

Abstract
A patient with Prader-Willi syndrome (PWS) was found to carry a de novo balanced reciprocal translocation, t(15;19)(q12;q13.41), which disrupted the small nuclear ribonucleoprotein N (SNRPN) locus. The translocation chromosome 15 was found to be paternal in origin. Uniparental disomy and abnormal DNA methylation were ruled out. The translocation breakpoint was found to have occurred between exon 0 (second exon) and 1 (third exon) of the SNRPN locus outside of the SmN open reading frame (ORF), which is intact. The transcriptional activities of ZNF127, IPW, PAR-1, and PAR-5 were detected with RT-PCR from fibroblasts of the patient, suggesting that these genes may not play a significant role in the PWS phenotype in this patient. Transcription from the first two exons and last seven exons of the SNRPN gene was also detected with RT-PCR; however, the complete mRNA (10 exons) was not detected. Thus, the PWS phenotype in the patient is likely to be the result of disruption of the SNRPN locus.
INTRODUCTION
Prader-Willi syndrome (PWS) is a complex genetic disorder and an example of genomic imprinting. The clinical manifestations include decreased fetal activity, neonatal hypotonia, neonatal feeding difficulties, hyperphagia with obesity, hypogonadism, short stature, small hands and feet, and mental retardation (1). About 60–70% of the individuals with PWS have been found to have an interstitial deletion at 15q11–13 (1,2). The deletion is of paternal origin (3). About 30% of PWS patients have maternal uniparental disomy of human chromosome 15 (4). The remainder have mutations in the imprinting process (5), and in a few rare cases, balanced reciprocal translocations involving chromosome 15 (1). However, no detailed molecular studies have been published on any of the balanced reciprocal translocation patients so far.
DNA markers specific for chromosomal region 15q11–13 have been developed and widely used for delineation of specific abnormalities in patients with PWS, including microsatellites, cosmids, and yeast artificial chromosomes (YACs) (6). Detailed physical mapping has demonstrated that the common deletion region encompasses D15S9 (ZNF127) to D15S12 (7). The smallest region of deletion overlap (SRO) for PWS has been suggested to include loci from D15S63 (PW71) to D15S174 (6,8,9). However, each of these deletions (8) affects the imprinting process over a 2 Mb region (5) (S. S. et al, submitted), suggesting that genes for PWS could lie anywhere in the 1.5 Mb domain from D15S9 to D15S174.
It has been shown that three imprinting phenomena are found in the PWS region (10,11). Several loci in this region show parent-specific DNA methylation, including D15S9(ZNF127) (8,12), D15S63 (PW71) (13), and SNRPN (5,14–16). DNA replication timing is reported to be asynchronous in this region (17,18). Multiple genes [SNRPN (14,16,19,20); IPW (21); ZNF127 (M. T. C. Jong, R. D. N., in preparation)] and transcripts [PAR-5 and PAR-1 (15)] within 15q11–13 are expressed monoallelically from the paternal chromosome. Each of these are thus candidates to play a role in the pathogenesis of PWS, thought from genetic evidence to involve at least two paternally expressed genes (e.g., reviewed in refs 11 and 29).
Recently, the SNRPN gene has been mapped to the PWS critical region within the smallest deletion overlap (22). It is functionally imprinted in mouse (23,24) and human (14,16,19,20). It has been suggested that the SNRPN gene may be a candidate gene for PWS (14,22). Two additional upstream exons were recently found for the SNRPN gene (15,16), and the first exon (designated −1 or α) lies within a CpG island likely to be the promoter (5,15,16).
Here we report a patient with the PWS phenotype and a balanced reciprocal translocation t(15;19)(q12;q13.41). The translocation chromosome 15 was found to be paternal in origin. Uniparental disomy and a large deletion were ruled out, and a DNA methylation study was normal for PW71 (D15S63) and SNRPN. Fluorescence in situ hybridization (FISH) with 15q11–13 probes, including subfragments of the SNRPN gene, together with Southern analysis with different exons from the SNRPN gene, established the breakpoint to be within the SNRPN gene. RNA expression studies by reverse transcription-polymerase chain reaction (RT-PCR) of multiple genes monoallelically expressed within 15q11–13 further implicate the SNRPN locus as playing a role in the PWS phenotype in this patient.
Clinical description
This 3 year 6 month old Caucasian male was referred because of obesity and behavioral problems. He was diagnosed with Prader-Willi syndrome (Fig. 1) on the basis of satisfying the consensus major diagnostic criteria (scored 8 points) and some minor diagnostic criteria (scored 2 points) for PWS (25). He was born at 42 weeks of gestation weighing 3720 g (50th centile) to a 36 year old mother following a pregnancy complicated by a 30 pound weight loss. Decreased fetal activity was also noted during the pregnancy. The child had a large cephalohematoma at birth after a prolonged difficult delivery. He was floppy and lethargic for the first 6 months of life and had a poor suck. He weighed 10 kg (97th centile) at 6 months of age and 15 kg (>97th centile) at 18 months. His development was delayed, for example, he walked at 18 months and spoke his first words at 2 1/2 years. His appetite increased drastically after 2 years of age, and by 31 months, his weight was 24 kg (≫ 97th centile). A head CT scan was normal. He had a history of hyperphagia. At 35 months of age, a Stanford Binet composite test score was at the 12th centile with below average short term memory and quantitative skills. Family history included a 5 year old sibling with cerebral palsy secondary to birth trauma and the mother had a dysplastic kidney. Physical examination at 42 months of age showed a weight of 33.8 kg (at the 50th centile for a 10 year child) with a height of 103 cm (75th centile for 42 months). He did not have hypopigmentation. His testicles were undescended. His hand length was at the 25th centile and finger length at the 10th centile. He had mild generalized hypotonia. Bone age was advanced by 9 months with a standard deviation of 6.7 months.
Figure 1.

Front facial view of the proband (3 years and 6 months old). Note narrow bifrontal diameter, almond-shaped eyes, and down-turned mouth.
RESULTS
Cytogenetic analysis and parental origin of the translocation chromosome 15
Chromosome analysis with high-resolution GTG banding showed a de novo balanced translocation, 46, XY, t(15;19) (q12;q13.41) in all cells analyzed from peripheral blood (Fig. 2). It has been demonstrated in PWS that the syndrome is caused by the absence of paternal gene(s) in 15q11–13, most commonly either by deletion (3) or by maternal uniparental disomy (10). Chromosome polymorphism has been used for elucidation of parental origin of chromosomes (3). Therefore, the parental origin of the translocation chromosome 15 was investigated with QFQ banding of metaphase chromosomes prepared from lymphoblast cell lines of the translocation patient and his parents (Fig. 3). The intensity and size of centromere and satellite of chromosome 15 were compared in the cells of the patient with those in cells of parents, and the der(15) was established as paternal in origin.
Figure 2.

Chromosome GTG high resolution banding (top) showing 46,XY,t(15;19)(q12;q13.41). Only the chromosomes 15 and 19 are shown. The ideogram (bottom) shows the translocation breakpoint indicated by arrows.
Figure 3.

Parental origin of the translocation chromosome. The normal chromosome 15 in the patient is inherited from his mother (C). The der(15) chromosome is inherited from the father (B). One of the chromosomes 15 of the father of the patient showed medium intensity centromere staining with a pale satellite. The other chromosome 15 in the father showed light centromere staining with a pale satellite. One of the mother’s chromosome 15 showed a pale centromere and a bright satellite. The other chromosome 15 in the mother showed medium to pale centromere staining and a negative satellite. The patient’s normal chromosome 15 showed a pale centromere and bright satellite, indicating it is maternal in origin. The der(15) showed a light centromere and pale satellite, indicating it is paternal in origin.
Analysis for uniparental disomy
Uniparental disomy accounts for about 30% of PWS patients and has been found in conjunction with chromosome 15 translocations (10,26) and inv dup(15) (27). Therefore, a number of microsatellite markers were used to analyze for uniparental disomy. PCR studies of DNA loci D15S541, D15S543, D15S63, D15S128, D15S10, GABRB3, and GABRA5 indicated heterozygosity, while MN1, D15S122, D15S97, D15S219 and D15S165 showed only one allele each, most likely indicating homozygosity for these loci. D15S128, GABRB3 and GABRA5 showed biparental inheritance (Table 1), ruling out uniparental disomy of chromosomes 15.
Table 1.
Haplotypes for three informative miscrosatellite loci
| Loci | Alleles | ||
|---|---|---|---|
|
| |||
| Father | Mother | t(15;19) proband | |
| D15S128 | 1–2 | 2–3 | 1–3 |
| GABRB3 | 3-1 | 1–2 | 3-2 |
| GABRA5 | 1–2 | 2–3 | 1–3 |
DNA methylation analysis
Recently, some PWS patients have been identified as having abnormal DNA methylation (imprinting mutation) of chromosome 15q11–13 (5,8,15,28,29) (S. S. et al, submitted) due to a microdeletion close to SNRPN in an element termed the imprinting center (IC) (5). Therefore, methylation analysis of loci PW71 and SNRPN was performed (Fig. 4). For PW71, two bands (6.6 kb and 4.4 kb) are detected using the PW71B probe (30) in a normal individual. Only one upper band (6.6 kb) is detected in a typical PWS patient. However, both bands are detected in our translocation patient (Fig. 4A). For the SNRPN promoter region, two bands (4.3 kb and 0.9 kb) are detected in a normal individual, only one band (0.9 kb) is detected in an Angelman syndrome patient, whereas in typical PWS patients only the upper 4.3 kb band is detected (5,16) (Fig. 4B). However, both bands were detected in our t(15;19) translocation patient. These data suggest that the t(15;19) translocation patient had a normal DNA methylation pattern and does not have an imprinting mutation.
Figure 4.
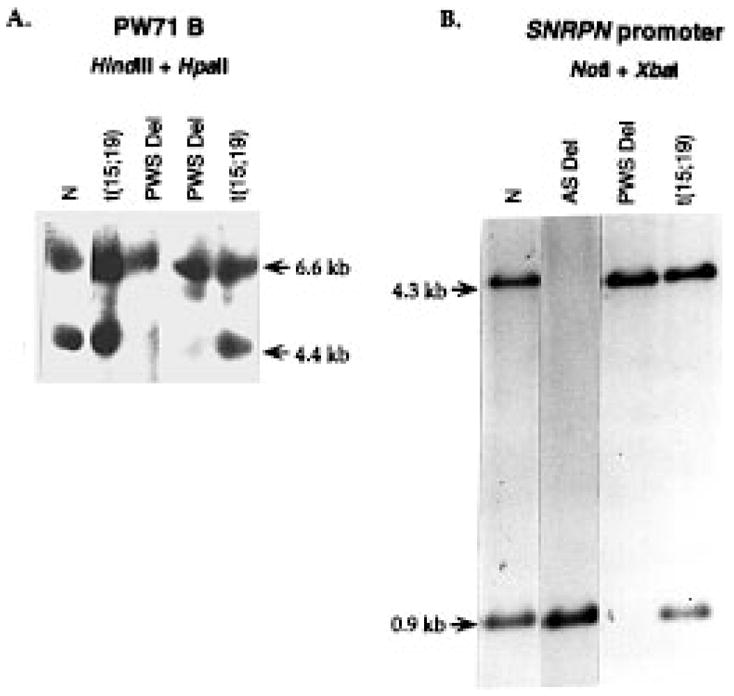
DNA methylation analysis. N, normal individual; t(15;19), the translocation patient described in this paper; PWS del, PWS deletion patient; AS del, Angelman deletion patient. (A) Leucocyte DNA was digested with HindIII and HpaII and hybridized with the PW71 B probe. The lower band (4.7 kb) is not detected in PWS deletion patients, but is present in normal individuals and the t(15;19) translocation patient. (B) The probe used was a cloned fragment from the SNRPN exon −1 region (16). DNA was digested with NotI and XbaI. The lower band is undetectable in PWS deletion patients, and for AS patients, the upper band is undetectable. Both bands are detectable in normal individuals and the t(15;19) translocation patient.
Breakpoint determination by FISH and Southern analysis
FISH analysis was performed with cosmid probes D15S10, D15S11, SNRPN and GABRB3 (Fig. 5A–D) to characterize the status of chromosome region 15q11–13. All metaphases produced hybridization signals in the cells studied from the patient. The probe D15S11 hybridized to the der(15) chromosome, whereas D15S10 and GABRB3 hybridized to the der(19) chromosome. Hybridization of the SNRPN probe resulted in signals on both der(15) and der(19) (Fig. 5). These data indicated that no deletion of these loci had occurred and that the breakpoint was within the SNRPN cosmid probe. This probe contains two overlapping cosmids which span approximately 40 kb. Since the SNRPN gene has been proposed to be a candidate gene for PWS (14,22), it is of importance to determine the breakpoints. To identify whether the translocation breakpoint was inside the SNRPN gene, two approaches were used, FISH and Southern analyses.
Figure 5.

FISH analysis. (A) Cosmid probe SNRPN; (B) D15S11; (C) GABRB3; (D) D15S10; (E) SNRPN subfragment F2 (Fig. 6A); (F) SNRPN subfragment F3 (Fig. 6A). In A–D, the labeled probes were purchased from Oncor, and a control locus PML cosmid probe was also included for identification of chromosome 15. It produces signals on 15q22. Arrows indicate der(15) (large arrow), der(19) (small arrow), or normal chromosome 15 (arrow head).
A contig of two SNRPN cosmids (A and B) was obtained from Oncor (Gaithersburg, MD). Restriction analysis with EcoRI, BamHI, and NotI and subsequent hybridization with a cDNA probe containing SNRPN exons 1–8 or a cDNA probe containing exons −1, 0 and 1 was performed to deduce which fragment contained which SNRPN exons (Fig. 6A). When digested with EcoRI (Fig. 6A), cosmid A generated four fragments larger than 5 kb: 11 (F2), 8.5 (F4), 6 (F3), and 5.8 kb (F1) (Fig. 6A). Digestion of cosmid B also produced four fragments larger than 5 kb: 14 (F5), 11 (F2), 6 (F3), 5.8 kb (F6) (Fig. 6A). The subfragments were isolated from an agarose gel and labeled with digoxigenin by sequence-independent PCR. The labeled probes were hybridized to metaphase chromosomes from the patient’s lymphoblasts. Fragments F1, F2 (Fig. 5E), and F5 detected signals on the der(15) while F3 (Fig. 5F), F4 and F6 detected signals on the der(19), suggesting the breakpoint is near the junction of F2 and F3 (Fig. 6A). According to the known genomic structure (Fig. 6A bottom) of SNRPN gene (5,15,16,22,31), the junction between fragments F2 and F3 is near exon 1 of the SNRPN gene. The results from this study indicate that the translocation breakpoint in chromosome 15 is within the SNRPN gene and located close to exon 1.
Figure 6.

Identification of the translocation breakpoint. (A) Restriction map showing area around the breakpoint. Restriction analysis of SNRPN cosmids A and B showed the contig contained the entire SNRPN gene. Each cosmid produced four subfragments larger than 5 kb after digestion with EcoRI (F1–F6). The normal restriction map around the breakpoint is shown at the bottom (5,16). Fragment F2 (and F1, F5) hybridizes to the der(15), while F3 (and F4, F6) hybridizes to the der(19). The arrow and oblong indicate the breakpoint region, and the ten exons are indicated by numbers −1 through 8. (B, BamHI; N, NotI; E, EcoRI; H, HindIII restriction sites). (B, C) Southern analysis for identification of the t(15;19) translocation breakpoint. DNA was digested with BamHI and hybridized with a cDNA probe (B) containing SNRPN exons 1–8 and a cDNA probe (C) containing exons −1, 0, and 1 (16). Lane 1, father; 2 and 7, t(15;19) proband; 3, mother; 4 and 6, PWS deletion patients; 5, PWS UPD patient; arrow, a novel fragments of 20 kb.
Southern blot analysis was performed by using a cDNA probe from the SNRPN gene exons 1–8. When genomic DNA was digested with BamHI, an extra novel band of approximately 20 kb was detected in the patient, but not in the parents (Fig. 6B). Southern analysis with a cDNA probe detecting exons −1, 0, and 1 uniquely was also performed. The exon 1 sequence of the probe again detected the extra 20 kb band, while the exon 0 sequence detected an abnormal extra band of approximately 17.5 kb (Fig. 6C). The exon −1 sequence of the probe detected a normal fragment of 15 kb. Abnormal fragments were also detected with EcoRI and HindIII (data not shown). These data indicate that the breakpoint in chromosome 15 is between exon 0 and exon 1 of the SNRPN gene (Fig. 6A).
RNA expression analysis
Recently, several genes and transcripts in 15q11–13 have shown allele-specific expression from the paternal chromosome only. Such genes include SNRPN (14,16,19,20), IPW (21), and ZNF127 (M. T. C. Jong, R. D. N., in preparation), and the transcripts PAR-5 and PAR-1 (15). These genes or transcripts are not expressed in PWS patients, while they are expressed in normal individuals, and are thus implicated in the PWS phenotype. In order to determine the expression pattern of these genes or transcripts in the translocation patient, expression of ZNF127, IPW, PAR-1, PAR-5, was performed with RT-PCR using total cellular RNA prepared from a fibroblast culture from the patient. Normal human brain mRNA and PCR in the absence of RT (RT−) were used as controls. Each of the latter four genes or transcripts showed expression in the control and in the t(15;19) translocation patient (Fig. 7A).
Figure 7.

RNA expression analysis with RT-PCR. (A) RT-PCR analysis of ZNF127, IPW, PAR-5, and PAR-1. The amplification was present in RT+ reactions (lane 4), but not in RT− reactions (lane 3). Lane 1, normal human brain mRNA for positive control; Lane 2, DNA derived from the translocation patient serving as positive control. It was omitted for IPW since RT-PCR spanned an intron. Lane 3, RNA derived from fibroblasts of the patient was incubated without reverse-transcriptase as control (RT−); Lane 4, RNA derived from the patient was incubated with reverse-transcriptase (RT+). (B) RT-PCR analysis for SNRPN exons proximal and distal to the translocation breakpoint. The amplification was present in RT+ reactions (lane 3), but not in RT− reactions (lane 2). Lane 1, RT-PCR product from mRNA derived from a normal human brain; Lane 2, RT-PCR product from RNA derived from the patient’s fibroblast culture without reverse-transcriptase (RT−); Lane 3, reaction product from RNA derived from the patient’s fibroblast culture with reverse-transcriptase (RT+).
Since the translocation breakpoint on chromosome 15 was found within the SNRPN gene, the expression of SNRPN was studied. Two primers (RN175 and RN97) were used to amplify exon −1 to 8 (the entire SNRPN gene). RT-PCR was performed with the total RNA prepared from the fibroblast culture of the patient (Fig. 7B). While expression of SNRPN in normal brain was detected, expression of SNRPN in the translocation patient was not detected. This result is expected, since this pair of primers span the translocation breakpoint, and also shows that the maternal allele remains silent.
Although no expression of SNRPN was detected, the expression of exons proximal and distal to the translocation breakpoint had not been established. It is reasonable to expect that the expression of exons proximal to the translocation might still occur, since the translocation did not disrupt the promoter sequences of the gene. Therefore, the expression of proximal and distal exons was studied with RT-PCR (Fig. 7B). The expression of exon −1 to 0 was detected as expected. Surprisingly, mRNA from exons 2 to 8 was also expressed. Both products were also detected in a normal human brain mRNA control.
DISCUSSION
We have described a patient with Prader-Willi syndrome and a balanced translocation involving chromosomes 15 and 19. The patient had neither maternal uniparental disomy, nor abnormal DNA methylation. The data presented in this paper indicate that the translocation disrupted the SNRPN locus and is likely to be responsible for the PWS phenotype in the t(15;19) patient. Translocation PWS patients are relatively rare. The majority of PWS patients have an interstitial deletion of chromosome 15q11-q13 (1,2). In translocation patients, the majority are unbalanced involving a deletion of chromosome 15 (1,32,33). Balanced reciprocal translocation patients with PWS are described in the literature (reviewed in ref. 1), but none were studied by molecular techniques. The patient presented in this paper is unique in that no deletion is associated with the translocation. The phenotype in our patient apparently resulted from the translocation, t(15;19)(q12;q13.41), which appears to have disrupted the SNRPN locus.
The disruption of the SNRPN locus is of significance and may be responsible for the PWS phenotype. SNRPN was the first gene to be mapped in the PWS minimum critical region. Recent studies have indicated that the SNRPN gene is a strong candidate for a role in PWS (5,14–16,20,22,28). However, no mutation has been reported in the coding region of the SNRPN gene in PWS or PWS-like patients. Nevertheless, the expression data for other genes within 15q11–13 suggest that the PWS phenotype in our patient did not result from the lack of expression of ZNF127, IPW, PAR-1, or PAR-5, if the same expression pattern occurred in brain, the critical tissue for the neurobehavioral Prader-Willi syndrome.
The level of transcription of SNRPN exons, ZNF127, IPW, PAR-1, and PAR-5 in fibroblast cells in the patient is not known. Whether these genes are expressed in brain cells and other tissues in the t(15;19) patient is also unknown. Thus, it is possible that an abnormal level of expression of these genes in critical tissues (e.g., brain) affects the patient’s phenotype. The developmental timing of expression of these genes in critical tissues may also be important in the development of the phenotype. In addition, in balanced translocations such as this, position effects in different tissues resulting from chromatin states of the translocation partner may also determine the status of imprinted genes from 15q11–13. This could result in gene silencing in different tissues.
The translocation in the patient presented here occurred between exons 0 and 1, outside of the SNRPN coding region. The SNRPN ORF which encodes the SmN splicing factor (31,34) is still intact, and RT-PCR analysis indicates that transcription of the ORF occurs. There are three possibilities to account for the PWS phenotype in the patient: (i) the SNRPN ORF is in a larger protein fusion product with a putative chromosome 19 sequence, both of whose original functions are lost; (ii) the fusion mRNA is not capable of translation and no SmN function is present; and (iii) the disruption of the upstream exons of the SNRPN resulted in the PWS phenotype. The SNRPN upstream 3 exons (exons −1, 0, and 1 of the SNRPN locus) have been suggested to encode an additional, independent reading frame, termed SNURF (35) (also R. D. N. et al, unpublished data). Since the translocation breakpoint on chromosome 15 of the patient was found to lie between exon 0 and 1, the putative SNURF sequence is disrupted in this patient. Thus, the disruption of SNURF may play a significant role in etiology of PWS phenotype in this patient.
Based on these findings, we propose a model for the expression of SNRPN in this patient (Fig. 8). It has been demonstrated that SNRPN is expressed only from the paternal allele (14,16,19,20). The maternal allele is inactive in transcription due to imprinting (Fig. 8A). The detected expression of SNRPN exons 2 to 8 and exons −1 to 0, but not exons −1 to 8, indicates expression from the chromosome 15 portion in the der(15) and der(19), respectively. As was demonstrated with chromosome 15 polymorphisms, the der(15) is paternal in origin, and the chromosome 15 portion in the der(19) can also be deduced to be paternal in origin. It is reasonable to assume that the transcription level of SNRPN exon −1 and 0 will be roughly at the same level as if no translocation had occurred (Fig. 8B). The expression of SNRPN exons 2 to 8 implies that the transcription may be driven by a promoter from an unknown gene on chromosome 19 (Fig. 8C), since the translocation separated exons 1–8 from the SNRPN promoter and moved them to the der(19).
Figure 8.

Model for expression of SNRPN exons proximal and distal to the translocation. The maternal SNRPN gene is inactive because of imprinting, and therefore no mRNA from exon −1 to 8 is produced (a). The translocation breakpoint maps between exon 0 and 1, and exons 1 to 8 from chromosome 15 distal to the breakpoint were translocated to chromosome 19 near q13.41 (b, right) forming the der(19). The region of chromosome 19 distal to the chromosome 19 breakpoint was translocated to chromosome 15 forming the der(15) (b, at left). The translocation may also have disrupted a gene on chromosome 19, thus forming two fusion genes (c). The promoter for the SNRPN gene remains active and drives transcription of exon −1 to 0, either extending into the putative chromosome 19 gene or terminating randomly (not shown). This product was detected by RT-PCR with exon −1 and 0 primers (Fig. 8B). The promoter for the putative chromosome 19 gene is also active in fibroblasts and transcription generates a fusion mRNA with SNRPN exons 1–8 that can be detected by RT-PCR with primers from exon 2 and 8 (Fig. 8B). An, polyadenylation signal.
The expression of all paternal-only expressed loci (SNRPN, ZNF127, IPW, PAR-5, and PAR-1) and a normal DNA methylation pattern at loci PW71 and the SNRPN promoter region suggest that paternal imprinting has been set prior to the translocation during paternal gametogenesis. It is thought that imprinting is established in the germ cells early during gametogenesis (36). Recently, it has been suggested that the translocation events in male gametogenesis most likely occur at the post-meiotic differentiating stage in spermatids (NO TAG). Therefore, it appears that the translocation in this t(15;19) patient probably occurred after imprinting was established in the paternal spermatid. Consistent with this, the loci IPW, PAR-5, and PAR-1 downstream (telomeric) to the SNRPN gene and the IC (5) continue to express presumably from paternal allele, since these genes have been previously demonstrated paternal-allele expression in fibroblasts (15,21). Alternatively, it is possible that the expression of the downstream loci simply reflects the positional effect from chromosome 19. It may be that the vicinity of the translocation breakpoint in chromosome 19 is a transcriptionally open domain, which could allow IPW, PAR-5, and PAR-1 to be transcribed in the translocation patient. In conclusion, the PWS phenotype in this patient may result from the disrupted expression of the SNRPN locus, rather than the lack of expression of downstream genes. Additional unique PWS patients and animal models may be necessary to definitively identify genes that play a role in the pathogenesis of Prader-Willi syndrome.
MATERIALS AND METHODS
Microsatellite analysis
PCR was undertaken for several loci from 15q11–13: D15S541, D15S543, D15S63, D15S128, MN1, D15S10, D15S122, GABRB3, D15S97, GABRA5, D15S219 and D15S165. PCR amplification and polyacrylamide gel electrophoresis of 32P end-labeled amplification products were analyzed according to modifications of the methods described by Weber and May (38). Oligonucleotide primer sequences for PCR were obtained from the NIH/DOE Genome Database (Johns Hopkins University) and from the report of the Second International Chromosome 15 Workshop (39).
FISH analysis
The hybridization and detection with probes D15S10, D15S11, SNRPN and GABRB3 (Oncor, MD), was carried out according to the manufacturer’s instruction. The SNRPN cosmid contig was provided by Oncor, Inc. (Gaithersburg, MD). SNRPN subfragments from EcoRI digestion of the SNRPN cosmid contig were labeled with digoxigenin by sequence-independent PCR (40). The fragments were cut out from an agarose gel, and purified with a Qiaex gel purification kit (Qiagen, CA). The purified fragments were labeled with digoxigenin by sequence-independent PCR (40). The labeled DNA was treated with DNase (200 pg/μl, Gibco BRL) for 15–20 min at room temperature. After inactivation of DNase at 65°C for 15 min, the DNA was precipitated with 100× human Cot-1 DNA (Gibco BRL). Hybridization was carried out in hybridization solution with 50% formamide/2× SSC at 37°C for 12–18 h. Posthybridization washes were 15 min each at 42°C in 50% formamide/2× SSC and 2× SSC. Detection was performed with a detection system for digoxigenin labeled probe (Oncor, MD). Signals were detected with a Leitz epifluorescence microscope equipped with a FITC filter and a triple-band pass filter.
Southern blot analysis
Southern blot analysis was performed according to standard methods (41). The SNRPN cDNA probe was synthesized by RT-PCR from total human brain mRNA. The primers were: forward 5′-GGATTTCCAGGCTGAACTGAGG-3′, reverse 5′-ACAAGA-CGCATTGCAGGGGA-3′. The PCR product was purified with the Qiaex gel purification kit, and confirmed by restriction analysis. Exon −1, 0 and 1-specific DNA probes were made by PCR from cDNA cloned in TA vectors (16).
RNA expression study
Total cellular RNA was isolated from fibroblast cultures of the patient with RNAgents™ total RNA isolation kit (Promega, WI). RT-PCR was performed essentially as described (14–16) with the exception of human normal brain mRNA. Human normal brain mRNA was a gift from Dr Lei Yu (Indiana University School of Medicine). One μg of human brain mRNA was used for reverse transcription (RT). Four to five μg of total RNA from the patient’s fibroblast cultures was used for first strand cDNA synthesis with GeneAmp® RNA PCR kit (Perkin Elmer). One-tenth of the reverse transcription products were used for PCR amplification. To rule out DNA contamination, control samples contained no reverse transcriptase (RT−). The primers were as follows: for SNRPN exons −1 to 0 (16): RN175 AGAGTGGAGCGGCCGC-CGG, RN134 GCTCAGTGAGGCAGTCCTTC; exons 2–8 (16): RN84 TGGTGGAACAGCAATCATG, RN97 GATTCCA-GAACCACCTGCG; for ZNF127, RN153 and DD29 (Jong, M. T. C., R. D. N. et al, in preparation); for PAR-1 and PAR-5, see Sutcliffe (15); for IPW, see Wevrick (21). PCR amplification was 40 cycles for 1 min each at 94°C, 60°C, and 72°C. For PAR-1, the PCR product was reamplified for another 30 cycles with the same conditions.
Acknowledgments
We thank Dr Lei Yu (Indiana University School of Medicine) for providing normal human brain mRNA, Susan Airhart (Oncor) for the unlabeled SNRPN cosmid contig, and Drs Ju Chen, Octavian Henegariu, and Nyla Heerema for suggestions and support. Funding for R. D. N. was provided by the National Institute of Health grant HD31491 and Pew Scholars Program in Biomedical Sciences.
References
- 1.Butler MG. Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet. 1990;35:319–332. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–329. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- 3.Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. 1983;i:1285–1286. doi: 10.1016/s0140-6736(83)92745-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mascari MJ, Gottlieb W, Rogan PK, Butler MG, Waller DA, Armour JA, Jeffreys AJ, Ladda RL, Nicholls RD. The frequency of uniparental disomy in Prader-Willi syndrome. Implications for molecular diagnosis. N Engl J Med. 1992;326:1599–1607. doi: 10.1056/NEJM199206113262404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nature Genet. 1995;9:395–400. doi: 10.1038/ng0495-395. [DOI] [PubMed] [Google Scholar]
- 6.Mutirangura A, Jayakumar A, Sutcliffe JS, Nakao M, McKinney MJ, Buiting K, Horsthemke B, Beaudet AL, Chinault AC, Ledbetter DH. A complete YAC contig of the Prader-Willi/Angelman chromosome region (15q11-q13) and refined localization of the SNRPN gene. Genomics. 1993;18:546–552. doi: 10.1016/s0888-7543(11)80011-x. [DOI] [PubMed] [Google Scholar]
- 7.Kuwano A, Mutirangura A, Dittrich B, Buiting K, Horsthemke B, Saitoh S, Niikawa N, Ledbetter SA, Greenberg F, Chinault AC, Ledbetter DH. Molecular dissection of the Prader-Willi/Angelman syndrome region (15q11–13) by YAC cloning and FISH analysis [erratum in Hum Mol Genet 1992;1:784] Hum Mol Genet. 1992;1:417–425. doi: 10.1093/hmg/1.6.417. [DOI] [PubMed] [Google Scholar]
- 8.Glenn CC, Nicholls RD, Robinson WP, Saitoh S, Niikawa N, Schinzel A, Horsthemke B, Driscoll DJ. Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet. 1993;2:1377–1382. doi: 10.1093/hmg/2.9.1377. [DOI] [PubMed] [Google Scholar]
- 9.Buiting K, Dittrich B, Gross S, Greger V, Lalande M, Robinson W, Mutirangura A, Ledbetter D, Horsthemke B. Molecular definition of the Prader-Willi syndrome chromosome region and orientation of the SNRPN gene. Hum Mol Genet. 1993;2:1991–1994. doi: 10.1093/hmg/2.12.1991. [DOI] [PubMed] [Google Scholar]
- 10.Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989;342:281–285. doi: 10.1038/342281a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholls RD. Genomic imprinting and candidate genes in the Prader-Willi and Angelman syndromes. Curr Opin Genet Dev. 1993;3:445–456. doi: 10.1016/0959-437x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- 12.Driscoll DJ, Waters MF, Williams CA, Zori RT, Glenn CC, Avidano KM, Nicholls RD. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics. 1992;13:917–924. doi: 10.1016/0888-7543(92)90001-9. [DOI] [PubMed] [Google Scholar]
- 13.Dittrich B, Robinson WP, Knoblauch H, Buiting K, Schmidt K, Gillessen-Kaesbach G, Horsthemke B. Molecular diagnosis of the Prader-Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11–13. Hum Genet. 1992;90:313–315. doi: 10.1007/BF00220089. [DOI] [PubMed] [Google Scholar]
- 14.Glenn CC, Porter KA, Jong MT, Nicholls RD, Driscoll DJ. Functional imprinting and epigenetic modification of the human SNRPN gene. Hum Mol Genet. 1993;2:2001–2005. doi: 10.1093/hmg/2.12.2001. [DOI] [PubMed] [Google Scholar]
- 15.Sutcliffe JS, Nakao M, Christian S, Örstavik KH, Tommerup N, Ledbetter DH, Beaudet AL. Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nature Genet. 1994;8:52–58. doi: 10.1038/ng0994-52. [DOI] [PubMed] [Google Scholar]
- 16.Glenn CC, Saitoh S, Jong MTC, Filbrandt MM, Swti U, Driscoll DJ, Nicholls RD. Gene structure, DNA methylation and imprinted expression of the human SNRPN gene. Am J Hum Genet. 1996 in press. [PMC free article] [PubMed] [Google Scholar]
- 17.Kitsberg D, Selig S, Brandeis M, Simon I, Keshet I, Driscoll DJ, Nicholls RD, Cedar H. Allele-specific replication timing of imprinted gene regions. Nature. 1993;364:459–463. doi: 10.1038/364459a0. [DOI] [PubMed] [Google Scholar]
- 18.Knoll JH, Cheng SD, Lalande M. Allele specificity of DNA replication timing in the Angelman/Prader-Willi syndrome imprinted chromosomal region. Nature Genet. 1994;6:41–46. doi: 10.1038/ng0194-41. [DOI] [PubMed] [Google Scholar]
- 19.Nakao M, Sutcliffe JS, Durtschi B, Mutirangura A, Ledbetter DH, Beaudet AL. Imprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E) Hum Mol Genet. 1994;3:309–315. doi: 10.1093/hmg/3.2.309. [DOI] [PubMed] [Google Scholar]
- 20.Reed ML, Leff SE. Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi syndrome. Nature Genet. 1994;6:163–167. doi: 10.1038/ng0294-163. [DOI] [PubMed] [Google Scholar]
- 21.Wevrick R, Kerns JA, Francke U. Identification of a novel paternally expressed gene in the Prader-Willi syndrome region. Hum Mol Genet. 1994;3:1877–1882. doi: 10.1093/hmg/3.10.1877. [DOI] [PubMed] [Google Scholar]
- 22.Özçelik T, Leff S, Robinson W, Donlon T, Lalande M, Sanjines E, Schinzel A, Francke U. Small nuclear ribonucleoprotein polypeptide N (SNRPN), an expressed gene in the Prader-Willi syndrome critical region. Nature Genet. 1992;2:265–269. doi: 10.1038/ng1292-265. [DOI] [PubMed] [Google Scholar]
- 23.Leff SE, Brannan CI, Reed ML, Özçelik T, Francke U, Copeland NG, Jenkins NA. Maternal imprinting of the mouse Snrpn gene and conserved linkage homology with the human Prader-Willi syndrome region. Nature Genet. 1992;2:259–264. doi: 10.1038/ng1292-259. [DOI] [PubMed] [Google Scholar]
- 24.Cattanach BM, Barr JA, Evans EP, Burtenshaw M, Beechey CV, Leff SE, Brannan CI, Copeland NG, Jenkins NA, Jones J. A candidate mouse model for Prader-Willi syndrome which shows an absence of Snrpn expression. Nature Genet. 1992;2:270–274. doi: 10.1038/ng1292-270. [DOI] [PubMed] [Google Scholar]
- 25.Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398–402. [PMC free article] [PubMed] [Google Scholar]
- 26.Woodage T, Deng ZM, Prasad M, Smart R, Lindeman R, Christian SL, Ledbetter DH, Robson L, Smith A, Trent RJ. A variety of genetic mechanisms are associated with the Prader-Willi syndrome. Am J Med Genet. 1994;54:219–226. doi: 10.1002/ajmg.1320540308. [DOI] [PubMed] [Google Scholar]
- 27.Robinson WP, Wagstaff J, Bernasconi F, Baccichetti C, Artifoni L, Franzoni E, Suslak L, Shih LY, Aviv H, Schinzel AA. Uniparental disomy explains the occurrence of the Angelman or Prader-Willi syndrome in patients with an additional small inv dup(15) chromosome. J Med Genet. 1993;30:756–760. doi: 10.1136/jmg.30.9.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reis A, Dittrich B, Greger V, Buiting K, Lalande M, Gillessen-Kaesbach G, Anvret M, Horsthemke B. Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader-Willi syndromes. Am J Hum Genet. 1994;54:741–747. [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholls RD. New insights reveal complex mechanisms involved in genomic imprinting. Am J Hum Genet. 1994;54:733–740. [PMC free article] [PubMed] [Google Scholar]
- 30.Dittrich B, Buiting K, Gross S, Horsthemke B. Characterization of a methylation imprint in the Prader-Willi syndrome chromosome region. Hum Mol Genet. 1993;2:1995–1999. doi: 10.1093/hmg/2.12.1995. [DOI] [PubMed] [Google Scholar]
- 31.Schmauss C, Brines ML, Lerner MR. The gene encoding the small nuclear ribonucleoprotein-associated protein N is expressed at high levels in neurons. J Biol Chem. 1992;267:8521–8529. [PubMed] [Google Scholar]
- 32.Rivera H, Zuffardi O, Gargantini L. Nonreciprocal and jumping translocations of 15q1→qter in Prader-Willi syndrome. Am J Med Genet. 1990;37:311–317. doi: 10.1002/ajmg.1320370304. [DOI] [PubMed] [Google Scholar]
- 33.Charrow J, Balkin N, Cohen MM. Translocations in Prader-Willi syndrome. Clin Genet. 1983;23:304–307. doi: 10.1111/j.1399-0004.1983.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 34.McAllister G, Amara SG, Lerner MR. Tissue-specific expression and cDNA cloning of small nuclear ribonucleoprotein-associated polypeptide N. Proc Natl Acad Sci USA. 1988;85:5296–5300. doi: 10.1073/pnas.85.14.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saitoh S, Seip J, Tommerup N, Greenswag L, Rogan PK, Nicholls RD. The imprinted SNRPN gene is associated with a polycistronic mRNA and an imprinting control element. Am J Hum Genet. 1994;55(Suppl):A146. [Google Scholar]
- 36.Monk M. Genomic imprinting. Genes Dev. 1988;2:921–925. doi: 10.1101/gad.2.8.921. [DOI] [PubMed] [Google Scholar]
- 37.Chandley AC. On the parental origin of de novo mutation in man. J Med Genet. 1991;28:217–223. doi: 10.1136/jmg.28.4.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44:388–396. [PMC free article] [PubMed] [Google Scholar]
- 39.Malcolm S, Donlon TA. Report of the second international workshop on human chromosme 15 mapping. Cytogenet Cell Genet. 1994;67:2–14. doi: 10.1159/000133717. [DOI] [PubMed] [Google Scholar]
- 40.Bohlander SK, Espinosa R, III, Fernald AA, Rowley JD, Le Beau MM, Diaz MO. Sequence-independent amplification and labeling of yeast artificial chromosomes for fluorescence in situ hybridization. Cytogenet Cell Genet. 1994;65:108–110. doi: 10.1159/000133612. [DOI] [PubMed] [Google Scholar]
- 41.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor; New York: 1982. [Google Scholar]