

Abstract
In light of repeated translational failures with preclinical neuroprotection based strategies, this pre-clinical study reevaluates brain swelling as an important pathological event in diabetic stroke and investigates underlying mechanism of the comorbidity-enhanced brain edema formation. Type 2 (mild), type 1 (moderate), and mixed type 1/2 (severe) diabetic mice were subjected to transient focal ischemia. Infarct volume, brain swelling, and IgG extravasation were assessed at 3 days post-stroke. Expression of vascular endothelial growth factor (VEGF)-A, endothelial specific molecule-1 (Esm1), and the VEGF receptor 2 (VEGFR2) was determined in the ischemic brain. Additionally, SU5416, a VEGFR2 inhibitor, was treated in the type 1/2 diabetic mice, and stroke outcomes were determined. All diabetic groups displayed bigger infarct volume and brain swelling compared to nondiabetic mice, and the increased swelling was disproportionately larger relative to infarct enlargement. Diabetic conditions significantly increased VEGF-A, Esm1, and VEGFR2 expressions in the ischemic brain compared to nondiabetic mice. Notably, in diabetic mice, VEGFR2 mRNA levels were positively correlated with brain swelling, but not with infarct volume. Treatment with SU5416 in diabetic mice significantly reduced brain swelling. The study shows that brain swelling is a predominant pathological event in diabetic stroke and that an underlying event for diabetes-enhanced brain swelling includes the activation of VEGF signaling. This study suggests consideration of stoke therapies aiming at primarily reducing brain swelling for subjects with diabetes.
Keywords: Brain swelling, Diabetes, Ischemic stroke, VEGF-A/VEGFR2
Introduction
Despite the importance of preclinical research in strategizing treatments for ischemic stroke, a myriad of neuroprotectants that effectively reduced lesion volume in animal models of stroke have not demonstrated clinical efficacy in humans [1, 2]. While publication bias, lack of statistical power, and inappropriate randomization and blinding may partially account for the translational failures [3], assessing the magnitude of neuroprotection by infarct size reduction without considering brain edema may be a contributing factor; indeed, infarct volume is typically reported after correcting brain swelling. However, post-stroke brain swelling has been recognized as a life-threatening complication during infarct development [4]. Moreover, clinical studies have demonstrated an association between swelling and increased morbidity and mortality in acute ischemic stroke [5, 6, 7] suggesting the need for a reevaluation of post-stroke swelling on ischemic stroke outcomes in preclinical studies. Another important issue hindering translational success is the relative paucity in studies addressing comorbidities in animal models of stroke, despite the fact that risk factors such as dyslipidemia, hypertension, diabetes, and obesity are prevailing conditions in stroke patients [4, 8]. Thus, strategies aiming to develop effective stroke therapies require the incorporation of these comorbidities in animal models of stroke.
While acute swelling stems from cytotoxicity due to cellular disruption of ionic balance and the associated inflammation, long-lasting brain edema is derived from increased vascular permeability often associated with new vessel formation (angiogenesis) [4, 9]. Among several factors that regulate vascular function, vascular endothelial growth factor (VEGF)-A is a well-known mediator that regulates angiogenesis and vascular permeability [10, 11]. VEGF-A mainly interacts with the receptor VEGFR2 to activate downstream signaling pathways regulating the expression of endothelial specific molecule-1 (Esm1), a dermatan sulfate proteoglycan localized in endothelial cells [12, 13, 14, 15]. A recent study showed that Esm1 is involved in VEGF-A-mediated vascular permeability and edema formation in stroke [16].
Elevated blood glucose and lipids in diabetic condition have been linked to endothelial dysfunction [17, 18], which leads to pathologically increased vascular permeability. By incorporating these prevalent comorbid conditions in an animal model of stroke, this study investigated the impact of diabetes on post-stroke brain swelling. Here we report that diabetic mice display disproportionately increased brain swelling over infarct size with elevated expression of VEGF-associated signaling molecules following large territorial infarction induced by proximal middle cerebral artery occlusion. In addition, a significant correlation between VEGFR2 expression with brain swelling, but not infarct size, in diabetic mice indicates that stroke-induced brain swelling may be an independent pathological event in diabetic stroke.
Methods
Animals
Procedures for the use of animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medicine in accordance with the IACUC, National Institutes of Health, and ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. Male C57BL/6 (C57) mice purchased from Jackson Laboratory (Bar Harbor, ME) were bred at the institute’s animal facility. The facility monitors and maintains temperature, humidity, and a 12 h light/dark cycle. A maximum of 5 mice were housed in a single cage with an individual ventilating system and irradiated bedding (The Anderson, Maumee, OH). Sterilized food and water were freely accessible in each cage.
In vivo study design
Mice were randomized to a specific diet, treatments, and surgery. In certain cases, surgeons could not be blinded to the identity of groups at the time of surgery due to apparent body weight gain from high fat diet. Animals’s identity and treatment were blinided to the persons who cryosectiones and evaluates outcomes. Sample size for stroke outcome measurements was calculated a priori with minimum 10 by predicting detectable differences to reach power of 0.80 at a significance level of <0.05, assuming a 40% difference in mean and a 30% standard deviation (SD) at the 95% confidence level. Sample size for the VEGF inhibition study was also calculated a priori (n=8/group) by predicting detectable differences to reach power of 0.80 at a significance level of <0.05, assuming a 45% difference in mean and a 30% SD at the 95% confidence level.
Animal models of diabetes
Six-week-old male C57 mice were fed a normal (ND, 5053, LabDiet, St. Louis, MO) or a diabetic high fat diet (DD, S3282, Bioserv, NJ) for 8 weeks [19]. Three weeks after the initiation of the diets, a low dose of streptozotocin (Sz, 40 mg/kg) or vehicle was intraperitoneally administered daily for 5 consecutive days. The diet intervention and/or Sz administration resulted in four groups of mice: normal diet with vehicle (ND), high fat diet with vehicle (DD, type 2), normal diet with stretpzotocin (Sz, type 1), and high fat diet with streptozotocin (DD/Sz, mixed type 1 and 2).
Glucose measurement, glucose tolerance test, and plasma insulin measurement
Fasting blood glucose measurement and glucose tolerance test (GTT) were performed as previously reported [19]. Briefly, fasting blood glucose levels were measured at 7 weeks of diet intervention by a glucometer (Ascensia Contour, Bayer, Germany). For GTT, overnight fasted mice were given an intraperitoneal injection of D-glucose (2 g/kg) and blood glucose levels were measured at pre, 15, 45, and 120 minutes after glucose injection. Plasma insulin levels were determined in overnight fasted mice using commercially available kits (insulin enzyme immunoassay kit, ALPCO Diagnostics, NH) according to the manufacturer’s instructions.
Transient middle cerebral artery occlusion (MCAO)
Mice were subjected to 30 min proximal MCAO using an intraluminal thread method according to previously reported methods [20, 21]. Briefly, mice were anesthetized with a mixture of isoflurane/oxygen/nitrogen and a 6-0 Teflon-coated black monofilament surgical suture (Doccol, Redland, CA) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the Circle of Willis to obstruct the origin of MCA. The filament was left in place during an entire ischemic period (30 min) and then withdrawn. Cerebral blood flow (CBF) was measured prior to stroke, during the entire 30 min of occlusion period, and 10 min after reperfusion. In order to monitor the CBF in the ischemic territory, a fiber optic probe was glued to the parietal bone (2 mm posterior and 5 mm lateral to the bregma) and connected to a Laser-Doppler Flowmeter (Periflux System 5010; Perimed, Järfälla, Sweden). Using a rectal probe controlled by a masterflex pump and thermistor temperature controller (Cole-Parmer, Vernon Hills, IL), animal’s body temperatures were maintained at 37±0.5°C during MCAO and 1h after post-ischemia. Mice exhibiting greater than 80% reduction of pre-ischemic baseline CBF during MCAO and CBF greater than 80% of baseline blood flow by 10 min of reperfusion were included in the study.
Tissue section strategy
As reported in a previous study [19], sections were collected from the brain regions spanning about 7 mm rostrocaudal (+3.1mm to −4.1mm from bregma). The brain block containing an entire infarct region was serially cryosectioned at 20 μm thickness at 600μm intervals, thus a total of 13 sections were used for infarct volume and swelling measurement. Tissues between the intervals were sectioned and collected for each hemisphere to perform molecular analyses. This unbiased sectioning strategy facilitated measurement of infarct/swelling and gene/protein in the same animals, thereby allowing correlation analyses between histological and molecular parameters.
Infarct volume and edema measurement
Infarct volume, corrected for brain swelling, was determined in serial sections using a phase contrast method and analyzed by Axiovision software (Zeiss, Germany) as we previously reported [19, 22]. Infarct volume was determined as integrating volume of each section obtained by infarct area (mm2) multiplied distance between adjacent sections (0.6mm). Final infarct volume was reported after subtracting the volume difference between contralateral and ipsilateral side to correct for swelling. Percent hemispheric swelling (% HS) was calculated using the difference in volume between two hemispheres and then dividing by contralateral hemisphere volume according to the following formula: % HS = [(ipsilateral volume-contralateral volume)/contralateral volume] x 100 [23].
IgG immunohistochemistry
Vascular permeability was determined by IgG immunohistochemistry [24]. Frozen brain sections (+1.9mm, +0.7mm, and −0.5mm from bregma) collected on gelatin-coated slides were fixed with 4% paraformaldehyde for 15 min. After incubation in methanol containing 0.3% H2O2 for 30 min to block endogenous hydroperoxidase, the sections were incubated in 1% bovine serum albumin and 10 % normal goat serum for 1h. The sections were labeled with anti-mouse IgG antibody (1:1000, Vector Lab., Burlingame, CA) for overnight at 4°C. After washing with phosphate-buffered saline, the sections were incubated with avidin/biotinylated peroxidase (ABC reagent, Vector Lab., Burlingame, CA) for 1h at room temperature and colorized by incubating in diaminobenzidine (Sigma, MO). The mean IgG intensity for each hemisphere was quantified by Image J software and results were presented as the ratio of ipsilateral over contralateral value.
Vegf-a, Esm1, Vegfr2 mRNA measurement
Relative mRNA levels were quantified with real- time reverse transcription-polymerase chain reaction (RT-PCR) using fluorescent TaqMan technology as described previously [21, 22]. Total RNA from brain tissue was extracted using Tri reagent (MRC, OH) and 1 μg of total RNA was reverse transcribed using QuantiTech reverse transcription kit (QIAGEN, Valencia, CA), according to the manufacturer’s protocol. PCR primers and probes specific for the genes in this study were obtained as TaqMan pre-developed assay reagents for gene expression (Life Technologies, Foster City, CA). Primers used were VEGF-A (Mm00437304_m1); VEGFR2 (Mm01222419_m2); Esm1 (Mm00469953_m1); and β-actin (Mm00607939_s1). β -Actin was used as an internal control for normalization of samples. The PCR reaction was performed in 20 μl total volume using FastStart Universal Probe Master Mix (Roche, Indianapolis, IN) by incubating at 95°C for 10 min followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. The results were analyzed using 7500 Fast Real-Time PCR System software (Life Technologies, Grand Island, NY).
VEGF-A and VEGFR2 protein measurement
For protein analyses, brain sections were homogenized in 200 μl of CeLytic buffer (Sigma, MO) containing protease inhibitors (Roche). Total protein concentrations were measured using NanoDrop (Thermo Scientific, DE). Brain VEGF-A levels were determined using a commercially available enzyme-linked immunosorbent assay kit according to manufacturer’s protocol (Boster Biological Technology, Fremont, CA). VEGF-A protein levels were normalized to total protein concentrations. Brain VEGFR2 levels were determined by western blot. Fifty microgram of protein was loaded on a NuPAGE 4-12% Bis-Tris Gel (Life Technology) and transferred onto polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA). The membrane was incubated in blocking buffer (Li-cor, Lincoln, NE) for 1h followed by overnight incubation at 4°C with VEGFR2 (2479S, 1:1000, Cell signaling, MA) or β-Actin (sc-1615, 1:10,000, Santa Cruz Biotechnology) antibody in blocking buffer and washed with tris-buffered saline containing 0.05% Tween-20 followed by incubation with appropriate secondary antibodies conjugated with Alexa Fluor 680 (A 21088, Life Technologies), or IRDye® 680RD (926-68071, Li-cor) in blocking buffer for 1h. Then each protein’s specific band was visualized using the Odyssey Imaging System (Li-cor). Western blots were performed in multiple gels. To normalize inter-blot variability, identical samples were loaded in each blot as an internal control and the density of the internal standard sample was used to standardize samples in multiple blots.
Administration of VEGFR2 inhibitor, SU5416
To inhibit the activation of the VEGF pathway, DD/Sz mice were treated with SU5416 (MedChem Express, NJ), a small molecule that blocks VEGFR2 phosphorylation. Severe diabetic mice (DD/Sz) randomly received a subcutaneous injection of either vehicle or 25mg/kg of SU5416 at 3h after transient MCAO. Stroke outcome was determined at 24h after stroke.
Data analyses
Infarct volume and % swelling were reported as mean ± 95% confidence intervals. Body weights, blood glucose levels, CBF changes, and all other molecular analyses were presented as mean ± standard error (sem). Comparison between two groups was statistically evaluated using Student’s t-test. Multiple comparisons were made using Analysis of Variance (ANOVA) followed by a post hoc Bonferroni’s multiple comparison tests. Correlation analyses between infarct volume and % swelling were made using linear regression, and slope differences among groups were further analyzed. Differences were considered statistically significant at p<0.05, p<0.01, and p<0.001.
Results
Establishment of experimental animal models of diabetes
Compared to ND group, DD, Sz, and DD/Sz goups showed mild, moderate, and severe hyperglycemia respectively (Fig. 1A). Compared to ND, body weight was significantly increased in DD, but not in Sz or DD/Sz groups (Fig. 1B). GTT showed delayed glucose clearance in all three diabetic groups with the largest impairment in DD/Sz mice, indicating mild, moderate or severe insulin resistance in relevant groups (Fig. 1C). Fasting plasma insulin level was significantly increased only in the DD group in comparison to the other groups (Fig. 1D). The results showed establishment of experimental animal models that reflect features of type 2 (DD), type 1 (Sz) and mixed type 1/2 (DD/Sz) diabetic conditions. The normal and diabetic mice were participated to histology and molecular analyses after being subjected to transient middle cerebral artery occlusion (MCAO). Stroke severity, indicated by CBF reduction during MCAO, and post-stroke mortality were similar among the four groups (Table 1).
Figure 1. Establishment of experimental diabetes models.
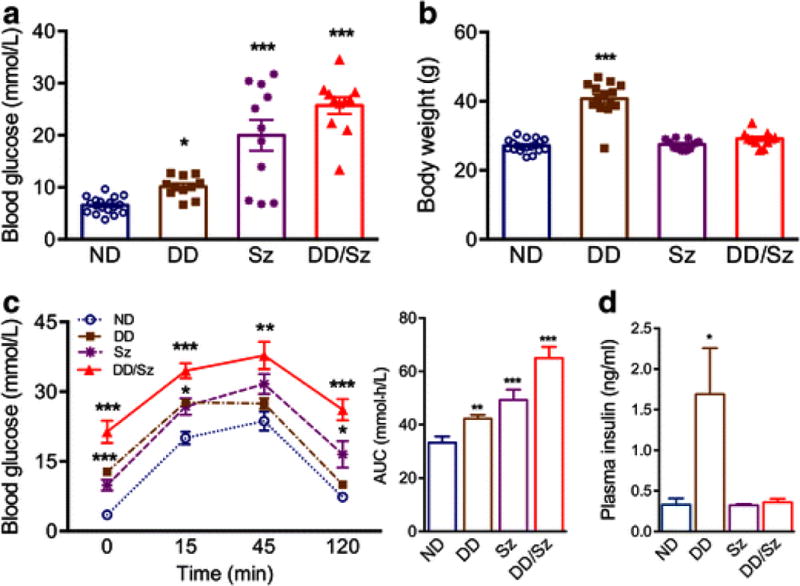
Different degrees of hyperglycemia were generated in C57BL mice by feeding a normal (ND) or high fat diet (DD) with or without streptozotocin (Sz). A&B, Fasting blood glucose levels at 7 weeks of diet (A) and body weight at the end of diet intervention (B), n=11-19/group C, GTT and the area under the curve (AUC) at 7 weeks of diet intervention. n=5/group, D, Plasma insulin levels in the overnight fasted animals. n=4/group, Data are expressed as mean ± sem, ND, normal; DD, Sz, DD/Sz, represent mild, moderate, and severe diabetic conditions. *, **,***p<0.05, 0.01, 0.001 vs. ND, one way ANOVA
Table 1.
Breakdown of the number of mice in the study
| ND | DD | Sz | DD/Sz | |
|---|---|---|---|---|
| Total number of animals | 34 | 33 | 19 | 40 |
| Failed MCAO | 3 | 7 | 1 | 3 |
| Successful MCAO | 31 | 26 | 18 | 37 |
| CBF during MCAO (%) | 16.7±0.7 | 13.9±0.9 | 16.6±1.3 | 14.5±0.7 |
| CBF reperfusion at 10 min (%) | 113.6±4.7 | 106.7±5.4 | 109.1±4.6 | 95.7±4.8 |
| Post-stroke death | 2 | 0 | 1 | 1 |
| % mortality | 5.9 | 0.0 | 5.3 | 2.5 |
| Animals entered in study | 29 | 26 | 17 | 36 |
Disproportionately large edema formation under diabetic condition
Infarct size and hemispheric swelling were determined at 3d post-ischemia. Compared to ND, Sz and DD/Sz mice had enlarged brain infarcts extending outside MCA territories (Fig. 2A and B). All three groups of diabetic mice displayed significantly enlarged brain swelling (Fig. 2C). Compared to ND, the intensity of IgG staining in moderate (Sz) and severe (DD/Sz) diabetic mice was significantly elevated, indicating increased vascular permeability in diabetic stroke (Fig. 2D).
Figure 2. Stroke outcomes in diabetic stroke.
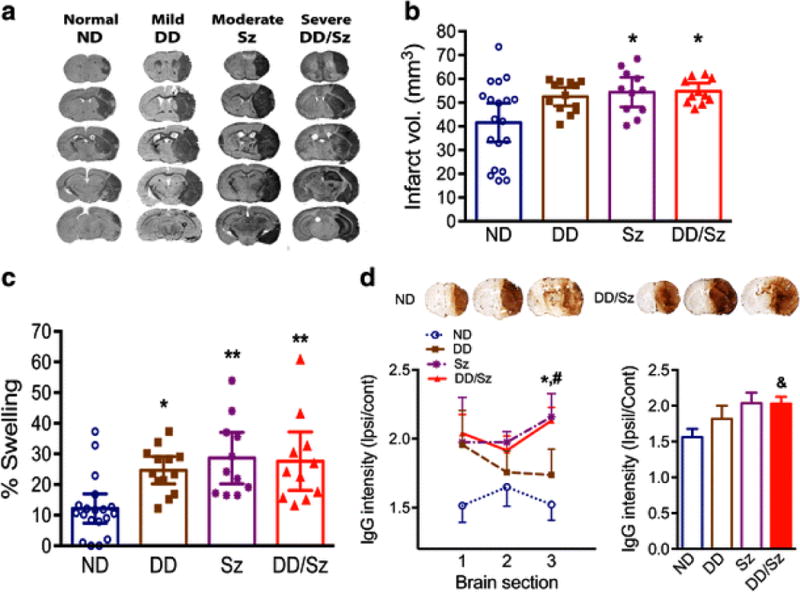
Infarct volume, % swelling, and IgG extravasation were determined at 3 days post-stroke A-C, Images of serially sectioned brains (A), infarct volume (B), and Percent hemispheric swelling (C), Data are expressed at the 95% confidence interval, n=11-19/group, *, **p<0.05, 0.01 vs. ND, one way ANOVA, D, IgG immunostaining using brain sections, The IgG intensity of each section, +1.9mm (1), +0.7mm (2), and −0.5mm (3) (left line graph) and average intensity of the three sections (right bar graph), Data are expressed as mean ± sem, n=5-11/group, *p<0.05 for Sz vs. ND, #p<0.01 for DD/Sz vs. ND, and &p<0.01 for DD/Sz vs. ND, one way ANOVA, ND, normal; DD, Sz, DD/Sz, represent mild, moderate, and severe diabetic conditions
To investigate the interplay between edema and infarct size under normal and diabetic conditions, correlation analyses were performed. Brain swelling was significantly correlated with the infarct size in ND mice (R2=0.487, p=0.0009, Fig. 3), but the correlation was absent in all diabetic groups. Importantly, slope elevations in the diabetic groups were significantly different from ND (p<0.05, Fig. 3). The absence of any correlation between brain swelling and infarct volume in diabetic mice suggests that brain swelling is a predominant and independent pathological component in diabetic stroke.
Figure 3. Correlation analyses between infarct volume and % swelling among normal and diabetic mice.
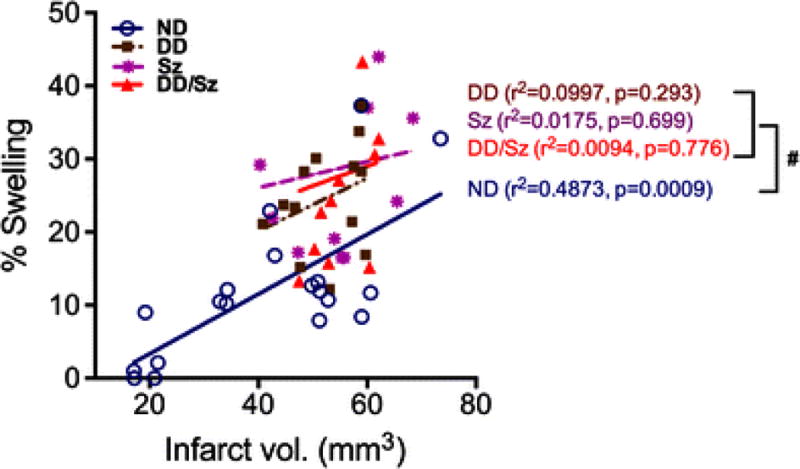
ND, normal; DD, Sz, DD/Sz, represent mild, moderate, and severe diabetic conditions, #p<0.05 in correlation slope vs. ND
Elevated VEGF-A and Esm1 expression in diabetic stroke
VEGF signaling regulates angiogenesis and vascular permeability through interaction of VEGF-A with the receptor, VEGFR2. The temporal profile in normal mice shows that stroke induced early VEGF-A mRNA expression at 6 h (Fig. 4A) and elevation of VEGFR2 mRNA expression sustained at 6 h and 3 days post-stroke (Fig. 4B). Based on the temporal profile results, we chose 6h and/or 3d-post stroke time points to measure the expression of VEGF-associated molecules between normal and diabetic conditions. In diabetic mice, VEGF-A gene expression tends to be increased in ischemic hemisphere compared to the non-stroked side at 6h-post ischemia, but there was no significant difference between normal and diabetic mice (Fig. 5A). However, significantly increased VEGF-A protein levels were observed in the 6h-post ischemic brain of most severe DD/Sz mice (Fig. 5B). There was a moderate increase in VEGF-A protein in the contralateral hemisphere in Sz and DD/Sz diabetic mice, suggesting the influence of chronic diabetic conditions on VEGF-A expression in the absence of stroke (Fig. 5B). A signaling molecule of VEGF downstream pathways, Esm1 mRNA was also significantly increased in diabetic condition(s) at both early (6h, Fig. 5C) and later (3d, Fig. 5D) time points after stroke.
Figure 4. Temporal gene expression of VEGF-A and VEGFR2 in non-diabetic stroked brain.
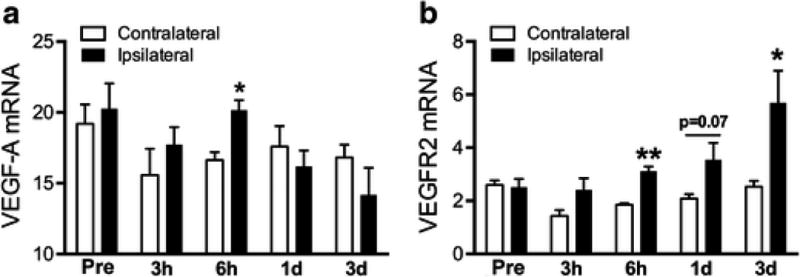
A&B, VEGF-A (A) and VEGFR2 (B) mRNA levels were measured in normal stroke brain at 0h (pre), 3h, 6h, 1d, and 3d-post. Data are expressed as mean ± sem, n=3-4/group, *,**p<0.05, 0.01vs. contralateral side, student t-test
Figure 5. VEGF-A and Esm1 expression in the stroked brain.
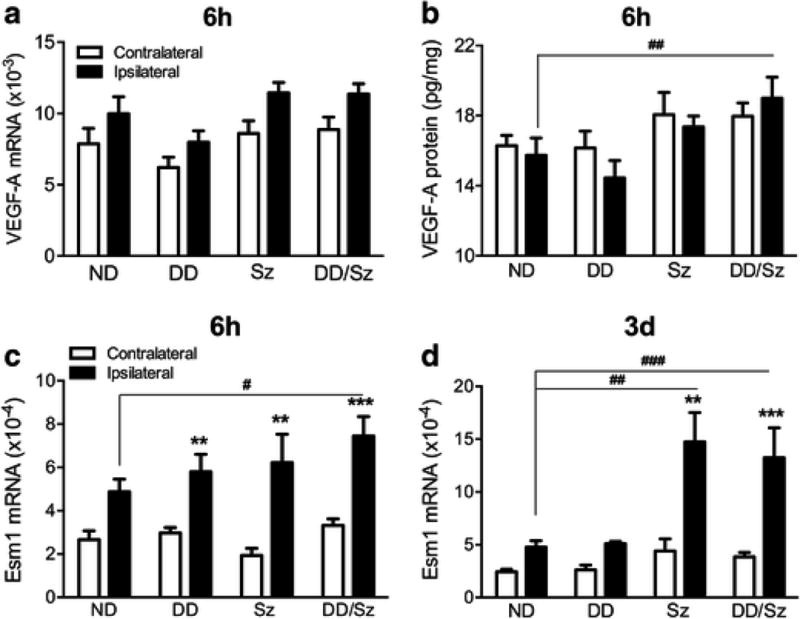
A&B, VEGF-A mRNA (A) and protein (B) levels in the brain at 6h-post ischemia, C&D, Esm1 mRNA expression levels in the brain at 6h and 3d post-ischemia. Data are expressed as mean ± sem, ND, normal; DD, Sz, DD/Sz, represent mild, moderate and severe diabetic conditions, n=5-12/group, **, ***p<0.01, 0.001 vs contralateral, #, ##, ###p<0.05, 0.01, 0.001 vs. ipsilateral of ND, two way ANOVA
A specific link between VEGFR2 pathway and brain swelling in diabetic stroke
The increased ligand VEGF-A and the downstream molecule Esm1 suggest the activation of VEGF signaling pathway. Thus we further assessed the expression of the receptor, VEGFR2 in normal and diabetic mice to confirm the activation of VEGF-A/VEGFR2 pathway. Stroke significantly increased VEGFR2 mRNA and protein levels at 3 days in diabetic mice with a statistical significance in DD/Sz mice compared to ND mice (Fig. 6A and B). To address the functional significance of VEGFR2 gene expression in diabetic stroke, stroke outcomes were correlated with VEGFR2 mRNA levels. As the three diabetic conditions showed significant elevation of swelling compared to ND mice, all diabetic mice were combined for the analyses. There was no correlation between VEGFR2 mRNA level and infarct size in normal and diabetic mice (Fig. 6C). However, there was a significant correlation between VEGFR2 mRNA levels and brain swelling in diabetic mice but not in normal mice (Fig. 6D). The results indicate a specific link between VEGF signaling and diabetes-exacerbated brain swelling in ischemic stroke. In order to determine causality, DD/Sz mice were treated with either vehicle or a VEGFR2 inhibitor, SU5416 at 3 h post-stroke. SU5416 did not change the infarct volume in the DD/Sz mice (Fig. 6E). However, the treatment significantly reduced swelling at 24h-post ischemia (Fig. 6F), indicating that VEGF signaling is involved in exacerbation of brain swelling in diabetic stroke.
Figure 6. A specific link of VEGFR2 pathway to brain swelling in diabetic stroke.
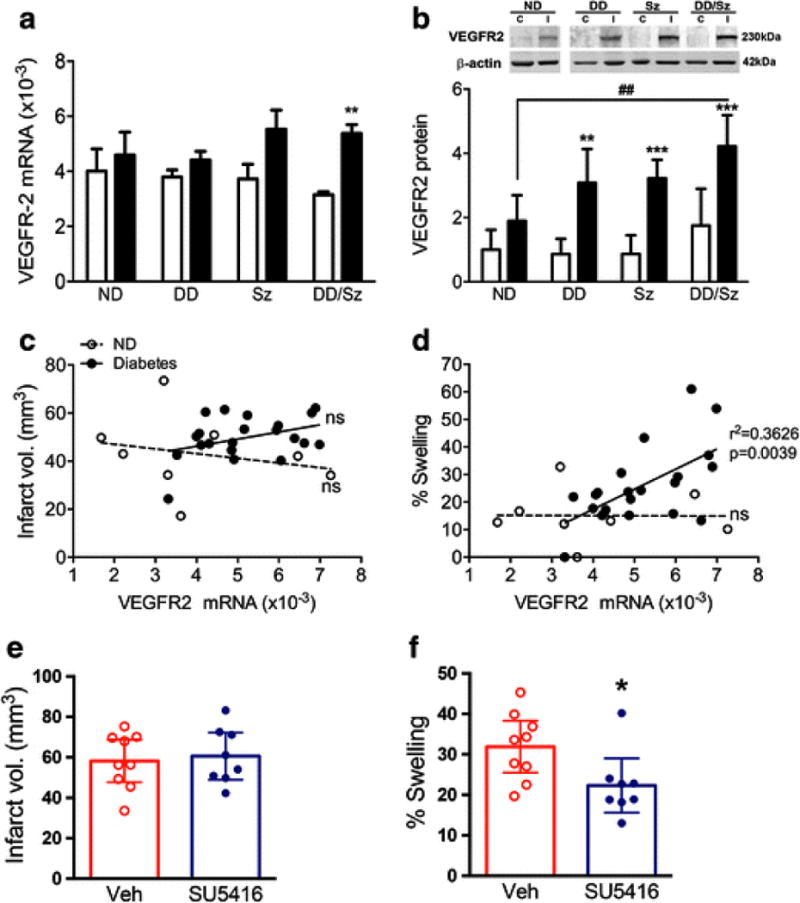
A & B, VEGFR2 mRNA (A) and protein (B) levels in the brain at 3d post-ischemia. Data are expressed as mean ± sem, C, contralateral; I, ipsilateral, ND, normal; DD, Sz, DD/Sz, represent mild, moderate, and severe diabetic conditions, n=4-12/group, **, ***p<0.01, 0.001 vs. contralateral, ##p<0.01 vs. ipsilateral of ND, two way ANOVA, C&D, Correlation analyses between VEGFR2 mRNA levels with infarct volume (C) or % brain swelling (D) in normal (ND) and diabetic groups (combined DD, Sz, DD/Sz mice). n=8-21/group, ns, non-significant, E&F, Infarct volume (E) and swelling (F) in the vehicle (Veh) or SU5416-treated DD/Sz mice at 24h-post ischemia. Data are expressed at the 95% confidence interval, n=8-9/group, *p<0.1 vs. Veh, Student t-test
Discussion
Infarct volume, an estimated final infarct size adjusted for brain swelling, has been a gold standard in reporting ischemic stroke outcomes. However, given multiple high-profile translational failures using acute neuroprotection strategies, more efforts need to be directed to increase translational efficacy. In this regard, this study addresses importance of post-stroke brain swelling, besides infarct volume, as an acute pathological outcome. Specifically, the study addressed the relative contribution of post-stroke brain swelling in a common comorbid condition, diabetes. With established experimental animal models of diabetes that reflect features of type 1 and 2 diabetes, the current study provided experimental evidence that brain swelling is a critical pathological component in diabetic stroke and showed a specific link of VEGF signaling to diabetes-exacerbated brain swelling. Despite the activation of VEGF signaling is an adaptive response following hypoxia and is associated with improved outcomes [25], the current finding showing that the same molecular pathway exacerbated brain swelling and negatively affected in diabetic stroke supports a novel view of difference on stroke pathology depending on peripheral metabolic status.
With the generation of mild (type 2), moderate (type 1), and severe diabetic phenotypes (type 1/2), we observed that these diabetic conditions increased infarct size, but exacerbated brain swelling to a greater extent (Fig. 2C). Stroke-induced brain swelling starts with cellular cytotoxicity leading to cytotoxic edema, which is followed by sustained vasogenic edema with increased blood brain barrier permeability [4, 26]. The compromised vascular function in diabetes has been implicated in pathologic angiogenesis and vasoregression after stroke [27]. It has been reported that vascular permeability increases in type 1 diabetic mice following photothrombotic stroke [28]. An intriguing observation from our current study is that the exacerbation of brain swelling occured across different types and sevierity of diabetes. Thus, the profound swelling observed at 3d post-stroke suggests that pathogenic angiogenesis coupled with leaky vessels, supported by the increased IgG staining (Fig. 2D), may underlie comorbidity-enhanced vasogenic edema.
Bigger lesions is normally accompanied by larger brain swelling in ischemic stroke. Consistantly, we observed a significant correlation between infarct size and brain swelling in normal mice, however, the correlation was not present in the metabolically compromised diabetic mice (Fig. 3). As diabetic mice displayed overall enlarged infarct size, it is possible that a relatively narrow range of injury volume in diabetic mice might be responsible for the absence of correlation between infarct volume and swelling. Nevetheless, the exacerbation of brain swelling in diabetic mice was significantly greater compared to normal mice, as displayed by prominent feature associated with diabetic conditions. We speculate that there may be intrinsic pathogenic mechanisms such as deregulated inflammatory responses [19] and endothelial dysfunction [29] that override normal stroke-induced injury development in diabetic conditions. Consistent with a clinical report showing that brain edema independently predict poor ischemic stroke outcomes [7], brain swelling thus may be an independent pathological component in diabetic stroke. Furthermore, stroke-induced brain edema has been associatiated with the neurological deficits [30, 31, 32]. Thus, whether the enhanced brain swelling in the diabetic mice negatively affects functional outcomes and the relevance of this finding in human patients remained to be addressed.
VEGF signaling through VEGFR2 has been shown to be beneficial by enhancing injury-induced angiogenesis in stroke uncomplicated by comorbidities [33, 34, 35]. The same VEGF signaling however negatively impacted stroke outcome under comorbid conditions leading to excessive vascular permeability [28, 35]. Consistent with literature, we observed general increase in the expression of VEGF-signaling associated molecules, VEGF-A and Esm1 in diabetic stroke. As a target and modulator of VEGF signaling, studies have demonstrated that Esm1 is required for VEGF-induced angiogenesis and vascular permeability [16, 36]. Since the Esm1 increases VEGF-A bioavailability by competing with VEGF-A to bind to endothelial matrix [16], the increased VEGF-A may be related to the elevated Esm1 expression in the diabetic conditions (Fig. 5). Accordingly, we found that the increase in ligand expression was also accompanied by upregulation at the receptor level. Notably, the significant correlation which is present between VEGFR2 expression and brain swelling, but not with injury size, in across various diabetic conditions implicates a specific link between molecules (VEGFR2) and pathology (swelling) in diabetic stroke. Moreover, the reduction of brain swelling, but not injury size, in SU5416 treated severe diabetic mice further implicates that VEGFR2 is a potential target to reduce edema in the diabetic condition. Reeson et al. reported the contributing role of VEGF signaling in local injury induced by photothrombotic stroke [28]. It is noteworthy in our study that the inhibition of VEGF signaling reduced diabetes-exacerbated brain swelling in animals with large territorial infarct, thereby suggesting that blocking VEGF signaling could be beneficial regardless of stroke severity. Understanding the impact of other comorbidities such as aging, hypertension, and hyperlipidemia on brain swelling and the effects of comorbidity-enhanced brain swelling on long-term stroke recovery warrant further investigation.
In summary, the current study showed that brain swelling is a predominant pathological event in diabetic stroke. The specific link between VEGFR2 expression and brain swelling indicate that an underlying event diabetes-enhanced brain swelling includes the activation of VEGF signaling. Our studyt identifies disparity of stroke pathology in normal and comorbid conditions, the study suggests consideration of therapeutic strategies for aiming at primarily reducing brain swelling for subjects with type 1 and 2 diabetes.
Acknowledgments
Research reported in this study was supported by the National Institute of Health awards, NINDS R01NS077897 (SC), R01NS095359-10 (SC) and the Burke Foundation (SC).
Funding
Research reported in this study was supported by the National Institute of Health awards, NINDS R01NS077897 (SC), R01NS095359-10 (SC), and the Burke Foundation (SC).
Footnotes
ORCID: Eunhee Kim, 0000-0002-7755-7291; Jiwon Yang, 0000-0003-4226-7224; Sunghee Cho, 0000-0002-9988-8227
Compliance with Ethical Standards
Author Contribution Statement
Eunhee Kim generated diabetic mice, characterized, performed MCAO, molecular analyses and wrote the manuscript, Jiwon Yang and Keun Woo Park contributed to molecular and biochemical assessment, and Sunghee Cho designed the study and wrote the manuscript.
Ethical Approval
This article does not contain any studies with human participants. The care and use of animals were followed by the ethical staindards of the Institutional Animal Care and Use Committee (IACUC) of Weill Cornell Medicine in accordance with the IACUC, National Institutes of Health, and ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Neuhaus AA, et al. Importance of preclinical research in the development of neuroprotective strategies for ischemic stroke. JAMA Neurol. 2014;71(5):634–9. doi: 10.1001/jamaneurol.2013.6299. [DOI] [PubMed] [Google Scholar]
- 2.O’Collins VE, et al. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59(3):467–77. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 3.Dirnagl U. Bench to bedside: the quest for quality in experimental stroke research. J Cereb Blood Flow Metab. 2006;26(12):1465–78. doi: 10.1038/sj.jcbfm.9600298. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg GA. Ischemic brain edema. Prog Cardiovasc Dis. 1999;42(3):209–16. doi: 10.1016/s0033-0620(99)70003-4. [DOI] [PubMed] [Google Scholar]
- 5.Schwab S, et al. The value of intracranial pressure monitoring in acute hemispheric stroke. Neurology. 1996;47(2):393–8. doi: 10.1212/wnl.47.2.393. [DOI] [PubMed] [Google Scholar]
- 6.Wijdicks EF, Diringer MN. Middle cerebral artery territory infarction and early brain swelling: progression and effect of age on outcome. Mayo Clin Proc. 1998;73(9):829–36. doi: 10.4065/73.9.829. [DOI] [PubMed] [Google Scholar]
- 7.Battey TW, et al. Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke. 2014;45(12):3643–8. doi: 10.1161/STROKEAHA.114.006884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto A, et al. Cerebrovascular risk factors and clinical classification of strokes. Semin Vasc Med. 2004;4(3):287–303. doi: 10.1055/s-2004-861497. [DOI] [PubMed] [Google Scholar]
- 9.Hatashita S, Hoff JT. Brain edema and cerebrovascular permeability during cerebral ischemia in rats. Stroke. 1990;21(4):582–8. doi: 10.1161/01.str.21.4.582. [DOI] [PubMed] [Google Scholar]
- 10.Keck PJ, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246(4935):1309–12. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 11.Leung DW, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 12.Bechard D, et al. Endocan is a novel chondroitin sulfate/dermatan sulfate proteoglycan that promotes hepatocyte growth factor/scatter factor mitogenic activity. J Biol Chem. 2001;276(51):48341–9. doi: 10.1074/jbc.M108395200. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara N, et al. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 14.Koch S, et al. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437(2):169–83. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- 15.Shin JW, et al. Transcriptional profiling of VEGF-A and VEGF-C target genes in lymphatic endothelium reveals endothelial-specific molecule-1 as a novel mediator of lymphangiogenesis. Blood. 2008;112(6):2318–26. doi: 10.1182/blood-2008-05-156331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rocha SF, et al. Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ Res. 2014;115(6):581–90. doi: 10.1161/CIRCRESAHA.115.304718. [DOI] [PubMed] [Google Scholar]
- 17.Okon EB, et al. Hyperglycemia and hyperlipidemia are associated with endothelial dysfunction during the development of type 2 diabetes. Can J Physiol Pharmacol. 2007;85(5):562–7. doi: 10.1139/y07-026. [DOI] [PubMed] [Google Scholar]
- 18.Hawkins BT, et al. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50(1):202–11. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- 19.Kim E, et al. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation. 2014;11:83. doi: 10.1186/1742-2094-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim E, et al. Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J Cereb Blood Flow Metab. 2014;34(8):1411–9. doi: 10.1038/jcbfm.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim E, et al. CD36 in the periphery and brain synergizes in stroke injury in hyperlipidemia. Ann Neurol. 2012;71(6):753–64. doi: 10.1002/ana.23569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim E, et al. Daidzein Augments Cholesterol Homeostasis via ApoE to Promote Functional Recovery in Chronic Stroke. J Neurosci. 2015;35(45):15113–26. doi: 10.1523/JNEUROSCI.2890-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin TN, et al. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24(1):117–21. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 24.ElAli A, et al. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke. 2011;42(11):3238–44. doi: 10.1161/STROKEAHA.111.615559. [DOI] [PubMed] [Google Scholar]
- 25.Yang JP, et al. VEGF promotes angiogenesis and functional recovery in stroke rats. J Invest Surg. 2010;23(3):149–55. doi: 10.3109/08941930903469482. [DOI] [PubMed] [Google Scholar]
- 26.Katzman R, et al. Report of Joint Committee for Stroke Resources. IV. Brain edema in stroke. Stroke. 1977;8(4):512–40. doi: 10.1161/01.str.8.4.512. [DOI] [PubMed] [Google Scholar]
- 27.Ergul A, et al. Cerebral neovascularization in diabetes: implications for stroke recovery and beyond. J Cereb Blood Flow Metab. 2014;34(4):553–63. doi: 10.1038/jcbfm.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reeson P, et al. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J Neurosci. 2015;35(13):5128–43. doi: 10.1523/JNEUROSCI.2810-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash R, et al. Vascularization pattern after ischemic stroke is different in control versus diabetic rats: relevance to stroke recovery. Stroke. 2013;44(10):2875–82. doi: 10.1161/STROKEAHA.113.001660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tominaga T, Ohnishi ST. Interrelationship of brain edema, motor deficits, and memory impairment in rats exposed to focal ischemia. Stroke. 1989;20(4):513–8. doi: 10.1161/01.str.20.4.513. [DOI] [PubMed] [Google Scholar]
- 31.Turner RC, et al. The role for infarct volume as a surrogate measure of functional outcome following ischemic stroke. J Syst Integr Neurosci. 2016;2(4) doi: 10.15761/JSIN.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoo AJ, et al. Infarct volume is a pivotal biomarker after intra-arterial stroke therapy. Stroke. 2012;43(5):1323–30. doi: 10.1161/STROKEAHA.111.639401. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y, et al. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111(12):1843–51. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang ZG, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106(7):829–38. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zechariah A, et al. Hyperlipidemia attenuates vascular endothelial growth factor-induced angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery via decreased pericyte coverage of brain endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(7):1561–7. doi: 10.1161/ATVBAHA.112.300749. [DOI] [PubMed] [Google Scholar]
- 36.Roudnicky F, et al. Endocan is upregulated on tumor vessels in invasive bladder cancer where it mediates VEGF-A-induced angiogenesis. Cancer Res. 2013;73(3):1097–106. doi: 10.1158/0008-5472.CAN-12-1855. [DOI] [PubMed] [Google Scholar]