs in the absence of E2, but when added in combination with E2, decreased the mineralizing activity of WT OBs. Importantly, PHTPP significantly reduced mineralization in Pyk2-KO OBs in the absence of E2, and had an even greater effect on Pyk2-KO OBs mineralization when combined with E2. Taken together, these data suggest that the ERβ receptor remains functional in Pyk2-KO OBs, and that E2 likely acts via ERβ to increase mineral deposition by Pyk2-KO cells.
Figure 7. The effects of an ERβ agonist and antagonist on OB mineralization.
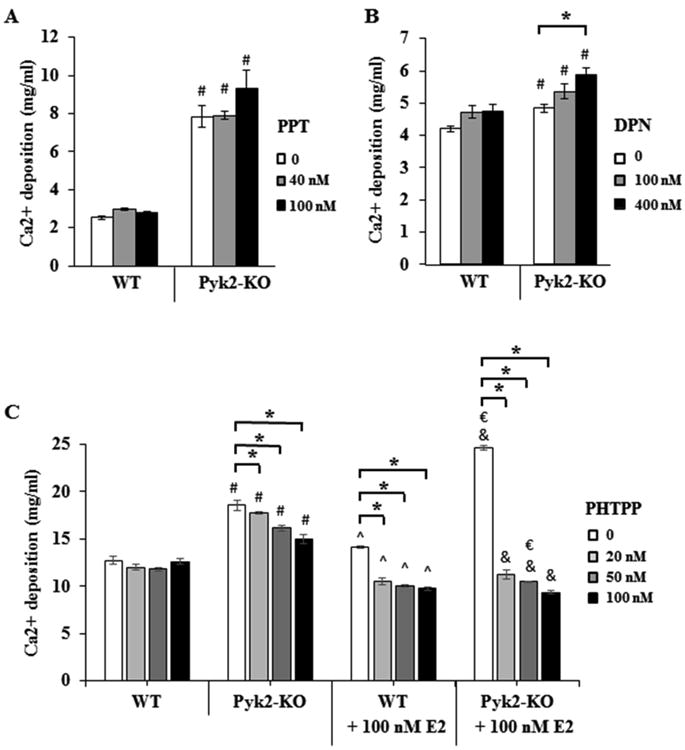
WT and Pyk2-KO OBs were cultured for 21 days under osteogenic conditions containing, A) the ERβ-specific agonist (DPN) or B) the ERα-specific agonist (PPT). C) Cells were cultured with the ERβ-specific antagonist (PHTPP) with or without E2 for 28 days. After culture, the cells were stopped and analyzed for mineral deposition using a quantitative Alizarin Red S mineralization assay. Experiments were performed in triplicate and replicated 2-3 times and representative data are shown. For A) and B), statistical significance of p<0.05 is indicated as (*) for drug effects within a genotype or (#) between genotypes. C) In the PHTPP graphs, the (#) indicates significance between WT (no E2) versus Pyk2-KO (no E2), whereas (&) indicates significant differences between WT (+E2) versus Pyk2-KO (+E2) for the same drug concentration. The symbols (^) and (€) are used to indicate WT (no E2) versus WT (+E2), and Pyk2-KO (no E2) versus Pyk2-KO (+E2), respectively.
3.6. Pyk2-deletion and E2-stimulation activate the ERK pathway
OB proliferation and differentiation are controlled by several signaling pathways, including mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPKs/ERKs) and protein kinase B (AKT). The ERK1 (44 kDa) and ERK2 (42 kDa) proteins are expressed in OBs and have important functions in bone metabolism (Ge et al., 2012,Lai et al., 2001). In addition, E2 binding promotes the rapid interaction of ERα and ERβ with the c-Src kinase, leading to ERK and AKT activation (Marino et al., 2006). Since Pyk2-KO OBs displayed significant increases in matrix maturation and mineralization, both of which were further increased with E2 or raloxifene, we investigated if Pyk2-deletion and/or E2 affected downstream activation of ERK. As shown in Figure 8, Pyk2-KO OBs exhibited higher levels of phosphorylated ERK compared to WT OBs. E2 further increased phosphorylated ERK (pERK) levels in both Pyk2-KO and WT OBs. In contrast, total ERK was similar between WT and Pyk2-KO OBs, and E2 increased total ERK only in Pyk2-KO OBs. The overall ratio of pERK/ERK was also higher in Pyk2-KO and was increased with E2 in Pyk2-KO as well as WT OBs (Figure 8 graphs). We also investigated if Pyk2-deletion and/or E2 stimulation affected ERK signaling in OBs cultured for 21 days (mature OBs) (data not shown). Our results revealed no differences in phosphorylated ERK or total ERK protein levels between WT and Pyk2-KO OBs, with or without E2 stimulation, suggesting that the increased mineralization of E2-treated Pyk2-KO OBs was associated with increased ERK signaling in early OBs (day 4). Since the activation of AKT is also associated with cell growth, proliferation and survival (Burr and Allen, 2013,Ge et al., 2012,Lai et al., 2001,Mandal et al., 2016,Ayala-Pena et al., 2013), we also examined phosphorylated and total AKT in our cells (supplementary Figure S5). However, no differences in phosphorylated AKT or total AKT levels between WT and Pyk2-KO OBs cultured for 4 days, with or without E2 treatment were observed. Taken together, these results suggest that Pyk2-deletion combined with E2-stimulation increases the mitogenic activity of early OBs through increased ERK phosphorylation and signaling.
Figure 8. The effect of Pyk2-deletion and estrogen on ERK phosphorylation in OBs.
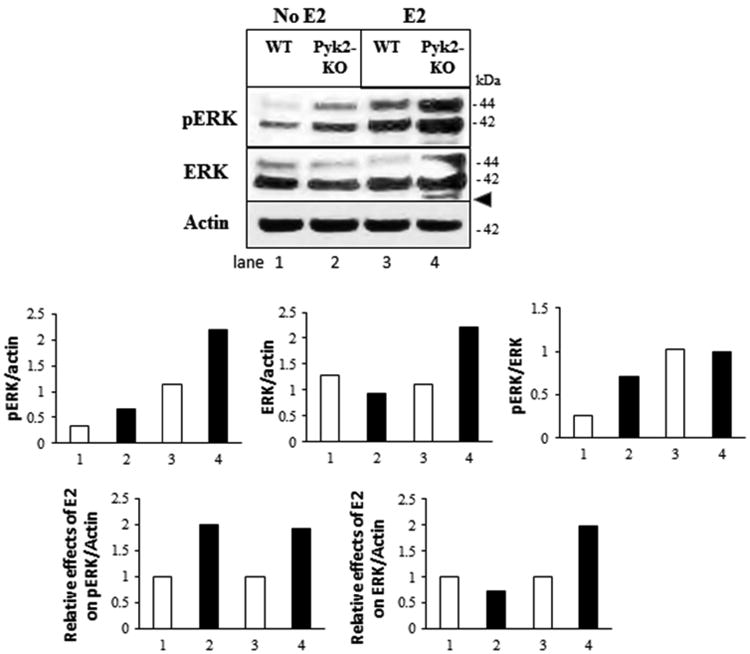
WT and Pyk2-KO OBs were cultured for 4 days with or without 100 nM E2. Cell lysates were blotted with antibodies to phosphorylated ERK (pERK) or total ERK as indicated. Phosphorylated-ERK runs as a doublet at 42 kDa and 44 kDa. An additional non-specific band approximating the molecular weight of phosphorylated p38 was also observed with the pERK antibody (arrowhead). Actin is used as the loading control. Protein bands were analyzed by densitometry (both p42 and p44 were used) and the ratio for effect differences is shown. The graphs for the ratio of pERK/actin, total ERK/actin and pERK/ERK are as indicated. Graphs showing the relative effects of E2 on either pERK or ERK are also shown. N=3 replicates.
4. Discussion
Bone formation by OBs is essential for bone remodeling as well as fracture repair. Our studies and others have shown that Pyk2-KO mice have higher bone mass compared to WT mice. OBs from these mice also show increased bone formation activity (Buckbinder et al., 2007,Gil-Henn et al., 2007). In the current study, we focused on the role of Pyk2 and estrogen on OB activity. We determined that Pyk2-KO OBs exhibit higher proliferation and differentiation than WT OBs as determined by increases in cell number, c-fos mRNA expression, ALP activity and mineralization. Although E2 supplementation for 4 days significantly increased the number of WT OBs, it had little effect on Pyk2-KO OBs number. However, Pyk2-KO OBs cultured with E2 still exhibited a higher cell number than E2-treated WT OBs, suggesting a basal increase in Pyk2-KO OB proliferation in our culture conditions. Of note, we previously reported that the proliferation of both WT and Pyk2-KO OBs is increased by megakaryocytes, which also promoted OB mineralization and increased bone mass (Cheng et al., 2013). However, in those studies, the basal proliferation rate between Pyk2-KO and WT OBs was similar. These differences are likely due to the different culture conditions; in our previous studies we used phenol-red containing media which is known to exert weak estrogenic effects (Berthois et al., 1986), whereas in the current studies we used phenol-free media as well as reduced serum levels.
During the early stages of differentiation as observed in 4 day cultures, we found that collagen type 1 mRNA expression in Pyk2-KO OBs was higher than in WT OBs. Runx2 was also higher in Pyk2-KO OBs, although osterix was unchanged. At later stages of the OB differentiation process (day 21-28), Pyk2-KO OBs also exhibited higher OCN mRNA expression, which is a marker of mature OBs. In addition, ALP activity and calcium deposition in Pyk2-KO OBs were elevated compared to WT OBs. These results confirm that Pyk2 is a negative regulator of OB activity, which is consistent with our previous data as well as other published studies (Buckbinder et al., 2007,Kacena et al., 2012,Cheng et al., 2013,Eleniste et al., 2016). Further, our current data suggest that E2 enhances Pyk2-KO OB differentiation to a greater extent than WT OBs. In support of this, E2 enhanced collagen type 1 in mature differentiated Pyk2-KO OBs (but not in WT OBs), which is necessary for OB mineralization. In addition, Runx2 levels were decreased by E2, which is consistent with reports that Runx2 levels are downregulated in mature osteoblasts (Komori, 2010). Our studies also demonstrated that E2 has an additive effect on ALP activity and calcium matrix deposition in Pyk2-KO OBs. These results were reproduced in media containing hormone-replete FBS or charcoal-stripped FBS, and using several E2 concentrations. Collectively, our data demonstrate that Pyk2-deficiency in OBs leads to accelerated differentiation compared to WT OBs, and that this process is further enhanced by E2.
In the current study, we investigated the mechanism of action of Pyk2 in the E2-signaling cascades by QPCR and Western blotting. Although ERα and ERα mRNA were similar, we found higher GPER1 mRNA in Pyk2-KO which was further increased by E2. GPER1 is a membrane estrogen receptor that binds E2 with high affinity. GPER1 also activates kinase cascades and calcium flux, and has been shown to phosphorylate ERα, suggesting crosstalk between these receptors (Heino et al., 2008,Carmeci et al., 1997,Kang et al., 2015). Although additional studies are required, it is possible that the effects of E2 on increasing mineralization in Pyk2-KO may be mediated in part through GPER1/GPR30.
Our finding that ERα protein levels were reduced in Pyk2-KO OBs, compared to WT OBs (Figure 5), prompted us to investigate the possible mechanism of degradation through the ubiquitin-proteosome degradation pathway. Consistent with published studies reports showing proteosome-mediated degradation (Petrel and Brueggemeier, 2003,Chai et al., 2015,Zhou and Slingerland, 2014), MG-132 increased ERα protein levels in Pyk2-KO OBs, and led to the accumulation of ERα in the cytosol (soluble fraction). Moreover, co-immunoprecipitation assays revealed a concentration-dependent increase in ERa-Ub protein complexes in Pyk2-KO OBs treated with MG-132. Although several publication indicate that ERβ can also undergo ubiquitin proteasome degradation in some cell types (Sanchez et al., 2013,Tateishi et al., 2006), we found similar levels of ERβ in Pyk2-KO OBs and WT OBs, suggesting only ERα is degraded in Pyk2-KO OBs. These findings support the idea that in the absence of Pyk2, ERα is more rapidly degraded in part via the ubiquitin-proteosome pathway. However, given that the ERα-Ub protein complexes were higher in Pyk2-KO OBs than in WT OBs, even in the absence of MG-132, other mechanisms regulating ERα protein complexes may also be involved. It is also known that ERα mRNA expression can vary temporally in OBs, and is higher during matrix maturation and then decreases during mineralization (Onoe et al., 1997,Wiren et al., 2002,Bord et al., 2003). In contrast, ERβ mRNA levels remain relatively constant throughout OB differentiation (Onoe et al., 1997,Wiren et al., 2002,Bord et al., 2003). Therefore, the reduced levels of ERα observed in Pyk2-KO OBs may reflect an increase in the differentiation phase of Pyk2-KO OBs compared to WT OBs. This is also supported by higher ALP and mineralizing activities observed in Pyk2-KO OBs.
Published reports largely attribute the anti-resorptive effects of estrogen-depletion on bone mass in vivo through its effects on reducing osteoclast number and activity (Imai et al., 2013,Vinel et al., 2016). However, it has been shown that E2 can stimulate the osteogenic differentiation of bone marrow cells derived from ERα-KO mice, as demonstrated by increases in collagen type 1 synthesis, ALP activity, and mineralization. Interestingly, similar to our findings in E2-treated Pyk2-KO OBs, in published reports, E2-treated ERα-KO OBs also showed an additive effect of E2 on ALP activity compared to control or E2-treated WT OBs (Parikka et al., 2005). In another study, it was reported that E2 significantly enhanced ALP activity and collagen type 1 mRNA expression in MG-63 osteosarcoma cells that had reduced levels of ERα, but not ERβ (Cao et al., 2003,McCauley et al., 2003). On the contrary, it was reported that the differentiation of OBs from ERαf/f; Prx-Cre and ERαf/f; Osx-Cre mice is significantly decreased when compared to controls (Almeida et al., 2013). This latter observation may result from a decrease in the ability of mesenchymal stem cells to differentiate into OB-lineage cells. Taken together, our findings suggest that the enhanced effect of E2 on ALP and mineralization in Pyk2-KO OBs, compared to WT OBs, may be mechanistically linked with the decrease in ERα in Pyk2-KO OBs. However, our studies also suggest that E2 signaling via ERβ contributes to the increased mineralization of Pyk2-KO OBs. Indeed, we found that DPN, an ERβ agonist, increased Pyk2-KO OBs mineralization, suggesting that ERβ is active in these cells. Importantly, the ERβ antagonist, PHTPP significantly reduced mineralization in Pyk2-KO OBs in the absence of E2, and had an even greater effect on Pyk2-KO OBs mineralization when combined with E2, confirming the critical role of ERβ in Pyk2-KO OBs. It is also possible that the increased osteogenic effect of E2 on Pyk2-KO OBs proceeds via a mechanism that does not require direct binding of E2 to either ERα or ERβ. Although more studies are needed, we found increased expression of GPER1 (GPR30) mRNA in Pyk2-KO OBs and with E2 stimulation, which is known to phosphorylate and active ERα, providing an alternative route by which mineralization might be increased in Pyk2-KO OBs. As another example, E2 signaling also occurs through the N-terminal truncated ERα isoform, or the orphan nuclear ER-related receptor α (ERRα) which is differentially expressed relative to ERα and ERβ in OBs and was reported to affect bone formation (Bonnelye and Aubin, 2002,Parikka et al., 2005,Bonnelye et al., 2001). In addition, a recent study found that Pyk2 promoted the expression and phosphorylation of the androgen receptor (Hsiao et al., 2016), suggesting a possible altered signaling of the androgen receptor in Pyk2-KO OBs.
The mechanism of action of raloxifene on target tissue is reported to occur through genomic and non-genomic pathways, involving both ERα and ERβ (Tee et al., 2004,Jordan et al., 2001). It has been shown that raloxifene preferentially binds ERα with approximately 3.5 fold higher affinity than ERβ (Zhu et al., 2006). Raloxifene exerts its estrogenic effects on bone by decreasing the remodeling rate, reducing osteoclast activity, and maintaining OB activity (Hegde et al., 2016). In addition to its anti-resorptive activity, several studies suggest that raloxifene may act as an osteogenic agent by increasing cell proliferation as well as Runx2 mRNA and collagen type 1 mRNA expression in primary human OBs. These effects were found to be partly mediated via ERK1 and ERK2 activation (Noda-Seino et al., 2013,Taranta et al., 2002). With respect to raloxifene in our studies, we found that raloxifene robustly enhances both ALP activity and mineral deposition in Pyk2-KO OBs, while exerting only a modest positive effect on the mineralizing activity of WT OB. Furthermore, the decreased levels of ERα but not ERβ in Pyk2-KO OBs, may suggest that the osteogenic effects of raloxifene may occur through ERβ, similar to our proposed mechanism for the actions of E2 in Pyk2-deficient OBs.
To examine if Pyk2-deletion affects signaling cascades that are common to ERα and/or ERβ, we examined the activity of the mitogenic protein, ERK, and the anti-apoptotic protein, AKT. Although AKT levels were similar in WT and Pyk2-KO OBs, Pyk2-KO OBs exhibited enhanced ERK phosphorylation compared to WT OBs (Figure 12). It is well-established that MAPKs are activated by phosphorylation of tyrosine/threonine residues though ERK, leading to the activation of nuclear transcription factors such as c-fos and c-jun during early OB differentiation (Marie, 2008). Consistent with this, ERK activation increases ALP activity (Jaiswal et al., 2000), OCN mRNA levels and mineralization during OB differentiation (Ge et al., 2007,Matsushita et al., 2009). Conversely, in mice that lack ERK1/ERK2, bone mineralization is substantially reduced when compared to WT mice (Matsushita et al., 2009). Published evidence suggests that E2 stimulation of MC3T3-E1 osteoblastic cells enhances ERK phosphorylation via ERα and prevents OB apoptosis (Yang et al., 2013). In our study, we also found that E2-stimulation further increases ERK phosphorylation in Pyk2-KO OBs. Given the osteogenic effects of Pyk2-deletion on OB proliferation and differentiation, we speculate that Pyk2-deletion leads to an increase in ERK phosphorylation which consequently increases OB differentiation markers, ALP activity and mineralizing activity. In addition Pyk2-deletion may affect other signaling pathways, such as p38 which is member of the MAPKs family of proteins, similar to ERK. It is also known that p38 promotes cell proliferation (Zhang and Liu, 2002) and plays an important role in the differentiation of calvarial OBs, bone marrow cells, and some OB cell lines (Wang et al., 2007,Rodriguez-Carballo et al., 2016,Hu et al., 2003), although this remains to be confirmed in our studies.
In summary, our results indicate that in normal OBs, Pyk2 acts a negative regulator of proliferation, differentiation and mineralization. Furthermore, Pyk2 appears to be an integral component of the E2-ERα/ERβ signaling cascade which regulates ERK signaling and consequently, OB proliferation and mineralizing activity. In addition, Pyk2 may stabilize ERα protein levels by preventing proteosome-mediated degradation of ERα. Thus, in the absence of Pyk2, ERα protein levels are decreased via ubiquitin-proteosome mediated degradation, which increases ERK signaling and OB activity. In the presence of E2, ERK signaling is further increased, most likely through ERβ, which promotes OB differentiation and mineralization (Figure 9). In conclusion, our studies suggest that Pyk2-deletion or chemical inhibitors of Pyk2 will promote bone formation and potentiate the effects of estrogen (or raloxifene) on OB matrix formation and mineralization. Given that Pyk2-deficient osteoclasts exhibit decreased osteoclast activity, and that estrogen and SERMs (e.g. raloxifene) are potent anti-resorptive agents, our studies also suggest that Pyk2-targeted inhibitors, in combination with anti-resorptive therapies, may have dual actions that prevent bone loss and increase skeletal mass in vivo.
Figure 9. Schematic representation of the proposed actions of Pyk2 and estrogen on OBs.
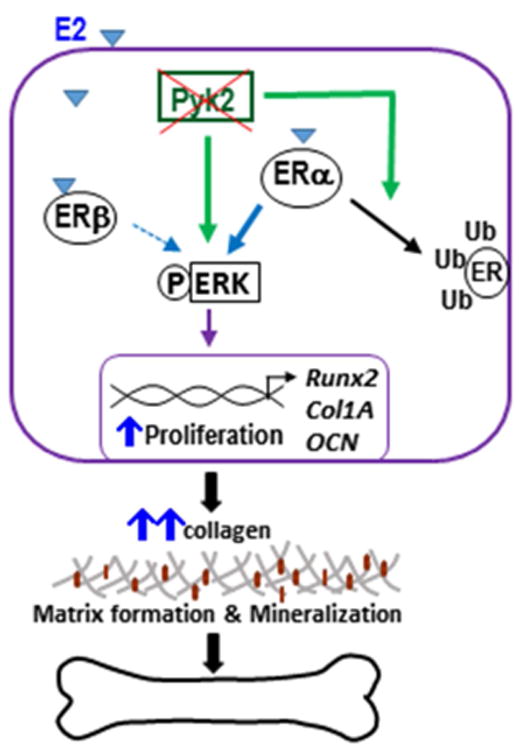
We postulate that inhibition of Pyk2 activity increases ERα degradation via the ubiquitin-proteosome pathway, which leads to an increase in ERK signaling and OB proliferation, and consequently promotes OB differentiation and mineralization activity. E2-stimulation, most likely acting through ERβ, has an additive effect on ERK signaling, which further promotes OB activity, leading to an increase in bone formation.
Supplementary Material
Figure S1. E2 stimulates mineralization in Pyk2-KO OBs cultured in hormone-depleted FBS. WT and Pyk2-KO OBs were cultured for 28 days in phenol-free αMEM supplemented with 2.5% charcoal-stripped FBS with ascorbic acid and β-glycerolphosphate to induce differentiation, in combination 0, 1, 10 or 100 nM E2. Cells were fixed and analyzed using a quantitative Alizarin Red S mineralization assay. The experiments were performed in triplicate and error bars represent mean ± SEM. Experiments were repeated 2 times. Statistical significance of p<0.05 is indicated (*) within a genotype and (#) between genotypes for the matched E2-treated groups.
Figure S2. Whole Western blots showing ERa protein levels. WT and Pyk2-KO OBs were cultured for 4 days (A) or 28 days (B) with or without 100 nM E2. Total cell lysates from WT and Pyk2-KO OBs were resolved by SDS-PAGE and blotted for anti-ERα as described in Figure 5. The blots in this figure represent expanded versions of the data shown in Figure 5, and confirm that ERα predominately migrates at ∼66 kDa, although other molecular weight species are also observed. The membrane for A) was cut in half and reblotted for actin. MW markers are indicated.
Figure S3. Short-term MG-132 does not affect OB viability. To confirm that MG-132 did not exert any negative effects on WT and Pyk2-KO OBs viability, cells were cultured for 4 days then treated for the last 4 hours with 0, 5, 10 or 20 μM MG-132. Cell viability/cytotoxicity was assayed using the Cell counting Kit-8 as per the manufacturer's instuctions. No change in cell viability was observed at the different MG-132 concentrations in either WT or Pyk2-KO mice, confirming that MG-132 was not cytotoxic at the dose and time used for the studies described in Figure 6 (20 μM, 3 hours).
Figure S4. Western blots showing immunoprecipitated ERα and Ub. As stated in Figure 6, WT and Pyk2-KO OBs were cultured with and without E2 and then treated for 3 hours with different concentrations of MG-132 (0, 5, 10, 20 μM). Cell lysates were subject to immunoprecipitation with a monoclonal antibody to ERα, followed by Western blotting using a polyclonal antibody to ubiquitin. Membranes were stripped and reblotted for ERα. The blots in this figure represent expanded versions of the data shown in Figure 6B, showing the Ub bands as well as other non-specific bands. MW markers are also shown.
Figure S5. Pyk2-deletion and estrogen do not effect AKT levels. WT and Pyk2-KO OBs were cultured for 4 days with or without 100 nM E2. Cell lysates were blotted for pAKT or total AKT as indicated. The graphs represent the ratio of pAKT/AKT and total AKT/actin and were determined by densitometry. No significant change in pAKT or total AKT were observed. N=3 replicates.
Highlights.
Pyk2 deletion increases osteoblast differentiation and mineralization
Pyk2 deletion promotes ERα degradation via the ubiquitin proteosome pathway
Inhibition of ERβ signaling inhibits the effects of estrogen on Pyk2-KO OBs
Estrogen increases ERK signaling as well as osteoblast differentiation and mineralization in Pyk2-deficient osteoblasts
Pyk2 targeted therapies in combination with estrogen receptor agonists may increase bone mass
Acknowledgments
Grant sponsors and numbers: National Institute of Health, NIAMS R01-AR060332; Indiana Clinical and Translational Science Institute, Collaboration in Biomedical/Translational Research (CBR/CTR) Pilot Program Grants, RR025761; IUPUI Office of the Vice Chancellor for Research (OVCR) Research Support Funds Grant (RSFG); IUPUI Biomechanics and Biomaterials Research Center (BBRC) Grant. We thank past and present members of our laboratories for technical assistance. We also thank Dr. Russell P. Main and Dr. Sarah H. Windahl for thoughtful suggestions and comments regarding these studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Compston JE. Sex steroids and bone. Physiol Rev. 2001;81:419–447. doi: 10.1152/physrev.2001.81.1.419. [DOI] [PubMed] [Google Scholar]
- Riggs BL, Hartmann LC. Selective estrogen-receptor modulators -- mechanisms of action and application to clinical practice. N Engl J Med. 2003;348:618–29. doi: 10.1056/NEJMra022219. [DOI] [PubMed] [Google Scholar]
- Imai Y, Youn MY, Inoue K, Takada I, Kouzmenko A, Kato S. Nuclear receptors in bone physiology and diseases. Physiol Rev. 2013;93:481–523. doi: 10.1152/physrev.00008.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinel A, Hay E, Valera MC, Buscato M, Adlanmerini M, Guillaume M, Cohen-Solal M, Ohlsson C, Lenfant F, Arnal JF, Fontaine C. Role of ERalpha in the Effect of Estradiol on Cancellous and Cortical Femoral Bone in Growing Female Mice. Endocrinology. 2016;157:2533–44. doi: 10.1210/en.2015-1994. [DOI] [PubMed] [Google Scholar]
- Clarke BL, Khosla S. Physiology of bone loss. Radiol Clin North Am. 2010;48:483–95. doi: 10.1016/j.rcl.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontiero O, Vidal C, Duque G. Aging and bone loss: new insights for the clinician. Ther Adv Musculoskelet Dis. 2012;4:61–76. doi: 10.1177/1759720X11430858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kondoh S, Kouzmenko A, Kato S. Minireview: osteoprotective action of estrogens is mediated by osteoclastic estrogen receptor-alpha. Mol Endocrinol. 2010;24:877–85. doi: 10.1210/me.2009-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum SA, Brown M. Unraveling estrogen action in osteoporosis. Cell Cycle. 2008;7:1348–52. doi: 10.4161/cc.7.10.5892. [DOI] [PubMed] [Google Scholar]
- Ren J, Wu JH. 17beta-estradiol rapidly activates calcium release from intracellular stores via the GPR30 pathway and MAPK phosphorylation in osteocyte-like MLO-Y4 cells. Calcif Tissue Int. 2012;90:411–9. doi: 10.1007/s00223-012-9581-x. [DOI] [PubMed] [Google Scholar]
- Kassem M. Cellular and molecular effects of growth hormone and estrogen on human bone cells. APMIS Suppl. 1997;71:1–30. [PubMed] [Google Scholar]
- Bonnelye E, Aubin JE. Differential expression of estrogen receptor-related receptor alphaand estrogen receptors alpha and beta in osteoblasts in vivo and in vitro. J Bone Miner Res. 2002;17:1392–400. doi: 10.1359/jbmr.2002.17.8.1392. [DOI] [PubMed] [Google Scholar]
- Arts J, Kuiper GG, Janssen JM, Gustafsson JA, Lowik CW, Pols HA, van Leeuwen JP. Differential expression of estrogen receptors alpha and beta mRNA during differentiation of human osteoblast SV-HFO cells. Endocrinology. 1997;138:5067–70. doi: 10.1210/endo.138.11.5652. [DOI] [PubMed] [Google Scholar]
- Roforth MM, Atkinson EJ, Levin ER, Khosla S, Monroe DG. Dissection of estrogen receptor alpha signaling pathways in osteoblasts using RNA-sequencing. PLoS One. 2014;9:e95987. doi: 10.1371/journal.pone.0095987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee MK, Rogatsky I, Tzagarakis-Foster C, Cvoro A, An J, Christy RJ, Yamamoto KR, Leitman DC. Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors alpha and beta. Mol Biol Cell. 2004;15:1262–72. doi: 10.1091/mbc.E03-06-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heino TJ, Chagin AS, Savendahl L. The novel estrogen receptor G-protein-coupled receptor 30 is expressed in human bone. J Endocrinol. 2008;197:R1–6. doi: 10.1677/JOE-07-0629. [DOI] [PubMed] [Google Scholar]
- Evans NJ, Bayliss AL, Reale V, Evans PD. Characterisation of Signalling by the Endogenous GPER1 (GPR30) Receptor in an Embryonic Mouse Hippocampal Cell Line (mHippoE-18) PLoS One. 2016;11:e0152138. doi: 10.1371/journal.pone.0152138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjimarkou MM, Vasudevan N. GPER1/GPR30 in the brain: Crosstalk with classical estrogen receptors and implications for behavior. J Steroid Biochem Mol Biol. 2017 doi: 10.1016/j.jsbmb.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Ford J, Hajibeigi A, Long M, Hahner L, Gore C, Hsieh JT, Clegg D, Zerwekh J, Oz OK. GPR30 deficiency causes increased bone mass, mineralization, and growth plate proliferative activity in male mice. J Bone Miner Res. 2011;26:298–307. doi: 10.1002/jbmr.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FP, Hsu T, Hu CH, Wang WD, Wang KC, Teng LF. Expression of estrogen receptors alpha and beta in human osteoblasts: identification of exon-2 deletion variant of estrogen receptor beta in postmenopausal women. Chang Gung Med J. 2004;27:107–15. [PubMed] [Google Scholar]
- Onoe Y, Miyaura C, Ohta H, Nozawa S, Suda T. Expression of estrogen receptor beta in rat bone. Endocrinology. 1997;138:4509–12. doi: 10.1210/endo.138.10.5575. [DOI] [PubMed] [Google Scholar]
- Wiren KM, Chapman Evans A, Zhang XW. Osteoblast differentiation influences androgen and estrogen receptor-alpha and -beta expression. J Endocrinol. 2002;175:683–94. doi: 10.1677/joe.0.1750683. [DOI] [PubMed] [Google Scholar]
- Teplyuk NM, Galindo M, Teplyuk VI, Pratap J, Young DW, Lapointe D, Javed A, Stein JL, Lian JB, Stein GS, van Wijnen AJ. Runx2 regulates G protein-coupled signaling pathways to control growth of osteoblast progenitors. J Biol Chem. 2008;283:27585–97. doi: 10.1074/jbc.M802453200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda-Seino H, Sawada K, Hayakawa J, Ohyagi-Hara C, Mabuchi S, Takahashi K, Nishio Y, Sakata M, Kurachi H, Kimura T. Estradiol and raloxifene induce the proliferation of osteoblasts through G-protein-coupled receptor GPR30. J Endocrinol Invest. 2013;36:21–7. doi: 10.3275/8301. [DOI] [PubMed] [Google Scholar]
- Bord S, Ireland DC, Beavan SR, Compston JE. The effects of estrogen on osteoprotegerin, RANKL, and estrogen receptor expression in human osteoblasts. Bone. 2003;32:136–41. doi: 10.1016/s8756-3282(02)00953-5. [DOI] [PubMed] [Google Scholar]
- Boutahar N, Guignandon A, Vico L, Lafage-Proust MH. Mechanical strain on osteoblasts activates autophosphorylation of focal adhesion kinase and proline-rich tyrosine kinase 2 tyrosine sites involved in ERK activation. J Biol Chem. 2004;279:30588–99. doi: 10.1074/jbc.M313244200. [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, Bonnette PC, Baumann AP, Hambor JE, Grasser WA, 3rd, Pan LC, Owen TA, Luzzio MJ, Hulford CA, Gebhard DF, Paralkar VM, Simmons HA, Kath JC, Roberts WG, Smock SL, Guzman-Perez A, Brown TA, Li M. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci U S A. 2007;104:10619–24. doi: 10.1073/pnas.0701421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Henn H, Destaing O, Sims NA, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camilli P, Baron R, Schlessinger J. Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(-/-) mice. J Cell Biol. 2007;178:1053–64. doi: 10.1083/jcb.200701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacena MA, Eleniste PP, Cheng YH, Huang S, Shivanna M, Meijome TE, Mayo LD, Bruzzaniti A. Megakaryocytes regulate expression of Pyk2 isoforms and caspase-mediated cleavage of actin in osteoblasts. J Biol Chem. 2012;287:17257–68. doi: 10.1074/jbc.M111.309880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzaniti A, Neff L, Sanjay A, Horne WC, De Camilli P, Baron R. Dynamin forms a Src kinase-sensitive complex with Cbl and regulates podosomes and osteoclast activity. Mol Biol Cell. 2005;16:3301–13. doi: 10.1091/mbc.E04-12-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleniste PP, Bruzzaniti A. Focal adhesion kinases in adhesion structures and disease. J Signal Transduct. 2012;2012:296450. doi: 10.1155/2012/296450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YH, Hooker RA, Nguyen K, Gerard-O'Riley R, Waning DL, Chitteti BR, Meijome TE, Chua HL, Plett AP, Orschell CM, Srour EF, Mayo LD, Pavalko FM, Bruzzaniti A, Kacena MA. Pyk2 regulates megakaryocyte-induced increases in osteoblast number and bone formation. J Bone Miner Res. 2013;28:1434–45. doi: 10.1002/jbmr.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleniste PP, Patel V, Posritong S, Zero O, Largura H, Cheng YH, Himes ER, Hamilton M, Baughman J, Kacena MA, Bruzzaniti A. Pyk2 and Megakaryocytes Regulate Osteoblast Differentiation and Migration via Distinct and Overlapping Mechanisms. J Cell Biochem. 2016 Jun;117(6):1396–406. doi: 10.1002/jcb.25430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okigaki M, Davis C, Falasca M, Harroch S, Felsenfeld DP, Sheetz MP, Schlessinger J. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci U S A. 2003;100:10740–5. doi: 10.1073/pnas.1834348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MZ, Rawlinson SC, Pitsillides AA, Zaman G, Mohan S, Baylink DJ, Lanyon LE. Human osteoblasts' proliferative responses to strain and 17beta-estradiol are mediated by the estrogen receptor and the receptor for insulin-like growth factor I. J Bone Miner Res. 2002;17:593–602. doi: 10.1359/jbmr.2002.17.4.593. [DOI] [PubMed] [Google Scholar]
- Taranta A, Brama M, Teti A, De luca V, Scandurra R, Spera G, Agnusdei D, Termine JD, Migliaccio S. The selective estrogen receptor modulator raloxifene regulates osteoclast and osteoblast activity in vitro. Bone. 2002;30:368–76. doi: 10.1016/s8756-3282(01)00685-8. [DOI] [PubMed] [Google Scholar]
- Zhou S, Zilberman Y, Wassermann K, Bain SD, Sadovsky Y, Gazit D. Estrogen modulates estrogen receptor alpha and beta expression, osteogenic activity, and apoptosis in mesenchymal stem cells (MSCs) of osteoporotic mice. J Cell Biochem Suppl. 2001;Suppl 36:144–55. doi: 10.1002/jcb.1096. [DOI] [PubMed] [Google Scholar]
- Machwate M, Jullienne A, Moukhtar M, Marie PJ. Temporal variation of c-Fos proto-oncogene expression during osteoblast differentiation and osteogenesis in developing rat bone. J Cell Biochem. 1995;57:62–70. doi: 10.1002/jcb.240570108. [DOI] [PubMed] [Google Scholar]
- Wagner EF. Functions of AP1 (Fos/Jun) in bone development. Ann Rheum Dis. 2002;61(2):ii40–2. doi: 10.1136/ard.61.suppl_2.ii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T. Regulation of osteoblast differentiation by Runx2. Adv Exp Med Biol. 2010;658:43–9. doi: 10.1007/978-1-4419-1050-9_5. [DOI] [PubMed] [Google Scholar]
- Burr D, Allen M. Basic and applied bone biology. 1st. Elsevier; China: 2013. [Google Scholar]
- Datta HK, Ng WF, Walker JA, Tuck SP, Varanasi SS. The cell biology of bone metabolism. J Clin Pathol. 2008;61:577–87. doi: 10.1136/jcp.2007.048868. [DOI] [PubMed] [Google Scholar]
- Soares CJ, Santana FR, Pereira JC, Araujo TS, Menezes MS. Influence of airborne-particle abrasion on mechanical properties and bond strength of carbon/epoxy and glass/bis-GMA fiber-reinforced resin posts. J Prosthet Dent. 2008;99:444–54. doi: 10.1016/S0022-3913(08)60106-7. [DOI] [PubMed] [Google Scholar]
- Russell RG. Pharmacological diversity among drugs that inhibit bone resorption. Curr Opin Pharmacol. 2015;22:115–30. doi: 10.1016/j.coph.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Hegde V, Jo JE, Andreopoulou P, Lane JM. Effect of osteoporosis medications on fracture healing. Osteoporos Int. 2016 Mar;27(3):861–871. doi: 10.1007/s00198-015-3331-7. [DOI] [PubMed] [Google Scholar]
- Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology. 2006;147:4132–50. doi: 10.1210/en.2006-0113. [DOI] [PubMed] [Google Scholar]
- Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–17. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- Kang WB, Deng YT, Wang DS, Feng D, Liu Q, Wang XS, Ru JY, Cong Y, Zhao JN, Zhao MG, Liu G. Osteoprotective effects of estrogen membrane receptor GPR30 in ovariectomized rats. J Steroid Biochem Mol Biol. 2015;154:237–44. doi: 10.1016/j.jsbmb.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Skaper SD, Dal Toso R, Petrelli L, Leon A. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19:514–20. doi: 10.1016/S0166-2236(96)10058-8. [DOI] [PubMed] [Google Scholar]
- Petrel TA, Brueggemeier RW. Increased proteasome-dependent degradation of estrogen receptor-alpha by TGF-beta1 in breast cancer cell lines. J Cell Biochem. 2003;88:181–90. doi: 10.1002/jcb.10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YM, Cho JY, Koo YD, Lee YJ. Effects of inhibiting the proteasomal degradation of estrogen receptor alpha on estrogen receptor alpha activation under hypoxic conditions. Biol Pharm Bull. 2009;32:2057–60. doi: 10.1248/bpb.32.2057. [DOI] [PubMed] [Google Scholar]
- Kretzer NM, Cherian MT, Mao C, Aninye IO, Reynolds PD, Schiff R, Hergenrother PJ, Nordeen SK, Wilson EM, Shapiro DJ. A noncompetitive small molecule inhibitor of estrogen-regulated gene expression and breast cancer cell growth that enhances proteasome-dependent degradation of estrogen receptor {alpha} J Biol Chem. 2010;285:41863–73. doi: 10.1074/jbc.M110.183723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail A, Nawaz Z. Nuclear hormone receptor degradation and gene transcription: an update. IUBMB Life. 2005;57:483–90. doi: 10.1080/15216540500147163. [DOI] [PubMed] [Google Scholar]
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci U S A. 1999;96:1858–62. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen D, Katzburg S, Sharon O, Grafi-Cohen M, Knoll E, Stern N. The effects of estrogen receptors alpha- and beta-specific agonists and antagonists on cell proliferation and energy metabolism in human bone cell line. J Cell Biochem. 2011;112:625–32. doi: 10.1002/jcb.22959. [DOI] [PubMed] [Google Scholar]
- Galea GL, Meakin LB, Sugiyama T, Zebda N, Sunters A, Taipaleenmaki H, Stein GS, van Wijnen AJ, Lanyon LE, Price JS. Estrogen receptor alpha mediates proliferation of osteoblastic cells stimulated by estrogen and mechanical strain, but their acute down-regulation of the Wnt antagonist Sost is mediated by estrogen receptor beta. J Biol Chem. 2013;288:9035–48. doi: 10.1074/jbc.M112.405456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. 2012;27:538–51. doi: 10.1002/jbmr.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CF, Chaudhary L, Fausto A, Halstead LR, Ory DS, Avioli LV, Cheng SL. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem. 2001;276:14443–50. doi: 10.1074/jbc.M010021200. [DOI] [PubMed] [Google Scholar]
- Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7:497–508. doi: 10.2174/138920206779315737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal CC, Das F, Ganapathy S, Harris SE, Choudhury GG, Ghosh-Choudhury N. Bone Morphogenetic Protein-2 (BMP-2) Activates NFATc1 Transcription Factor via an Autoregulatory Loop Involving Smad/Akt/Ca2+ Signaling. J Biol Chem. 2016;291:1148–61. doi: 10.1074/jbc.M115.668939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Pena VB, Scolaro LA, Santillan GE. ATP and UTP stimulate bone morphogenetic protein-2,-4 and -5 gene expression and mineralization by rat primary osteoblasts involving PI3K/AKT pathway. Exp Cell Res. 2013;319:2028–36. doi: 10.1016/j.yexcr.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS. Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci U S A. 1986;83:2496–500. doi: 10.1073/pnas.83.8.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai F, Liang Y, Bi J, Chen L, Zhang F, Cui Y, Jiang J. REGgamma regulates ERalpha degradation via ubiquitin-proteasome pathway in breast cancer. Biochem Biophys Res Commun. 2015;456:534–40. doi: 10.1016/j.bbrc.2014.11.124. [DOI] [PubMed] [Google Scholar]
- Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer. 2014;14:26–38. doi: 10.1038/nrc3622. [DOI] [PubMed] [Google Scholar]
- Sanchez M, Picard N, Sauve K, Tremblay A. Coordinate regulation of estrogen receptor beta degradation by Mdm2 and CREB-binding protein in response to growth signals. Oncogene. 2013;32:117–26. doi: 10.1038/onc.2012.19. [DOI] [PubMed] [Google Scholar]
- Tateishi Y, Sonoo R, Sekiya Y, Sunahara N, Kawano M, Wayama M, Hirota R, Kawabe Y, Murayama A, Kato S, Kimura K, Yanagisawa J. Turning off estrogen receptor beta-mediated transcription requires estrogen-dependent receptor proteolysis. Mol Cell Biol. 2006;26:7966–76. doi: 10.1128/MCB.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikka V, Peng Z, Hentunen T, Risteli J, Elo T, Vaananen HK, Harkonen P. Estrogen responsiveness of bone formation in vitro and altered bone phenotype in aged estrogen receptor-alpha-deficient male and female mice. Eur J Endocrinol. 2005;152:301–14. doi: 10.1530/eje.1.01832. [DOI] [PubMed] [Google Scholar]
- Cao L, Bu R, Oakley JI, Kalla SE, Blair HC. Estrogen receptor-beta modulates synthesis of bone matrix proteins in human osteoblast-like MG63 cells. J Cell Biochem. 2003;89:152–64. doi: 10.1002/jcb.10486. [DOI] [PubMed] [Google Scholar]
- McCauley LK, Tozum TF, Kozloff KM, Koh-Paige AJ, Chen C, Demashkieh M, Cronovich H, Richard V, Keller ET, Rosol TJ, Goldstein SA. Transgenic models of metabolic bone disease: impact of estrogen receptor deficiency on skeletal metabolism. Connect Tissue Res. 2003;44(1):250–63. [PubMed] [Google Scholar]
- Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, Onal M, Xiong J, Weinstein RS, Jilka RL, O'Brien CA, Manolagas SC. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest. 2013;123:394–404. doi: 10.1172/JCI65910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnelye E, Merdad L, Kung V, Aubin JE. The orphan nuclear estrogen receptor-related receptor alpha (ERRalpha) is expressed throughout osteoblast differentiation and regulates bone formation in vitro. J Cell Biol. 2001;153:971–84. doi: 10.1083/jcb.153.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao YH, Huang YT, Hung CY, Kuo TC, Luo FJ, Yuan TC. PYK2 via S6K1 regulates the function of androgen receptors and the growth of prostate cancer cells. Endocr Relat Cancer. 2016;23:651–63. doi: 10.1530/ERC-16-0122. [DOI] [PubMed] [Google Scholar]
- Jordan VC, Gapstur S, Morrow M. Selective estrogen receptor modulation and reduction in risk of breast cancer, osteoporosis, and coronary heart disease. J Natl Cancer Inst. 2001;93:1449–57. doi: 10.1093/jnci/93.19.1449. [DOI] [PubMed] [Google Scholar]
- Marie PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys. 2008;473:98–105. doi: 10.1016/j.abb.2008.02.030. [DOI] [PubMed] [Google Scholar]
- Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, Pittenger MF. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem. 2000;275:9645–52. doi: 10.1074/jbc.275.13.9645. [DOI] [PubMed] [Google Scholar]
- Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol. 2007;176:709–18. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol. 2009;29:5843–57. doi: 10.1128/MCB.01549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Chen K, Li B, Chen JW, Zheng XF, Wang YR, Jiang SD, Jiang LS. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis. 2013;18:1363–75. doi: 10.1007/s10495-013-0867-x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- Wang X, Goh CH, Li B. p38 mitogen-activated protein kinase regulates osteoblast differentiation through osterix. Endocrinology. 2007;148:1629–37. doi: 10.1210/en.2006-1000. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Carballo E, Gamez B, Ventura F. p38 MAPK Signaling in Osteoblast Differentiation. Front Cell Dev Biol. 2016;4:40. doi: 10.3389/fcell.2016.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chan E, Wang SX, Li B. Activation of p38 mitogen-activated protein kinase is required for osteoblast differentiation. Endocrinology. 2003;144:2068–74. doi: 10.1210/en.2002-220863. [DOI] [PubMed] [Google Scholar]
- Lin Y, Liu LJ, Murray T, Sodek J, Rao L. Effect of raloxifene and its interaction with human PTH on bone formation. J Endocrinol Invest. 2004;27:416–23. doi: 10.1007/BF03345284. [DOI] [PubMed] [Google Scholar]
- Matsumori H, Hattori K, Ohgushi H, Dohi Y, Ueda Y, Shigematsu H, Satoh N, Yajima H, Takakura Y. Raloxifene: its ossification-promoting effect on female mesenchymal stem cells. J Orthop Sci. 2009;14:640–5. doi: 10.1007/s00776-009-1357-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. E2 stimulates mineralization in Pyk2-KO OBs cultured in hormone-depleted FBS. WT and Pyk2-KO OBs were cultured for 28 days in phenol-free αMEM supplemented with 2.5% charcoal-stripped FBS with ascorbic acid and β-glycerolphosphate to induce differentiation, in combination 0, 1, 10 or 100 nM E2. Cells were fixed and analyzed using a quantitative Alizarin Red S mineralization assay. The experiments were performed in triplicate and error bars represent mean ± SEM. Experiments were repeated 2 times. Statistical significance of p<0.05 is indicated (*) within a genotype and (#) between genotypes for the matched E2-treated groups.
Figure S2. Whole Western blots showing ERa protein levels. WT and Pyk2-KO OBs were cultured for 4 days (A) or 28 days (B) with or without 100 nM E2. Total cell lysates from WT and Pyk2-KO OBs were resolved by SDS-PAGE and blotted for anti-ERα as described in Figure 5. The blots in this figure represent expanded versions of the data shown in Figure 5, and confirm that ERα predominately migrates at ∼66 kDa, although other molecular weight species are also observed. The membrane for A) was cut in half and reblotted for actin. MW markers are indicated.
Figure S3. Short-term MG-132 does not affect OB viability. To confirm that MG-132 did not exert any negative effects on WT and Pyk2-KO OBs viability, cells were cultured for 4 days then treated for the last 4 hours with 0, 5, 10 or 20 μM MG-132. Cell viability/cytotoxicity was assayed using the Cell counting Kit-8 as per the manufacturer's instuctions. No change in cell viability was observed at the different MG-132 concentrations in either WT or Pyk2-KO mice, confirming that MG-132 was not cytotoxic at the dose and time used for the studies described in Figure 6 (20 μM, 3 hours).
Figure S4. Western blots showing immunoprecipitated ERα and Ub. As stated in Figure 6, WT and Pyk2-KO OBs were cultured with and without E2 and then treated for 3 hours with different concentrations of MG-132 (0, 5, 10, 20 μM). Cell lysates were subject to immunoprecipitation with a monoclonal antibody to ERα, followed by Western blotting using a polyclonal antibody to ubiquitin. Membranes were stripped and reblotted for ERα. The blots in this figure represent expanded versions of the data shown in Figure 6B, showing the Ub bands as well as other non-specific bands. MW markers are also shown.
Figure S5. Pyk2-deletion and estrogen do not effect AKT levels. WT and Pyk2-KO OBs were cultured for 4 days with or without 100 nM E2. Cell lysates were blotted for pAKT or total AKT as indicated. The graphs represent the ratio of pAKT/AKT and total AKT/actin and were determined by densitometry. No significant change in pAKT or total AKT were observed. N=3 replicates.