

Abstract
Recent developments in serial crystallography at X-ray free electron lasers (XFELs) and synchrotrons have been driven by two scientific goals in structural biology – first, static structure determination from nano or microcrystals of membrane proteins and large complexes that are difficult for conventional cryocrystallography, and second, direct observations of transient structural species in biochemical reactions at near atomic resolution. Since room-temperature diffraction experiments naturally demand a large quantity of purified protein, sample economy is critically important for all steps of serial crystallography from crystallization, crystal delivery to data collection. Here we report the development and applications of “crystal-on-crystal” devices to facilitate large-scale in situ serial diffraction experiments on protein crystals of all sizes – large, small, or microscopic. We show that the monocrystalline quartz as a substrate material prevents vapor loss during crystallization and significantly reduces background X-ray scattering. These devices can be readily adopted at XFEL and synchrotron beamlines, which enable efficient delivery of hundreds to millions of crystals to the X-ray beam, with an overall protein consumption per dataset comparable to that of cryocrystallography.
Keywords: crystallization, in situ diffraction, Laue diffraction, serial crystallography, structural dynamics
Introduction
Cryocrystallography has propelled rapid and lasting growth of structural knowledgebase of biological macromolecules in the past decades (rcsb.org/pdb/statistics/contentGrowthChart.do?content=explMethod-xray). Although observing dynamic structures at the atomic resolution has been highly desired since the dawn of protein crystallography, the current knowledgebase of protein structures mostly contains static snapshots of macromolecules, which are best represented by the T and R structural states of hemoglobin. The dynamic behaviors during the interconversion among several discrete states of a functional protein are largely absent at cryogenic temperatures such as 100 K – perhaps the only drawback of cryocrystallography (Fraser et al., 2011). As a result, the atomic details of a biochemical reaction or biological process are often inferred from comparisons of these static structures. Direct observation of protein structural dynamics near the atomic resolution is a challenge that greatly hinders the advance in our understanding of the operating mechanisms of protein functions.
Cryocrystallography protocols have been highly optimized and streamlined over the last two decades at the synchrotron beamline facilities, which not only prolong the crystal lifetime during X-ray diffraction but also simplify many logistics in crystal storage, transportation, and mounting (Pflugrath, 2015). Serial crystallography, conducted on a massive scale at X-ray free electron lasers (XFELs) and later at synchrotrons in recent years, represents an entirely different strategy from crystallization to data collection (Martin-Garcia et al., 2016). Serial crystallography makes no attempt to complete a dataset from a single crystal. Rather, it considers an experimental specimen as a batch of crystals consisting of millions of nano- or micro-crystallites, in which each crystal has a chance to contribute a small slice of data to a computationally synthesized dataset. Cryoprotection of protein crystals is no longer needed, as the complicated X-ray radiation damage process is expected to take place after an ultrashort XFEL pulse hits a protein crystal (Caleman et al., 2011). Such a large-scale serial strategy offers new opportunities for structural studies of biomolecules at (near) atomic resolution beyond the original purpose for structural determination of the hard-to-crystallize proteins such as membrane proteins. Specifically, serial diffraction techniques, if properly implemented at synchrotron beamlines (Stellato et al., 2014), have the potential to transform how diffraction experiments are conducted at room temperature, which is critically important for capturing experimentally induced functional relevant protein dynamics on the time scale ranging from seconds to 100 ps (> 10−10 s). This endeavor requires new method development and reengineering of protocols for crystallization, crystal storage, transportation, and mounting. For example, macroscopic crystals ranging from tens to hundreds of microns demand protocols very different from those developed for nano and microcrystals. Accordingly, new algorithms and software tools are needed to properly scale and merge a large number of data fragments from many individual crystals during data processing.
For conventional diffraction experiments at room temperature, protein crystals are individually mounted and sealed in thin-walled glass or plastic capillaries to prevent dehydration. Such manual operations are effective for a small-scale experiment as long as a protein crystal lasts long enough in the X-ray beam to produce a substantial fraction of a dataset. However, due to severe X-ray radiation damage at room temperature, a serial protocol is often needed to acquire a complete dataset for most projects by combining data from a large number of crystals as many as 103-106, which are introduced into an X-ray beam either by injection of a liquid or paste stream (Sierra et al., 2012; Weierstall et al., 2014), or by a carrier device (Baxter et al., 2016; Fuller et al., 2017; Oghbaey et al., 2016), or a mounting raster (Cohen et al., 2014; Zarrine-Afsar et al., 2012). In situ diffraction experiments at room temperature have also been conducted at synchrotrons with both standard and special crystallization plates (Axford et al., 2012; Bingel-Erlenmeyer et al., 2011; Jacquamet et al., 2004; Kisselman et al., 2011; le Maire et al., 2011). Serial Laue diffraction using polychromatic X-rays was first demonstrated by Perry et al. (2014), while a variety of microfluidic platforms were designed to carry out both crystallization and diffraction without the need for crystal manipulation (Maeki et al., 2016; Pawate et al., 2015; Perry et al., 2014; Schieferstein et al., 2018; Sui et al., 2016). Alternatively, protein crystals can be loaded onto a porous silicon chip for diffraction experiments (Meents et al., 2017; Roedig et al., 2016).
Here we report a new generation of crystallization devices based on a “crystal-on-crystal” concept (Ren, 2017). Our strategy is to crystallize proteins on a monocrystalline quartz substrate made from an intact piece of single crystal, which contrasts with amorphous fused silica, with or without chemically etched patterns. We show that such monocrystalline material contributes virtually no background scattering to protein diffraction images, which presents a significant advantage over other widely used substrate materials such as glass and plastics. Traditionally, single crystal materials were not favored as substrate for in situ devices due to a concern that strong Bragg reflections may interfere with protein diffraction patterns. We have shown that strong reflections from the substrate materials can be effectively eliminated. These monocrystalline devices can be used to deliver hundreds to millions of protein crystals grown on-chip into an X-ray beam at synchrotrons or XFELs for in situ diffraction in a wide range of applications such as room temperature static crystallography, Laue diffraction for time-resolved crystallography, as well as femtosecond serial crystallography at XFELs. In this work, we conduct in situ Laue diffraction experiments using two different protein crystals, a plant UV-B photoreceptor UVR8 (Christie et al., 2012; Wu et al., 2012; Zeng et al., 2015) and a light driven DNA repair photolyase PhrB (Zhang et al., 2013, 2017). Our crystallographic results demonstrate that such crystal-on-crystal chips are well suited for room temperature time-resolved studies at both synchrotrons and XFELs.
Results
Most protein crystals at room temperature are fragile samples highly susceptible to various perturbations to its native state. For example, temperature change, pressure change, strong vibration, dehydration, and physical manipulations like touching and stirring may affect the crystallinity thus degrade the diffraction quality. For photosensitive crystals, near infrared (NIR) light is often the only choice for crystal visualization in order to avoid unintended light activation. Some redox sensitive crystals may be affected by air exposure. In addition, procedures involving manual handling of individual crystals such as loop mounting in cryocrystallography and capillary mounting at room temperature are not compatible with a large-scale serial protocol. Since protein crystals vary widely in property and quality, a broadly applicable serial crystallography technology must consider the most stringent requirements. Therefore, a chief consideration in our design and implementation is how to achieve large-scale in situ diffraction of protein crystals in their most native state, without using pressurized injection, acoustic injection, screening, drying, and protection from humidified airflow.
The most common choices of non-crystalline materials for in situ diffraction so far have been glass or plastics, for example, poly(methylmethacrylate) (PMMA), cyclic olefin copolymer (COC), polyethylene terephthalate (Mylar), and polyimide (Kapton). Since thin-film polymers (< 100 μm in thickness) are not adequate vapor barriers required for crystallization over days, weeks, or even longer, materials thicker than protein crystals are often used (Axford et al., 2012; Bingel-Erlenmeyer et al., 2011; Dhouib et al., 2009; Kisselman et al., 2011; le Maire et al., 2011). As a result, many of these devices suffer from high background scattering because strong X-ray scattering from thicker materials may overwhelm weak, high-resolution diffraction spots from protein crystals. Although digital subtraction of background scattering from diffraction images is possible (Jacquamet et al., 2004), there is no statistical gain in signal-to-noise ratio. Other strategies used to reduce background scattering, for example, use of microbeams or low-divergence beams, large area detectors, and long distances from sample to detector, would not change the root cause of high background. Another important but less recognized factor that may compromise the signal-to-noise ratio is the X-ray absorption by the chip material after the diffracted X-rays exit a crystal sample and before reaching the detector. To address this problem, graphene has been proposed as an alternative substrate material (Sui et al., 2016).
Monocrystalline quartz as device substrate
Monocrystalline quartz as a substrate material has several advantages. First, due to its molecular structure, intact monocrystalline quartz is not permeable to water that enables reliable crystallization and crystal storage. Like glass, single crystal quartz is an excellent vapor barrier even with a very small thickness. Our devices are able to retain solution samples in μL volume indefinitely without any measurable vapor loss over a time period relevant to protein crystallization. Second, quartz is transparent in a wide wavelength range from mid ultraviolet (UV) to infrared (IR). It therefore offers an optical window for crystal visualization and photoexcitation. Third, monocrystalline quartz produces minimal background X-ray scattering compared to other non-crystalline materials of the same thickness due to the lack of solvent and disordered structure in its crystal lattice. We here compare the scattering images obtained from several commonly used substrate materials (Fig. 1). The resulting cylindrically averaged scattering curves support that the crystal quartz material has virtually zero contribution to the background of protein diffraction images. We further demonstrate that strong Bragg reflections from the single crystal quartz can be avoided completely if the device is aligned at certain orientations relative to the X-ray beam at which no discrete Bragg conditions are satisfied. This consideration has been incorporated in our design (see below). In addition, these devices are compatible with the common crystallization methods such as vapor diffusion and microbatch, with protein consumption comparable to conventional cryocrystallography.
Figure 1.
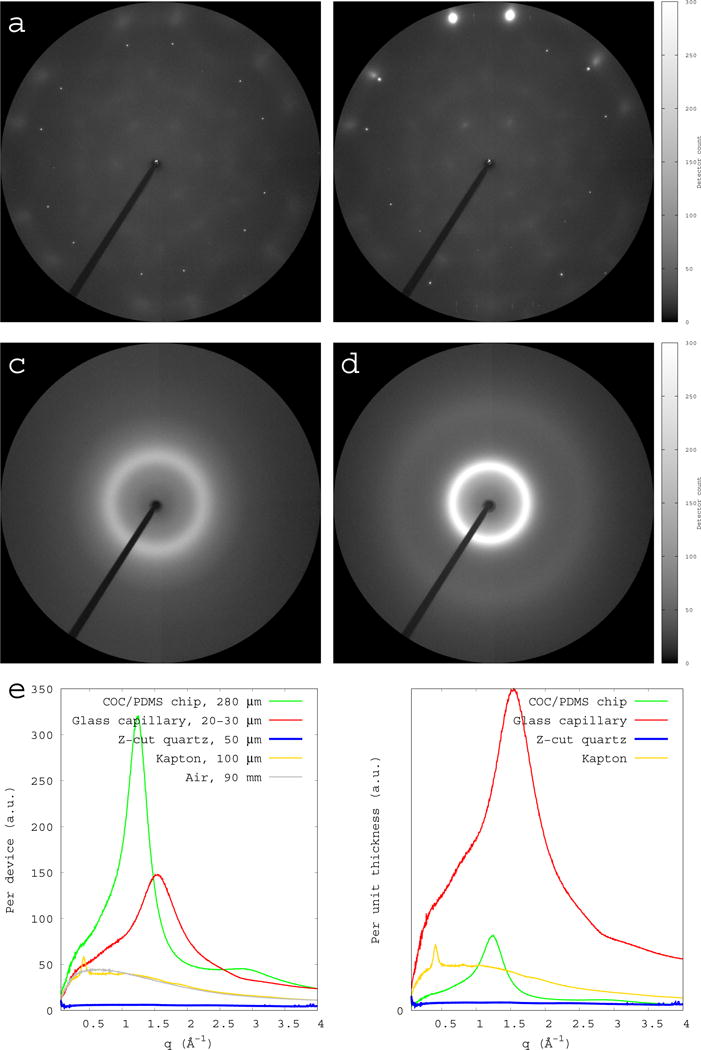
Comparisons of background scattering from single crystal quartz and other devices. All X-ray scattering images are collected under the same condition, including exposure time and sample-to-detector distance of 90 mm. All images are rendered using the same software settings. The diffraction resolution at the edge of the detector is 1.4 Å. (a) Scattering image from a single crystal quartz with Z-axis aligned with the X-ray beam. (b) Scattering image from a single crystal quartz with Z-axis offset from the X-ray beam. Strong reflections are observed at a high Bragg angle, but the diffuse scattering from the quartz crystal is still minimal. (c) Scattering image from an empty thin-wall glass capillary of a wall thickness of 10 μm (Hampton Research Corp.). The beam path through glass is ~20 μm. (d) Scattering image from an empty COC chip of 280 μm with no crystallization solution. (e) Cylindrically averaged scattering intensities I of the single crystal quartz and other devices are plotted as function of scattering vector q. The thickness of each device is labeled in the key. The air scattering over a 90-mm distance is also plotted in grey as a comparison. (f) All scattering intensities are normalized to unity in thickness.
To promote spreading of crystallization solutions in a vapor diffusion setup (Fig. 2cd) or to facilitate liquid flow through chambers or channels like microfluidics control devices, we also experimented fabrication of several desired patterns on Z-cut quartz wafers based on the published protocols of wet-etching on monocrystalline quartz substrate (Hedlund et al., 1993; Rangsten et al., 1998; Methods). The etching process closely follows that predicted by theoretical simulations (Fig. 2ab). We have achieved a thickness of 30 μm for a well bottom with manual control over an etching reaction. Such on-chip flow-cell constructs open possibilities for studying ligand-induced processes in future applications. However, etching is not necessary for the microbatch crystallization method.
Figure 2.
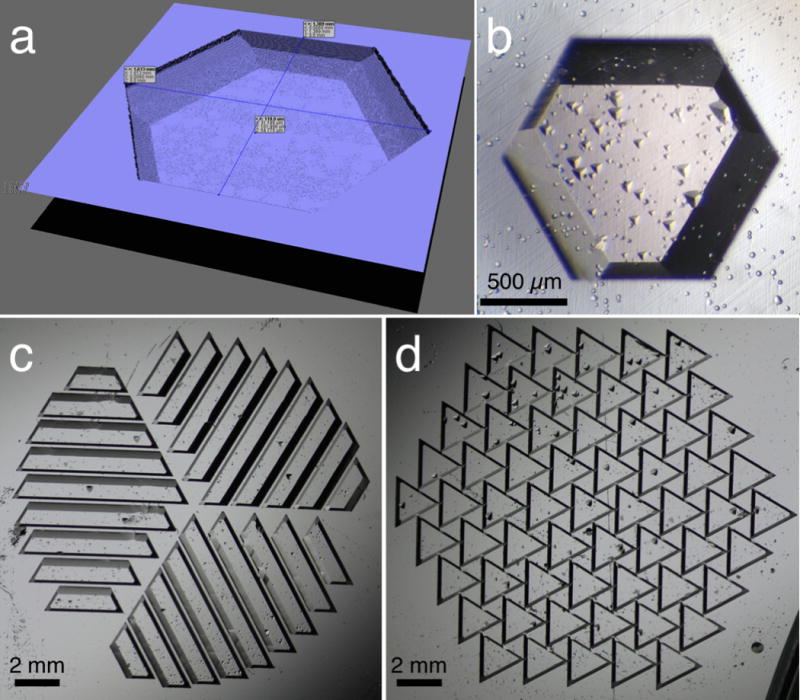
Computer simulation (a) and micrographs (bcd) of various etched wells on Z-cut monocrystalline quartz. The patterns are designed to accommodate the trigonal symmetry of the quartz crystal. The well capacity in (ab) is 0.16 μL per 100 μm thickness. (c) and (d) are designed to promote solution spreading in a hanging-drop vapor-diffusion method. Small triangular shapes inside wells are due to fabrication defects.
Crystallization and visualization
We have successfully tested crystallization of several proteins using such devices based on both the hanging drop vapor diffusion and microbatch methods (Fig. 3). UVR8 is a plant UV-B photoreceptor with a molecular weight of ~40 kDa. Using a batch method, we grew hundreds to millions of UVR8 crystals of various sizes on one such device (Fig. 3abc), given the same protein consumption as in the hanging drop vapor diffusion method. We found that microbatch crystallization can be readily achieved by using 1.5× precipitant concentration as that for vapor diffusion.
Figure 3.

Micrographs of protein crystals grown on the monocrystalline quartz chips. Each micrograph shows a small portion of the entire device. All crystals are obtained from the microbatch method except (l), which is based on vapor diffusion. All micrographs were taken using the same magnification. (abc) UV-B photoreceptor UVR8 crystals of large, small, and micron sizes are grown on chip in a controlled manner. (de) Large and small crystals of a bilin-binding photoreceptor photographed under the blue safety light. (f) A flavin-binding protein from plant. (g) A dual-sensor cyanobacterial photoreceptor. (f) and (g) are photographed under the red safety light. (h) A DNA repair enzyme photolyase PhrB. (i) A direct oxygen sensor protein. (j) A DNA-polymerase complex. (k) A prokaryotic sensory histidine kinase fragment. (l) UVR8 crystals obtained by vapor diffusion.
Crystals on such devices can be visualized or photographed during and after crystallization using a digital microscope equipped with a translation stage. For photosensitive crystals, safety light or near IR light shall be used to prevent unintended illumination or activation (Fig. 3d-g). We tested a light source of 0.3 mW IR LED at 1,085 nm for visualization of crystals on chip. The heating effect due to absorption by water is minimized at this IR wavelength.
Device assembly
Each device consists of a base and a cover layer with sealant in between (Fig. 4a), whether it is pre-assembled in a microbatch setup or assembled following vapor diffusion crystallization. Crystallization solution is pipetted onto one layer of the chip in the same way for vapor diffusion crystallization. Vacuum grease, oil of a suitable viscosity, or a silicon rubber made from a thin sheet of polydimethylsiloxane (PDMS) can be used as a sealant. The desired device capacity can be achieved by adjusting the thickness of the sealant and shim. For example, we routinely set up devices of 10 μL using shim of 50 μm thickness in microbatch crystallization. Grease and oil are excellent sealants to prevent vapor loss. High-quality, flat PDMS sheets can be used to combine the functions of shim and sealant. With these commonly used sealants, we found that the evaporative loss from a device made of monocrystalline quartz, if assembled properly, is too small to quantify. Specifically, a grease or oil sealed quartz chamber is able to retain a small amount of solution (10 – 50 μL) longer than the time scale relevant to protein crystallization, that is, weeks to months.
Figure 4.
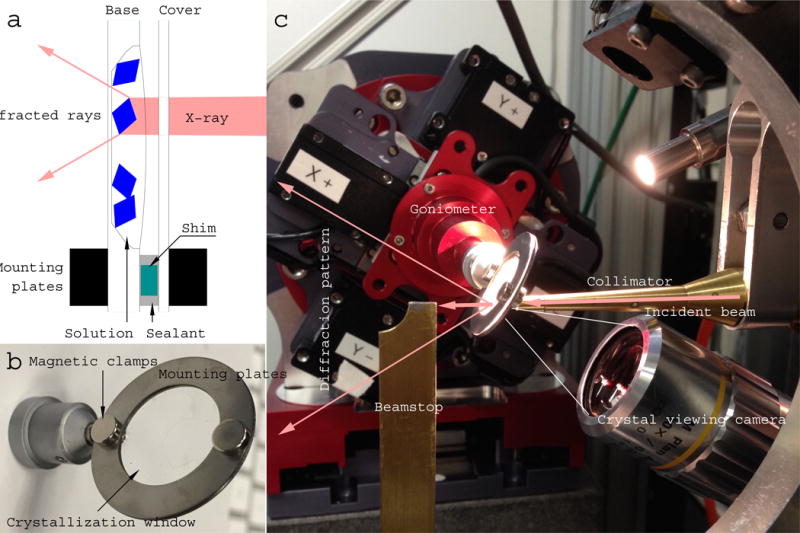
An assembled device and its beamline environment. (a) A schematic drawing of an X-ray beam path through an assembled chip (not drawn to scale). The base of an etched well is 10 – 50 μm in thickness, and faces downstream of the X-ray beam. The cover chip facing upstream is 50 μm thick. Both layers of the chip, a protein crystal, and the crystallization solution are along the X-ray path. Background scattering comes from disordered solvent that is inside and around a protein crystal, but not from the crystalline chip itself (Fig. 1). The base layer of the chip and the solution, but not the cover layer of the chip, contribute to absorption of diffracted X-rays. Use of etched wells or shim of various thickness are optional depending on specific applications. (b) Photograph of a circular crystallization device of 25 mm (1”) in diameter with a double-layered single crystal quartz window of 16 – 19 mm (5/8” – 3/4”) in diameter. (c) A crystallization device mounted on the goniometer set at the viewing mode on the 14-ID-B beamline of the Advanced Photon Source, where in situ Laue diffraction experiments were conducted.
Crystals grown on chip using microbatch method are ready for in situ diffraction without further manipulation. A quartz wafer, preferably with etched features on one side to promote spreading of crystallization solution (Fig. 2cd), can be used as a cover slip for vapor diffusion. The reservoir solution is stored in the wells of a crystallization plate, for example, the commonly used Linbro plate. Each quartz chip after vapor diffusion must be assembled with a cover chip prior to diffraction experiments. This procedure is considered quasi in situ since it requires opening of the original crystallization chamber for device assembly. Nevertheless, it does not involve direct manipulations of individual crystals such as drying and physical touch of any kind.
Diffraction experiments
An assembled device is mounted with an adaptor to a standard goniometer at synchrotron beamlines for crystal positioning and diffraction experiment (Fig. 4). The device is designed to stay perpendicular to the X-ray beam at all time during data collection, while crystals on this device are individually centered in the X-ray beam by two-dimensional translation of the device.
We have carried out diffraction experiments with the quartz devices using the monochromatic oscillation and polychromatic Laue methods. In both cases, we were able to obtain excellent protein diffraction patterns and completely avoid the quartz diffraction spots (Fig. 5). Since the rhombohedral α quartz has unit cell lengths of ~5 Å, only a few Bragg reflections of the α quartz crystal have chance to fall within the common resolution range of protein diffraction. They can be completely avoided when the three-fold axis of the Z-cut quartz wafer is aligned with the X-ray beam, which is perpendicular to the wafer plane. Consistent with our calculations, no Bragg reflections from a Z-cut quartz crystal are observed given X-rays at an energy of ~12 keV or wavelengths from 0.95 to 1.12 Å (Fig. 5). Similarly, X-rays of ~15 keV in the wavelength range from 0.75 to 0.82 Å are not expected to produce any Bragg reflections from this quartz crystal. We estimate that the angular tolerance of the Z-cut quartz wafer in an X-ray beam without detectable diffraction spots is on the order of several degrees given a wavelength centered at the midpoints of these ranges. Thus, a crude chip alignment by eyeballing would suffice. In other words, any interference from quartz crystal diffraction can be avoided with tunable X-ray at typical synchrotron beamlines. However, the rotation range of such device is limited to a few degrees in monochromatic oscillation before strong Bragg reflections from the quartz crystal enter a diffraction pattern.
Figure 5.
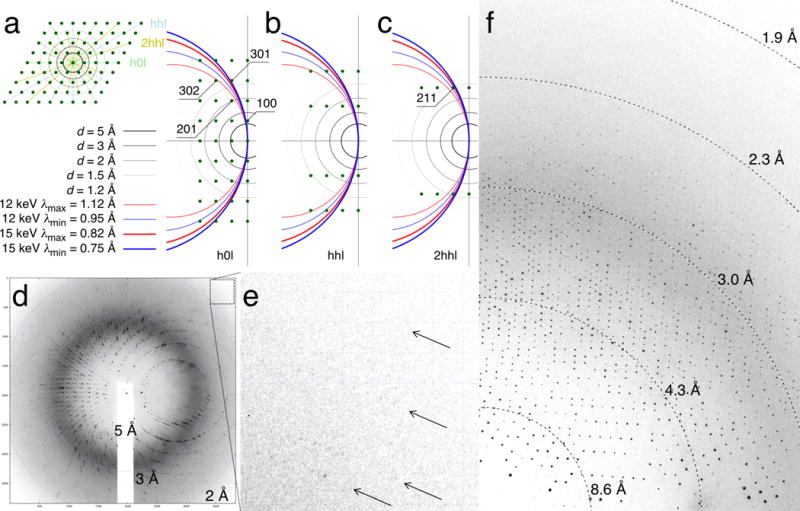
Diffraction from protein and quartz crystals. (a) Ewald construction of zone h0l with the Z-axis of α quartz along the X-ray beam. The reciprocal lattice perpendicular to the three-fold Z-axis is shown in the inset. Three major zones h0l, hhl, and 2hhl are colored in green, blue and yellow, respectively. The Ewald spheres corresponding to the minimum and maximum wavelengths of the Laue beam are colored in blue and red, respectively. Both 12 and 15 keV X-rays are considered. Only the reciprocal lattice points between these two Ewald spheres satisfy the diffraction condition. A few reflections near the space of diffraction condition are labeled. (b) Ewald construction of zone hhl. (c) Ewald construction of zone 2hhl. (d) A Laue diffraction image from a UVR8 crystal grown on the quartz chip. Note that no quartz diffraction is observed although both layers of the quartz crystals are in the X-ray beam during diffraction. (e) A zoom-in view of the upper right corner of the diffraction image in (d). (f) A monochromatic in situ diffraction image from a crystal of a flavin-binding photoreceptor on the quartz chip.
Serial Laue data collection
We conducted Laue diffraction experiments on two different types of crystals, UVR8 and PhrB, at BioCARS 14-ID-B using a polychromatic beam of ~6% bandwidth measured as the full-width at half-maximum (Fig. 6e). Due to non-uniform X-ray intensities in the polychromatic Laue beam, wavelength normalization is necessary to correct systematic variations in beam intensity across the entire incident spectrum. This procedure does not require any experimentally measured spectrum of beam intensity. Rather, wavelength normalization, also known as the λ-curve, is numerically determined based on relative intensities of redundant and symmetry-related reflections recorded at different wavelengths (Ren and Moffat, 1995a; Ren et al., 1999). Therefore, the outcome of wavelength normalization serves as a direct indicator of the Laue data quality. Both datasets collected from hundreds of UVR8 and PhrB crystals on-chip give rise to accurate wavelength normalization curves (Fig. 6e). Small changes in peak position and bandwidth can be attributed to the run-to-run variations in undulator settings and beam focusing.
Figure 6.
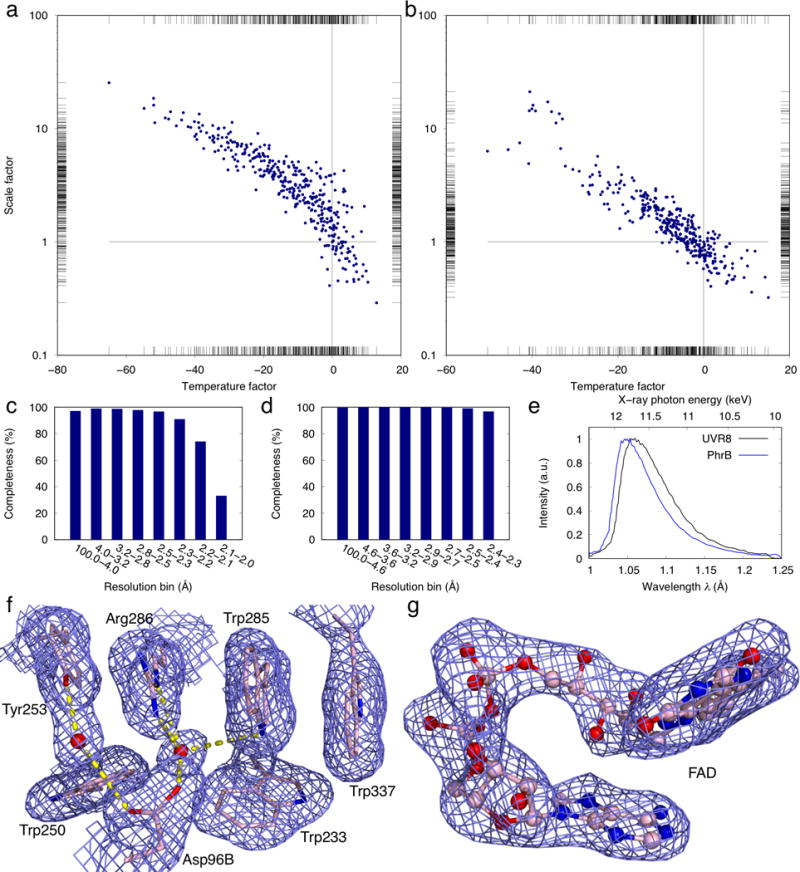
Results from serial crystallography datasets. (ab) Correlation plots of frame-to-frame scale factors versus temperature factors of the UVR8 (a) and PhrB (b) datasets. The reference frame at the cross point has a scale factor of 1 and temperature factor of 0. The scale and temperature factors are refined for individual frames before merging. Strong and weak diffraction images are located at the lower right and upper left ends, respectively. (cd) Plots of completeness as function of resolution in the final data sets of UVR8 and PhrB. (e) The refined wavelength normalization curves for the UVR8 and PhrB datasets. (f) 2Fo-Fc electron density map near the active site of the refined UVR8 structure. The map is contoured at 1σ showing well-defined protein side chains and water molecules. (g) Simulated annealing omit map (contoured at 2σ) of the refined flavin adenine dinucleotide (FAD) in the PhrB structure.
The UVR8 dataset was processed from 362 Laue images selected from a total of 874 frames collected from 11 devices at room temperature. Among them, 218, 88, and 56 frames are from the first, second, and third X-ray exposures of the same crystal, respectively. Each exposure of polychromatic X-rays lasts a few μs (see Methods for detail). The overall diffraction quality of the second and third exposures deteriorates quickly from the initial exposure due to radiation damage. The standard deviation of the refined unit cell lengths a and c among all images is restricted to 2% and that of the β angle is 0.5°. Due to the lack of a reference length scale in the geometry of Laue diffraction, not all unit cell lengths can be refined. Therefore, the cell length b is fixed as a constant. These deviations of cell parameters indicate an imperfect isomorphism among all crystals used. A total of 808,932 diffraction spots are measured. 749,321 integrated intensities for 99,141 unique reflections are accepted in the final dataset. Therefore, each diffraction image contains more than 2,000 spots on average (Fig. 5d), and the overall redundancy for each unique reflection is 7.6. Since the first frame from a fresh crystal and subsequent frames from a deteriorating crystal have very different diffraction quality, the scale factors and temperature factors vary significantly from frame to frame (Fig. 6a). Therefore, we performed joint data scaling in a large-scale minimization with the interdependent wavelength normalization (Ren and Moffat, 1995a). The resulting dataset is 85.9% complete to 2 Å resolution with Rmerge = 22.2%, with 6.6% bandwidth in the wavelength normalization curve (Fig. 6ce). The slight incompleteness despite the deconvolution of harmonic overlaps (Ren and Moffat, 1995b) is attributed to the monoclinic P21 space group of the UVR8 crystals. Such low-symmetry space groups often require more frames to completely survey the reciprocal space (see Discussion below). Subsequent structural refinement of UVR8 (phenix.refine) against the Laue dataset results in Rwork = 23.5% and Rfree = 29.8%, respectively. The 2Fo-Fc map is of excellent quality with well-defined electron densities for the active site and water molecules (Fig. 6f).
The Laue dataset from the PhrB crystals grown on chip consists of 458,622 observed diffraction spots in 334 frames selected from the 593 first exposures collected from seven devices. 422,221 integrated intensities are accepted in the final merged dataset. On average, each frame contributes 1,264 observed reflections. A total of 27,730 unique reflections extend to 2.3 Å resolution with an overall redundancy greater than 15. This high redundancy results in a dataset of 99.3% completeness and Rmerge = 12.6% (Fig. 6d). The standard deviation of the refined unit cell lengths a and c among all images is 0.5% with several outliers removed. All PhrB crystals seem to exhibit better isomorphism. The bandwidth calculated by wavelength normalization is 5.6% (Fig. 6e). Structural refinement (phenix.refine) shows the final R-factor and Rfree of 16.6% and 23.5%, respectively. The map quality is evidenced by well-resolved electron densities in both the protein moiety and cofactors (Fig. 6g).
Discussion
Data completeness
Our results demonstrate that the quartz devices enable room temperature in situ diffraction experiments at synchrotron beamlines. Compared to the previous reports on serial Laue diffraction (Meents et al., 2017; Pawate et al., 2015; Perry et al., 2014; Sui et al., 2016), we are able to increase the number of frames from a few tens to more than 300, which result in nearly complete datasets for both UVR8 and PhrB. Other than the wider source bandwidth, a major difference between the Laue diffraction and monochromatic oscillation methods is the use of stationary crystals. Without crystal motions, the reciprocal space at high resolution is severely under-sampled, because the effective X-ray bandwidth near the resolution limit is significantly narrower than that at low resolution (Ren et al., 1999). It has been shown that a complete Laue dataset must have at least 6-fold redundancy to that resolution. Therefore, tens or even hundreds of frames from randomly orientated crystals are often not sufficient to complete a dataset at high resolution. We expect to achieve thousands of frames per dataset with further development on automation in data collection and processing.
Exposures per crystal
In serial crystallography, one fresh crystal at a random orientation on chip is expected to contribute one frame to a dataset, that is, a hit. The second and subsequent exposures from the same crystal are often excluded from merging into the final dataset due to their inferior diffraction quality, even if they are possible. For injector-based serial crystallography, a second exposure is not an option. While such one-crystal-one-frame scenario is adequate for collecting a static dataset, it would be ideal in time-resolved and other dynamic experiments if two frames can be obtained from the same crystal at the same orientation so that systematic errors such as shot-to-shot fluctuations and crystal-to-crystal differences are self-canceled for an improved accuracy of difference signals. In a pump-probe experiment, the first frame obtained before a pump pulse is a reference; the second frame recorded after the pump serves as a probe. Due to severe X-ray radiation damage at room temperature, most protein crystals cannot sustain back-to-back exposures thus difficult to produce a reference-probe pair of diffraction patterns of comparable quality. With a few exceptions such as photoactive yellow protein that is not very sensitive to X-ray radiation damage, typical protein crystals such as UVR8 and PhrB crystals, which diffract to resolutions of 1.6 and 1.5 Å respectively (Zeng et al, 2015; Zhang et al., 2013, 2017), are expected to face challenges in collecting such pairwise frames for dynamic studies. Alternatively, one can minimize systematic errors between the “reference” and “probe” frames by collecting pairwise frames from two separate volumes of the same crystal. While a second exposure from the same crystal is not an option in injector-based technology, the second and subsequent exposures from the same crystal can be easily planned with our crystal-on-crystal chip. With automation by crystal recognition and shot planning software, these chips are expected to produce thousands of such pairwise diffraction patterns in high throughput in situ diffraction experiments.
Hit rate versus yield
Currently, there are two major categories of sample delivery systems for large-scale serial crystallography experiments: injector-based and fixed target. A well-known problem with injector-based systems is the low hit rate. That is, only a small percentage of all recorded images actually contain any diffraction signals while the majority are blank images. The fixed target methods perform far better than injectors in this aspect (Chapman, 2015; Oghbaey et al., 2016). However, the current implementations in both categories perform poorly in terms of “yield”, a metrics we propose to evaluate the sample economy in serial crystallography. Specifically, the yield is defined as the percentage of signal-producing crystals over the total number of crystals prepared. The numerators are the same for hit rate and yield – the number of images that contain diffraction signals, but their denominators are different. The denominator of hit rate is the total number of images taken, while that of the yield is the total number of sample crystals successfully grown. Thus, hit rate measures the efficiency of X-ray pulses; while yield measures the efficiency of protein crystals. While the value of yield is not reported for the current delivery systems, we estimate that the yield is diminishingly small for injectors, evidenced by their extremely high protein consumption in recent reports. Protein consumption as measured by the yield is one of the most important factors that determine the feasibility of a biophysical experiment.
In both cases presented in this work, no crystal is left unexposed on a chip, which gives rise to a yield of nearly 100%. Since not all crystals produce usable data, we include 41.4% and 56.3% of the total images in the final datasets of UVR8 and PhrB, respectively. These numbers are equivalent to the commonly reported hit rate. In other words, approximately half of the images taken are useful while almost all crystals on the chip can produce diffraction images. It is also worthy to point out that each dataset consumes less than 1 mg of protein (Methods).
Crystal individuality
In conventional cryocrystallography, a complete dataset is usually collected from a single frozen crystal. In some cases where radiation damage is a major concern, several subsets of data collected from multiple crystals are merged to obtain a complete dataset. In serial crystallography conducted at room temperature, a dataset is obtained by combining thousands to millions of small slices of data, each represents a partial measurement from a specimen that is assumed to be identical to others in a large pool. If the specimen distribution in a pool is not known, it is hard to judge to what extent this assumption holds true. The merging statistics and refinement statistics (R-factors in Table S1) suggest that a pool of crystals present significant individuality and distributions as indicated by their imperfect isomorphism. However, it is not clear which parameters and how they differ from crystal to crystal. A static crystal structure is a result of a spatial average over an ensemble of molecules and a temporal average over the elapsed exposure time of data collection, during which many molecular events including radiation damage may occur. In this sense, a structure determined from a serial protocol of data collection represents such an average over a much broader base. How to address the effect of crystal individuality is a challenge in future development of serial crystallography when it comes to accurately determine the static and dynamic conformational distributions.
Conclusions
This work demonstrates that a monocrystalline substrate can offer several important advantages for room temperature diffraction experiments at both synchrotrons and XFELs, such as stabilized crystallization, enhanced signal-to-noise ratio, compatibility with macroscopic crystals and vapor diffusion and microbatch, in situ diffraction of thousands of crystals with a hit rate and yield of nearly 100%, and a superb optical window for crystal visualization and photoexcitation. With the concept, design, and implementation presented in this work, the crystal-on-crystal chip is potentially transformative for serial crystallography.
Methods
Etching on monocrystalline quartz
Quartz crystals are widely used for their piezoelectric and optical properties. Produced under controlled temperature and pressure, synthetic quartz crystals are commercially available as thin wafers of various thicknesses, which are sawed and polished at specific orientations with respect to the crystal lattice. In our applications, we apply well-established protocols of chemical wet etching, which is based on hydrofluoric acid HF (50%) buffered by ammonium fluoride NH4F (40%), to etch shallow triangular or hexagonal wells on a Z-cut quartz wafer (Hedlund et al., 1993). The etchant of HF : NH4F = 2 : 3 is kept in a water bath at a constant temperature of 55°C. Different surfaces of quartz crystal exhibit very anisotropic behaviors in terms of the etching rate (Rangsten et al., 1998), with the fastest rate of about 30 μm/hr along the Z-axis. It has been demonstrated that a remaining membrane as thin as 3.5 μm could be achieved (Rangsten et al., 1998). A large, flat well bottom of 10 – 50 μm in thickness is desired in our applications to minimize the substrate absorption of scattered X-rays from protein crystals, which is comparable to traditional crystal mounting in glass or silica capillaries. Polishing thin wafer and etching large well bottom below 25 μm are technically achievable but become increasingly expensive due to falling success rate. Here we demonstrate that an exit quartz window of 30 – 50 μm in thickness is adequate for protein diffraction. Thinner windows will be advantageous for diffraction from microcrystals.
Crystallization
UVR8 protein is purified as previously described (Zeng et al., 2015). In a microbatch setup, 25 μL crystallization solution is loaded into a crystallization chamber with a thickness of 125 μm. The thickness is controlled by a washer-like shim cut out from a plastic sheet using an electronic cutting machine (Silhouette, Inc.). The initial condition of microbatch contains 3.5 mg/mL protein, 7-10% polyethylene glycol (PEG) 8000, 50 mM MgCl2, and 50 mM tris buffer at pH 9.2. Microcrystals appeared within hours after the microbatch assembly. With lower PEG concentration, fewer and larger crystals appeared and grow to their full size of > 100 μm in a few days. The total consumption of protein for 11 devices is less than 1 mg.
PhrB protein is purified as previously described (Zhang et al., 2013). PhrB crystallization is set up using the microbatch method. The crystallization chamber is about 50 μm in thickness. On each device, 10 μL of crystallization solution contains 1.7 mg/mL protein, 9-13% PEG 4000, 6% isopropanol, and 50 mM sodium citrate buffer at pH 5.6. PhrB crystals of typical size of 200 × 50 × 50 μm3 appear on chips within a few days. Altogether, seven devices consume only 0.12 mg of protein. All devices are stored in dark, and crystal growth is monitored under microscope with a near IR light source.
Laue data collection and processing
Laue diffraction experiments are carried out at the BioCARS beamline 14-ID-B at Advanced Photon Source (APS), Argonne National Laboratory. Two collinear undulators U23 and U27 produce the X-rays (Graber et al., 2011). The X-ray beam is focused in two stages by the primary and secondary pairs of Kirkpatrick-Baez (KB) mirrors. The focal spot at the sample is 20 (horizontal) × 21 (vertical) μm2 measured as full-width at half-maximum. The footprint of the full beam is about 50 × 50 μm2. The exposure time of each image is 7.5 μs, which consists of 48 consecutive single bunches of 100 ps each during the 24-bunch standard operation mode of APS. All Laue diffraction images are recorded on a Rayonix MX 340 X-ray detector.
Hundreds of Laue images are indexed frame-by-frame using the software package Precognition/Epinorm™ (Renz Research, Inc.). Since diffraction images are obtained from many randomly orientated crystals on-chip, the orientation matrix determined and refined from one diffraction image does not apply to other images except for those taken from subsequent exposures from the same crystal. Integrated intensities for about half million of reflections from hundreds of images are scaled and merged via a joint minimization procedure, in which the frame-by-frame scale factors, temperature factors, and most importantly, wavelength normalization curves are determined (Fig. 6).
Structure refinement
Laue datasets are used to refine the crystal structures of UVR8 and PhrB using the initial models 4naa and 4dja from PDB, respectively. All model-building tasks are carried out using Coot. PyMOL is used to illustrate the crystal structures and electron density maps. Crystal structures of UVR8 and PhrB at room temperature are deposited to the Protein Data Bank under the accession IDs of 6dd7 and 6dd6, respectively.
Supplementary Material
Acknowledgments
Use of Advanced Photon Source, an Office of Science User Facility operated for US Department of Energy by Argonne National Laboratory, was supported by contract DE-AC02-06CH11357. Use of BioCARS was supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number R24GM111072. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Use of LS-CAT Sector 21 was supported by Michigan Economic Development Corporation and Michigan Technology Tri-Corridor grant 085P1000817. We acknowledge the technical support and use of instrumentation in Research Resources Center and Nanotechnology Core Facility, University of Illinois at Chicago. This work is supported in part by grants from the University of Illinois at Chicago and National Institutes of Health (R01EY024363) to XY. ZR is the inventor of the crystal-on-crystal chips on US patent 9632042 granted to Renz Research, Inc.
Footnotes
Author contributions
ZR & XY designed the research, performed structural refinement, and wrote the paper. ZR invented the crystal-on-crystal chips. ZR & KB analyzed the Laue diffraction data. MA, SB, KB, IK, SG, HS, CW, and XZ purified proteins and performed on-chip crystallization.
References
- Axford D, Owen RL, Aishima J, Foadi J, Morgan AW, Robinson JI, Nettleship JE, Owens RJ, Moraes I, Fry EE, et al. In situ macromolecular crystallography using microbeams. Acta Crystallogr D Biol Crystallogr. 2012;68:592–600. doi: 10.1107/S0907444912006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter EL, Aguila L, Alonso-Mori R, Barnes CO, Bonagura CA, Brehmer W, Brunger AT, Calero G, Caradoc-Davies TT, Chatterjee R, et al. High-density grids for efficient data collection from multiple crystals. Acta Crystallogr Sect Struct Biol. 2016;72:2–11. doi: 10.1107/S2059798315020847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingel-Erlenmeyer R, Olieric V, Grimshaw JPA, Gabadinho J, Wang X, Ebner SG, Isenegger A, Schneider R, Schneider J, Glettig W, et al. SLS crystallization platform at beamline X06DA - A fully automated pipeline enabling in situ X-ray diffraction screening. Cryst Growth Des. 2011;11:916–923. [Google Scholar]
- Caleman C, Huldt G, Maia FRNC, Ortiz C, Parak FG, Hajdu J, van der Spoel D, Chapman HN, Timneanu N. On the Feasibility of Nanocrystal Imaging Using Intense and Ultrashort X-ray Pulses. ACS Nano. 2011;5:139–146. doi: 10.1021/nn1020693. [DOI] [PubMed] [Google Scholar]
- Chapman HN. Serial Femtosecond Crystallography. Synchrotron Radiat News. 2015;28:20–24. [Google Scholar]
- Christie JM, Arvai AS, Baxter KJ, Heilmann M, Pratt AJ, O’Hara A, Kelly SM, Hothorn M, Smith BO, Hitomi K, et al. Plant UVR8 photoreceptor senses UV-B by tryptophan-mediated disruption of cross-dimer salt bridges. Science. 2012;335:1492–1496. doi: 10.1126/science.1218091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AE, Soltis SM, González A, Aguila L, Alonso-Mori R, Barnes CO, Baxter EL, Brehmer W, Brewster AS, Brunger AT, et al. Goniometer-based femtosecond crystallography with X-ray free electron lasers. Proc Natl Acad Sci. 2014;111:17122–17127. doi: 10.1073/pnas.1418733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhouib K, Malek CK, Pfleging W, Gauthier-Manuel B, Duffait R, Thuillier G, Ferrigno R, Jacquamet L, Ohana J, Ferrer JL, et al. Microfluidic chips for the crystallization of biomacromolecules by counter-diffusion and on-chip crystal X-ray analysis. Lab Chip. 2009;9:1412–1421. doi: 10.1039/b819362b. [DOI] [PubMed] [Google Scholar]
- Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller FD, Gul S, Chatterjee R, Burgie ES, Young ID, Lebrette H, Srinivas V, Brewster AS, Michels-Clark T, Clinger JA, et al. Drop-on-demand sample delivery for studying biocatalysts in action at X-ray free-electron lasers. Nat Methods. 2017;14:443–449. doi: 10.1038/nmeth.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber T, Anderson S, Brewer H, Chen YS, Cho HS, Dashdorj N, Henning RW, Kosheleva I, Macha G, Meron M, et al. BioCARS: a synchrotron resource for time-resolved X-ray science. J Synchrotron Radiat. 2011;18:658–670. doi: 10.1107/S0909049511009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund C, Lindberg U, Bucht U, Soderkvist J. Anisotropic etching of Z-cut quartz. J Micromechanics Microengineering. 1993;3:65. [Google Scholar]
- Jacquamet L, Ohana J, Joly J, Borel F, Pirocchi M, Charrault P, Bertoni A, Israel-Gouy P, Carpentier P, Kozielski F, et al. Automated analysis of vapor diffusion crystallization drops with an X-ray beam. Structure. 2004;12:1219–1225. doi: 10.1016/j.str.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Kisselman G, Qiu W, Romanov V, Thompson CM, Lam R, Battaile KP, Pai EF, Chirgadze NY. X-CHIP: an integrated platform for high-throughput protein crystallization and on-the-chip X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 2011;67:533–539. doi: 10.1107/S0907444911011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeki M, Yamazaki S, Pawate AS, Ishida A, Tani H, Yamashita K, Sugishima M, Watanabe K, Tokeshi M, Kenis PJA, et al. A microfluidic-based protein crystallization method in 10 micrometer-sized crystallization space. CrystEngComm. 2016;18:7722–7727. [Google Scholar]
- le Maire A, Gelin M, Pochet S, Hoh F, Pirocchi M, Guichou JF, Ferrer JL, Labesse G. In-plate protein crystallization, in situ ligand soaking and X-ray diffraction. Acta Crystallogr D Biol Crystallogr. 2011;67:747–755. doi: 10.1107/S0907444911023249. [DOI] [PubMed] [Google Scholar]
- Martin-Garcia JM, Conrad CE, Coe J, Roy-Chowdhury S, Fromme P. Serial femtosecond crystallography: A revolution in structural biology. Arch Biochem Biophys. 2016;602:32–47. doi: 10.1016/j.abb.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meents A, Wiedorn MO, Srajer V, Henning R, Sarrou I, Bergtholdt J, Barthelmess M, Reinke PYA, Dierksmeyer D, Tolstikova A, et al. Pink-beam serial crystallography. Nat Commun. 2017;8 doi: 10.1038/s41467-017-01417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oghbaey S, Sarracini A, Ginn HM, Pare-Labrosse O, Kuo A, Marx A, Epp SW, Sherrell DA, Eger BT, Zhong Y, et al. Fixed target combined with spectral mapping: approaching 100% hit rates for serial crystallography. Acta Crystallogr Sect Struct Biol. 2016;72:944–955. doi: 10.1107/S2059798316010834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawate AS, Šrajer V, Schieferstein J, Guha S, Henning R, Kosheleva I, Schmidt M, Ren Z, Kenis PJA, Perry SL. Towards time-resolved serial crystallography in a microfluidic device. Acta Crystallogr Sect F Struct Biol Commun. 2015;71:823–830. doi: 10.1107/S2053230X15009061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SL, Guha S, Pawate AS, Henning R, Kosheleva I, Srajer V, Kenis PJA, Ren Z. In situ serial Laue diffraction on a microfluidic crystallization device. J Appl Crystallogr. 2014;47:1975–1982. doi: 10.1107/S1600576714023322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugrath JW. Practical macromolecular cryocrystallography. Acta Crystallogr Sect F Struct Biol Commun. 2015;71:622–642. doi: 10.1107/S2053230X15008304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangsten P, Hedlund C, Katardjiev IV, Bäcklund Y. Etch rates of crystallographic planes in Z-cut quartz - experiments and simulation. J Micromechanics Microengineering. 1998;8:1. [Google Scholar]
- Ren Z. Single crystal quartz chips for protein crystallization and X-ray diffraction data collection and related methods. 9632042 US patent. 2017
- Ren Z, Moffat K. Quantitative analysis of synchrotron Laue diffraction patterns in macromolecular crystallography. J Appl Crystallogr. 1995a;28:461–481. [Google Scholar]
- Ren Z, Moffat K. Deconvolution of energy overlaps in Laue diffraction. J Appl Crystallogr. 1995b;28:482–494. [Google Scholar]
- Ren Z, Bourgeois D, Helliwell JR, Moffat K, Srajer V, Stoddard BL. Laue crystallography: coming of age. J Synchrotron Rad. 1999;6:891–917. [Google Scholar]
- Roedig P, Duman R, Sanchez-Weatherby J, Vartiainen I, Burkhardt A, Warmer M, David C, Wagner A, Meents A. Room-temperature macromolecular crystallography using a micro-patterned silicon chip with minimal background scattering. J Appl Crystallogr. 2016;49:968–975. doi: 10.1107/S1600576716006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieferstein JM, Pawate AS, Varel MJ, Guha S, Astrauskaite I, Gennis RB, Kenis PJA. X-ray transparent microfluidic platforms for membrane protein crystallization with microseeds. Lab Chip. 2018;18:944–954. doi: 10.1039/c7lc01141e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra RG, Laksmono H, Kern J, Tran R, Hattne J, Alonso-Mori R, Lassalle-Kaiser B, Glöckner C, Hellmich J, Schafer DW, et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr D Biol Crystallogr. 2012;68:1584–1587. doi: 10.1107/S0907444912038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellato F, Oberthür D, Liang M, Bean R, Gati C, Yefanov O, Barty A, Burkhardt A, Fischer P, Galli L, et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ. 2014;1:204–212. doi: 10.1107/S2052252514010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui S, Wang Y, Kolewe KW, Srajer V, Henning R, Schiffman JD, Dimitrakopoulos C, Perry SL. Graphene-based microfluidics for serial crystallography. Lab Chip. 2016;16:3082–3096. doi: 10.1039/c6lc00451b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JCH, Bruce Doak R, Nelson G, Fromme P, et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun. 2014;5 doi: 10.1038/ncomms4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Hu Q, Yan Z, Chen W, Yan C, Huang X, Zhang J, Yang P, Deng H, Wang J, et al. Structural basis of ultraviolet-B perception by UVR8. Nature. 2012;484:214–219. doi: 10.1038/nature10931. [DOI] [PubMed] [Google Scholar]
- Zarrine-Afsar A, Barends TRM, Müller C, Fuchs MR, Lomb L, Schlichting I, Miller RJD. Crystallography on a chip. Acta Crystallogr D Biol Crystallogr. 2012;68:321–323. doi: 10.1107/S0907444911055296. [DOI] [PubMed] [Google Scholar]
- Zeng X, Ren Z, Wu Q, Fan J, Peng PP, Tang K, Zhang R, Zhao KH, Yang X. Dynamic crystallography reveals early signaling events in ultraviolet photoreceptor UVR8. Nat Plants. 2015;1:14006. doi: 10.1038/nplants.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Scheerer P, Oberpichler I, Lamparter T, Krauss N. Crystal structure of a prokaryotic (6-4) photolyase with an Fe-S cluster and a 6,7-dimethyl-8-ribityllumazine antenna chromophore. Proc Natl Acad Sci. 2013;110:7217–7222. doi: 10.1073/pnas.1302377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ma H, Bowatte K, Kwiatkowski D, Mittmann E, Qasem H, Krauß N, Zeng X, Ren Z, Scheerer P, et al. Crystal Structures of Bacterial (6-4) Photolyase Mutants with Impaired DNA Repair Activity. Photochem Photobiol. 2017;93:304–314. doi: 10.1111/php.12699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.