

Abstract
Background and Purpose
The cannabinoid system exerts functional regulation of neural stem cell (NSC) proliferation and adult neurogenesis, yet not all effects of cannabinoid‐like compounds seen can be attributed to the cannabinoid 1 (CB1) or CB2 receptor. The recently de‐orphaned GPR55 has been shown to be activated by numerous cannabinoid ligands suggesting that GPR55 is a third cannabinoid receptor. Here, we examined the role of GPR55 activation in NSC proliferation and early adult neurogenesis.
Experimental Approach
The effects of GPR55 agonists (LPI, O‐1602, ML184) on human (h) NSC proliferation in vitro were assessed by flow cytometry. Human NSC differentiation was determined by flow cytometry, qPCR and immunohistochemistry. Immature neuron formation in the hippocampus of C57BL/6 and GPR55−/− mice was evaluated by immunohistochemistry.
Key Results
Activation of GPR55 significantly increased proliferation rates of hNSCs in vitro. These effects were attenuated by ML193, a selective GPR55 antagonist. ML184 significantly promoted neuronal differentiation in vitro while ML193 reduced differentiation rates as compared to vehicle treatment. Continuous administration of O‐1602 into the hippocampus via a cannula connected to an osmotic pump resulted in increased Ki67+ cells within the dentate gyrus. O‐1602 increased immature neuron generation, as assessed by DCX+ and BrdU+ cells, as compared to vehicle‐treated animals. GPR55−/− animals displayed reduced rates of proliferation and neurogenesis within the hippocampus while O‐1602 had no effect as compared to vehicle controls.
Conclusions and Implications
Together, these findings suggest GPR55 activation as a novel target and strategy to regulate NSC proliferation and adult neurogenesis.
Abbreviations
- 2‐AG
2‐arachidonoylglycerol
- ACSF
artificial cerebral spinal fluid
- AEA
N‐arachidonoylethanolamine
- bFGF
basic FGF
- BrdU
5‐bromo‐2′‐deoxyuridine
- CB1 receptor
cannabinoid receptor 1
- CBD
cannabidiol
- CNR1
CB1 receptor gene
- CNR2
CB2 receptor gene
- DCX
doublecortin
- eCB
endocannabinoid
- FGFR1
fibroblast growth factor receptor 1
- GFAP
glial fibrillary acidic protein
- GRM1
mGlu1 receptor gene
- LPI
L‐α‐lysophosphatidylinositol
- MAP2
microtubule associated protein 2
- NSC
neural stem cell
- SGZ
subgranular zone
- SVZ
subventricular zone
- THC
Δ9‐tetrahydrocannabinol
- WT
wild type
Introduction
Adult neurogenesis, or the generation of new neurons in the adult brain, was first reported by Joseph Altman in 1965 (Altman and Das, 1965). Since this discovery, it is now well established that multipotent and self‐renewing adult neural stem cells (NSCs) reside within the adult brain and divide to produce neurons and glia. These cells are restricted to two major neurogenic niches: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) within the dentate gyrus of the hippocampus. Upon neuronal differentiation, these cells integrate into functioning neuronal circuits and promote plasticity by making synaptic connections with mature neurons (Ming and Song, 2011). Proper integration of NSCs into the existing circuitry is critical for processes such as learning, memory and pattern separation (Deng et al., 2010; Sahay et al., 2011a; Sahay et al., 2011b; Christian et al., 2014). These processes are tightly regulated by numerous intrinsic factors including growth factor availability, inflammatory regulators and endogenous signalling mechanisms.
The cannabinoid system has recently been recognized for its role in NSC biology and adult neurogenesis. The cannabinoid system is made up of endocannabinoids (eCBs), plant‐derived cannabinoids (phytocannabinoids) and their main target receptors, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56&familyId=13&familyType=GPCR and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=57&familyId=13&familyType=GPCR, G‐protein coupled cannabinoid receptors. The two best‐characterized eCBs are N‐arachidonoylethanolamine (anandamide, AEA) and 2‐arachidonoylglycerol (2‐AG) while the most prominent phytocannabinoids are Δ9‐tetrahydrocannabinol (THC, the main psychoactive component of Cannabis sativa) and cannabidiol (CBD) (Elsohly and Slade, 2005; Luchicchi and Pistis, 2012; Pertwee, 2012). Recent studies suggest a crucial role of eCBs on adult neurogenesis and NSC maintenance. NSCs express both CB1 and CB2 receptors, and as NSCs begin to differentiate CB1 receptor expression increases while CB2 expression is lost as these cells become mature neurons (Begbie et al., 2004; Palazuelos et al., 2006). Signalling through the CB2 receptor is implicated in increased NSC proliferation rates and the ability of these cells to self‐renew through the mTORC‐1 pathway (Molina‐Holgado et al., 2007; Palazuelos et al., 2012) while NSCs deficient in CB2 receptors have impaired proliferation both in vitro and within the mouse hippocampus (Palazuelos et al., 2006). The CB1 receptor has been associated with NSC differentiation and neurogenesis in that chronic treatment with the cannabinoid compound HU‐210 increased rat hippocampal neurogenesis while CB1 −/− mice display reduced rates of hippocampal neurogenesis (Jin et al., 2004; Jiang et al., 2005; Xapelli et al., 2013). Interestingly, it has recently been reported that there is an interaction between CB1 and CB2 receptor regulation of rat NSC proliferation and neurogenesis (Rodrigues et al., 2017). Of note, data presented by Rodrigues et al. demonstrate that SVZ and SGZ neurogenic niches respond differently to the same cannabinoid pharmacological treatment. However, activation of either CB1 or CB2 receptors does not account for all effects seen after cannabinoid treatment of NSC populations, which suggests that cannabinoid‐like ligands may activate multiple receptors within these niches (Prenderville et al., 2015). Consequently, other targets of eCBs need to be studied to fully elucidate the mechanisms behind cannabinoid signalling and neurogenesis. One such potential target is the recently de‐orphaned GPCR, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=109&familyId=114&familyType=GPCR.
GPR55 has been considered a possible third cannabinoid receptor since the discovery that certain cannabinoid‐like compounds activate this receptor even though the binding pocket of GPR55 does not share a ‘functional fingerprint’ with CB1 or CB2 receptors (Petitet et al., 2006), and the receptor itself shares a relatively low amino acid sequence homology with CB1 (13.5%) and CB2 receptors (14.4%) (Ryberg et al., 2007; Elbegdorj et al., 2013). Recent evidence has demonstrated that GPR55's endogenous ligand is http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4028 (LPI), a non‐cannabinoid lipid signalling molecule (Oka et al., 2007). GPR55 is also thought to be activated by a number of endo‐, phyto‐ and synthetic cannabinoid ligands including AEA, 2‐AG, THC, CB1 receptor antagonists AM251 and SR141716A (Rimonabant®), and the synthetic compounds, abnormal CBD (Abn‐CBD), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5525 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5565 (CID‐2440433; Johns et al., 2007; Ryberg et al., 2007; Henstridge et al., 2010; Heynen‐Genel et al., 2010; Zhang et al., 2010). Activation of GPR55 by LPI induces a sustained, oscillatory Ca2+ release pathway which is dependent on Gα13 and requires RhoA activation (Henstridge et al., 2009; Sharir and Abood, 2010). Further study has suggested that ligand binding to GPR55 also phosphorylates ERK1/2, CREB, activates the NFκB pathway and translocates NFAT to the nucleus, yet these responses are ligand‐dependent, which further complicates the pharmacology of this receptor (Henstridge et al., 2009; Henstridge et al., 2010). Differences in signalling mechanisms may be due to a wide distribution of GPR55 in the body. Outside the CNS, GPR55 is implicated in gastrointestinal inflammation and colitis (Schicho et al., 2011), osteoclast function (Whyte et al., 2009) and modulation of immune responses and nociception (Schuelert and McDougall, 2011; Li et al., 2013; Stancic et al., 2015). However, the role GPR55 plays in the physiology and pathophysiology of the CNS is poorly understood. GPR55 expression has been found in numerous regions of the human and rodent brain including the hippocampus (Sawzdargo et al., 1999; Wu et al., 2013; Hurst et al., 2017). Studies elucidating the role of GPR55 in the hippocampus have demonstrated microglial‐dependent neuroprotective effects and LPI‐mediated increases in hippocampal CA1 and CA3 LTP (Kallendrusch et al., 2013; Hurst et al., 2017). However, the effects of GPR55 activation on NSC proliferation and differentiation within the hippocampus have never been examined.
These data, along with studies showing a regulatory role of GPR55 in growth cone morphology and axon guidance (Cherif et al., 2015), urge further examination of GPR55 as a necessary facet of NSC physiology. In the current study, we examined selective GPR55 agonist treatment on NSC proliferation and subsequent neuronal differentiation in vitro and in vivo. We found that treatment of a human NSC culture in vitro with three separate GPR55 agonists increased the number of cells progressing through S‐phase of the cell cycle, while differentiating cultures had significant increases in the number of cells becoming neurons compared to vehicle control. Analysis of the dentate gyrus of C57BL/6 mice treated for 14 days with O‐1602 displayed significantly increased numbers of proliferating NSCs and a significant increase in the population of immature neurons within this region, while GPR55−/− mice showed no effect of treatment. Of note, GPR55−/− mice exhibited reduced basal numbers of proliferating NSCs and neurogenesis rates compared to wild‐type (WT) animals. Together, these observations point to a novel mechanism regulating hippocampal neurogenesis and provide evidence that GPR55 may be a potential target for future therapeutics.
Methods
Cell culture
In vitro experiments utilized primary human NSCs (ReNcell VM) commercially obtained from Millipore Inc. (Billerica, MA, USA; Cat# SCC008, RRID:CVCL_E921). The ReNcell VM cell line is an immortalized human NSC line derived from the ventral mesencephalon region of human fetal brain. These cells can differentiate into dopaminergic neurons, astrocytes and oligodendrocytes after growth factor withdrawal (Donato et al., 2007). The hNSCs were regularly characterized by flow cytometry for the molecular markers nestin and Sox2. Undifferentiated cells were maintained and cultured in ReNcell NSC maintenance medium (Millipore) supplemented with EGF (20 ng·mL−1; Millipore) and basic FGF (bFGF; 20 ng·mL−1; Millipore). The cells were grown as an adherent monolayer on laminin‐coated (mouse laminin, Millipore, 10 μg·mL−1 in DMEM/F12 w/o HEPES, w/L‐glutamine) 75 cm2 cell culture flasks, six‐well plates or cover slips at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were passaged when flasks were ~80–90% confluent. Media was changed every 2–3 days. Differentiation of hNSCs was induced by removal of growth factors (bFGF, EGF) from the culture medium. HEK293 cells overexpressing human GPR55 (HEK293 hGPR55, Kerafast, Boston, MA, USA; Cat# EIU003, from the laboratory of Dr. Ken Mackie, Indiana University) were used as a positive control for human GPR55 expression. HEK293 cells without hGPR55 were used as a negative control. HEK cells were grown in DMEM containing 10% FBS and 1% penicillin/streptomycin as an adherent monolayer at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were passaged when ~80–90% confluent. Media were changed every 2–3 days.
Flow cytometric analysis of NSC markers
For flow cytometric analysis, hNSCs were detached using Accutase (Millipore), washed and resuspended at 1 × 106 .100 μL‐1 in PBS containing 2% BSA (Sigma‐Aldrich). Cells were fixed using intracellular (IC) fixation buffer (eBiosciences, San Diego, CA, USA). For intracellular markers, cells were permeabilized after fixation using permeabilization buffer (eBiosciences) following the manufacturer's instructions and incubated at room temperature for 30 min in antibodies against human Sox2 (BD Biosciences, San Jose, CA, USA, Cat# 561610 Lot# RRID:AB_10712763), nestin (BD Biosciences Cat# 560393 Lot#3305985 RRID:AB_1645170), S100β (Abcam, Cambridge, UK Cat#ab196442 Lot#GR206303‐4 RRID:AB_2722596) or βIII‐tubulin (BD Biosciences Cat# 560394 Lot# 7019589 RRID:AB_1645400). Cytometric acquisition was performed using a BD FACS Canto II flow cytometer and analysed with FlowJo software (Tree Star, Inc., Ashland, OR, USA; RRID:SCR_008520).
Pharmacological treatments
To investigate the effects of GPR55 activation on hNSC proliferation, cells were plated on laminin‐coated 6‐well plates. Cells were allowed to adhere overnight and then treated with LPI (1 μM), the endogenous ligand for GPR55, or synthetic agonists, O‐1602 (1 μM) or ML184 (1 μM), in a reduced growth factor media (5% growth factor). Reduced growth factor medium was utilized to better mimic a less proliferative phenotype while still maintaining a ‘stemness’ state. Analysis by flow cytometry showed no significant reduction of nestin+ or Sox2+ populations after 48 h (data not shown). Cells treated with the selective GPR55 antagonist ML193 (5 μM) were pretreated for 30 min prior to addition of agonist. Vehicle‐treated cells received 0.1% DMSO in 5% growth factor media. For differentiation studies, cells were treated with either vehicle, ML184 (1 μM), ML193 (5 μM), or a combination of ML184 (1 μM) and ML193 (5 μM) in ReNcell medium that did not contain growth factors.
Flow cytometric analysis of hNSC proliferation
The FITC BrdU Flow Kit (BD Biosciences Cat# 559619 Lot#6097660 RRID:AB_2617060) was used to determine the proportion of S‐phase hNSCs. Cells were incubated with BrdU (10 μM) for the final 1 h of a 48 h exposure to GPR55 selective agonist/antagonist treatment. Cells were detached using Accutase (Millipore), fixed with BD Cytofix/Cytoperm buffer and treated with DNase to expose incorporated BrdU. A FITC‐conjugated anti‐BrdU antibody and 7‐amino‐actinomycin D (7‐AAD) were used to determine cell cycle phases (G0/G1, S, G2 + M). Fluorescent signals from FITC and 7‐AAD/DNA complexes were detected on a BD FACS Canto II and analysed using FlowJo (Tree Star, Inc.) software. hNSCs were first gated by cell area and width, and S‐phase cells were defined as being FITC‐BrdU+ and 7‐AAD+. Results are displayed as % of results of vehicle‐treated cells (control) to account for variability between separate experiments.
RT‐PCR
Total RNA from human NSCs was extracted using RNeasy® Mini kit (Qiagen, Valencia, CA, USA) as per the manufacturer's instructions, utilizing QIAshredder spin columns and DNase to reduce the possibility of PCR amplification of genomic DNA. Total RNA (2 mg) was converted to cDNA using high‐capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, Waltham, MA, USA). Human GPR55 was amplified using primers: 5′‐ATC CAT GGC TTC AGC ACC TT‐3′ and 5′‐ATG GTG CAG ATC CCA AA‐3′, which yielded a product of 311 bp. Human GAPDH was amplified using primers: 5′‐TCC ACC CAT GGC AAA TTC CA‐3' and 5′‐TGG TTC ACA CCC ATG ACG AA‐3′ which yielded a product of 260 bp. Mouse GPR55 was amplified using primers: 5′‐AGA CCT TTG GGA TCT GCT GC‐3′ and 5′‐AGC TGA TGC CCT GCT TCA TT‐3′ which yielded a product of 420 bp. Mouse GAPDH was amplified using primers: 5′‐CTC ACT GGC ATG GCC TTC CG‐3′ and 5′‐ACC ACC CTG TTG CTG TAG CC‐3′ which yielded a product of 292 bp. The template was first denatured at 95°C for 2 min followed by 35 cycles (95°C for 45 s, 57°C for 30 s and 72°C for 30 s), then at 72°C for 2 min in an Eppendorf vapo protect thermal cycler. Aliquots (20 μL) of the PCR products were run on a 2% agarose gel containing 0.5 mg.mL‐1 ethidium bromide.
qPCR
Total RNA from human NSCs was extracted using RNeasy Mini kit (Qiagen) as per manufacturer's instructions. Total RNA (2 mg) was converted to cDNA using high‐capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific). Specific primers and probes (TaqMan) for human and mouse genes were purchased from Life Technologies (Thermo Fisher Scientific), and analyses were performed using the StepOnePlus real‐time PCR system (Thermo Fisher Scientific). For a full list of primer and probe sets, see Table 1 (human) and Table 2 (mouse). Data for human samples were normalized to GAPDH while data for mouse samples were normalized to Rn18s. Characterization data utilizing qPCR (Figure 1, Figure 4) are reported as ΔCT [threshold cycle (CT) of the gene of interest minus the CT of the reference gene] in which a ΔCT of −1 corresponds to a doubling of the mRNA. qPCR analysis for in vitro differentiation studies is represented as fold change using the 2−ΔΔCt method. Statistics for these studies were performed on ΔCT values. Samples were normalized to GAPDH. Differentiation samples treated with vehicle were used as controls for mRNA levels for each gene of interest.
Table 1.
Human qPCR primer/probe sets
| Target | Assay ID | Amplicon length |
|---|---|---|
| CNR1 (CB1 receptor) | Hs00275634_m1 | 83 |
| CNR2 (CB2 receptor) | Hs00952004_m1 | 108 |
| EGFR | Hs01076090_m1 | 57 |
| FGFR1 | Hs00241111_m1 | 81 |
| GAPDH | Hs02786624_g1 | 157 |
| GFAP | Hs00909236_m1 | 59 |
| GPR55 | Hs00271662_s1 | 80 |
| GRM1 (mGlu1 receptor) | Hs00168250_m1 | 102 |
| MAP2 | Hs00258900_m1 | 98 |
| NES (nestin) | Hs00707120_s1 | 81 |
| S100β | Hs00902901_m1 | 96 |
| Sox2 | Hs01053049_s1 | 91 |
| TUBB3 (βIII tubulin) | Hs00801390_s1 | 134 |
Table 2.
Mouse qPCR primer/probe sets
| Target | Assay ID | Amplicon length |
|---|---|---|
| Cnr1 (CB1 receptor) | Mm01212171_s1 | 66 |
| Cnr2 (CB2 receptor) | Mm00438286_m1 | 91 |
| Egfr | Mm01187858_m1 | 101 |
| Fgfr1 | Mm00438930_m1 | 76 |
| Gpr55 | Mm02621622_s1 | 102 |
| Grm5 (mGlu5 receptor) | Mm00690332_m1 | 97 |
| NES (nestin) | Mm00450205_m1 | 72 |
| SOX2 | Mm03053810_s1 | 86 |
| Rn18s | Mm04277571_s1 | 115 |
Figure 1.
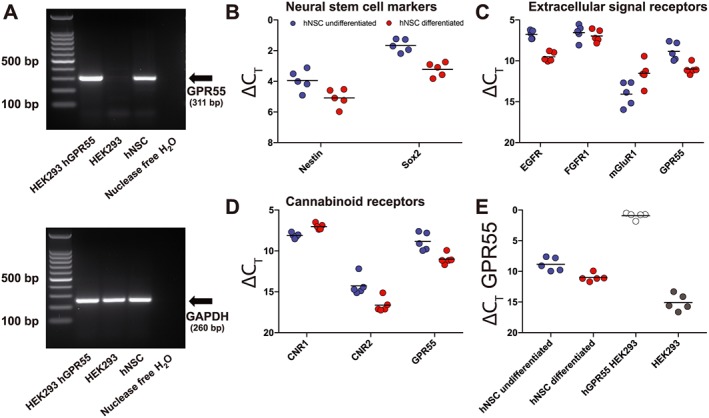
Expression of GPR55 in human NSCs. RNA from hNSCs was extracted and cDNA and PCR amplification were performed. GPR55 was amplified and yielded a product of 311 bp. GAPDH was used as control and yielded a product of 260 bp (A). HEK293 cells overexpressing human GPR55 were used as a positive control while HEK293 cells were used as a negative control. RNA for qPCR analysis (B–E) was isolated from whole cells of cultures either in undifferentiation conditions or after 10 days of differentiation conditions (removal of growth factors from culture medium). Shown are individual ΔCt values (n = 5, each individual point refers to RNA extracted from a separate culture) for NSC markers (B), extracellular signal receptors (mGluR1 is mGlu1 receptor) (C) and CB1 (CNR1) and CB2 (CNR2) receptors (D). Expression of GPR55 in undifferentiated and differentiation conditions (10 days) was compared to expression of GPR55 by HEK293 hGPR55 overexpressing cells and HEK293 cells (E).
Figure 4.
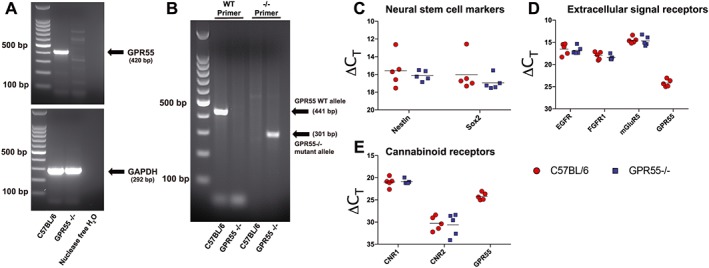
Expression of GPR55 in primary murine hippocampal NSCs. RNA from primary hippocampal NSC cultures from C57BL/6 and GPR55−/− was extracted and cDNA and PCR amplification were performed. GPR55 was amplified and yielded a product of 420 bp. GAPDH was used as a control and yielded a product of 292 bp (A). DNA was also extracted from tail clips of C57BL/6 and GPR55−/− mice to determine genotype (B). Two sets of primers were used to detect amplification of the WT GPR55 allele (441 bp) or GPR55−/− mutant allele (301 bp). RNA for qPCR analysis was isolated from whole cells of primary hippocampal NSC cultures derived from C57BL/6 and GPR55−/− animals. Individual data points represent separate NSC harvests; n = 5. Shown are individual ΔCt values of NSC markers (C), extracellular signal receptors (mGluR5, mGlu5 receptor) (D) and CB1 (CNR1) and CB2 (CNR2) receptors (E) for samples from C57BL/6 and GPR55−/− cultures.
Animals
GPR55−/− mice were obtained from the Texas A&M Institute for Genomic Medicine (Dr. Andrei Golovko). The generation of these animals is described in detail by Wu et al. (2010). These animals have been backcrossed onto a C57BL/6 backbone for at least 10 generations. In short, mice heterozygous for the GPR55 deletion were intercrossed to produce homozygous C57BL/6 GPR55−/− pups. A colony of all homozygous GPR55−/− animals was maintained at Temple University. All wild type (WT) C57BL/6 mice used in this study were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). We did not detect significant differences in NSC markers (Nestin, Sox2), extracellular signalling receptors [http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1797 Egfr ), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1808(Fgfr1 ), the metabotropic glutamate receptor, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=293] or CB1 and CB2 receptors (Cnr1 and Cnr2) as presented in Figure 4. All mice were male, 12–15 weeks of age at the beginning of the study and single‐housed on a 12:12 light/dark cycle (7:00 am–7:00 pm) at 22.1 ± 1°C with ad libitum access to food and water. Animals were randomly assigned to treatment groups. Based on power analyses from previous experiments conducted by the laboratory, we determined that a minimum of six animals per group would provide sufficient power to detect a significant difference with 95% confidence. Experimental design and analyses were carried out in accord with the guidelines laid out by the editorial board of BJP. All in vivo experiments were approved by the Temple University Institutional Animal Care and Use Committee in accordance with guidelines based on the National Institutes of Health (NIH) guide for care and use of laboratory animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Animal genotyping
For genotyping, DNA extraction from tail clips was obtained using the REDExtract‐N‐Amp tissue PCR kit (Sigma‐Aldrich) as per the manufacturer's instructions. Samples were stored at −20°C. Supernatants were used as DNA templates for PCR reactions. Samples were tested for two separate sets of primers to detect either the WT GPR55 allele (Forward 5′‐GCC ATC CAG TAC CCG ATC C‐3′, Reverse 5′‐GTC CAA GAT AAA GCG GTT CC‐3′; 441 bp product) or the GPR55 mutant allele (Forward 5′‐TCA AGC TAC GTT TTG GGT T‐3′, Reverse 3′‐GCA GCG CAT CGC CTT CTA TC‐3′; 301 bp product). PCR cycle conditions were: 5 min at 95°C, 35 cycles of 30 s at 95°C, 45 s at 58°C, 1 min at 68°C, then 2 min at 68°C using standard PCR reagents.
Intrahippocampal injections
Animals were anaesthetized with isoflourane and placed in a stereotaxic frame. Isoflurane was vaporized and a concentration of 4–5% at induction and maintenance at 1.5–2%. Stereotactic coordinates were calculated from Bregma (in mm) −2.0 anterio‐posterior, +1.5 medial‐lateral, −1.5 dorso‐ventral. Vehicle [artificial cerebral spinal fluid (ACSF, Tocris Bioscience), 0.05% EtOH] or O‐1602 (O‐1602 diluted in 100% EtOH, then further diluted in ACSF with a final concentration of no more than 0.05% EtOH) was injected via a stainless‐steel cannula (Alzet Durect, Cupertino, CA, USA; brain infusion kit 3) connected through a polyvinyl tube to an osmotic mini‐pump (Alzet Durect, model 1002). Pumps were primed in sterile saline overnight at 37°C to ensure infusion would begin upon implantation. The infusion period was a continuous 14 days. Infusion doses were set to 4 μg·kg−1·day−1. O‐1602 was chosen for treatment in vivo due to ML184 being a piperazine and structurally similar to another benzoylpiperazine GPR55 agonist from GlaxoSmithKline (GSK494581A), which is active at human GPR55 but not rodent GPR55 (Brown et al., 2011).
BrdU injections
Animals were injected with 100 mg·kg−1 5‐bromo‐2′‐deoxyuridine (BrdU; Sigma‐Aldrich) i.p. twice per day for days 1 and 2 of the experimental schedule and once per day for days 3 and 4, a total of six injections.
Primary mouse NSC culture
Primary hippocampal NSCs were obtained from C57BL/6 and GPR55−/− mice at 4–6 weeks of age. Briefly, whole brains were removed and placed in ice‐cold wash buffer (30% glucose, 1M HEPES, penicillin/streptomycin in HBSS) on ice. Brains were washed in wash buffer and transferred to a coronal brain matrix where brains were cut into 1 mm sections. The hippocampus was carefully sectioned out and cut into small pieces. Pieces were then digested in a collagenase solution (collagenase type IV, Worthington Biochemical, Lakewood, NJ, USA) for 30 min, further digested with trypsin for 10 min and triturated. Dissociated cells were cultured at a density of 2 × 105 cells. mL‐1 in proliferation medium [DMEM/F12 (1:1) supplemented with 0.2% heparin, 1xB27 supplement (Gibco), 20 ng.mL‐1 mouse EGF, 10 ng.mL‐1 mouse bFGF, Pen/Strep and L‐glutamine]. Neurospheres were allowed to grow for 7 days with half the medium changed every other day. For expansion as an adherent monolayer, neurospheres were dissociated with Accutase and plated on Matrigel (Corning, Corning, NY, USA)‐coated six‐well plates. Proliferation medium was changed every other day.
Immunofluoresence
Hippocampal samples were sectioned coronally on a Leica CM1860 cryostat (Leica Biosystems, Wetzlar, Germany) into 30 μm sections. Samples were serially sectioned into six wells. All groups of sections were washed three times with PBS to remove residual cryoprotectant. In vitro hNSCs attached to laminin on coverslips were fixed with 4% paraformaldehyde solution for 15 min and washed three times with PBS to remove residual paraformaldehyde. All samples were permeabilized with 0.1% Triton X‐100 (Sigma‐Aldrich) in PBS and blocked in 1% BSA (Sigma‐Aldrich), 5% normal donkey serum (Jackson ImmunoResearch, West Grove, PA, USA) in PBS/0.1% Triton X‐100 for 2 h at room temperature before they were incubated with primary antibodies. Primary antibodies were diluted in blocking solution, and samples were incubated overnight at 4°C. Sections or cells were then washed four times with PBS/0.1% Triton X‐100 and incubated with Alexa Fluor chromophore conjugated secondary antibodies (1:400, Thermo Fisher Scientific) in blocking solution for 2 h at room temperature in the dark. Samples were then washed three times in PBS/0.1% Triton X‐100, once in PBS and mounted onto slides using antifade reagent with DAPI (4′,6‐diamidino‐2‐phenylinodole, Dilactate; Vector Laboratories, Burlingame, CA, USA Cat# H‐1200 Lot#ZC0814 RRID:AB_233679). For BrdU labelling, free floating brain sections were pretreated with ice‐cold 1 N HCl at 4°C for 10 min, 2 N HCl at 37°C for 30 min then washed twice in 0.1 M borate buffer (pH 8.5) for 5 min each before beginning wash steps and blocking. Antibodies used were anti‐Ki67 (Abcam; Cat# ab15580 Lot# GR264777 RRID:AB_443209), anti‐doublecortin (DCX; Millipore Cat# AB2253 Lot# 2828588 RRID:AB_1586992), anti‐BrdU (Abcam Cat# ab6326 Lot# GR267766‐1 RRID:AB_305426) and anti‐βIII‐tubulin (Cell Signaling Technology Cat# 5568S Lot#6 RRID:AB_10694505). Secondary antibodies used were Alexa‐488® goat anti‐guinea pig (Thermo Fisher Scientific Cat# A‐11073 Lot#1841755 RRID:AB_2534117), Alexa‐594® donkey anti‐rat (Thermo Fisher Scientific Cat# A‐21209 Lot#45081A RRID:AB_2535795), Alexa‐488 donkey anti‐rabbit (Thermo Fisher Scientific Cat# A‐21206 Lot#1608521 RRID:AB_2535792). Imaging for hNSCs was performed using a CoolSNAP EZ CCD camera (Photometrics, Tucson, AZ, USA) coupled to a Nikon i80 Eclipse (Nikon Instruments Inc., Melville, NY, USA). Confocal images were taken on a Nikon A1R confocal microscope (Nikon).
Cell counts and quantification
Based on a modified unbiased stereology protocol (Malberg et al., 2000; Rossi et al., 2006; Efstathopoulos et al., 2015), one out of every six adjacent sections was chosen and processed for immunofluorescence staining. The number of Ki67+, BrdU+ or DCX+ cells was then counted under a fluorescent microscope (CoolSNAP EZ CCD camera (Photometrics) coupled to a Nikon i80 Eclipse (Nikon) in the area of the SGZ (defined as a two cell layer in the borders of the granular cell layer, omitting those in the outermost focal planes) for a total of 12 coronal sections through the hippocampus. The total number of positive cells in the SGZ of the hippocampal dentate gyrus was estimated by multiplying the total number of positive cells per section by six and taking the sum of all 12 sections. All counts were performed by a person blinded to treatment group or genotype. The percentage of BrdU+ cells also expressing DCX was determined by dividing the total number of DCX+/BrdU+ cells per animal by the total number of BrdU+ cells and multiplying by 100.
Data and statistical analysis
All values are expressed as mean ± SEM. Analyses were performed using GraphPad Prism 7.0 software (San Diego, CA, USA; RRID:SCR_002798). The significance of the differences between groups was evaluated by one‐way ANOVA, followed by Dunnet's post hoc test or Tukey's post hoc test as necessary. Post hoc tests were only performed when F achieved P < 0.05 and there was no significant variance in the homogeneity. P < 0.05 values were determined to be statistically significant. Statistics for qPCR data were performed on ΔCT values. Confidence intervals and P values for each are denoted for each statistically significant event. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Materials
5‐Methyl‐4‐[(1R,6R)‐3‐methyl‐6‐(1‐cyclohexen‐1‐yl]‐1,3‐benzenediol (O‐1602, analogue of CBD and potent GPR55 agonist; Tocris Bioscience, Bristol, UK), 3‐[[4‐(2,3‐dimethylphenyl)‐1‐piperazinyl]carbonyl]‐N,N‐dimethyl‐4‐(1‐pyrrolidinyl)‐benzenesulfonamide (ML184 [CID‐2440433], synthetic GPR55 agonist; Cayman Chemical, Ann Arbor, MI, USA), LPI (endogenous GPR55 agonist; Sigma‐Aldrich, St. Louis, MO, USA), N‐[4‐[[(3,4‐dimethyl‐5‐isoxazolyl)amino]sulfonyl]phenyl]‐6,8‐dimethyl‐2‐(2‐pyridinyl)‐4‐quinolinecarboxamide [ML193 (CID‐1261822), GPR55 antagonist; Cayman Chemical]. Master stock solutions of O‐1602, ML184, LPI and ML193 were prepared in 100% DMSO according to their solubility and manufacturer's instructions. Working concentrations were generated in culture media and further diluted to final concentrations on the day of use. For in vivo experiments, master stock solutions of O‐1602 were prepared in 100% ethanol (EtOH). All master stock solutions were aliquoted and stored at −20°C.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
Expression of GPR55 in human NSCs
First, we examined the expression of GPR55 mRNA in hNSCs by RT‐PCR (Figure 1A). hNSCs were found to express GPR55 mRNA. HEK293 cells with stable overexpression of human GPR55 were used as a positive control while HEK293 without overexpression were used as a negative control. Expression of GAPDH was used as a loading control to determine that equal amounts of cDNA were generated. We then sought to determine GPR55 mRNA expression from hNSCs under both undifferentiating and differentiation conditions in comparison to NSC markers nestin and Sox2 (Figure 1B), extracellular signal receptors EGFR, FGFR1, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=289 (mGlu1 receptor) (Figure 1C), and cannabinoid receptors CB1 (CNR1) and CB2 (CNR2) (Figure 1D). hNSC GPR55 mRNA was also analysed in comparison to both hGPR55 HEK293 overexpressing cells and HEK293 without hGPR55 (Figure 1E). GPR55 mRNA expression did become detectable within HEK293 samples but it should be noted that all raw CT values were above 36 (36 cycles). GPR55 mRNA within undifferentiated hNSCs was found to be expressed at lower levels than EGFR, FGFR1 and CNR1 and at higher levels than GRM1 and CNR2. After 10 days of culture in differentiating conditions, cells showed reduced expression of nestin, Sox2, EGFR, CNR2 and GPR55. Expression of CNR1 and GRM1 was increased after 10 days of differentiation. These data suggest that hNSCs express GPR55 and that levels of expression of GPR55 mRNA are reduced upon differentiation.
Effects of GPR55 agonists on hNSC proliferation
Since cannabinoid compounds increase NSC proliferation rates (Molina‐Holgado et al., 2007; Palazuelos et al., 2012; Xapelli et al., 2013), we sought to determine if selective activation of GPR55 by its endogenous ligand LPI or synthetic cannabinoid compounds O‐1602 and ML184 (CID2440433) would elicit similar responses. hNSCs were treated with 1 μM LPI, O‐1602 or ML184 for 48 h either with or without the GPR55 antagonist ML193 (5 μM). During the last hour of treatment, cells were pulsed with BrdU (10 μM) to ascertain the total number of cells actively in the S‐phase of the cell cycle. Cells were collected, stained for BrdU and 7‐AAD, run through a BD FACS Canto II flow cytometer and analysed using FlowJo software. Cells were determined to be actively within the S‐phase if they showed positive staining for BrdU. G0/1, S‐phase and G2 populations were determined by 7‐AAD staining (Figure 2A). Activation of GPR55 by ML184 (Figure 2B), O‐1602 (Figure 2C) and LPI (Figure 2D) significantly increased proliferation rates of hNSCs as compared to vehicle control. These effects were attenuated by concurrent treatment with the selective GPR55 antagonist ML193. ML193 treatment alone, or in combination with agonist treatment, did not significantly alter proliferation rates as compared to vehicle control.
Figure 2.
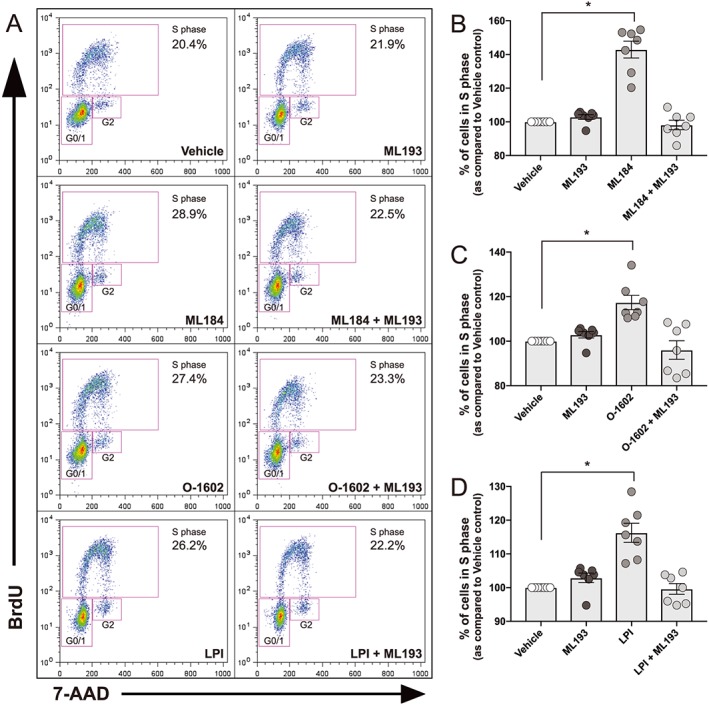
Effects of GPR55 agonists on hNSC proliferation. hNSCs were plated on T25 flasks coated with laminin. Cells were allowed to adhere overnight prior to vehicle, agonist or antagonist treatment. Vehicle refers to medium with 0.01% DMSO. Cells were then treated for 48 h with GPR55 agonists (O‐1602 1 μM, LPI 1 μM, ML184 1 μM), GPR55 antagonist ML193 (5 μM) or both. ML193 was added to cells 30 min prior to addition of agonists. BrdU (10 μM) was added for the final 1 h of treatment. Cells were then collected and stained for BrdU incorporation and total DNA (7‐AAD) for flow cytometric analysis. (A) Representative dot plots of vehicle, agonist and antagonist treatment. Treatment of hNSCs with GPR55 agonists ML184 (B), O‐1602 (C) and LPI (D) increased proliferation rates while pretreatment with ML193 attenuated these effects. ML193 treatment alone did not alter proliferation rates. Data are displayed as percentage of cells in S phase for the last 1 h of treatment as compared to vehicle control. Results are expressed as mean ± SEM; n = 7 experiments. *P<0.05, one‐way ANOVA with Dunnet's post hoc test was used to detect statistical significance.
Effects of GPR55 agonists on hNSC differentiation
Next, we sought to determine the effects of GPR55 activation on hNSC differentiation in vitro. Previous reports indicate that cannabinoid signalling can induce neuronal differentiation through the CB1 or CB2 receptors (Jin et al., 2004; Jiang et al., 2005; Rodrigues et al., 2017). hNSCs were cultured in differentiation medium with either vehicle, ML184 (1 μM), ML193 (5 μM) or ML184 + ML193 for 10 days. Representative images of βIII‐tubulin positive staining are shown in Figure 3 [vehicle (A), ML184 (B), ML193 (C), ML184 + ML193 (D), undifferentiated (E)]. Cells were also collected and stained for flow cytometric analysis. Results showed a significant increase in the number of cells expressing βIII‐tubulin after treatment with ML184. These effects were mediated through GPR55 as concurrent treatment with the GPR55 antagonist ML193 attenuated the increases in neuronal differentiation by ML184. Interestingly, treatment with ML193 alone significantly reduced βIII‐tubulin expression as compared to vehicle control. To determine effects of GPR55 activation on glial differentiation, cells were analysed via flow cytometry for the astrocytic marker S100β. Treatment with ML184 showed no significant changes in S100β from vehicle. Treatment with ML193 showed a slight increase in S100β as did the combination treatment of ML184 with ML193, yet these results were not significant. Representative histograms and quantitative analysis of these experiments are shown in Figure 3F–I. Treatment with ML184 during differentiation also resulted in significant increases in expression of βIII‐tubulin (CI 0.075–1.622; P = 0.029, n = 5) and microtubule associated protein 2 (MAP 2) (CI 0.225–1.586; P = 0.007, n = 5) mRNA (Figure 3J–K), while treatment with ML193 or the combination ML184 plus ML193 showed no differences compared to vehicle control. Treatment with ML184 was also significantly different from ML193 treatment for βIII‐Tubulin (CI −1.602 to −0.055; P = 0.034, n = 5) and MAP 2 (CI −1.738 to −0.377; P = 0.002). ML184 treated samples also had significantly increased MAP 2 expression as compared to ML184 plus ML193 samples (CI −1.763 to −0.402; P = 0.001, n = 5). Treatments did not significantly alter astrocytic marker mRNA expression [glial fibrillary acidic protein (GFAP), S100β; n = 5]. These results suggest that signalling through GPR55 plays a significant role in the formation of new neurons from the NSC population without affecting glial differentiation.
Figure 3.
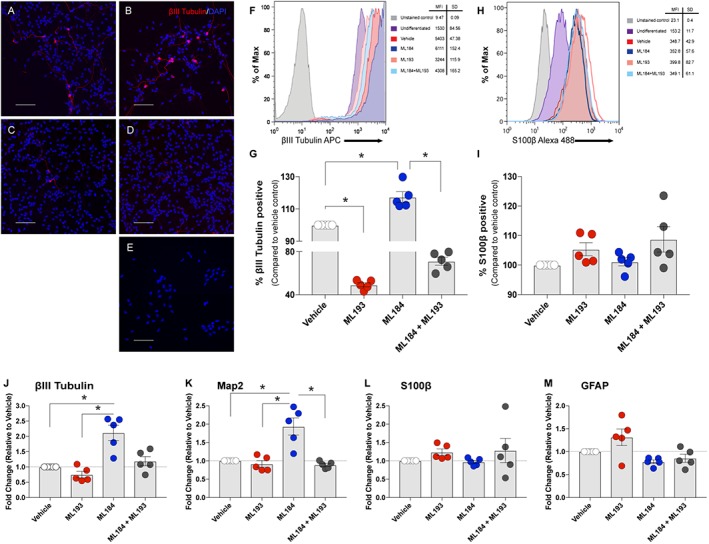
Effects of GPR55 agonists on hNSC differentiation. hNSCs were cultured under differentiating conditions (maintenance medium without bFGF and EGF) and treated with either vehicle (0.01% DMSO), the selective GPR55 agonist ML184 (1 μM), selective antagonist ML193 (5 μM) or a combination of ML184 and ML193 for 10 days. Representative images of vehicle‐treated (A), ML184 (B), ML193 (C), ML184 + ML193 (D) and undifferentiated (E) cells stained for βIII‐tubulin (red) and DAPI (blue). Scale bars are 100 μm. (F, H) Representative histograms of flow cytometric analysis of βIII‐tubulin and S100β respectively. For flow cytometric analysis, cells were incubated with antibodies against human βIII‐tubulin (APC) and S100β (Alexa‐fluor 488). Cytometric acquisition was performed using a BD FACS Canto II flow cytometer and analysed with FlowJo software. Treatment with ML184 increased the expression of βIII‐tubulin after 10 days, while ML193 attenuated these effects. ML193 treatment decreased the formation of neurons as compared to differentiated vehicle control. Quantitative analysis of βIII‐tubulin+ cells (G) and S100β + cells (I) as compared to differentiated control from flow cytometric analysis of five experiments. mRNA was derived from whole cells collected from each experiment and analysed via qPCR for neuronal markers (βIII‐tubulin, J; Map2, K) and astrocytic markers (S100β, L; GFAP, M). Data are presented as fold change of vehicle‐treated cells under differentiating conditions for 10 days. Individual data points represent separate, individual experiments. *P<0.05, one‐way ANOVA and Dunnet's post hoc test.
Primary murine hippocampal NSCs express GPR55
The expression of GPR55 mRNA in hippocampus‐derived primary NSCs was confirmed by RT‐PCR (Figure 4A). NSCs derived from the hippocampus of GPR55−/− mice showed no expression of GPR55 and were used as a negative control. GAPDH was used as a loading control for RT‐PCR reactions. We further confirmed the lack of GPR55 in GPR55−/− mice using DNA derived from tail clips of both C57BL/6 and GPR55−/− mice (representative image in Figure 4B). Genotyping data utilized a primer set for the WT GPR55 allele and GPR55−/− mutant allele as describe by Wu et al. (2010). C57BL/6 mice displayed expression of the WT GPR55 allele while GPR55−/− mice did not. The opposite was found for expression of the mutant GPR55 allele. We subsequently characterized primary hippocampal‐derived NSC cultures to determine any differences between cultures from C57BL/6 and GPR55−/− mice. qPCR analysis of GPR55−/− NSCs displayed no differences in NSC markers (nestin, Sox2; Figure 4C), extracellular signal receptors [Egfr, Fgfr1, Grm5; Figure 4D] or cannabinoid receptors (Cnr1, Cnr2; Figure 4E) as compared to C57BL/6 cultures (n = 5 separate NSC harvests). GPR55 was not found via qPCR in samples derived from GPR55−/− animals.
Effect of O‐1602, a GPR55 agonist, on murine NSC proliferation in vivo
Twelve to 15‐week‐old C57BL/6 (WT) and GPR55−/− mice were treated for 14 days with either vehicle (0.05% EtOH in ACSF) or O‐1602 (4 μg·kg−1·day−1) directly administered into the left hippocampus via a cannula attached to an osmotic pump. Experimental time points are presented in Figure 5A. NSC proliferation within the SGZ of the dentate gyrus in the hippocampus after 14 days of continuous agonist treatment was determined by positive staining for Ki67, a nuclear antigen expressed during all stages of the cell cycle except G0 (Fisher et al., 2002). The total number of Ki67+ cells in WT animals averaged 2567 ± 119.8 (n = 8). Treatment with the GPR55 agonist O‐1602 significantly increased the total number of Ki67 positive cells in WT mice. Vehicle‐treated GPR55−/− mice displayed a significantly reduced rate of proliferation as compared to vehicle‐treated WT animals. Agonist treatment had no effect on GPR55−/− mice as total Ki67 positive cells were not significantly different from vehicle‐treated GPR55−/− animals. Quantitative analysis is shown in Figure 5B. Representative images of in vivo Ki67 staining are shown in Figure 5C. These data suggest that chronic treatment with O‐1602 increases NSC proliferation within the dentate gyrus of the hippocampus after 14 days and that this effect is mediated by GPR55.
Figure 5.
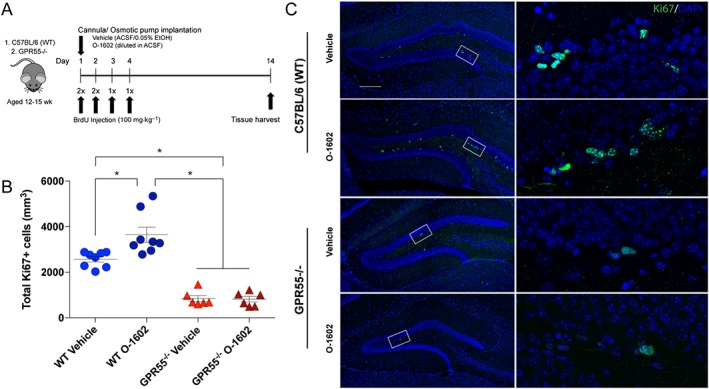
Effects of O‐1602, a GPR55 agonist, on murine NSC proliferation in vivo. C57BL/6(WT) and GPR55−/− mice were implanted with subdermal osmotic pumps set for a dispersion time of 14 days. Pumps were connected to brain infusion kits for direct infusion into the left hippocampus via continuous cannulation. Mice were treated with ACSF +0.05% EtOH (vehicle) or O‐1602 (4 μg·kg−1·day−1). BrdU (100 mg·kg−1) was injected i.p. twice a day for the first 2 days and once a day for days 3 and 4. Brain tissue was harvested at day 14. Schematic representation of experimental time points is displayed in (A). (B) Quantitative analysis of total Ki67 positive cells per dentate gyrus per mouse. Statistical analysis showed significant increases in Ki67+ cells within the SGZ of the dentate gyrus in O‐1602‐treated WT animals as compared to vehicle controls; n = 8 per group. Vehicle‐treated GPR55−/− animals (n = 6) displayed significantly lower Ki67+ cells in the SGZ as compared to WT vehicle‐treated animals. O‐1602 treatment had no significant effect in GPR55−/− animals; n = 7; *P<0.05, one‐way ANOVA and Tukey's multiple comparisons test. (C) Representative confocal microscopy images depicting localization of Ki67 (green) within the dentate gyrus from 30 μm brain sections of WT and GPR55−/− mice. Scale bar = 200 μm.
Effect of O‐1602 on adult murine hippocampal immature neuron generation in vivo
To investigate if GPR55 agonist treatment can increase adult hippocampal immature neuron formation in vivo, 12‐ to 15‐week‐old C57BL/6 and GPR55−/− mice were treated with either vehicle (0.05% EtOH in ACSF) or O‐1602 (4 μg·kg−1·day−1) as outlined in the proliferation study. A series of BrdU injections (100 mg·kg−1) were given for the first 4 days of the treatment schedule. Injections were given twice a day for the first 2 days and once a day for days 3 and 4. After 14 days, immature neuronal markers and proliferating NSC survival were measured in the SGZ of the dentate gyrus in the hippocampus. Representative images of DCX+ and BrdU+ staining within the dentate gyrus are shown in Figure 6A. As hypothesized, the total number of DCX‐positive cells was significantly increased after continuous treatment with O‐1602 as compared to vehicle‐treated WT mice (Figure 6B). BrdU+ cells were significantly increased in WT animals treated with O‐1602 as compared to vehicle control mice (Figure 6C). Analysis of double labelled DCX+/BrdU+ cells in WT animals demonstrated a significant increase due to O‐1602 treatment as compared to vehicle (Figure 6D). The percentage of total BrdU+ cells also expressing DCX, although not significant, was trending to increase as well (Figure 6E). Meanwhile, vehicle‐treated GPR55−/− mice displayed significantly reduced BrdU+, DCX+ and DCX+/BrdU+ cells as compared to WT vehicle treated animals. The percentage of BrdU+ cells also expressing DCX was also significantly reduced in GPR55−/− mice. O‐1602 treatment had no significant effect on the generation of immature neurons within GPR55−/− animals nor the percentage of BrdU+ cells also expressing DCX. These data suggest that activation of GPR55 in vivo increases adult NSC differentiation along a neuronal lineage within the hippocampus.
Figure 6.
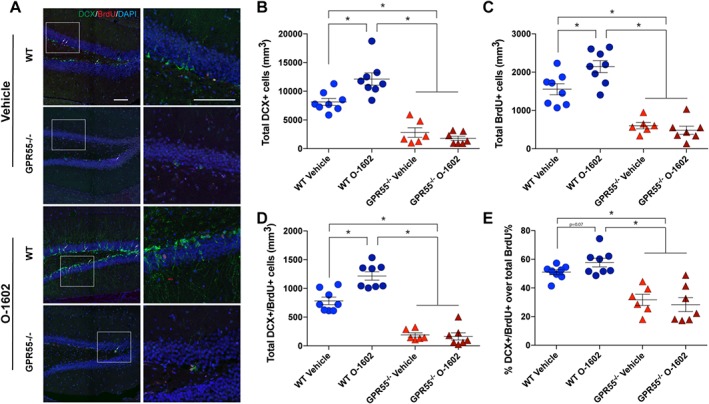
Effects of O‐1602 on adult murine hippocampal neurogenesis in vivo. C57BL/6(WT) and GPR55−/− mice were implanted with subdermal osmotic pumps set for a dispersion time of 14 days. Experimental methods were the same as described for Figure 5. (A) Representative confocal microscopy images depicting localization of BrdU (red) and DCX (green) within the dentate gyrus of the hippocampus from 30 μm brain sections of WT and GPR55−/− mice. Scale bar = 100 μm. Arrows indicate BrdU+/DCX+ double labelled cells. Quantitative analysis of DCX (B), BrdU (C), DCX/BrdU (D) total positive cells per dentate gyrus per mouse. Analysis shows significant increases in DCX+, BrdU+ and DCX+/BrdU+ cells within the SGZ of the dentate gyrus after treatment with O‐1602 as compared to vehicle control; n = 8 per group. Vehicle‐treated GPR55−/− animals (n = 6) exhibit a significant reduction in the number of DCX+, BrdU+ and DCX+/BrdU+ cells in the SGZ as compared to WT vehicle‐treated control animals. Treatment with O‐1602 in GPR55−/− animals had no significant effect on the total number of DCX+, BrdU+ or DCX+/BrdU+ cells in the SGZ. (E) Analysis also determined that the percentage of total BrdU+ cells that were also DCX+ was increased in WT animals treated with O‐1602, yet results were not significant; P > 0.05. GPR55−/− animals demonstrated a significantly reduced rate of BrdU+ cells becoming DCX+ as compared to WT vehicle‐treated control. O‐1602 treatment had no effect on this percentage as compared to vehicle‐treated GPR55−/− mice. Bars represent mean ± SEM; *P<0.05, one‐way ANOVA and Tukey's multiple comparisons test.
Discussion
Recent studies have shown novel roles for cannabinoid receptors and cannabinoid‐like ligands on NSC physiology and pathophysiology (Aguado et al., 2005; Karanian et al., 2005; Palazuelos et al., 2006; Xapelli et al., 2013; Avraham et al., 2014; Rodrigues et al., 2017). Expression of GPR55, an emerging potential member of the cannabinoid family (Yang et al., 2016), has been demonstrated within numerous regions of the adult brain including the hippocampus, a known NSC niche (Wu et al., 2013). However, comprehensive studies concerning GPR55 on NSC biology and function are lacking.
Our study shows for the first time that human and murine NSCs do in fact express GPR55. Our results indicate that GPR55 mRNA levels are comparable to other known extracellular signal receptors [EGFR, FGFR1, GRM1 (human), Grm5 (mouse)] as well as cannabinoid receptors (CNR1, CNR2) within the NSC population. Interestingly, levels of GPR55 mRNA decreased after culture of hNSC samples for 10 days in differentiating conditions suggesting that as NSCs differentiate the levels of GPR55 expression are reduced. Further studies are needed to ascertain exact expression characteristics of GPR55 as NSCs differentiate along neuronal or glial lineages. To further assess GPR55 in the murine hippocampal NSC population, we harvested primary NSCs from the hippocampus of GPR55−/− mice and compared these samples to those of C57BL/6 animals. GPR55−/− NSCs showed no difference in NSC markers, extracellular signal receptors or cannabinoid receptors as compared to C57BL/6 NSCs. These results suggest that GPR55 may be a potent target for pharmacological activation on NSCs with possible therapeutic potential.
Regarding hNSC proliferation, our results indicate that activation of GPR55 by endogenous (LPI) and synthetic agonists (ML184, O‐1602) increases NSC proliferation in a low‐proliferating culture of human NSCs. These actions were attenuated by concurrent treatment with the selective GPR55 antagonist ML193 suggesting that the increase in proliferation seen with GPR55 agonists is dependent on signalling through GPR55. Several reports show that activation of the cannabinoid system through both the CB1 and CB2 receptors increases NSC proliferation in primary murine NSC cultures. The synthetic cannabinoids HU210 and WIN‐55212‐2 promote NSC proliferation through the CB1 receptor yet, interestingly, did not increase differentiation of these cells (Aguado et al., 2005; Jiang et al., 2005). CB1 receptor activation has also recently been found to induce self‐renewal of murine‐derived SVZ NSC cultures and increase proliferation via ERK‐ and PI3K‐dependent mechanisms indicating a necessary role for CB1 receptors in the maintenance of NSC self‐renewal and proliferation (Xapelli et al., 2013). Further study of cannabinoid receptor signalling on NSC proliferation showed differences between cultures of murine SVZ and DG NSCs; SVZ cells had a significant increase in proliferation with CB1 receptor activation while DG cells only had increases with a combination of CB1 and CB2 receptor activation (Rodrigues et al., 2017). Interestingly, all proliferative effects seen by Rodrigues et al. were blocked by either CB1 or CB2 receptor antagonism suggesting that both CB1 and CB2 receptors are necessary for NSC proliferation, possibly through the formation of a CB1/CB2 receptor heteromer. It may be that GPR55 activation and signalling also has similar actions as GPR55 has also been shown to form heteromers with both CB1 and CB2 receptors (Balenga et al., 2014; Martinez‐Pinilla et al., 2014). Of note, treatment of hNSCs with ML184 showed the greatest increase in actively proliferating hNSCs during BrdU administration. It is noteworthy that all three GPR55 agonists used in the proliferation experiments have different pharmacological structures: ML184 is a piperazine; O‐1602 is an atypical cannabinoid (derivative of abnormal‐cannabidiol); LPI is a phospholipid. Recent studies have begun to elucidate specific amino acids critical for activation of GPR55 by ML184 and LPI (Lingerfelt et al., 2017). The study by Lingerfelt et al. found conserved amino acid residues necessary for both ML184 and LPI activation of GPR55, yet they also found another residue (F6.55) that was only necessary for ML184. It may be that activation of GPR55 within hNSCs by these compounds and subsequent different ligand interactions within the receptor binding pocket is facilitating altered responses which may explain differences in proliferation data.
In fact, it has also been reported that there are species differences concerning some compounds that activate GPR55. It was found that two benzoylpiperazines (GSK598945A, GSK522372A) failed to activate mouse GPR55 while GSK494581A failed to activate rat GPR55 (Brown et al., 2011). It was determined that there are two residues that vary between human and rodent GPR55 and that this alteration precludes benzoylpiperazines from interacting with a residue (K2.60) necessary for GPR55 activation and is therefore a confounding mutation (Lingerfelt et al., 2017). Therefore, our in vivo experiments utilized O‐1602 as a synthetic GPR55 ligand as O‐1602 has been shown to activate mouse GPR55. Our results indicate that chronic O‐1602 treatment directly into the hippocampus elevates the number of Ki67+ cells, a marker for cell proliferation, within the SGZ in comparison to vehicle treatment after 14 days. These results suggest that chronic administration of O‐1602 increases the proliferation of NSCs within the hippocampus. Furthermore, GPR55−/− animals displayed reduced numbers of Ki67+ cells within the SGZ as compared to WT controls. Treatment with O‐1602 in these animals had no effect on Ki67+ NSCs suggesting a necessary role of GPR55 activation in NSC proliferation, similar to the findings in the CB1 and CB2 receptor studies. Of note, studies utilizing CB1 −/− or CB2 −/− mice have also demonstrated reduced rates of hippocampal NSC proliferation as compared to WT controls (Jin et al., 2004; Palazuelos et al., 2006). Interestingly, our data may help to explain some of the enigmatic results seen in recent studies with the CB1 receptor and TRPV1 antagonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=743. This antagonist has been shown to increase NSC proliferation both in the SGZ and SVZ of the mouse brain, yet CB1 −/− mice also showed similar increases in proliferation rates suggesting that this compound has another target (Jin et al., 2004). Intensive studies into the pharmacology of GPR55 have recently revealed that SR141716A acts as an antagonist at GPR55 at low concentrations (2 μM) yet produces robust agonist activity at higher concentrations (10–30 μM) implying that GPR55 may play a yet unknown role in the effects seen in studies utilizing SR141716A (Lauckner et al., 2008; Kapur et al., 2009).
An up‐regulation of neuronal differentiation has also been reported as a key aspect of cannabinoid action on the NSC population. Recent reports have suggested that activation of either the CB1 or CB2 receptor up‐regulates neuronal differentiation. CB1 receptor activation by the eCB AEA significantly promoted NSC differentiation into neurons while decreasing the percentage of astrocytes and having no effect on the oligodendrocyte population in culture (Compagnucci et al., 2013). Therefore, we sought to determine if activation of GPR55 had an impact on the differentiation of hNSCs. We chose to utilize only ML184 for differentiation studies in vitro due to this compound having the most robust results in the proliferation studies discussed earlier. Our results indicate that activation of GPR55 promotes neuronal differentiation in hNSC culture as evidenced by increases in βIII‐tubulin‐positive cells treated with ML184 for 10 days under differentiating conditions as compared to vehicle‐treated samples. Interestingly, we also observed a significant reduction in βIII‐tubulin‐positive cells after treatment with ML193 as compared to vehicle‐treated samples. Concurrent treatment of both ML184 and ML193 still showed a reduction in βIII‐tubulin‐positive cells yet not to the same degree as ML193 alone. To further assess alterations in differentiation upon GPR55 activation, we determined population differences in S100β, a marker for astrocytic differentiation. Flow cytometric analysis showed no significant differences between samples treated with ML184, ML193 or concurrent treatment of ML184 with ML193 as compared to vehicle‐treated samples. To confirm these results, we determined the mRNA expression of neuronal markers (βIII‐tubulin, MAP 2) and astrocytic markers (S100β, GFAP). Similar to flow cytometric analysis, we observed significant increases in βIII‐tubulin and MAP 2 mRNA from cells treated for 10 days under differentiating conditions with ML184 as compared to vehicle‐treated samples. ML193 treatment decreased mRNA expression as compared to vehicle but results did not reach significance. S100β and GFAP mRNA was not significantly changed yet there was a trend of increased expression by samples treated with ML193. Based on these data, we assert that activation of GPR55 plays a key role in neuron formation in vitro. To our knowledge, this is the first report of GPR55 activation having direct involvement in the differentiation state of NSCs.
Moreover, we observed an increase in the number of immature neurons (DCX+) and BrdU+ cells within the hippocampus of C57BL/6 animals chronically treated with O‐1602 for 14 days. In addition, we also determined that the number of surviving, proliferating cells at the start of treatment (BrdU+) that subsequently differentiated along a neuronal lineage was also increased with O‐1602 treatment as compared to vehicle‐treated animals (BrdU+/DCX+). These data are the first to demonstrate a role for GPR55 activation in hippocampal NSC differentiation and survival of proliferating cells. Furthermore, we found that GPR55−/− animals have diminished basal rates of immature neuron generation within the hippocampus, implying that signalling through GPR55 enhances early neurogenesis in vivo. We also observed that GPR55−/− animals have a significantly reduced percentage of BrdU+ cells differentiating along a neuronal lineage as compared to WT controls, suggesting that a lack of GPR55 impairs neuronal differentiation in vivo in terms of the total rate of neurogenesis and percentage of proliferating cells actually differentiating along a neuronal lineage. Importantly, recent evidence suggests that GPR55 activation by LPI or O‐1602 transiently increases the frequency of CA1 excitatory postsynaptic currents and enhances hippocampal CA1 LTP and synaptic integrity (Sylantyev et al., 2013; Hurst et al., 2017). It may be possible that GPR55 ligands are strengthening the synaptic connections of newly generated progenitors into the hippocampal circuitry and allowing these cells to survive and integrate into the existing circuitry at a higher rate.
It should be noted that we have not yet performed studies regarding the mechanistic aspects as to how GPR55 signalling influences neurogenesis. Interestingly, the effects of GPR55 activation on NSCs are similar to those seen by targeting CB1 and CB2 receptors, yet the known downstream signalling cascades between GPR55 and CB1/CB2 receptors differ vastly. GPR55 is known to utilize Gq or G12/13, as opposed to Gi/O in CB1/CB2 receptor, for signal transduction ultimately leading to increased intracellular Ca2+, ERK1/2 phosporylation, NFAT activation, nuclear translocation of NFκB and activation of CREB (Lauckner et al., 2008; Henstridge et al., 2009; Kapur et al., 2009; Henstridge et al., 2010). Each of these downstream signalling targets has been implicated in adult neurogenesis, necessitating further studies to fully elucidate the signalling mechanisms mediated by GPR55 on NSCs.
In summary, our findings support the hypothesis that NSCs express functional GPR55 and that targeting GPR55 with selective agonists increases NSC proliferation and early neurogenesis both in vitro and in vivo. A better understanding of the role GPR55 plays during neurogenesis may provide new prospects for therapeutic treatments utilizing this receptor as a target.
Author contributions
J.D.H. designed and performed experiments, wrote the manuscript and supervised the project. V.Z.R. designed animal experiments. S.G. and M.W. performed in vitro experiments and histology. Y.P. supervised the project and refined the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The authors would like to thank Dr. Mary Abood and Dr. Linda Console‐Bram for their help, insight and assistance with this project. This study was supported in part by NIH grants T32DA007237‐28 (NIDA) (J.H.), R37AA015913 and U01AA023552 (NIAAA) (Y.P.).
Hill, J. D. , Zuluaga‐Ramirez, V. , Gajghate, S. , Winfield, M. , and Persidsky, Y. (2018) Activation of GPR55 increases neural stem cell proliferation and promotes early adult hippocampal neurogenesis. British Journal of Pharmacology, 175: 3407–3421. 10.1111/bph.14387.
Contributor Information
Jeremy D Hill, Email: tue64030@temple.edu.
Yuri Persidsky, Email: yuri.persidsky@tuhs.temple.edu.
References
- Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B et al (2005). The endocannabinoid system drives neural progenitor proliferation. FASEB J 19: 1704–1706. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–s129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J, Das GD (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124: 319–335. [DOI] [PubMed] [Google Scholar]
- Avraham HK, Jiang S, Fu Y, Rockenstein E, Makriyannis A, Zvonok A et al (2014). The cannabinoid CB(2) receptor agonist AM1241 enhances neurogenesis in GFAP/Gp120 transgenic mice displaying deficits in neurogenesis. Br J Pharmacol 171: 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balenga NA, Martinez‐Pinilla E, Kargl J, Schroder R, Peinhaupt M, Platzer W et al (2014). Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br J Pharmacol 171: 5387–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begbie J, Doherty P, Graham A (2004). Cannabinoid receptor, CB1, expression follows neuronal differentiation in the early chick embryo. J Anat 205: 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Daniels DA, Kassim M, Brown S, Haslam CP, Terrell VR et al (2011). Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed‐activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J Pharmacol Exp Ther 337: 236–246. [DOI] [PubMed] [Google Scholar]
- Cherif H, Argaw A, Cecyre B, Bouchard A, Gagnon J, Javadi P et al (2015). Role of GPR55 during axon growth and target innervation. eNeuro 2: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian KM, Song H, Ming GL (2014). Functions and dysfunctions of adult hippocampal neurogenesis. Annu Rev Neurosci 37: 243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnucci C, Di Siena S, Bustamante MB, Di Giacomo D, Di Tommaso M, Maccarrone M et al (2013). Type‐1 (CB1) cannabinoid receptor promotes neuronal differentiation and maturation of neural stem cells. PloS one 8: e54271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11: 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S et al (2007). Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathopoulos P, Kourgiantaki A, Karali K, Sidiropoulou K, Margioris AN, Gravanis A et al (2015). Fingolimod induces neurogenesis in adult mouse hippocampus and improves contextual fear memory. Translational psychiatry 5: e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbegdorj O, Westkaemper RB, Zhang Y (2013). A homology modeling study toward the understanding of three‐dimensional structure and putative pharmacological profile of the G‐protein coupled receptor GPR55. J Mol Graph Model 39: 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsohly MA, Slade D (2005). Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci 78: 539–548. [DOI] [PubMed] [Google Scholar]
- Fisher BJ, Naumova E, Leighton CC, Naumov GN, Kerklviet N, Fortin D et al (2002). Ki‐67: a prognostic factor for low‐grade glioma? Int J Radiat Oncol Biol Phys 52: 996–1001. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Ford LA, Ross RA, Waldhoer M, Irving AJ (2009). The GPR55 ligand L‐alpha‐lysophosphatidylinositol promotes RhoA‐dependent Ca2+ signaling and NFAT activation. FASEB J 23: 183–193. [DOI] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Schroder R, Kargl JK, Platzer W, Martini L et al (2010). GPR55 ligands promote receptor coupling to multiple signalling pathways. Br J Pharmacol 160: 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heynen‐Genel S, Dahl R, Shi S, Milan L, Hariharan S, Sergienko E , et al (2010). Screening for selective ligands for GPR55 – antagonists. In Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information (US): Bethesda (MD). [PubMed]
- Hurst K, Badgley C, Ellsworth T, Bell S, Friend L, Prince B et al (2017). A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 27: 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Zhang Y, Xiao L, Van Cleemput J, Ji SP, Bai G et al (2005). Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic‐ and antidepressant‐like effects. J Clin Invest 115: 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Xie L, Kim SH, Parmentier‐Batteur S, Sun Y, Mao XO et al (2004). Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol 66: 204–208. [DOI] [PubMed] [Google Scholar]
- Johns DG, Behm DJ, Walker DJ, Ao Z, Shapland EM, Daniels DA et al (2007). The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br J Pharmacol 152: 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallendrusch S, Kremzow S, Nowicki M, Grabiec U, Winkelmann R, Benz A et al (2013). The G protein‐coupled receptor 55 ligand l‐alpha‐lysophosphatidylinositol exerts microglia‐dependent neuroprotection after excitotoxic lesion. Glia 61: 1822–1831. [DOI] [PubMed] [Google Scholar]
- Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS et al (2009). Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem 284: 29817–29827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanian DA, Brown QB, Makriyannis A, Bahr BA (2005). Blocking cannabinoid activation of FAK and ERK1/2 compromises synaptic integrity in hippocampus. Eur J Pharmacol 508: 47–56. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K (2008). GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A 105: 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Feng JY, Li YY, Yuece B, Lin XH, Yu LY et al (2013). Anti‐inflammatory role of cannabidiol and O‐1602 in cerulein‐induced acute pancreatitis in mice. Pancreas 42: 123–129. [DOI] [PubMed] [Google Scholar]
- Lingerfelt MA, Zhao P, Sharir HP, Hurst DP, Reggio PH, Abood ME (2017). Identification of crucial amino acid residues involved in agonist signaling at the GPR55 receptor. Biochemistry 56: 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Pistis M (2012). Anandamide and 2‐arachidonoylglycerol: pharmacological properties, functional features, and emerging specificities of the two major endocannabinoids. Mol Neurobiol 46: 374–392. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20: 9104–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Pinilla E, Reyes‐Resina I, Onatibia‐Astibia A, Zamarbide M, Ricobaraza A, Navarro G et al (2014). CB1 and GPR55 receptors are co‐expressed and form heteromers in rat and monkey striatum. Exp Neurol 261: 44–52. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70: 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina‐Holgado F, Rubio‐Araiz A, Garcia‐Ovejero D, Williams RJ, Moore JD, Arevalo‐Martin A et al (2007). CB2 cannabinoid receptors promote mouse neural stem cell proliferation. Eur J Neurosci 25: 629–634. [DOI] [PubMed] [Google Scholar]
- Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T (2007). Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 362: 928–934. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzman M, Galve‐Roperh I (2006). Non‐psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J 20: 2405–2407. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Ortega Z, Diaz‐Alonso J, Guzman M, Galve‐Roperh I (2012). CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J Biol Chem 287: 1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2012). Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos Trans R Soc Lond B Biol Sci 367: 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitet F, Donlan M, Michel A (2006). GPR55 as a new cannabinoid receptor: still a long way to prove it. Chem Biol Drug Des 67: 252–253. [DOI] [PubMed] [Google Scholar]
- Prenderville JA, Kelly AM, Downer EJ (2015). The role of cannabinoids in adult neurogenesis. Br J Pharmacol 172: 3950–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues RS, Ribeiro FF, Ferreira F, Vaz SH, Sebastiao AM, Xapelli S (2017). Interaction between cannabinoid type 1 and type 2 receptors in the modulation of subventricular zone and dentate gyrus neurogenesis. Front Pharmacol 8: 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi C, Angelucci A, Costantin L, Braschi C, Mazzantini M, Babbini F et al (2006). Brain‐derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur J Neurosci 24: 1850–1856. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J et al (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152: 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS et al (2011a). Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Wilson DA, Hen R (2011b). Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron 70: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawzdargo M, Nguyen T, Lee DK, Lynch KR, Cheng R, Heng HH et al (1999). Identification and cloning of three novel human G protein‐coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res Mol Brain Res 64: 193–198. [DOI] [PubMed] [Google Scholar]
- Schicho R, Bashashati M, Bawa M, McHugh D, Saur D, Hu HM et al (2011). The atypical cannabinoid O‐1602 protects against experimental colitis and inhibits neutrophil recruitment. Inflamm Bowel Dis 17: 1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelert N, McDougall JJ (2011). The abnormal cannabidiol analogue O‐1602 reduces nociception in a rat model of acute arthritis via the putative cannabinoid receptor GPR55. Neurosci Lett 500: 72–76. [DOI] [PubMed] [Google Scholar]
- Sharir H, Abood ME (2010). Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther 126: 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancic A, Jandl K, Hasenohrl C, Reichmann F, Marsche G, Schuligoi R et al (2015). The GPR55 antagonist CID16020046 protects against intestinal inflammation. Neurogastroenterol Motil 27: 1432–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylantyev S, Jensen TP, Ross RA, Rusakov DA (2013). Cannabinoid‐ and lysophosphatidylinositol‐sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci U S A 110: 5193–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ et al (2009). The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo . Proc Natl Acad Sci U S A 106: 16511–16516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Chen H, Sun H, Zhu J, Jew CP, Wager‐Miller J et al (2013). GPR55, a G‐protein coupled receptor for lysophosphatidylinositol, plays a role in motor coordination. PloS one 8: e60314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Zhu J, Wager‐Miller J, Wang S, O'Leary D, Monory K et al (2010). Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci 32: 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xapelli S, Agasse F, Sarda‐Arroyo L, Bernardino L, Santos T, Ribeiro FF et al (2013). Activation of type 1 cannabinoid receptor (CB1R) promotes neurogenesis in murine subventricular zone cell cultures. PloS one 8: e63529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhou J, Lehmann C (2016). GPR55 – a putative "type 3" cannabinoid receptor in inflammation. J Basic Clin Physiol Pharmacol 27: 297–302. [DOI] [PubMed] [Google Scholar]
- Zhang X, Maor Y, Wang JF, Kunos G, Groopman JE (2010). Endocannabinoid‐like N‐arachidonoyl serine is a novel pro‐angiogenic mediator. Br J Pharmacol 160: 1583–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]