

Abstract
Background and Purpose
Antioxidants provide a promising therapeutic effect for the cardiovascular disease. Luteolin, a polyphenolic bioflavonoid, is known to confer cardioprotection, although the underlying mechanisms, especially the role of luteolin on the antioxidant enzymes, such as the peroxiredoxin family, remain unknown.
Experimental Approach
We measured the effects of luteolin on myocardial ischaemia/reperfusion (MI/R) injury in vivo (Sprague‐Dawley rats) and in vitro, together with the underlying mechanisms, with a focus on signalling by peroxiredoxins. H9c2 cells were used to assess the changes in peroxiredoxins and the other antioxidant enzymes. Oxidative stress, cardiac function, LDH release, ROS and infarct size were also assayed.
Key Results
Luteolin exerted significant cardioprotective effects in vivo and in vitro via improving cardiac function, increasing the expression of anti‐apoptotic protein Bcl‐2 and decreasing the pro‐apoptotic protein Bax and active caspases 3 and 9, associated with MI/R. Mechanistically, luteolin markedly enhanced expression of peroxiredoxin II, without significant effects on other forms of peroxiredoxin, catalase or SOD1. Molecular docking showed that luteolin could indeed bind to the enzymic active pocket of peroxiredoxin II. Furthermore, down‐regulation of peroxiredoxin II by peroxiredoxin II‐antisense, administered by adenovirus infection of H9c2 cardiomyocytes, and inhibition of peroxiredoxin II in vivo significantly reversed the cardioprotective effects of luteolin.
Conclusions and Implications
Our findings, for the first time, demonstrate that luteolin protects against MI/R injury through promoting signalling through the endogenous antioxidant enzyme, peroxiredoxin II, indicating the important beneficial role of this antioxidant system in the heart.
Abbreviations
- AST
aspartate transaminase
- CHD
coronary heart disease
- CK‐MB
creatine kinase – muscle and brain (subunits)
- DHE
dihydroethidium
- EF
ejection fraction
- FS
fractional shortening
- LAD
left anterior descending coronary artery
- LV dP/dtmax
maximum rates of developed left ventricular pressure
- LV dP/dtmin
minimum rates of developed left ventricular pressure
- LVEDP
left ventricular end‐diastolic pressure
- LVSP
left ventricular systolic pressure
- MDA
malondialdehyde
- MI/R
myocardial ischaemia/reperfusion
- TTC
2,3,5‐triphenyltetrazolium chloride
Introduction
Coronary heart disease (CHD) is a widespread public health problem with high morbidity and mortality (Moran et al., 2014). Myocardial ischaemia/reperfusion (MI/R) injury is a major clinical outcome of CHD (Rodriguez‐Porcel et al., 2006; Monassier, 2008). The overproduction of ROS and neutrophil infiltration, together with intracellular calcium overload, are major causative factors of MI/R injury (Turer and Hill, 2010). Particularly, accumulation of intracellular ROS can lead to apoptotic cell death (Zorov et al., 2000) and has direct effects on cellular structure and function in heart tissue (Tsutsui et al., 2006). Therefore, agents that support the endogenous antioxidant defence systems could ameliorate MI/R injury.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5215 (3′,4′,5,7‐tetrahydroxylflavone), is a bioflavonoid found in food, as a constituent of many types of plants, including fruits, vegetables and medicinal herbs (Li et al., 2009; Chen et al., 2016). Preclinical studies have shown that luteolin possesses a variety of biological and pharmacological activities, such as anti‐neoplastic (Kwon et al., 2015; Lin et al., 2015), anti‐hepatotoxic, anti‐allergic (Kritas et al., 2013) and antioxidant effects (Chen et al., 1990). Epidemiological studies have revealed that a high dietary intake of luteolin can significantly reduce the risk of acute myocardial infarction and decrease mortality (Marniemi et al., 2005; Mink et al., 2007). However, the molecular target for the beneficial effect of luteolin requires further investigation.
The peroxiredoxins, a ubiquitous family of antioxidant enzymes, help to control intracellular peroxide levels (Hall et al., 2009; Shi et al., 2016) and maintain the intracellular reducing milieu via redox reactions. There are indications that the activity of peroxiredoxin II reduced apoptosis and improved renal cell survival rates (Hsu et al., 2011). Peroxiredoxin II is also a unique antioxidant in the cardiac system and may represent a potential target for cardiac protection from oxidative stress‐induced injury (Zhao et al., 2009). Our previous investigation demonstrated that the level of peroxiredoxin II, apart from the other five peroxiredoxins forms (peroxiredoxins I and III–VI), was markedly decreased in an ex vivo model of cardiac ischaemia/reperfusion (I/R) or in cardiomyocytes following H2O2 exposure in vitro. These findings indicated the important effects of peroxiredoxin II in MI/R injury. Luteolin acts not only by correcting oxidant/antioxidant imbalance and preventing impaired antioxidant system in H9c2 cells subjected to H2O2 treatment (Zhao et al., 2009) but also by attenuating ROS production accumulation in HepG2 cells caused by palmitate (Liu et al., 2010). However, the cellular and molecular mechanisms of luteolin‐mediated cardiovascular protection, especially the relationship between the effects of this compound and specific antioxidant enzymes, including peroxiredoxin II, still needs to be defined.
Therefore, the present study was designed to determine the therapeutic activities of luteolin and the involvement of the specific antioxidant systems in mediating its protection, using a model of MI/R injury in rats and a cellular H2O2‐induced oxidative stress injury model. Our results provided evidence that luteolin protects hearts from MI/R injury in vivo and in vitro specifically through activating peroxiredoxin II, which should be provide a better understanding of the beneficial effect of luteolin in cardiac MI/R. Our data also highlight the role of peroxiredoxin II as a major component of endogenous cardioprotection.
Methods
Cell culture and treatment
The H9c2 cell line (https://www.atcc.org/Products/All/CRL-1446.aspx) was purchased from the American Type Culture Collection (Shanghai, China). Cells were cultured in DMEM: Nutrient Mixture F‐12 (DMEM/F12) with 10% FBS. Cells were cultured at 37°C in a humidified atmosphere of 5% CO2 and were synchronized by serum starvation before treatment with luteolin or hydrogen peroxide (H2O2). Cells were fed every 2–3 days and subcultured when they reached 70–80% confluence.
Luteolin and H2O2 treatment of H9c2 cells
The cells were used when they reached 70–80% confluence. As luteolin was dissolved in DMSO and diluted to give a final concentration of 0.1% DMSO in cell culture media, we first measured the effect of DMSO (0.1%, v/v) on H9c2 cells and found that incubation with this level of DMSO showed no significant alterations in the cells, compared cells incubated without any DMSO. Therefore, cells in the control cultures were routinely exposed to media containing DMSO at 0.1% (v/v). For the experiments performed in the presence of luteolin , the compound was added to the cells for 2 h prior to treatment with H2O2. The exact group size for each experimental group in vitro was 5.
Adenoviral‐mediated gene transfer
Adenovirus containing antisense to peroxiredoxin II (Ad.Prx II AS) or as a control, adenovirus containing GFP (Ad.GFP) were obtained as previously reported (Zhao et al., 2009). Adenoviruses were amplified in HEK293 cells, purified with ViraKit from Virapur and tittered, according to the standard procedure of AdenoXTM rapid titer kit from BIOMIGA (San Diego, CA, USA). After 2 h of plating, H9c2 cells were infected with Ad.Prx II AS or the control Ad.GFP at a multiplicity of infection (MOI) of 200 for 2 h before the addition of a suitable volume of complete DMEM medium. The efficiency of adenoviral gene transfer was evaluated in cultured H9c2 cells by fluorescence microscopy (Nikon Eclipse Ti‐S; Nikon Ltd., Tokyo, Japan). Nearly 100% of cells appeared infected at 200 MOI by 48 h. The cell phenotype and morphology remained similar among non‐infected and adenoviral‐infected groups after 48 h of infection. The myocytes were then treated with luteolin or H2O2 for indicated time, washed with PBS and harvested for quantitative immunoblotting, or used in the experiments outlined in the results.
Cell viability and LDH assays
To measure cell viability, H9c2 cells seeded in 96‐well plates (5000 cells per well) were grown at 37°C for 24 h until confluence reached ≥80%. Then cells were starved for 12 h in DMEM/F12 supplemented with 0.5% FBS. After serum starvation, H9c2 cells were cultured with different concentration of luteolin (0.1–100 μM) for 24 h. In another set of cells, different concentrations of H2O2 (50, 250, 500 and 1000 μM) were added to the cardiomyocytes and incubated for 2 h. To evaluate the protective effects of luteolin, H9c2 cells were first exposed different concentrations of luteolin for 2h and then cells were exposed to 250 μM H2O2 for 2 h. Model groups were incubated under the same conditions, while cells in control groups were treated with the same volume of PBS. After the indicated time, cell viability was evaluated by MTT assay (Zheng et al., 2013; Ma et al., 2015). The LDH release was measured using the cytotoxicity detection kit, according to the manufacturer's instructions (Jiancheng Bioengineering Institute, Nanjing, China) and quantified by absorbance at 490 nm using a BioTek plate reader. Results were normalized to the control group and expressed as %control; the LDH release of control cells was set to 100%.
Detection of ROS
Intracellular levels of ROS were assessed using conversion of non‐fluorescent dihydroethidium (DHE) to fluorescent ethidium bromide (Arduini et al., 1988). Cells were plated in a six‐well plate (2.5 × 105 cells per well) and incubated with 5 μM DHE at 37°C for 30 min after incubation with 250 μM H2O2 for 2 h. Ethidium fluorescence (excitation at 488 nm and emission at 525 nm) was examined by flowcytometry (Accuri C6; BD, San Jose, CA, USA) immediately. Ten thousand cells were collected and analysed. The superoxide anion levels were calculated by the FlowJo software. Additionally, morphological analysis was performed through fluorescence microscopy (Nikon Eclipse Ti‐S; Nikon Ltd.). For calculation of relative changes in ROS level, values of individual samples were divided by the mean value of samples from the Ad.GFP control group.
TUNEL staining
Analysis of cardiomyocyte apoptosis was performed by TUNEL staining using the http://www.so.com/link?url=http%3A%2F%2Fwww.biomart.cn%2Finfosupply%2F12096243.htm&q=%E8%AF%BA%E7%BB%B4%E8%B5%9E+TUNEL&ts=1492156746&t=4010e373467f8088120f1175d64fb34&src=haosou_blank (Vazyme Biotech, Nanjing, China). Briefly, H9c2 cardiomyocytes were cultured in 24‐well plates for 24 h. After exposure to H2O2 for 2 h, H9c2 cardiomyocytes were fixed by incubation in 10% neutral buffered formalin solution for 25 min at 4°C. The cells were then incubated for 30 min with 0.3% hydrogen peroxide in methanol. Thereafter, cells were treated with a permeabilizing solution (0.1% sodium citrate and 0.2% Triton X‐100) for 5 min at 4°C and then incubated in TUNEL reaction mixture for 60 min at 37°C. Morphological analysis was performed with fluorescence microscopy (Nikon Eclipse Ti‐S; Nikon Ltd.). Five fields were randomly selected from each sample, and at least 100 cells were counted to calculate apoptosis.
Molecular docking studies
Surflex‐Dock (Sybyl‐X2.1 program) was used for the docking studies. The crystal structure of dimeric peroxiredoxin II (PDB:1QMV, 1.7 Å) was retrieved from protein data bank (PDB) (source: http://www.rcsb.org/pdb/externObjLink) based on the previous report (Arduini et al., 1988).
Animals
All animal care and experimental studies were approved by the Ethical Committee of Zhengzhou University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 110 specific pathogen‐free (SPF) male Sprague–Dawley rats weighing 200–250 g, 7–8 weeks old, were obtained from the Experimental Animal Center of Henan Province (Zhengzhou, China). Rats were housed at three to four per cage and kept under standard diurnal lighting conditions (12/12 h), at a controlled ambient temperature of 25 ± 2°C for 7 days before the experiment. All efforts were made to minimize the suffering of the animals and the number of animals needed to obtain reliable results based on the rule of the replacement, refinement or reduction (the 3Rs).
Preparation of rat myocardial I/R model
The rats were anaesthetized with sodium pentobarbital (50 mg·kg−1; i.p.). Then, following tracheal intubation and midline thoracotomy, a 5–0 silk thread was passed behind the left coronary artery approximately 2–3 mm from its origin. A silicone rubber venous cannula was placed between the ends. After a stabilization period of 5–10 min, myocardial ischaemia was initiated by complete ligation of the left anterior descending coronary artery (LAD) together with the venous cannula. Then, 30 min later, the heart was allowed 24 h of reperfusion, by carefully drawing out the venous cannula. Sham‐operated control rats experienced the same protocol without vascular occlusion (Wei et al., 2014). A lead II electrocardiograph was monitored throughout the study. During the entire surgical process, the body temperature of the rats was maintained at 37°C using a heating blanket. Interrupted sutures were made to close wounds. After the surgery, the rats received 0.05 mg·kg−1 buprenorphine (Sigma‐Aldrich, St. Louis, MO, USA) immediately by i.m. injection to minimize pain and distress (Birnbaum et al., 2005) and moved to a separate and quiet room under red lighting. Once they regained purposeful locomotion, they were returned to their home cages. The rats were monitored frequently to make sure they appeared active, normally responsive, calm, awake and resting normal without appearing agitated. All animals were offered softened diet as a complementary and more easily obtainable food source. To confirm the correct LAD ligation in all of our experiments, we checked the location of ligation sutures for each animal post mortem.
Experimental protocols
The experimental timelines are shown in Figure 1.
Rats were randomly divided into five groups (n = 11) using random number generator (https://www.random.org) (Figure 1A): sham, MI/R, LH + MI/R, LM + MI/R and LL + MI/R groups. Saline containing 1% DMSO (2 mL) was injected i.p. as vehicle in sham and I/R groups. In the treatment groups, 20, 10 and 5 mg·kg−1 luteolin were injected i.p., (Qiao et al., 2012), 15 min before vascular ligation (Lin et al., 2016).
Conoidin A has been shown to be a specific covalent inhibitor of peroxiredoxin II (Haraldsen et al., 2009; Nguyen et al., 2013). Therefore, rats were randomly divided into five groups (n = 11) using a random number generator (https://www.random.org) (Figure 1B): sham, MI/R, luteolin + MI/R, conoidin A + MI/R and conoidin A + luteolin + MI/R groups. Saline containing 1% DMSO (2mL) was injected i.p. as vehicle in sham and I/R groups. Conoidin A (5 mg·kg−1) was administered i.p. daily for three successive days before MI/R injury. Luteolin (10 mg·kg−1) was injected i.p., 15 min before vascular ligation.
Figure 1.
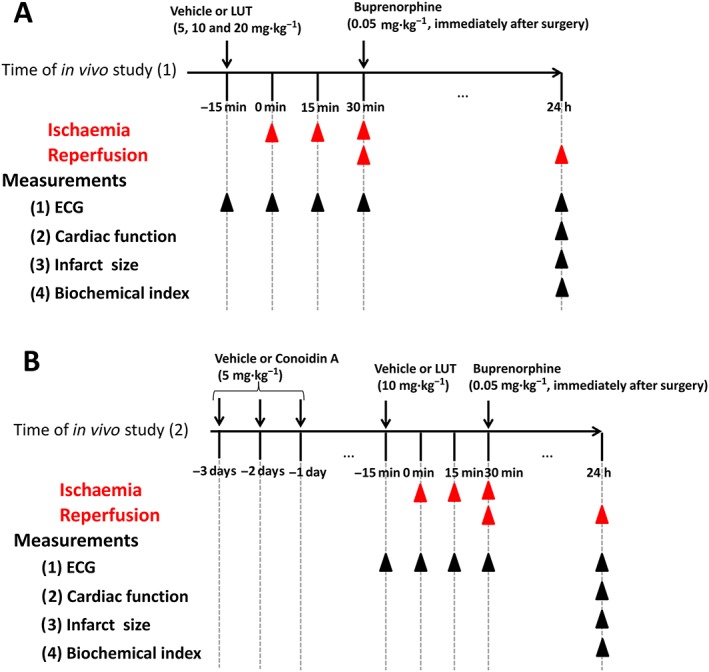
Experimental protocols to investigate the effects of luteolin (LUT) on MI/R injury in rats. (A) Protocol of in vivo study (1). (B) Protocol of in vivo study (2).
ECG
Electrocardiographic recordings were made in anaesthetized rats and calculated as lead II electrocardiograph by RM6240 multi‐channel physiological signal acquisition and processing system (Chengdu Instrument Factory, Chengdu, China). The types of alterations (ST‐segment elevation or depression) in experimental rats were recorded.
Echocardiographic assessment of cardiac function
Left ventricular function was assessed 24 h after MI/R injury using an MS‐250, 16.0–21.0 MHz imaging transducer connected to an ultrasonic echocardiographic system (FUJIFILM VisualSonics Vevo 2100, Inc., Toronto, Ontario, Canada), specifically designed for small animals. Under anaesthesia, the chest of the rat was shaved, and two‐dimensional echocardiography was performed. Images were obtained by identifying the interventricular spectrum and the left ventricular posterior wall. The left ventricular fractional shortening (FS, %) and ejection fraction (EF, %) were automatically calculated by the echocardiographic system. Each parameter was evaluated by calculating average of five cardiac cycles.
Cardiac catheter study for haemodynamic measurements
Haemodynamic measurements were also performed for the estimation of cardiac function. Briefly, the carotid artery was exposed and cannulated with a polyethylene 90 catheter filled with heparinized saline (500 U·mL−1) (Wei et al., 2014). Cardiac function parameters measured included heart rate, left ventricular end‐diastolic pressure (LVEDP), left ventricular systolic pressure (LVSP) and rate of contractility and relaxation LVdP/dtmax and LVdP/dtmin respectively. All the parameters were continuously recorded using a RM6240 multi‐channel physiological signal acquisition and processing system.
Blood sample and tissue processing
At the end of the experimental period, all the rats were anaesthetized with urethane (1 g·kg−1; i.p.) and then killed with a lethal i.p. dose of sodium pentobarbital (3%; 80 mg·kg−1). Blood was collected in dry test tubes without anticoagulant for serum. Heart tissues were excised immediately and were dissected quickly on ice. Some samples were rinsed in ice‐chilled normal saline for evaluating the infarct size and were stored in liquid nitrogen for Western blotting analysis, and others were homogenized immediately in appropriate buffer for ROS determination. All the biochemical assays were performed immediately.
Detecting of myocardium infarct size
Assessment of area at risk and infarct size was performed as described previously (Hall et al., 2016). Briefly, at the end of the 24 h reperfusion, the heart was perfused with 1% Evans' blue dye (Sigma‐Aldrich) to delineate the ischaemic area at risk and then was quickly frozen at −20°C for 30 min. Thereafter, the heart was cut into five equal pieces at a line parallel to the coronary sulcus and below the ligation of the heart. All slices were incubated in 1% 2,3,5‐triphenyltetrazolium chloride (TTC, Sigma Co., St. Louis, MO, USA) in pH 7.4 buffer at 37°C in the dark for 15 min. The infarcted tissues remained unstained (white or pale), whereas normal tissues were stained red. The area at risk and the infarct zone was demarcated and analysed by ImageJ software. Risk area% was measured as the ratio of risk area to myocardium × 100. Infarct size was measured as the ratio of infarcted myocardium to risk region × 100.
Detecting myocardial enzymes
Activities of creatine kinase – muscle and brain (subunits) (CK‐MB), aspartate transaminase (AST) and LDH were assayed in serum using commercial kits purchased from Jiancheng Institute of Biotechnology (Nanjing, China).
Detection of oxidative parameters
Heart tissue was assayed for the endogenous anti‐oxidant enzymes, such as SOD activity, and reduced GSH using commercial kits obtained from Jiancheng Institute of Biotechnology. Lipid peroxidation was determined by the formation of malondialdehyde (MDA)–thiobarbituric acid reactive substances adduct according to the method of Ledwozyw et al. (1986). The released MDA served as the index of lipid peroxidation. Additionally, I/R regions in each group were homogenized in 10 weight/volume of ice‐cold potassium phosphate buffer (50 mM, pH 7.4) containing 1 mM EDTA and centrifuged at 12 000× g for 15 min at 4°C. The supernatants were separated and used for protein determination and ROS assay. The ROS generation and protein level of the supernatant were determined by kits from Jiancheng Biotech (Nanjing, China). For calculation of relative changes in ROS level, values of individual samples were divided by the mean value of samples from the sham group.
Histopathological analysis of heart
Heart tissues were fixed in 10% formalin solution and embedded in paraffin. For the histological examinations, paraffin‐embedded tissue sections of heart (4 μm) were stained with haematoxylin–eosin and examined by light microscopy (×200) for observation of structural abnormality.
TUNEL staining of heart section
TUNEL assay was performed by using the TUNEL Apoptosis Detection Kit (KeyGen Biotech, Nanjing, China) according to the manufacturer's instructions. Both positive (DNase treated) and negative (no addition of TdT) control tissue sections were incorporated in each assay. TUNEL‐positive (brown) cardiac myocytes were regarded as apoptotic cells, and the TUNEL‐positive cells were expressed as a percentage of the total cells. The experiment was repeated on five different sections for each specimen. Ten random fields (×400) per section were analysed.
Western blotting assay
Western blots were performed as described in our previous studies (Wei et al., 2013; Wei et al., 2014). For cellular samples, cell lysates were obtained using commercial RIPA (Beyotime, Shanghai, China). For heart samples, heart tissue was homogenized with commercial RIPA (Beyotime). The protein content was determined by using the BCA kit (Beyotime). Equal amounts of 50 μg per lane of protein were analysed by SDS‐PAGE and transferred to PVDF membranes (Millipore Corporation, Billerica, MA, USA). Blots were blocked for 2 h in 5% non‐fat dry milk–TBS–0.1% Tween 20 and then washed. Primary antibodies were incubated overnight at 4°C followed by a HRP‐conjugated secondary anti‐rabbit antibody (1:10 000; Cell Signaling Technology Co., Ltd., Minneapolis, MN, USA) for 2 h. Immunoreactivity was detected by the Enhanced Chemiluminescence Detection Reagents SuperSignal West Pico Chemiluminescent reagent (34079, Pierce; Thermo Scientific, Rockford, IL, USA) by a gel imaging system (Protein Simple, Santa Clara, CA, USA). The primary antibodies used were polyclonal antibodies against peroxiredoxins I ‐ VI (http://www.ptgcn.com/products/PRDX1-Antibody-15816-1-AP.htm, http://www.ptgcn.com/products/PRDX2-Antibody-10545-2-AP.htm, http://www.ptgcn.com/products/PRDX3-Antibody-55087-1-AP.htm, http://www.ptgcn.com/products/PRDX4-Antibody-10703-1-AP.htm, http://www.ptgcn.com/products/PRDX5-Antibody-17724-1-AP.htm and 13585‐1‐AP, respectively; Proteintech Biotech, Wuhan, China), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2844 (BS70205) and Bax (BS6420) from Bioworld (Minneapolis, MN, USA) and active http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619) (#9661), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1625 (#9507), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2979 (#14097) and SOD1 (#2770) from Cell Signaling Technology Co., Ltd. Protein expression levels were normalized against levels of β‐actin (AP0060) from Bioworld, which was used as a loading control. For calculation of relative changes in protein expression, values of individual samples were divided by the mean value of samples from the control, Ad.GFP control or sham group.
Randomization and blinding
Laboratory rats were randomly assigned to experiments and treatment conditions using a random number generator (https://www.random.org). The specimens of experiments were coded using a random number generator (https://www.random.org). The biochemical assays and analysis of the results was carried out without knowledge of the treatment groups (blinded).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Data are expressed as mean ± SD. Comparisons between two groups were performed by Student's t‐test (from Microsoft Office, Excel), while one‐way ANOVA with Tukey's post hoc test or nonparametric Kruskal–Wallis test followed by the Bonferroni test (from GraphPad Prism 5.0) were used for multi‐group comparison. Results were considered statistically significant at P < 0.05.
Materials
Luteolin (purity >98%, http://www.aladdin-e.com/zh_cn/l107329.html) was purchased from Aladdin Chemistry Co., Ltd., Shanghai, China. Conoidin A (purity >98%, C4293) was purchased from ApexBio Technology LLC, Houston, TX, USA. CK‐MB, AST, LDH, SOD, MDA and GSH commercial kits were obtained from Jiancheng Institute of Biotechnology. TUNEL http://www.so.com/link?url=http%3A%2F%2Fwww.biomart.cn%2Finfosupply%2F12096243.htm&q=%E8%AF%BA%E7%BB%B4%E8%B5%9E+TUNEL&ts=1492156746&t=4010e373467f8088120f1175d64fb34&src=haosou_blank was purchased from Vazyme Biotech. All the other reagents used were of analytical grade.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Luteolin inhibited H2O2‐induced cell death in H9c2 cells through activating peroxiredoxin II
To determine the role of luteolin on cell viability in H2O2‐treated H9c2 cells, MTT and LDH leakage assays were chosen. We found that luteolin at up to 100 μM did not exhibit any significant cytotoxicity in H9c2 cells (Figure 2A). However, treatment of cardiomyocytes with 250, 500 and 1000 μM H2O2 for 2 h resulted in a significant reduction of cell viability, compared with the control cells (Figure 2B). Therefore, luteolin concentrations lower than 100 μM and a concentration of 250 μM H2O2 were chosen for the subsequent experiments. Pretreatment with luteolin (10, 15 and 20 μM) for 2 h significantly increased cell viability in H9c2 cells exposed to H2O2 (Figure 2C). Membrane integrity was monitored by measuring the release of LDH. Treatment with H2O2 markedly elevated LDH release and pretreatment with luteolin (20 or 15 μM) reduced this increased LDH release, concentration‐dependently (Figure 2D). Compared with the control group, the morphology of H9c2 cells showed shrinkage and death due to H2O2 treatment, whereas luteolin markedly restored such H2O2‐induced damage (Figure 2E).
Figure 2.
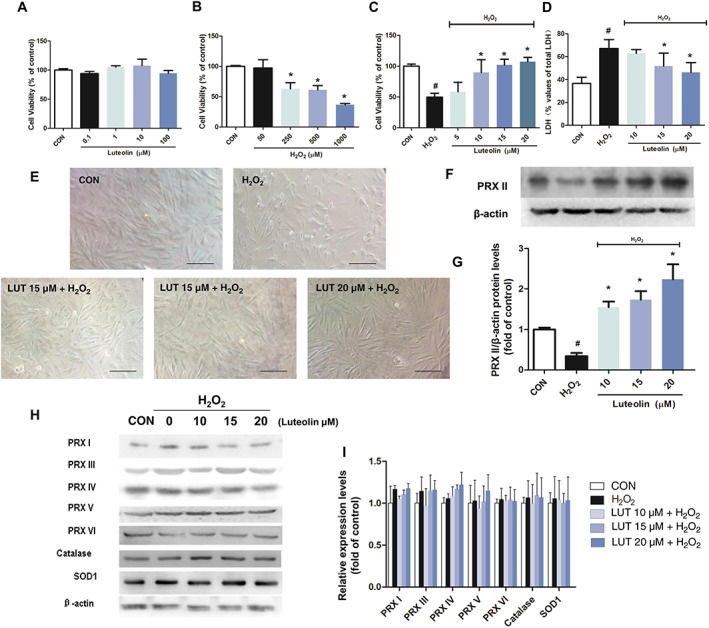
Luteolin prevented H2O2‐induced injury in H9c2 cardiomyocytes. Cardiomyocytes were treated with (A) luteolin (LUT) only (0.1–100 μM) for 24 h, or (B) increasing concentrations of H2O2 (50, 250, 500 and 1000 μM) for 2 h, and (C) luteolin (10–20 μM) followed by H2O2 (250 μM) for 2 h. Cell viability was measured using the MTT assay. (D) The release of LDH at the end of the incubation with H2O2 was determined. (E) Cell morphology was observed after 2 h of H2O2 exposure. Abnormal cell morphology was induced by H2O2, whereas pretreatment with luteolin resulted in dose‐dependent protection from the H2O2‐induced morphological changes (×200, bar = 100 μm). Effects of luteolin on the protein levels of the peroxiredoxins (PRX), catalase and SOD1 in H2O2‐exposed H9c2 cells (F–I). Cells were cultured in six‐well plates until confluent, and the medium was replaced with serum‐free medium, with or without luteolin (10, 15 and 20 μM) for 2 h. The cells were then stimulated with 250 μM H2O2 for 2 h. Results were expressed as percentages of the control values (CON). Data shown are means ± SD for five independent experiments. Data in (G) and (I) were normalized against levels of β‐actin, which was used as a loading control. For calculation of relative changes in protein expression, values of individual samples were divided by the mean value of samples from the control group. Microscopic images are representative of five independent experiments. H2O2, simulated H2O2‐treated only. # P < 0.05, significantly different from control; *P < 0.05, significantly different from H2O2.
To elucidate the mechanism(s) underlying these effects of luteolin, we then investigated the effect of luteolin on the expression of the peroxiredoxin family, following H2O2‐induced injury in H9c2 cells. We found that H9c2 cells exposed to 250 μM H2O2 for 2 h showed markedly decreased levels of peroxiredoxin II, compared with the controlgroup (Figure 2F, G), without significant alterations in the expressions of catalase, SOD1 and the other peroxiredoxins (i.e. peroxiredoxins I and III–VI) (Figure 2H, I). Interestingly, pre‐treatment with luteolin (10, 15 or 20 μM) significantly reversed the H2O2‐induced depressed expression of peroxiredoxin II, compared with the control cultures (Figure 2F, G). These findings indicate that peroxiredoxin II may be associated with the cardioprotective effect of luteolin on H2O2‐induced cell injury.
The protective effect of luteolin on rat MI/R in vivo
To investigate the influence of luteolin on MI/R in vivo, rats were subjected to 30 min LAD ligation followed by 24 h reperfusion. Throughout the whole MI/R surgery procedure, ECG was monitored and recorded at the baseline, during ischaemia and from starting point to 30 min after reperfusion, as well as at 24 h after reperfusion. During the ischaemia, marked ST‐segment elevation in the ECG, was observed in both MI/R and luteolin‐treated groups, compared with the sham group. In order to assess the successful reperfusion of ischaemic region, a significant ST‐segment resolution in our ECG data was observed for each animal (Nai et al., 2015) (Figure 3A). After 24 h reperfusion, ST segment remained elevated in MI/R group; however, in the groups treated with luteolin, the ST‐segment elevation was significantly ameliorated (Figure 3A). Additionally, the infarct size of individual rat hearts was evaluated by the Evans blue‐TTC double staining assay (Figure 3B) and the area at risk was found to be similar under all conditions (Figure 3C). An increase in myocardial infarction size was observed in MI/R rats, compared with the sham group. However, pretreatment with luteolin (20, 10 or 5 mg·kg−1) clearly reduced the infarct size, compared with the untreated MI/R group (Figure 3D). Furthermore, three sensitive indexes of the myocardial zymogram in serum were analysed at 24 h post‐reperfusion injury (Figure 3E–G). In the MI/R group, the activities of CK‐MB, AST or LDH in serum were significantly increased, compared with those data in the sham group. These values were significantly reduced in the MI/R rats that had been rpre‐treated with luteolin (20, 10 or 5 mg·kg−1), as shown in Figures 3E, F. Luteolin (20 or 10 mg·kg−1) also markedly reduced the release of LDH after MI/R (Figure 3G). However, the lowest dose of luteolin (5 mg·kg−1) only showed a trend to reverse the increased LDH activity in serum, and no statistical difference was found.
Figure 3.
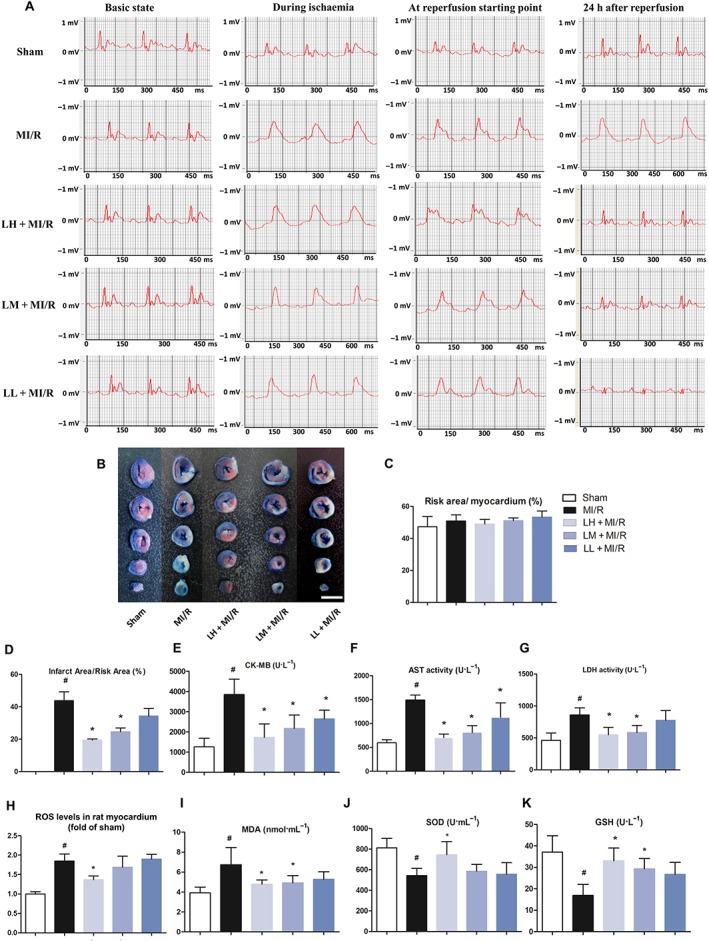
Luteolin protects hearts from MI/R injury in vivo. Representative ECG traces in each group (recorded from lead II with recording speed 50 ms per division) (A). Sham rats revealed normal ECG pattern. MI/R rats showed marked elevation of the ST segment and decreased R amplitude. Treatment with luteolin (20 mg·kg−1, LH or 10 mg·kg−1; LM), but not luteolin (5 mg·kg−1; LL) considerably ameliorated these changes in MI/R rats. Additionally, luteolin reduced myocardial infarct size (B, bar = 10 mm) and ratio of infarct area to risk area (D) in MI/R rats. (C) Ratio of risk area to myocardium in MI/R rats. Luteolin reduces serum marker enzymes CK‐MB (E), AST (F) and LDH (G) levels. Data were presented as mean ± SD (n = 6, C–G). Additionally, luteolin reduces cardiac ROS (H) in heart of MI/R rats. For calculation of relative changes in ROS level, values of individual samples were divided by the mean value of samples from the sham group. Data are presented as mean ± SD (n = 5). Luteolin also decreases the MDA (I) content and enhances endogenous antioxidant enzyme systems including SOD (J) and GSH (K) levels in serum of MI/R rats. Data are presented as mean ± SD (n = 6, I–K). # P < 0.05, significantly different from sham group; *P < 0.05, significantly different from MI/R group.
During MI/R, severe oxidative stress leads to lipid peroxidation. Compared with the sham group, there was a significant elevation of MDA in serum of MI/R rats (Figure 3I), a decrease in GSH levels (Figure 3K) and decreased activity of SOD (Figure 3J). Notably, administration with luteolin dramatically enhanced the activities of these enzymatic antioxidant defences systems in a dose‐dependent manner. Specifically, luteolin increased levels of GSH to ~2‐fold and activities of SOD to ~1.4‐fold of those in MI/R rats (Figure 3K). Moreover, cardiac ROS contents were clearly raised above sham levels in the rats subjected to MI/R. However, pre‐treatment with luteolin (20 mg·kg−1) reversed this increased ROS content to near sham levels (Figure 3H).
Effect of luteolin on the cardiac function after MI/R injury in rats
Left ventricular function was evaluated by echocardiography (Figure 4A). All echocardiographic parameters were similar among the five groups of animals at baseline (data not shown). Compared with the sham group, heart function at 24 h post‐MI/R was compromised, as demonstrated by significant decreases in EF (Figure 4B) and FS (Figure 4C). However, pre‐treatment with luteolin (20, 10 or 5 mg·kg−1) significantly improved both EF and FS, after MI/R (Figure 4B, C).
Figure 4.
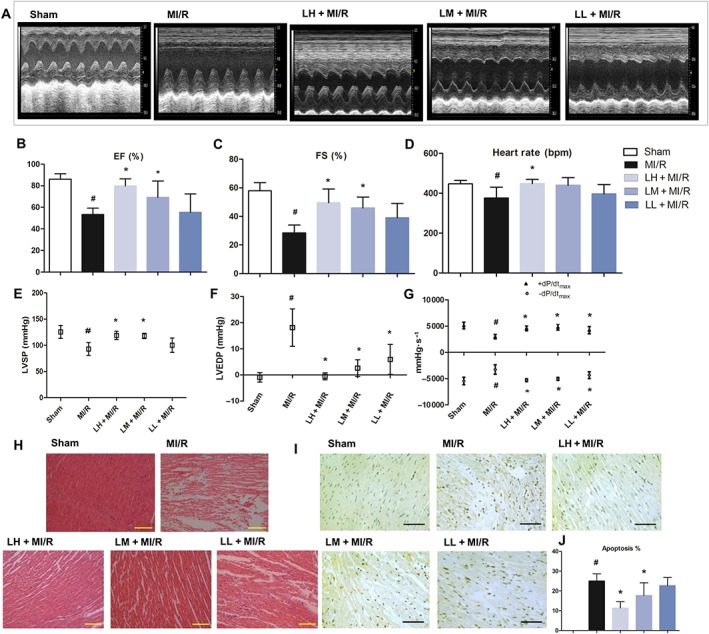
Luteolin protects cardiac function from MI/R injury in vivo. Representative traces of M‐mode echocardiography performed 24 h after MI/R injury in rats (sham, MI/R and luteolin, in mg·kg−1: 20, LH; 10, LM and 5, LL). MI/R rats showed dramatic left ventricular functional impairment as indicated by two‐dimensional M‐mode tracing of wall motion, compared with sham‐operated animals (A). Luteolin treatment significantly improved cardiac functions. EF (B). FS (C). Additionally, luteolin improves cardiac function in MI/R rats as shown by effects on heart rate (D), LVSP (E), LVEDP (F) and maximum and minimum rates of developed left ventricular pressure (LVdP/dtmax and LVdP/dtmin) (G). Data are presented as mean ± SD (n = 6). Histopathological changes in rat cardiac apexes (haematoxylin–eosin) are shown in (H). Sham group showed normal cardiac fibres without any infarction, inflammatory infiltration and cardiac necrosis; MI/R group showed large area of infarction with inflammatory infiltration, cardiac necrosis and splitting of muscle bundles. Treatment with luteolin showed focal area of infarction with less infiltration of inflammatory cells and necrosis in a dose‐dependent manner (200×, bar = 100 μm). In addition, representative photomicrographs of TUNEL staining in myocardial layers within the ischaemic/reperfused area at risk were also examined (400×, bar = 200 μm, I and J). Data are presented as mean ± SD (n = 6). # P < 0.05, significantly different from sham group; *P < 0.05, significantly different from MI/R group.
The MI/R injury was also confirmed by a decreased recovery of haemodynamic function. Heart rate was decreased in the MI/R group, compared with that in sham rats, and pre‐treatment with luteolin reversed this alteration back to normal values (Figure 4D). The markedly reduced LVSP (Figure 4E) and LVEDP (Figure 4F) and a considerable fall in the values of left ventricular maximal and minimal rates of pressure (LV dP/dtmax and LV dP/dtmin) (Figure 4G) were clearly observed in the MI/R group, relative to those in sham group, further signs of left ventricular dysfunction. Here also, the protective effect of treatment with luteolin was shown by the restoration of the post‐reperfusion haemodynamic parameters (Figure 4E–G). All these results clearly demonstrated the cardioprotective effect of luteolin on MI/R injury in vivo.
Microscopic examinations of histological sections of hearts from different experimental groups disclosed very clear differences, as shown in Figure 4H. Hearts from the sham group exhibited clear integrity of myocardial membrane, normal cardiac fibres without any infarction, inflammatory infiltration or cardiac necrosis. However, in hearts from the MI/R group of rats, there were clear signs of leukocyte infiltration, large areas of cardiac necrosis and serious oedema, compared with the sham group. All these pathological changes were diminished after pre‐treatment with luteolin (Figure 4H).
As shown in Figure 4I, the number of TUNEL‐positive cardiomyocyte nuclei was significantly increased after MI/R and the cardiac tissues of these rats showed many more dark brown, apoptotic cells, than the samples from the sham group. Pre‐treatment with luteolin, dose‐dependently, decreased the number of TUNEL‐positive cells following MI/R (Figure 4I, J). Furthermore, the expression levels of key apoptotic‐related proteins were quantitatively assessed. MI/R injury was associated with significant decreases in the expression of Bcl‐2 and marked increases in Bax expression, compared with those data in the sham group. Moreover, in MI/R rats, the pro‐apoptotic, active caspases 3 and 9 were significantly increased to ~2‐fold and 2.3‐fold of that in sham (Figure 5A, B). However, these MI/R‐related alterations were prevented by pre‐treatment with luteolin (20 mg·kg−1) (Figure 5A, B).
Figure 5.
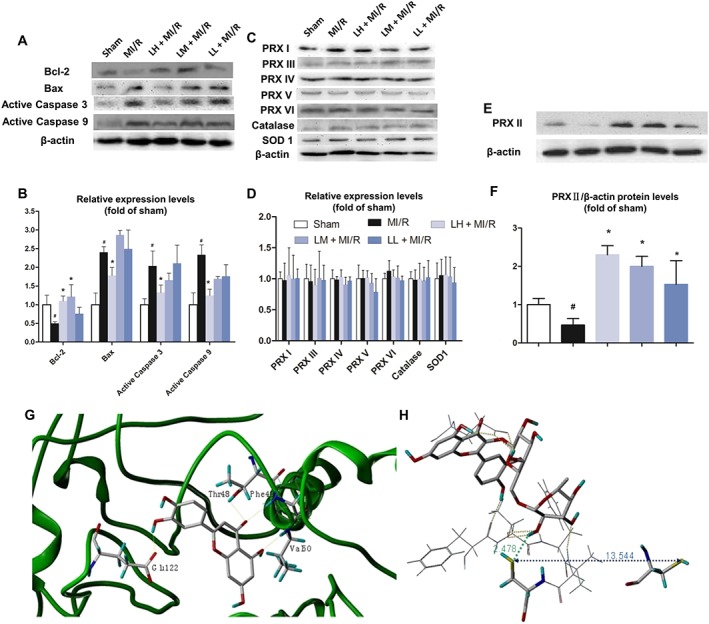
Effect of luteolin on protein levels in cardiac tissue after MI/R in rats. The levels of the apoptosis‐related proteins (Bcl‐2, Bax and active caspases 3 and 9) (A and B), peroxiredoxins (PRX I ‐ VI) and the two representative antioxidant enzymes (catalase and SOD1) (C–F) were measured. Expression levels were normalized against levels of β‐actin, which was used as a loading control. For calculation of relative changes in protein expression, values of individual samples were divided by the mean value of samples from the sham group. In G and H, molecular modelling was used to dock luteolin into the binding pocket of peroxiredoxin II. Data are expressed relative to the mean value for sham group and were presented as mean ± SD (n = 5). # P < 0.05, significantly different from sham group; *P < 0.05, significantly different from MI/R group.
Effect of luteolin on the expression of peroxiredoxins in vivo
As the peroxiredoxin enzymes may play a role in mediating the antioxidant effects in the heart (Dost et al., 2008), we also looked for changes in the other members of the peroxiredoxin family (peroxiredoxins I and III–VI), as well as two representative antioxidant enzymes, catalase and SOD1, in MI/R injured hearts. We found that the MI/R procedure, with or without luteolin pre‐treatment, changed none of these proteins, compared with those in sham group (Figure 5C, D). However, only one of the antioxidant enzymes in the heart, peroxiredoxin II, was significantly decreased by MI/R, compared with the sham group. Pre‐treatment with 20, 10 or 5 mg·kg−1 luteolin significantly increased peroxiredoxin II after MI/R, compared with the sham group (Figure 5E, F). Our results strongly indicated that luteolin may protect hearts from MI/R injury by increasing peroxiredoxin II activity.
Luteolin could bind to the enzymic active pocket of peroxiredoxin II
To determine how luteolin interacts with peroxiredoxin II, computer‐based molecular modelling was performed (Figure 5G, H). The crystal structure (PDB:1QMV, 1.7 Å)1 of human decameric peroxiredoxin II purified from erythrocytes was used and modelling carried out with the Surflex‐Dock (Sybyl‐X2.1 program). The enzyme structure is a toroid comprising five dimers (chain: A–J) linked end‐on through predominantly hydrophobic interactions. However, the residue Cys51 in the site of peroxide reduction of this crystal structure is oxidized to cysteine sulphinic acid, which lies approximately 10 Å away from residue Cys172. Therefore, the comparison with two previously reported dimeric peroxiredoxin structures reveals that the catalytic cycle of 2‐Cys peroxiredoxin requires significant conformational changes that include the unwinding of the active‐site helix and the movement of four loops.
Here, only chains A and B were chosen in the molecular simulation study. After one cysteine sulphinic acid (CSD51) was mutated back to the normal residue Cys51, the peroxiredoxin II protein was prepared with the module of Biopolymer. The geometry of the small molecule, luteolin, was optimized, using Tripos force field and Gasteiger–Hückel charge, with the convergence criteria 0.05 kcal per (mol·Å). Then, luteolin was manually docked into the pocket of the chain B of dimeric peroxiredoxin II, respectively, using the module of Surflex‐Dock and considering the lowest 10 conformers in energy. All the calculations were performed in a Mac workstation. In the modelled luteolin‐peroxiredoxin II complex, the following favourable interactions between luteolin and peroxiredoxin II chain B were observed: for the terminal glycosyl group of the small molecule luteolin, there are three hydrogen bonds formed between the OH group of the C2 and the Thr48 side‐chain OH group or Phe49 main‐chain NH group and Cys51 side‐chain SH group, while another hydrogen bond formed between the O atom of the C3 OH group and NH group of Val50. For the carboxyl group of Glu122 in the biomolecule peroxiredoxin II, there are three hydrogen bonds formed by OH of the middle glycosyl and phenyl group respectively. Similarly, there are nonpolar interactions between the small molecule luteolin and Pro44, Leu45, Pro147, Thr48, Cys51, Val50 and Phe49 of peroxiredoxin II (Figure 5G). These computer‐based calculations and findings further confirmed the possibility of direct interaction between luteolin and peroxiredoxin II.
Effects of Ad.Prx II‐AS‐induced down‐regulation of peroxiredoxin II on the protective effect of luteolin in H9c2 cells
To further determine the important role of peroxiredoxin II in cardiomyocyte death induced by H2O2, H9c2 cells were infected with Ad.GFP or Ad.Prx II‐AS for 48 h, and the levels of peroxiredoxin II were examined. In the cells infected with Ad.Prx II‐AS, the level of peroxiredoxin II was decreased by 60%, compared with the Ad.GFP control group (Figure 6A, B). However, no apparent morphological alterations or differences in the number of adherent cells were observed among the two groups.
Figure 6.
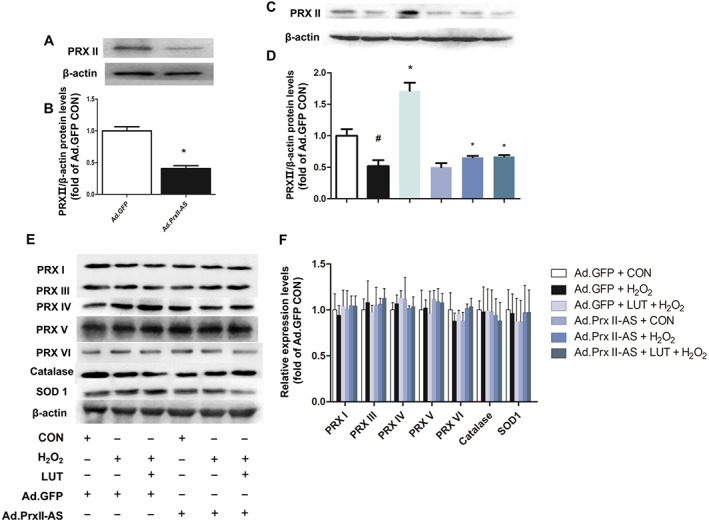
Effect of peroxiredoxin II down‐regulation on protein levels of catalase, SOD1 and peroxiredoxins and on the protective effects of luteolin in H9c2 cells. Cardiomyocytes were infected with adenoviruses, either Ad.GFP or Ad.Prx II‐AS (antisense).In A and B, quantitative immunoblotting of peroxiredoxin II (PRXII). Data are means ± SD for five independent experiments. *P < 0.05, significantly different from Ad.GFP control group. Quantitative immunoblotting of peroxiredoxin II, then catalase, SOD1 and the other peroxiredoxins in infected cardiomyocytes were also examined (C–F). Expression levels were normalized against levels of β‐actin, which was used as a loading control. For calculation of relative changes in protein expression, values of individual samples were divided by the mean value of samples from the Ad.GFP control group. Data are means ± SD for five independent experiments. # P < 0.05, significantly different from control (without H2O2 treatment). *P < 0.05, significantly different from H2O2 (H9c2 cells treated with H2O2 only).
The protein level of peroxiredoxin II was markedly reduced in H9c2 cells infected by Ad.Prx II‐AS, compared with that in the Ad.GFP control group (Figure 6C). However, the expression of the other peroxiredoxins (I and III–VI) and the representative antioxidant enzymes, catalase and SOD1, showed no significant differences in Ad.Prx II‐AS group, compared with those in the GFP control group (Figure 6E, F). In the H9c2 cells with GFP infection, pre‐treatment with luteolin significantly up‐regulated the expression of peroxiredoxin II without any alterations of the other peroxiredoxins, after H2O2 injury. However, the peroxiredoxin II levels in Ad‐PrxII‐AS groups remained decreased (Figure 6D). These findings suggest that peroxiredoxin II may play a key role in mediating the cardioprotective effects of luteolin.
Protection by luteolin against H2O2‐induced cell death in H9c2 cells is associated with peroxiredoxin II
To determine the role of peroxiredoxin II in the beneficial effects of luteolin in H9c2 cells, the infected cardiomyocytes were treated with H2O2, and cell viability as well as release of LDH were examined. After H2O2 treatment, cell viability was significantly decreased in both Ad.GFP‐infected and Ad.Prx II‐AS‐infected cells, compared with the Ad.GFP + control group (Figure 7A). The LDH release was significantly higher in the cells of Ad.Prx II‐AS + H2O2 than that in Ad.GFP + H2O2 group (Figure 7B). Pretreatment with luteolin (20 μM) alone significantly restored these effects caused by H2O2 in H9c2 cells infected with Ad.GFP, whereas in Ad.Prx II‐AS‐infected cells, the protective action of luteolin was significantly blocked (Figure 7A, B). These findings indicate that peroxiredoxin II indeed plays a key role in the beneficial effect of luteolin against H2O2‐induced injury in H9c2 cells.
Figure 7.
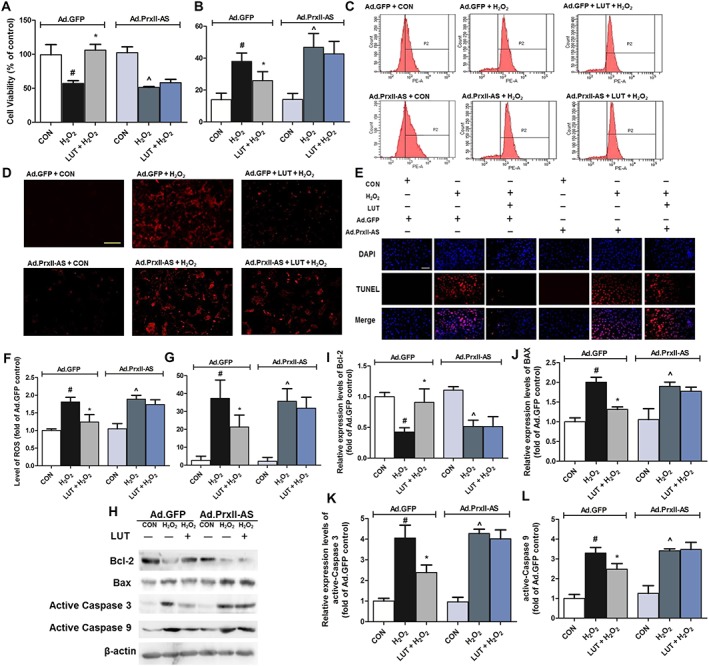
Peroxiredoxin II mediated the protective effect of luteolin on H2O2‐induced oxidative stress in H9c2 cardiomyocytes. Cells were treated in the presence or absence of luteolin (20 μM) and exposed to H2O2 (250 μM) for 2 h after 48 h Ad.GFP or Ad.Prx II‐AS infection. Thereafter, cell viability (A), LDH release (B) and intracellular ROS generation were measured using the DHE assay (200×, bar = 400 μm) (C and D). Additionally, cardiomyocytes were treated in the presence or absence of luteolin (20 μM) and exposed to H2O2 (250 μM) for 2 h after 48 h viral infection (Ad.GFP or Ad.Prx II‐AS). Apoptotic cells were examined under a fluorescence microscope after TUNEL staining (400×, bar = 200 μm) (E). (F) and (G) show quantitative analysis of the results of intracellular ROS and apoptosis respectively. Total number of cells in a given area was determined by DAPI nuclear staining. The apoptotic index was determined as the number of DAPI‐stained TUNEL‐positive cells counted. Microscopic images are representative of five independent experiments. Protein levels of apoptosis‐related proteins (Bcl‐2, Bax and active caspases 3 and 9) were also assayed (H–L). Protein expression levels were normalized against levels of β‐actin, which was used as a loading control. For calculation of relative changes in protein expression or ROS level, values of individual samples were divided by the mean value of samples from the Ad.GFP+ CON group. Data are means ± SD (n = 5). # P < 0.05, significantly different from Ad.GFP + CON (control cells infected by Ad.GFP). *P < 0.05, significantly different from Ad.GFP + H2O2 (Ad.GFP‐infected cells treated with H2O2). ^P < 0.05, significantly different from Ad.Prx II‐AS + CON (control cells infected by Ad.Prx II‐AS).
ROS levels were also measured using a fluorescent probe, DHE. H2O2 treatment induced ROS accumulation in both Ad.GFP‐infected and Ad.Prx II‐AS‐infected cells, compared with Ad.GFP + CON group (Figure 7C, D). Luteolin significantly reduced the ROS accumulation in Ad.GFP‐infected cells. More importantly, down‐regulation of peroxiredoxin II by the antisense almost completely blocked the anti‐oxidative action of luteolin (Figure 7F).
TUNEL staining was performed in H9c2 cells to determine whether peroxiredoxin II was involved in the anti‐apoptotic action of luteolin. The numbers of TUNEL‐positive cardiomyocytes were increased, to the same extent, in both Ad.GFP‐infected and Ad.Prx II‐AS‐infected cells, after treatment with H2O2. Notably, in Ad.Prx II‐AS‐infected cells, these numbers were higher levels than that in Ad.GFP cells (Figure 7E, G). Pretreatment with luteolin (20 μM) alone for 2 h significantly reduced TUNEL‐positive cardiomyocytes induced by H2O2 in Ad.GFP‐infected cells. By contrast, in the cells transfected with Ad.Prx II‐AS, such beneficial effects of luteolin were blocked. Additionally, quantitative immunoblotting to determine the expression levels of Bcl‐2, Bax and active caspases 3 and 9 was also examined (Figure 7H). Ad.GFP + H2O2 and Ad.Prx II‐AS + H2O2 groups were associated with significant decreases in the Bcl‐2 and marked increases in Bax expression, compared with the Ad.GFP + control group. Treatment of luteolin (20 μM) further reversed such alterations as observed in Ad.GFP + luteolin + H2O2 group (Figure 7I, J). Moreover, the active caspases 3 and 9 were significantly increased in Ad.GFP + H2O2 and Ad.Prx II‐AS + H2O2 groups, relative to the levels in the Ad.GFP + control group (Figure 7K, L). These increased values were also significantly prevented by pre‐treatment with luteolin. However, in Ad.Prx II‐AS‐infected cells, the anti‐apoptotic action of luteolin was significantly blocked. These results suggest that luteolin could indeed reduce the H2O2‐induced apoptosis, mainly through activation of peroxiredoxin II.
Luteolin protects against MI/R injury in rats through its effects on peroxiredoxin II
To further determine the involvement of peroxiredoxin II on cardioprotection by luteolin in vivo, we used a specific covalent inhibitor (conoidin A) of this anti‐oxidant enzyme (Haraldsen et al., 2009; Nguyen et al., 2013). Compared with the sham group, the ST‐segment of the ECG was markedly elevated, at 24 h reperfusion in rats with MI/R (Figure 8A) and treatment with luteolin (10 mg·kg−1) reduced this ST‐segment elevation. However, pretreatment with conoidin A (5 mg·kg−1) blocked the effect of luteolin on the ST‐segment elevation. Furthermore, there was an increase in the infarct size in the MI/R group which was reduced by luteolin (Figure 8B–D). Pre‐treatment with conoidin A abolished this effect of luteolin on infarct size (Figure 8B). We also measured the myocardial zymogram in serum and found that MI/R markedly elevated the activities of CK‐MB, AST and LDH. Treatment with luteolin only reduced the activities of these enzymes after MI/R (Figure 8E–G) and these effects of luteolin were prevented by pre‐treatment with conoidin A (Figure 8E–G).
Figure 8.
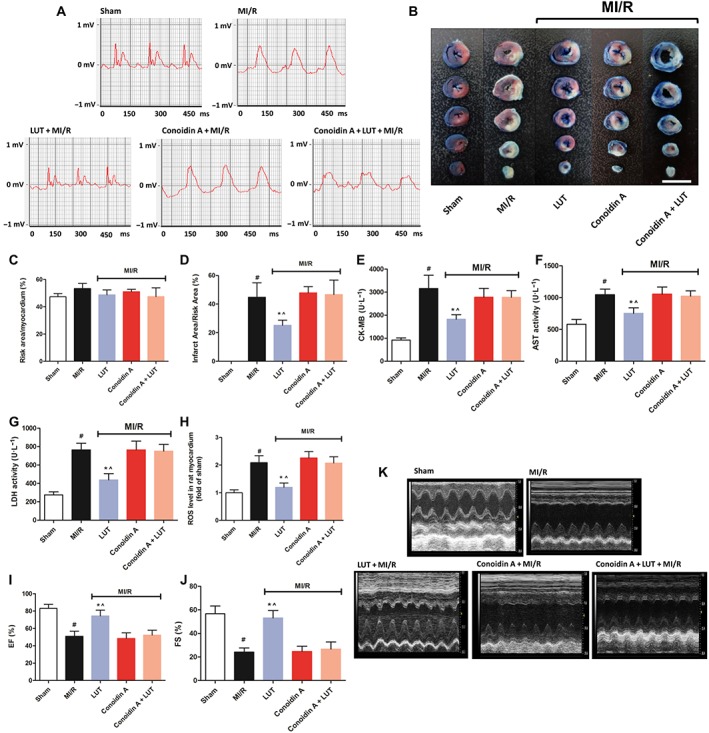
Luteolin ameliorated MI/R injury in rats associated with peroxiredoxin II. Rats were divided into five groups, including sham, MI/R, luteolin (10 mg·kg−1) + MI/R, conoidin A (5 mg·kg−1) + MI/R and conoidin A + luteolin + MI/R. Representative electrocardiograms (A) were measured 24 h after MI/R injury. Myocardial infarct size (B, bar = 10 mm), ratio of risk area to myocardium (C) and infarct area to risk area (D) in each group were also measured. Data (A to D) are presented as mean ± SD (n = 6 per group). The levels of serum marker enzymes, including CK‐MB (E), AST (F) and LDH (G), were examined in each group. Additionally, cardiac ROS were also determined (H). For calculation of relative changes in ROS level, values of individual samples were divided by the mean value of samples from the sham group. Data (E to H) are presented as mean ± SD (n = 5 per group). Representative traces of M‐mode echocardiography (I) performed 24 h after MI/R injury in each group. EF (J). FS (K). Data (I to K) are presented as mean ± SD (n = 6 per group). # P < 0.05, significantly different from sham group. *P < 0.05, significantly different from MI/R group. ^P < 0.05, significantly different from conoidin A + MI/R.
Additionally, compared with those in the sham group, cardiac ROS levels were significantly increased in the MI/R group. Luteolin alone significantly reduced these ROS values (Figure 8H) and conoidin A pretreatment significantly reversed the antioxidative effect of luteolin. Furthermore, at 24 h post‐MI/R, compromised heart function was observed in the MI/R animals, demonstrated by significant decreases in EF (Figure 8I) and FS (Figure 8J), compared with the sham group. Treatment with luteolin alone provided significant improvement in EF and FS. Here also, conoidin A pretreatment impaired the protective effects of luteolin. It should be noted that treatment with conoidin A alone did not change any of the parameters mentioned above, compared with the MI/R group (Figure 8A–K). Our findings strongly indicated that the cardioprotective action of luteolin in vivo is closely dependent on the activity of peroxiredoxin II.
Discussion
In this study, we demonstrated that the cardioprotective effects of luteolin are mainly exerted through its elevation of cardiac peroxiredoxin II expression and suppressing apoptosis in infarcted myocardium both in vivo and in vitro (Figure 9). To the best of our knowledge, this is the first study of the detailed mechanism of the cardioprotective effects of luteolin on endogenous oxidative and redox systems, especially through its up‐regulating effects on peroxiredoxin II.
Figure 9.
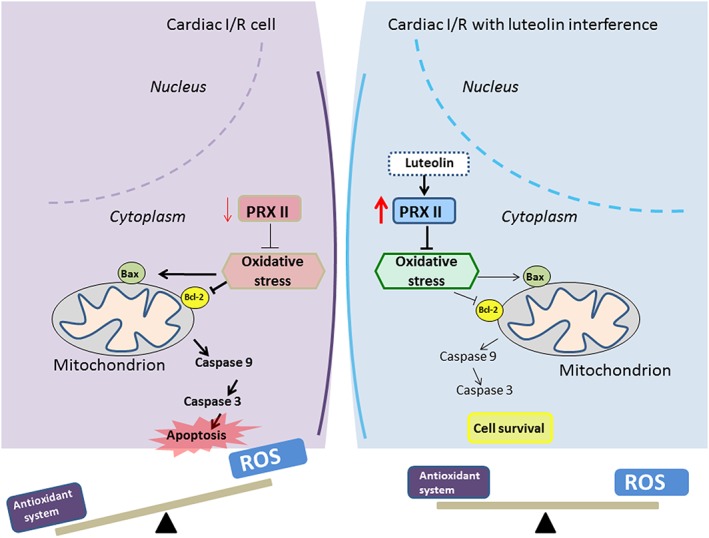
Scheme summarizing the possible mechanisms of the effects of luteolin against MI/R injury by activating cardiac peroxiredoxin II (PRXII). Left: cardiac I/R injury; right: cardiac I/R injury with luteolin interference.
Several epidemiological studies have demonstrated that higher intake of flavonoids, including luteolin, could have protective effects on heart disorders (Amrani et al., 1993; Konorev et al., 1993; McGrath et al., 2010; Xu et al., 2012). Recently, many reports have shown that luteolin could improve I/R‐induced cardiomyocyte contractile function both ex vivo and in vitro (Fang et al., 2011; Qi et al., 2011; Xin et al., 2013; Bian et al., 2015). Sun et al. (2012) reported that luteolin limited infarct size and improved cardiac function after MI/R injury in diabetic rats. The underlying mechanism(s) of cardioprotection by luteolin may be related to anti‐apoptosis via the PI3k/Akt pathway, decreased endoplasmic reticulum stress response, improved contractile function and decreased miR‐208b‐3p and increased Ets1 expression, indicating the multi‐target functional effects of luteolin (Fang et al., 2011; Qi et al., 2011; Sun et al., 2012; Xin et al., 2013; Bian et al., 2015). However, the exact mechanism by which luteolin exerts cardioprotection against MI/R injury in vivo has not been fully elucidated.
Qi et al. (2011) demonstrated that luteolin could improve the contractile function of isolated hearts subjected to I/R. Similarly, we found that luteolin not only improved left ventricular contractile function but also decreased infarcted size in heart tissue. A possible mechanism underlying the effects of luteolin on the elevated ST segment, is through the sulfonylurea receptor‐containing ATP‐sensitive potassium channel which is involved in the ST‐segment elevation (Stoller et al., 2010). Additionally, luteolin has been shown to activate the potassium channels in rat thoracic aorta (Jiang et al., 2005). Therefore, we hypothesize that luteolin may decrease the elevated ST segment through its effects on potassium channels. However, the detailed mechanism remains not completely understood so far. Mounting evidence demonstrates that release of ROS could exacerbate ventricular dysfunction, resembling the pathophysiological changes in MI/R patients (Antonella et al., 1999). Therefore, we evaluated the oxidative stress induced by MI/R injury in our rat model. We found that luteolin could decrease cardiac oxidative stress, as shown by a fall in ROS concentrations, both in cardiocmyocytes and heart tissue, as well as increased antioxidant enzyme/substrate in vivo after 24 h reperfusion, which is consistent with the earlier report that luteolin could decrease ROS levels in rats subjected to MI/R (30 min ischaemia followed by 1 h reperfusion) (Kilkenny et al., 2010). However, our findings indicated a longer‐lasting protective effect of luteolin.
The family of peroxiredoxin enzymes have been suggested to play a pivotal role in mediating antioxidant effects in the heart (Dost et al., 2008; Liu et al., 2008). Notably, increases in the levels of peroxiredoxin II could protect the cardiomyocytes against oxidative stress‐induced apoptosis and cell death by H2O2, the major source of ROS in cardiomyocytes under stress conditions (Kim et al., 2000). Our previous study has also demonstrated that the decreased expression of peroxiredoxin II contributes to MI/R injury (Zhao et al., 2009). In addition, the antioxidant properties of peroxiredoxin II in the heart could be independent of other antioxidant enzymes, suggesting a unique effect of peroxiredoxin II in the process of MI/R injury‐induced cell death (Zhao et al., 2009). Consistent with these findings, in the current study, the expression of peroxiredoxin II was significantly decreased in rat ischaemic‐reperfused heart in vivo, or following H2O2 insult in vitro. Because the other peroxiredoxins, I and III ‐ VI, as well as two representative antioxidant enzymes (catalase and SOD1), are present in cardiac tissue, levels of these proteins were simultaneously measured. Catalase and SOD1 may be the predominant scavengers of H2O2 in the heart and many studies have revealed the important role of catalase and SOD1 in protecting the heart from I/R injury (Triana et al., 1991). However, several studies did not observe such effects of these two antioxidant enzymes on the recovery of contractile function during reperfusion (Hamilton et al., 2003; Robin et al., 2007; Venardos et al., 2007). In the present study, protein expression of catalase and SOD1 was not altered in MI/R rats or following H2O2 injury to cardiomyocytes in vitro, which is compatible with the earlier reports (Arduini et al., 1988; Amrani et al., 1993; Konorev et al., 1993; Hamilton et al., 2003; Robin et al., 2007; Venardos et al., 2007). Additionally, our current data has confirmed that luteolin stimulated the expression of peroxiredoxin II, without altering protein levels of catalase, SOD1 and the other isoforms of the peroxiredoxin, in MI/R rats or following H2O2 injury in vitro. Moreover, pre‐treatment with luteolin markedly promoted the cardiac expression of peroxiredoxin II, reduced cardiac ROS levels and therefore increased the expression of Bcl‐2 and decreased Bax protein expression, further inhibiting apoptosis and cell death following MI/R injury in vivo. These data demonstrated the beneficial effects of luteolin on cardiac function and on the size of the myocardial infarct.
Using transfection with antisense to peroxiredoxin II in adenoviruses, we further confirmed that these beneficial effects of luteolin in H2O2‐induced H9c2 cell injury were dependent on peroxiredoxin II. Additionally, the specific inhibitor of peroxiredoxin II (conoidin A) significantly blocked the beneficial effect of luteolin against MI/R injury in vivo, further supporting a critical role of peroxiredoxin II in the cardioprotective actions of luteolin. Therefore, taking together the results obtained both in vivo and in vitro, it is clear that up‐regulation of peroxiredoxin II was a major contributor to the cardioprotective effect of luteolin. The docking results also revealed the potential binding sites and direct interaction of luteolin with peroxiredoxin II, which could further stabilize the function and structure of peroxiredoxin II.
In conclusion, the inhibitory effects of luteolin on oxidative stress and apoptosis were mainly exerted through its ability to augment cardiac peroxiredoxin II expression both in vivo and in vitro. Our findings suggest that modulation of cardiac peroxiredoxin II presents a novel strategy to improve antioxidant efficacy in ischaemic heart disease.
Author contributions
W.Z. conceived the project. W.Z., B.W., Q.L. and Y.‐G.J. designed the experiments and secured funding. B.W., Q.L., Y.‐G.J. and Y.‐C.Z. performed the experiments. Y.‐C.Z., L.‐N.D., W.‐J.Z. and L.‐H.Z. analysed the data. B.W. wrote the manuscript. W.Z., B.W. and C.‐Y.G. provided critical discussion, editing and final approval of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the Chinese Minister of Science and Technology grant (2016YFA0501800 by W.Z.), the National Natural Science Foundation of China (no. 81470524 by W.Z., 81603114 by B.W. and U1604184 by C.‐Y.G.) and the China Postdoctoral Science Foundation (2016M592316 by B.W.) respectively.
Wei, B. , Lin, Q. , Ji, Y.‐G. , Zhao, Y.‐C. , Ding, L.‐N. , Zhou, W.‐J. , Zhang, L.‐H. , Gao, C.‐Y. , and Zhao, W. (2018) Luteolin ameliorates rat myocardial ischaemia–reperfusion injury through activation of peroxiredoxin II. British Journal of Pharmacology, 175: 3315–3332. 10.1111/bph.14367.
Contributor Information
Chuan‐Yu Gao, Email: gaocy6802@163.com.
Wen Zhao, Email: zhaowen100@139.com, Email: zhaowen@zzu.edu.cn.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani M, Allen NJ, O'Shea J, Corbett J, Dunn MJ, Tadjkarimi S et al (1993). Role of catalase and heat shock protein on recovery of cardiac endothelial and mechanical function after ischemia. Cardioscience 4: 193–198. [PubMed] [Google Scholar]
- Antonella B, Claudio C, Anna C, Palmira B, Salvatore C, Roberto F (1999). Oxidative stress and ventricular dysfunction in ischemic heart disease. Heart Fail Rev 4: 1–10. [Google Scholar]
- Arduini A, Mezzetti A, Porreca E, Lapenna D, Dejulia J, Marzio L et al (1988). Effect of ischemia and reperfusion on antioxidant enzymes and mitochondrial inner membrane proteins in perfused rat heart. Biochimica et Biophysica Acta (BBA) Molecular Cell Research 970: 113–121. [DOI] [PubMed] [Google Scholar]
- Bian C, Xu TD, Zhu H, Pan DF, Liu Y, Luo YY et al (2015). Luteolin inhibits ischemia/reperfusion‐induced myocardial injury in rats via downregulation of microRNA‐208b‐3p. Plos One 10: e0144877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum Y, Ye Y, Rosanio S, Tavackoli S, Hu ZY, Schwarz ER et al (2005). Prostaglandins mediate the cardioprotective effects of atorvastatin against ischemia–reperfusion injury. Cardiovasc Res 65: 345–355. [DOI] [PubMed] [Google Scholar]
- Chen L, Tian G, Tang WD, Luo W, Liu P, Ma ZQ (2016). Protective effect of luteolin on streptozotocin‐induced diabetic renal damage in mice via the regulation of RIP140/NF‐κB pathway and insulin signalling pathway. J Funct Foods 22: 93–100. [Google Scholar]
- Chen YT, Zheng RL, Jia ZJ, Ju Y (1990). Flavonoids as superoxide scavengers and antioxidants. Free Radical Biology and Medicine 9: 19–21. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dost T, Cohen MV, Downey JM (2008). Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning . Basic Res Cardiol 103: 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Li DY, Pan HJ, Chen D, Qi LL, Zhang RQ et al (2011). Luteolin inhibits apoptosis and improves cardiomyocyte contractile function through the PI3K/Akt pathway in simulated ischemia/reperfusion. Pharmacology 88: 149–158. [DOI] [PubMed] [Google Scholar]
- Hall A, Karplus P, Poole L (2009). Typical 2‐Cys peroxiredoxins – structures, mechanisms and functions. FEBS J 276: 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TM, Gordon C, Roy R, Schwenke DO (2016). Delayed coronary reperfusion is ineffective at impeding the dynamic increase in cardiac efferent sympathetic nerve activity following myocardial ischemia. Basic Res Cardiol 111: 35. [DOI] [PubMed] [Google Scholar]
- Hamilton KL, Staib JL, Phillips T, Hess A, Lennon SL, Powers SK (2003). Exercise, antioxidants, and HSP72: protection against myocardial ischemia/reperfusion. Free Radic Biol Med 34: 800–809. [DOI] [PubMed] [Google Scholar]
- Haraldsen JD, Liu G, Botting CH, Walton JGA, Storm J, Phalen TJ et al (2009). Identification of conoidin A as a covalent inhibitor of peroxiredoxin II. Org Biomol Chem 7: 3040–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HH, Di Marco GS, Endlich N, Peter‐Katalinic J, Weide T, Pavenstadt H (2011). Downregulation of the antioxidant protein peroxiredoxin 2 contributes to angiotensin II‐mediated podocyte apoptosis. Kidney Int 80: 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Xia Q, Wang X, Song J, Bruce IC (2005). Luteolin induces vasorelaxion in rat thoracic aorta via calcium and potassium channels. Die Pharmazie‐An International J Pharm Sci 60: 444–447. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE Guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee TH, Park ES, Suh JM, Park SJ, Chung HK et al (2000). Role of peroxiredoxins in regulating intracellular hydrogen peroxide and hydrogen peroxide‐induced apoptosis in thyroid cells. J Biol Chem 275: 18266–18270. [DOI] [PubMed] [Google Scholar]
- Konorev EA, Struck AT, Baker JE, Ramanujam S, Thomas JP, Radi R et al (1993). Intracellular catalase inhibition does not predispose rat heart to ischemia–reperfusion and hydrogen peroxide‐induced injuries. Free Radic Res Commun 19: 397–407. [DOI] [PubMed] [Google Scholar]
- Kritas S, Saggini A, Varvara G, Murmura G, Caraffa A, Antinolfi P et al (2013). Luteolin inhibits mast cell‐mediated allergic inflammation. J Biol Regul Homeost Agents 27: 955–959. [PubMed] [Google Scholar]
- Kwon EY, Jung UJ, Park T, Yun JW, Choi MS (2015). Luteolin attenuates hepatic steatosis and insulin resistance through the interplay between the liver and adipose tissue in mice with diet‐induced obesity. Diabetes 64: 1658–1669. [DOI] [PubMed] [Google Scholar]
- Ledwozyw AS, Michalak J, Stepień A, Kadziołka A (1986). The relationship between plasma TG, cholesterol, total lipid peroxidation product during human atherosclerosis. Clin Chim Acta 155: 275–283. [DOI] [PubMed] [Google Scholar]
- Li L, Henry G, Seeram N (2009). Identification and bioactivities of resveratrol oligomers and flavonoids from Carex folliculata seeds. J Agric Food Chem 57: 7282–7287. [DOI] [PubMed] [Google Scholar]
- Lin CW, Lai GM, Chen KC, Lin TH, Fan JJ, Hsu RL et al (2015). RPS12 increases the invasiveness in cervical cancer activated by c‐Myc and inhibited by the dietary flavonoids luteolin and quercetin. J Funct Foods 19: 236–247. [Google Scholar]
- Lin TY, Lu CW, Wang SJ (2016). Luteolin protects the hippocampus against neuron impairments induced by kainic acid in rats. Neurotoxicology 55: 48–57. [DOI] [PubMed] [Google Scholar]
- Liu JF, Ma Y, Wang Y, Du ZY, Shen JK, Peng HL (2010). Reduction of lipid accumulation in HepG2 cells by luteolin is associated with activation of AMPK and mitigation of oxidative stress. Phytother Res 25: 588–596. [DOI] [PubMed] [Google Scholar]
- Liu Y, Yang XM, Iliodromitis EK, Kremastinos DT, Dost T, Cohen MV et al (2008). Redox signaling at reperfusion is required for protection from ischemic preconditioning but not from a direct PKC activator. Basic Res Cardiol 103: 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LY, Zheng YC, Wang SQ, Wang B, Wang ZR, Pang LP et al (2015). Design, synthesis, and structure–activity relationship of novel LSD1 inhibitors based on pyrimidine–thiourea hybrids as potent, orally active antitumor agents. J Med Chem 58: 1705–1716. [DOI] [PubMed] [Google Scholar]
- Marniemi J, Alanen E, Impivaara O, Seppänen R, Hakala P, Rajala T et al (2005). Dietary and serum vitamins and minerals as predictors of myocardial infarction and stroke in elderly subjects. Nutr Metab Cardiovasc Dis 15: 188–197. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, Mclachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink PJ, Scrafford CG, Barraj LM, Harnack L, Hong CP, Nettleton JA et al (2007). Flavonoid intake and cardiovascular disease mortality: a prospective study in postmenopausal women. American Journal of Clinical Nutrition 85: 895–909. [DOI] [PubMed] [Google Scholar]
- Monassier JP (2008). Reperfusion injury in acute myocardial infarction. From bench to cath lab. Part I: basic considerations. Arch Cardiovasc Dis 101: 491–500. [DOI] [PubMed] [Google Scholar]
- Moran A, Roth G, Narula J, Mensah G (2014). 1990–2010 global cardiovascular disease atlas. Glob Heart 9: 3–16. [DOI] [PubMed] [Google Scholar]
- Nai C, Xuan H, Zhang YY, Shen MX, Xu TD, Pan DF et al (2015). Luteolin exerts cardioprotective effects through improving sarcoplasmic reticulum Ca2+‐ATPase activity in rats during ischemia/reperfusion in vivo. Evid Based Complement Alternat Med 2015: 365854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JB, Pool CD, Wong CY, Treger RS, Williams DL, Cappello M et al (2013). Peroxiredoxin‐1 from the human hookworm Ancylostoma ceylanicum forms a stable oxidized decamer and is covalently inhibited by conoidin A. Chem Biol 20: 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao HM, Dong LP, Zhang XJ, Zhu XL, Wang LN, Liu ZJ et al (2012). Protective effect of luteolin in experimental ischemic stroke: upregulated SOD1, CAT, Bcl‐2 and claudin‐5, down‐regulated MDA and Bax expression. Neurochem Res 37: 2014–2024. [DOI] [PubMed] [Google Scholar]
- Qi LL, Pan HJ, Li DY, Fang F, Chen D, Sun H (2011). Luteolin improves contractile function and attenuates apoptosis following ischemia–reperfusion in adult rat cardiomyocytes. Eur J Pharmacol 668: 201–207. [DOI] [PubMed] [Google Scholar]
- Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD et al (2007). Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J Biol Chem 282: 19133–19143. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Porcel M, Zhu XY, Chade AR, Amores‐Arriaga B, Caplice NM, Ritman EL et al (2006). Functional and structural remodeling of the myocardial microvasculature in early experimental hypertension. Am J Physiol Heart Circ Physiol 290: 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XJ, Ding L, Zhou W, Ji Y, Wang J, Wang H et al (2016). Pro‐apoptotic effects of JDA‐202, a novel natural diterpenoid, on esophageal cancer through targeting peroxiredoxin I. Antioxid Redox Signal 27: 73–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoller DA, Fahrenbach JP, Chalupsky K, Tan BH, Aggarwal N, Metcalfe J et al (2010). Cardiomyocyte sulfonylurea receptor 2‐KATP channel mediates cardioprotection and ST segment elevation. Am J Physiol Heart Circ Physiol 299: 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun DD, Huang J, Zhang Z, Gao HK, Li JY, Shen M et al (2012). Luteolin limits infarct size and improves cardiac function after myocardium ischemia/reperfusion injury in diabetic rats. Plos One 7: e33491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triana JF, Li XY, Jamaluddin U, Thornby JI, Bolli R (1991). Postischemic myocardial “stunning”. Identification of major differences between the open‐chest and the conscious dog and evaluation of the oxygen radical hypothesis in the conscious dog. Circ Res 69: 731–747. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Ide T, Kinugawa S (2006). Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal 8: 1737–1744. [DOI] [PubMed] [Google Scholar]
- Turer A, Hill J (2010). Pathogenesis of myocardial ischemia–reperfusion injury and rationale for therapy. Am J Cardiol 106: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venardos KM, Perkins A, Headrick J, Kaye DM (2007). Myocardial ischemia–reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem 14: 1539–1549. [DOI] [PubMed] [Google Scholar]
- Wei B, You MG, Ling JJ, Wei LL, Wang K, Li WW et al (2013). Regulation of antioxidant system, lipids and fatty acid β‐oxidation contributes to the cardioprotective effect of sodium tanshinone IIA sulphonate in isoproterenol‐induced myocardial infarction in rats. Atherosclerosis 230: 148–156. [DOI] [PubMed] [Google Scholar]
- Wei B, Li WW, Ji J, Hu QH, Ji H (2014). The cardioprotective effect of sodium tanshinone IIA sulfonate and the optimizing of therapeutic time window in myocardial ischemia/reperfusion injury in rats. Atherosclerosis 235: 318–327. [DOI] [PubMed] [Google Scholar]
- Xin W, Xu TD, Li DY, Zhu SS, Chen QP, Hu WJ et al (2013). ERK/PP1a/PLB/SERCA2a and JNK pathways are involved in luteolin‐mediated protection of rat hearts and cardiomyocytes following ischemia/reperfusion. Plos One 8: e82957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Li DY, Jiang DH (2012). Targeting cell signaling and apoptotic pathways by luteolin: cardioprotective role in rat cardiomyocytes following ischemia/reperfusion. Nutrients 4: 2008–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Fan G, Zhang Z, Bandyopadhyay A, Zhou X, Kranias E (2009). Protection of peroxiredoxin II on oxidative stress‐induced cardiomyocyte death and apoptosis. Basic Res Cardiol 104: 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YC, Duan YC, Ma JL, Xu RM, Zi XL, Lv WL et al (2013). Triazole–dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J Med Chem 56: 8543–8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000). Reactive oxygen species (ROS)‐induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192: 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]