

Abstract
Background and Purpose
Non‐alcoholic steatohepatitis (NASH) is the most severe form of non‐alcoholic fatty liver disease and is a serious public health problem around the world. There are currently no approved treatments for NASH. FGF21 has recently emerged as a promising drug candidate for metabolic diseases. However, the disadvantages of FGF21 as a clinically useful medicine include its short plasma half‐life and poor drug‐like properties. Here, we have explored the effects of PsTag600‐FGF21, an engineered long‐acting FGF21 fusion protein, in mice with NASH and describe some of the underlying mechanisms.
Experimental Approach
A long‐acting FGF21 was prepared by genetic fusion with a 600 residues polypeptide (PsTag600). We used a choline‐deficient high‐fat diet‐induced model of NASH in mice. The effects on body weight, insulin sensitivity, inflammation and levels of hormones and metabolites were studied first. We further investigated whether PsTag600‐FGF21 attenuated inflammation through the Th17‐IL17A axis and the associated mechanisms.
Key Results
PsTag600‐FGF21 dose‐dependently reduced body weight, blood glucose, and insulin and lipid levels and reversed hepatic steatosis. PsTag600‐FGF21 enhanced fatty acid activation and mitochondrial β‐oxidation in the liver. The profound reduction in hepatic inflammation in NASH mice following PsTag600‐FGF21 was associated with inhibition of IL17A expression in Th17 cells. Furthermore, PsTag600‐FGF21 depended on adiponectin to exert its suppression of Th17 cell differentiation and IL17A expression.
Conclusions and Implications
Our data have uncovered some of the mechanisms by which PsTag600‐FGF21 suppresses hepatic inflammation and further suggest that PsTag600‐FGF21 could be an effective approach in NASH treatment.
Abbreviations
- ACSL
acyl CoA synthetase
- ALT
alanine aminotransaminase
- AST
aspartate aminotransaminase
- CD
choline‐deficient
- CD‐HFD
choline‐deficient with a high‐fat diet
- DIO
diet‐induced obese
- FATP
fatty acid transport protein
- HCC
hepatocellular carcinoma
- HFD
high‐fat diet
- MCD
methionine‐ and choline‐deficient
- NAFLD
non‐alcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
non‐alcoholic steatohepatitis
- OGTT
oral glucose tolerance test
- SD
standard diet
- siRNA
short interfering RNA
- WAT
white adipose tissue
Introduction
Non‐alcoholic fatty liver disease (NAFLD), characterized by hepatic steatosis, is a very common chronic liver disease worldwide (Ma et al., 2016; Liu et al., 2016b). Hepatic steatosis combined with other pathological changes, such as lipotoxicity, oxidative stress and mitochondrial dysfunction, induces non‐alcoholic steatohepatitis (NASH) from NAFLD (Michelotti et al., 2013; Bashiardes et al., 2016). The prevalence of NASH is estimated to be 2–5% in the general population (Liu et al., 2016a). NASH is the most severe category of NAFLD and is a significant risk factor for liver cirrhosis and hepatocellular carcinoma (HCC) (Tanaka et al., 2012). NASH‐associated cirrhosis is predicted to surpass chronic hepatitis C as the leading cause of liver transplantation in the USA within the next 3 years (Wree et al., 2013; Cassidy and Syed, 2016). However, currently, there is no established treatment for NASH.
The growth factor FGF21 is a member of the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=968 that is secreted mainly from tissues with high metabolic activity, such as liver, pancreas, muscle and adipose tissue, and acts in many tissues (Gimeno and Moller, 2014; Chen et al., 2017). In humans, the circulating levels of FGF21 are elevated in individuals with both NAFLD and NASH, suggesting that FGF21 may have a role in the progression of fatty liver disease (Liu et al., 2015). Furthermore, a range of studies have shown a positive correlation between serum FGF21 and typically metabolic parameters, such as free fatty acids and triglycerides, in patients who developed NAFLD and NASH (Liu et al., 2014). The major risk factors for NAFLD are insulin insensitivity, obesity and dyslipidemia, which are improved by FGF21 (Zhang and Li, 2014; Talukdar et al., 2016). In FGF21‐KO mice fed a diet deficient in methionine and choline (MCD), the lack of FGF21 is associated with markedly worsened hepatic steatosis, apoptosis, fibrosis, inflammation and thus the progression to severe NASH (Reitman, 2013; Fisher et al., 2014; Fisher and Maratos‐Flier, 2016). Importantly, FGF21 treatment attenuates or eliminates the MCD diet‐induced progression to NASH (Ju et al., 2016). The first clinical studies using an FGF21 analogue appear to redirect the therapeutic utility of FGF21 to lipids and pathological conditions, such as NASH (Kharitonenkov and Dimarchi, 2015; Talukdar et al., 2016). These findings support the development of FGF21 as a novel drug candidate for the treatment of NASH, although the mechanisms of FGF21 actions in NASH are mostly unknown (Inagaki, 2015; Musso et al., 2016). Recently, several studies suggested that inflammation preceded steatosis and determined the long‐term prognosis of NASH (Wolf et al., 2014; Machado and Diehl, 2016). Recent findings have suggested that systemic inflammation may be orchestrated by Th17 cells in NASH pathogenesis (Harley et al., 2014; Gomes et al., 2016). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4982 induces neutrophil infiltration in white adipose tissue (WAT) and liver causing NASH, and HCC. Additionally, the effects and mechanisms of FGF21 on reducing inflammation in NASH have not been fully addressed.
We have syntheiszed a novel recombinant polypeptide, PsTag, that could be useful in the development of bio‐drugs with properties comparable to those achievable by PEGylation, but with potentially fewer side effects (Podust et al., 2015; Yin et al., 2016). PsTag600‐FGF21 was a fusion protein engineered by fusing PsTag600 polypeptides to human FGF21. This fusion protein showed not only prolonged but also greater pharmacological effects in mice and could therefore be a more effective drug candidate than the unmodified FGF21. PsTag600‐FGF21 not only markedly improved the serum half‐life to 12.93 h in mice but also prevented the rapid aggregation of native FGF21 (Yin et al., 2016). The strong correlation between Type 2 diabetes, obesity and NASH provides a logical justification for therapies such as PsTag600‐FGF21 that reduce body weight and enhance insulin sensitivity. However, the therapeutic effects of PsTag600‐FGF21 on high‐fat diet (HFD) ‐induced obese (DIO) mice have only been studied in terms of reducing blood glucose and lipid levels and reversing hepatic steatosis (Yin et al., 2016). The effects on steatohepatitis, especially its inflammatory aspects, need to be assessed for PsTag600‐FGF21 to be a therapeutic candidate for NASH.
In mice, NASH‐like hepatic pathology can be induced by a choline‐deficient (CD) diet or an MCD diet (Takahashi et al., 2012). However, the CD or MCD diet does not promote insulin resistance, obesity or metabolic syndrome but rather induces weight loss and even cachexia (Wolf et al., 2014; Nakagawa, 2015). Therefore, these dietary models do not completely reproduce NASH and its consequences in humans. However, mice fed with a diet deficient in choline and with a high fat content (CD‐HFD) exhibit insulin resistance and obesity, as well as a human NASH‐like pathology (Takahashi et al., 2012; Wolf et al., 2014). We therefore used a CD‐HFD‐induced NASH model, because the metabolic characteristics of this model closely resemble those of human NASH and could therefore provide a broader measurement of a range of relevant pharmacodynamic parameters. In this study, our objective was to evaluate the effects of PsTag600‐FGF21 in NASH and to provide mechanistic insights into its underlying anti‐inflammatory effects.
Methods
Animals
All animal care and experimental protocols complied with the Laboratory Animal Management Regulations in China and the ethical committee at China Pharmaceutical University. All animals were used in accordance with the procedures approved by the College of Veterinary Medicine Yangzhou University and Use Committee (licence No. SCXK (Su) 2012–0004). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
In pharmacological studies, male C57BL/6 mice at 3 weeks of age were used. All animals were provided by the Animal Facility at the College of Veterinary Medicine, Yangzhou University. The mice were kept as follows: 4 per cage for a single‐dose pharmacodynamics study in NASH mice; 5 per cage for a 15 day pharmacological study in NASH mice. All animals were housed in a temperature‐controlled (22–24°C) environment with a relative humidity of 60–70% and a 12 h light and 12 h dark cycle. Apart from the NASH mice, all animals received water and food ad libitum. Mice were maintained on their respective diets during the treatment period. After 12 weeks of a diet period, either a standard diet (SD) (XIETONG, China) or a CD‐HFD (44.9 kcal% fat, 35.1 kcal% carbohydrates and 20.0 kcal% protein, without added choline) (D05010402, Research diets, New Brunswick, NJ, USA), the mice were randomly assigned to the treatment or vehicle group.
General procedures
We chose the CD‐HFD‐induced NASH mouse model because the metabolic characteristics of this model more closely resemble those of human NASH (Wolf et al., 2014; Chackelevicius et al., 2016) and this model provides measurements of a broader range of relevant metabolic parameters than those available for CD or MCD diet‐induced NASH.
Randomization was conducted by an individual other than the operator. The animals were selected randomly from the pool of all cages eligible for inclusion in the study and were randomly divided into groups.
The group sizes were as follows: 8 per group for a single‐dose pharmacodynamics study in NASH mice and 10 per group for a 15 days pharmacological study in NASH mice. We used the minimum possible number of mice that would achieve statistical significance.
Experimental procedures in mice
In vivo biology
In a single‐dose pharmacodynamics study, mice (n = 32) were randomly divided into four groups, which were injected i.p. with vehicle, FGF21 (1 mg·kg−1; 51 nmol·kg−1) or PsTag600‐FGF21 (0.37 and 3.7 mg·kg−1; 3.7 mg·kg−1 PsTag600‐FGF21 is equivalent to 51 nmol·kg−1 FGF21). The coding sequence of human FGF21 (NCBI accession no. AAQ89444.1) and PsTag600 (NCBI accession no KT964028) was synthesized by GenScript (Nanjing, China). Native FGF21 and PsTag600‐FGF21 were purified as described (Yin et al., 2016). Body weight was measured every day for 7 days after the single injection. The serum concentrations of FGF21 were measured with a sandwich elisa (DF2100, R&D Systems, Minneapolis, MN, USA).
In the 15 day pharmacological study, i.p. injections of vehicle, 0.37 or 3.7 mg·kg−1 PsTag600‐FGF21, were administered every 3 days for 15 days. Body weight and food and water intakes were recorded daily throughout the study. Body composition was measured on day 12 using a MesoMR23‐060H‐I imaging instrument (Shanghai Niumag Corporation, China). On day 13, plasma was collected from the retro‐orbital sinus of each mouse to measure blood glucose, glucagon, insulin, triglyceride, cholesterol, free fatty acid, adiponectin, alanine aminotransaminase (ALT) and aspartate aminotransaminase (AST) levels. An oral glucose tolerance test (OGTT) was performed on day 14 with an intragastric administration of a glucose solution (2 g·kg−1) after a 12 h fast. Tissues were collected for lipid analyses, mRNA and protein expression analyses and histological examinations.
For co‐administration of PsTag600‐FGF21 and rIL17A, mice were injected i.p. with 3.7 mg·kg−1 PsTag600‐FGF21 every 3 days and with 25 μg·kg−1 recombinant mouse IL17A (Z03031, GenScript, Nanjing, China) twice a day for 15 days. For rIL17A treatment, mice were injected i.p. with 25 μg·kg−1 recombinant mouse IL17A (Z03031, GenScript, Nanjing, China) twice a day for 15 days. To neutralize adiponectin in vivo, mice were injected i.p. (100 μg per mouse) with anti‐adiponectin (ab3455, Abcam, Cambridge, MA, USA) or control IgG once a week and 3.7 mg·kg−1 PsTag600‐FGF21 every 3 days for 15 days. Body weight and food and water intakes were recorded every day. Tissues were collected for histological and immunohistochemical analyses.
T 1 ‐weighted magnetic resonance imaging (MRI)
T 1‐weighted MRI was measured as previously described (Yin et al., 2016) and performed using the MesoMR23‐060H‐I imaging instrument (Shanghai Niumag Corporation).
Evaluation of hormones and metabolites
Plasma insulin, adiponectin, glucagon (Millipore Billerica, MA, USA) and IL17A (Abcam, Cambridge, MA, USA) levels were determined with elisa kits, and elisa was conducted following the protocol provided by the manufacturer. Plasma cholesterol, triglyceride, free fatty acids, ALT and AST were measured with an autoanalyser (Hitachi 7020; Hitachi, Tokyo, Japan). The lipids were measured with a cholesterol kit (A111–1, Jiancheng, Nanjing, China) and a triglyceride kit (A110–1, Jiancheng, Nanjing, China). Long‐chain acyl CoAs were measured using liquid chromatography and tandem mass spectrometry by the Key Laboratory of Drug Quality Control and Pharmacovigilance at China Pharmaceutical University. Blood glucose was measured at 0, 15, 30, 60, 90 and 120 min with a One Touch glucometer (Sannuo, Changsha, China). The glucose AUC during the OGTT from 0 to 120 min was calculated by the linear trapezoidal method (GraphPad Prism 5, GraphPad Software Inc., San Diego, CA, USA).
Histological and immunohistochemical analyses
Freshly harvested livers were fixed immediately in 10% (v/v) zinc‐formalin. Tissue samples were embedded in paraffin, cut to a thickness of 4 μm and stained with haematoxylin and eosin (H&E) and Masson's trichrome. For Oil‐red O staining, liver samples were frozen in liquid nitrogen and sectioned at 8 μm thickness using a cryostat. Liver samples were graded based on the NAFLD activity score (NAS) (Kleiner et al., 2005). For the immunohistochemical analysis, the liver sections were incubated with a primary anti‐CD3 (ab16669, Abcam, Cambridge, MA, USA) or anti‐F4/80 antibody (ab6640, Abcam, Cambridge, MA, USA), followed by incubation with the corresponding secondary antibody.
Gene expression analysis
Total RNA was extracted from the mouse livers, differentiated 3T3‐L1 adipocytes and naïve CD4+ T cells by using the EasyPure™ RNA Kit (TransGen, Beijing, China), and complementary DNA was synthesized using the GoTaq® 2‐Step RT qPCR System (Promega, Madison, Wisconsin, USA). Quantitative RT‐PCR was performed using SYBR Green (Applied Biosystem, Foster City, CA, USA) according to the manufacturer's instructions (StepOne Plus, Applied Biosystem). The data were analysed by using the ΔΔCt method and normalized to the internal control, β‐actin mRNA. Primers were designed and synthesized by GenScript (Nanjing, China) and are listed in Supporting Information Table S1.
Preparation of cells and flow cytometry analysis
Mononuclear cells in the liver were isolated and labelled as previously described (Gomes et al., 2016) and then stimulated with ionomycin and PMA (BD Biosciences, San Jose, CA, USA) for another 4–6 h after initial activation for 72 h. The cells were also treated with 2 μL·mL−1 Leukocyte Activation Cocktail, with GolgiPlug (BD Biosciences, San Jose, CA, USA). The cells were collected and washed after 4–6 h and then incubated with anti‐mouse CD16/CD32 (Biolegend, San Diego, CA, USA) for 30 min to block non‐specific Fc interactions. Then, the cells were stained with APC‐anti‐mouse CD4 (Biolegend, San Diego, CA, USA) for 20 min at 4°C in the dark. After washing twice with staining buffer (eBioscience, San Diego, CA, USA), the cells were fixed and permeabilized using Cytofix/Cytoperm solution (eBioscience, San Diego, CA, USA) for 20 min at 4°C in the dark, and the intracellular cytokine was stained using PE‐anti‐mouse IL17A (Biolegend, San Diego, CA, USA) for 40 min at 4°C in the dark. Then, the cells were centrifuged and washed to remove unbound antibodies. All protocols were according to the manufacturer's instructions. FACS analysis was performed on a Becton‐Dickinson FACS Calibur (BD Biosciences, San Jose, CA, USA), and data were analysed using FlowJo software (Tree Star Inc., USA).
In vitro assays
The 3T3‐L1 preadipocytes, AML12 cells and C2C12 myoblasts were obtained from the American Type Culture Collection (ATCC, Manassas, USA). Induction of preadipocyte differentiation was performed according to the method described previously (Yin et al., 2016). Briefly, the differentiation of 3T3‐L1 fibroblasts was induced 2 days later in DMEM supplemented with 10% FBS, 1 μmol·L−1 dexamethasone, 0.5 mmol·L−1 3‐isobutyl‐1‐methylxanthine and 1 μg·mL−1 insulin (48 h); the medium was then changed to DMEM/10% FBS/1 μg·mL−1 insulin (48 h).
The spleens were dissected from C57BL/6 mice and passed through sterilized 70 μm cell strainers (BD Biosciences, San Jose, CA, USA) to obtain single‐cell suspensions in IMDM containing 10% FBS (Invitrogen, Carlsbad, CA, USA). Red blood cells were lysed with RBC lysis buffer (eBioscience, San Diego, CA, USA). Naïve CD4+ T cells were purified by using anti‐CD4 magnetic beads (Miltenyi Biotech, Bergisch Gladbach, Germany).
For mouse Th17 differentiation, naïve CD4+ T cells were stimulated with plate‐bound anti‐mCD3 (5 μg·mL−1, eBioscience, San Diego, CA, USA) and soluble anti‐mCD28 (1 μg·mL−1, eBioscience, San Diego, CA, USA). The following were used to stimulate the Th17 polarized condition: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998 (10 ng·mL−1, R&D Systems, Minneapolis, MN, USA), mTGF‐β (1 ng·mL−1, R&D Systems, Minneapolis, MN, USA) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4978 (10 ng·mL−1, R&D Systems, Minneapolis, MN, USA). Recombinant mouse http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3726 (Z03148, GenScript, Nanjing, China) was absent or present in parallel wells to show its effect on Th17 cell differentiation. For intracellular cytokine detection, cells were stimulated with PMA and ionomycin for another 4–6 h after initial activation for 72 h.
Differentiated 3T3‐L1 adipocytes and naïve CD4+ T cells were cultured in IMDM supplemented with 10% FBS. Coculture of adipocytes and naïve CD4+ T cells was performed using a transwell system (Corning, Acton, MA, USA) with a 0.4 μm porous membrane to separate the upper and lower chambers. In the lower chamber, 2.5 × 105 differentiated 3T3‐L1 adipocytes were cultured, whereas 5 × 105 naïve CD4+ T cells were cultured in the upper chamber. The cells were treated with the indicated concentrations of PsTag600‐FGF21 in the presence or absence of the PPARγ antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3442 (HY‐16578, MCE, NJ, USA) or short interfering RNA (siRNAs) for the indicated times.
Immunoblotting
Western blot analyses were performed using standard methods with commercially available antibodies. Anti‐CCL2 (ab8101), anti‐TNFα (ab6671), anti‐adiponectin (ab22554), anti‐adiponectin receptor 1 (ab126611) and anti‐adiponectin receptor 2 (ab77612) antibodies were obtained from Abcam (Cambridge, MA, USA). Anti‐mouse IL‐17/IL‐17A (MAB421) and anti‐mouse klotho β (AF2619) antibodies were obtained from R&D Systems (Minneapolis, MN, USA). Anti‐β‐actin antibody was obtained from ZSGB‐Bio (Beijing, China). The corresponding secondary antibodies were obtained from Sangon Biotech (Shanghai, China). The protein concentration was determined using the Bradford Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). The images were calculated by densitometric scanning using Quantity One® (Bio‐Rad, Hercules, CA, USA). The signal intensity of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4406, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074 and IL17A was normalized to the one of β‐actin to control for unwanted variations.
RNA interference
Accell siRNAs against adiponectin (SMARTpool) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=649 (SMARTpool) and non‐targeting control siRNA were obtained from GE Dharmacon. All transfections were performed using Accell siRNA reagents following the manufacturer's instructions (GE Dharmacon). Naïve CD4+ T cells were cultured with anti‐CD3 along with anti‐mCD28 antibody for specific activation for 36 h. Then, the cells were centrifuged at 1000× g for 10 min to exchange the Th17‐polarizing medium (mIL‐6, mTGF‐β and mIL‐23) in the presence of 1 μmol·L−1 siRNA. The gene expression levels of adiponectin and Adipo1 receptor were quantified by RT‐PCR as described above.
Data and statistical analysis
The data and statistical analysis comply with the recommendations for experimental design and analysis in pharmacology (Curtis et al., 2018). All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). Data were compared by two‐way ANOVA, followed by post hoc Student–Newman–Keuls tests, and all data were expressed as the mean ± SD. Post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. A P value below 0.05 (P < 0.05) was considered significantly different.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Results
The effect of single injections of FGF21 and PsTag600‐FGF21 on body weight in NASH mice
The systemic effects of single i.p. injections of native FGF21 and PsTag600‐FGF21 on body weight were assessed in CD‐HFD‐induced NASH mice. The CD‐HFD induced a more than 16‐fold increase in serum FGF21 (Supporting Information Figure S1). Meanwhile, the single injection of PsTag600‐FGF21 (3.7 mg·kg−1) significantly increased serum FGF21 level lasting until day 5. As shown in Figure 1A, body weight was decreased by 2.3 g at 3 days after treatment with the high dose of PsTag600‐FGF21 (3.7 mg·kg−1) and lasted until day 7, whereas no loss of body weight was detected after injection of native FGF21. Treatment with the low dose of PsTag600‐FGF21 (0.37 mg·kg−1) also reduced body weight until day 3. It was worth noting that the serum half‐life of native FGF21 in mice was very short (approximately 15–30 min) (Yin et al., 2016; Kharitonenkov and Dimarchi, 2017), which could account for its lack of effect on body weight. Significant reductions in liver cholesterol and triglycerides were observed in the PsTag600‐FGF21 (3.7 mg·kg−1) treated group compared with the vehicle control at the end of the study (Supporting Information Figure S2).
Figure 1.
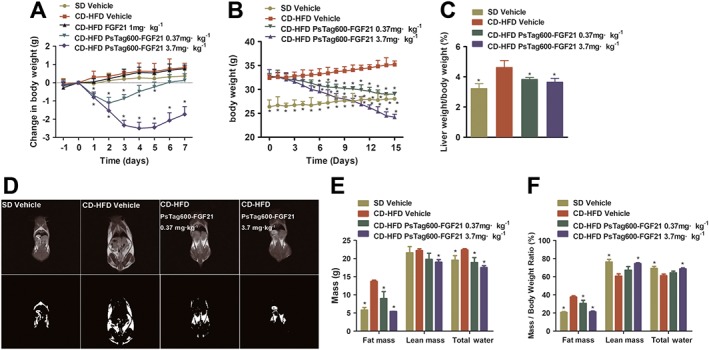
Physiological effects of PsTag600‐FGF21 in CD‐HFD‐induced NASH mice. (A) Body weight change was measured daily for 7 days. NASH mice were injected i.p. with a single dose on day 0 (n = 8 per group). (B) In a 15 day pharmacological study, body weight was monitored every day (n = 10 per group). (C) Liver weight/body weight (%) of mice after 15 days of treatment. (D) T 1‐weighted MRI images (top) and the fat highlight images (bottom) of mice were shown. (E) Body compositions were analysed. (F) Change in body composition adjusted to body weight. Data shown are means ± SD. *P < 0.05, significantly different from vehicle‐treated NASH mice).
A long‐acting human FGF21 fusion protein, PsTag600‐FGF21, attenuates the development of steatohepatitis
To evaluate the effect of PsTag600‐FGF21 on NASH, we treated CD‐HFD‐induced NASH mice for a period of 15 days with PsTag600‐FGF21 (0.37 and 3.7 mg·kg−1 i.p. every 3 days). Treatment with PsTag600‐FGF21 (0.37 and 3.7 mg·kg−1) reduced body weight by 11.0 and 26.2% at the end of 15 days of study (Figure 1B) and was associated with 17.0 and 21.2% reductions in liver‐to‐body‐weight ratios respectively (Figure 1C). PsTag600‐FGF21 also dose‐dependently reduced liver, inguinal WAT and brown adipose tissue weight compared with vehicle‐treated NASH mice (Table 1).
Table 1.
Metabolic parameters in mice fed a Standard diet (SD) or the CD‐ HFD and treated with vehicle and PsTag600‐FGF21
| Parameters | SD | CD‐HFD | ||
|---|---|---|---|---|
| Vehicle | Vehicle | PsTag600‐FGF21 0.37 mg·kg−1 | PsTag600‐FGF21 3.7 mg·kg−1 | |
| Body weight (g) | 27.6 ± 0.6a | 36.6 ± 0.9 | 29.0 ± 1.2a | 24.2 ± 1.0a |
| Liver weight (g) | 0.89 ± 0.11a | 1.69 ± 0.21 | 1.11 ± 0.08a | 0.88 ± 0.10a |
| iWAT weight (g) | 0.38 ± 0.15a | 1.68 ± 0.20 | 1.08 ± 0.21a | 0.45 ± 0.26a |
| BAT weight (g) | 0.10 ± 0.02 | 0.13 ± 0.02 | 0.10 ± 0.04 | 0.08 ± 0.02a |
| Plasma insulin (nmol·L−1) | 2.5 ± 1.1a | 13.7 ± 4.5 | 2.6 ± 0.4a | 1.3 ± 0.4a |
| Plasma fed glucose (mmol·L−1) | 6.63 ± 0.90a | 8.90 ± 0.35 | 6.53 ± 0.32a | 5.67 ± 0.75a |
| Plasma fasting glucose (mmol·L−1) | 5.20 ± 0.41a | 6.33 ± 0.75 | 3.73 ± 0.69a | 4.5 ± 0.45a |
| Plasma glucagon (pg·mL−1) | 279 ± 28 | 283 ± 43 | 223 ± 13 | 235 ± 16 |
| Plasma free fatty acid (mmol·L−1) | 1.06 ± 0.15 | 1.15 ± 0.26 | 0.72 ± 0.04a | 0.58 ± 0.10a |
n = 10 per group. Data are means ± SD. BAT, brown adipose tissue; iWAT, inguinal WAT.
P < 0.05, significantly different from vehicle‐treated NASH mice.
After 12 days of treatment, T 1‐weighted MR images were used to evaluate whole‐body fat distribution in NASH mice associated with obesity. PsTag600‐FGF21 significantly reduced signal (dark zones) in the whole‐body scan, compared with vehicle‐treated NASH mice (Figure 1D). In terms of body composition, treatment with PsTag600‐FGF21 (0.37 and 3.7 mg·kg−1) was associated with 34.6 and 60.5% reductions in body fat mass respectively (Figure 1E). Similarly, lean mass and total water in NASH mice were also significantly reduced (Figure 1E). Body weight loss mirrored reductions in body fat content (Figure 1F).
We next measured the effects of PsTag600‐FGF21 on glucose and lipid metabolism. A glucose tolerance test was performed on study day 14 after a 12 h fast (Figure 2A). PsTag600‐FGF21 protected against CD‐HFD‐induced glucose tolerance. The AUC for blood glucose after treatment with PsTag600‐FGF21 (3.7 mg·kg−1) was reduced by 35.9% (Figure 2A) compared with vehicle‐treated NASH mice. Treatment with either dose of PsTag600‐FGF21 reduced circulating levels of cholesterol (Figure 2B), triglycerides (Figure 2C), AST (Figure 2D) and ALT (Figure 2E) on day 15 of treatment in comparison with the vehicle‐treated CD‐HFD group. Hepatic cholesterol and triglycerides content after PsTag600‐FGF21 treatment were also significantly reduced (Figure 2F, G). Meanwhile, there was a profound increase in total long‐chain acyl CoA content in the livers (Figure 2H). Changes in glucose homeostasis were also detected, including reduced insulin, fed glucose, fasting glucose and free fatty acids (Table 1).
Figure 2.
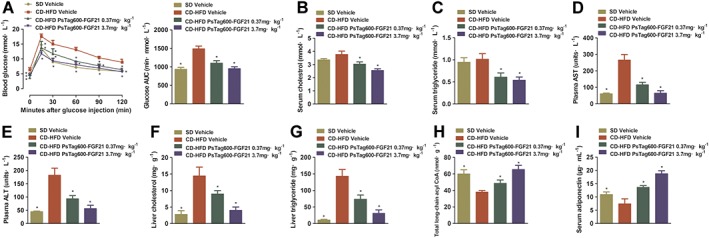
Effects on glucose tolerance, levels of hormones and metabolites. (A) For OGTT, blood glucose and the glucose AUC from 0 to 120 min were measured. (B) Serum cholesterol. (C) Serum triglyceride. (D) Plasma AST. (E) Plasma ALT. (F) Liver cholesterol. (G) Liver triglyceride. (H) Liver total long‐chain acyl CoA. (I) Serum adiponectin. Data shown are means ± SD (n = 10 per group). *P < 0.05, significantly different from vehicle‐treated NASH mice.
Histological examination of hepatic sections stained with H&E (Figure 3A) and Oil‐red‐O (Figure 3B, G) showed that there were extensive microvesicular cellular vacuoles, reflecting intrahepatic fat accumulation in CD‐HFD‐induced NASH mice. The average NAS in vehicle‐treated NASH groups was 5.2 (Figure 3F), indicating that these scores were largely considered diagnostic of NASH (Kleiner et al., 2005). In contrast, hepatocellular vacuolization and lipid droplets were seldom observed in hepatic sections from PsTag600‐FGF21‐treated NASH mice (Figure 3A, B, G). The NAS of PsTag600‐FGF21‐treated group was also significantly decreased (Figure 3F). As shown in Figure 3C, Masson's trichrome staining of liver revealed that PsTag600‐FGF21 treatment resulted in a significant reversal effect of pericellular fibrosis. We next investigated whether PsTag600‐FGF21 treatment affected the infiltration of hepatic immune cells in CD‐HFD‐induced NASH mice. Strong increases in intrahepatic immune cells comprised CD3+ T cells and F4/80+ macrophages in CD‐HFD compared with SD mice (Figure 3D, E, H, I). PsTag600‐FGF21 treatment significantly reduced CD3+ T cells and F4/80+ macrophages levels (Figure 3D, E, H, I).
Figure 3.
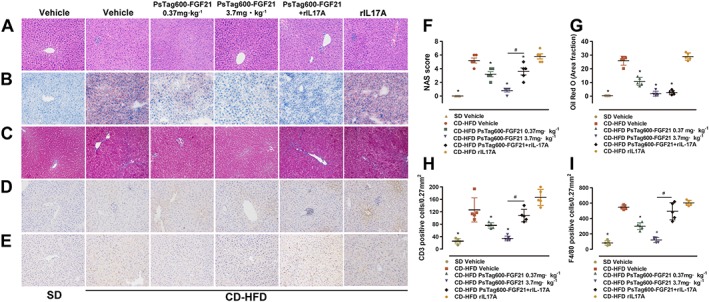
Tissue histological and immunohistochemical analyses. Representative histology and IHC on liver sections after the treatment of PsTag600‐FGF21, PsTag600‐FGF21 (3.7 mg·kg−1) + rIL17A or rIL17A. From A to E: H&E, Oil‐red‐O, Masson, CD3 and F4/80 (200× magnification). (F) NAS. (G–I) Quantification analysis of Oil‐red‐O, CD3 and F4/80 using image analytical software Image‐pro plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA). Data shown are means ± SD (n = 5 per group). *P < 0.05 significantly different from vehicle‐treated NASH mice. # P < 0.05, significantly different from PsTag600‐FGF21‐treated NASH mice.
To investigate the related gene expression by which PsTag600‐FGF21 exerted its effects on NASH, we first studied the transcription of key genes involved in fatty acids activation and mitochondrial β‐oxidation in the liver. Long‐chain fatty acids are activated to acyl CoAs by several acyl CoA synthetases (ACSLs) and several fatty acid transport proteins (FATPs) (Ellis et al., 2010; Li et al., 2010). Treatments with PsTag600‐FGF21 markedly increased the expression of ACSL1, ACSL5 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=214#1112 compared with vehicle‐treated NASH mice (Figure 4A). PsTag600‐FGF21 treatment led to a significant increase in the mRNA for PPARγ coactivator 1α to enhance mitochondrial β‐oxidation (Figure 4A). Next, in addition to its potent anti‐steatosis actions, PsTag600‐FGF21 also suppressed hepatic inflammation in NASH mice as indicated by reduced mRNA levels of several hepatic inflammatory markers (Figure 4B). Fifteen days of treatment with PsTag600‐FGF21 significantly reduced the expression of CCL2, TNFα, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974, F4/80 and CD68 (Figure 4B).The http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=50601 gene involved in fibrosis was down‐regulated after PsTag600‐FGF21 treatment (Figure 4B). Reduced CCL2 and TNFα protein levels were also observed in the livers of PsTag600‐FGF21‐treated NASH mice (Figure 4C, D).
Figure 4.

Gene analysis of liver. (A) Relative mRNA levels of key genes associated with hepatic metabolism in livers. ACSL; FATP; Pgc‐1α, PPARγ coactivator 1α. (B) Relative mRNA levels of proinflammatory genes in livers. CCL2; TNFα; IL‐6; IL‐1β; F4/80; CD68. (C, D) Western blot analysis of CCL2 and TNFα protein levels in the livers and quantitation of results of Western blot using Quantity One® (Bio‐Rad). Data shown are means ± SD (n = 10 per group). *P < 0.05, significantly different from vehicle‐treated NASH mice.
PsTag600‐FGF21 suppressed infiltration of Th17 cells and IL17A expression in NASH mice
To investigate how PsTag600‐FGF21 suppressed hepatic inflammation, we examined whether PsTag600‐FGF21 had the potential to modulate IL17A expression. Flow cytometry analysis of CD‐HFD‐induced NASH mice showed significant increase in IL17A‐expressing Th17 cells in livers (Figure 5A). The number of Th17 cells was lower in PsTag600‐FGF21‐treated NASH mice than that in the untreated NASH mice (Figure 5A). Decreased gene expressions of IL17A and the IL17A target lipocalin2 were observed after PsTag600‐FGF21 treatment (Figure 5B). Reduced levels of IL17A protein were also measured in liver and blood of PsTag600‐FGF21‐treated NASH mice (Figure 5C, D).
Figure 5.

The roles of IL17A in CD‐HFD‐induced NASH mice. (A) Flow cytometry analysis and quantification of IL17A expression on CD4+ T cells in the livers. (B) Relative mRNA levels of IL17A and Lcn2 in livers. (C) Western blot analysis of IL17A protein levels in the livers and quantitation of results of Western blot using Quantity One® (Bio‐Rad). (D) Serum IL17A was measured in CD‐HFD‐induced NASH mice with/without PsTag600‐FGF21. Data shown are means ± SD (n = 10 per group). *P < 0.05, significantly different from vehicle‐treated NASH mice.
Recombinant IL17A reversed the anti‐inflammatory effect of PsTag600‐FGF21 in NASH mice
We next tested whether recombinant IL17A (rIL17A) injection in CD‐HFD‐induced NASH mice would reverse the anti‐inflammatory effect of PsTag600‐FGF21. One interesting finding was that co‐administration of PsTag600‐FGF21 + rIL17A tended to further reduce body weight, compared with the single administration of PsTag600‐FGF21 (Figure 6A). However, liver‐to‐body‐weight ratios were not significantly different between the PsTag600‐FGF21 + rIL17A group and PsTag600‐FGF21 group (Figure 6B). Glucose tolerance tests revealed that rIL17A had no effect on glucose tolerance (Figure 6C). Moreover, mice treated with PsTag600‐FGF21 alone showed an increase in mRNA levels of key genes associated with hepatic metabolism, which were not altered in mice receiving co‐administration with PsTag600‐FGF21 + rIL17A (Supporting Information Figure S3). As shown in Figure 3C–3E, 3H, 3I, co‐administration of PsTag600‐FGF21 + rIL17A failed to reduce liver inflammation and fibrosis, in contrast to mice treated with PsTag600‐FGF21 alone. Compared to vehicle‐treated NASH mice, both combined administration of PsTag600‐FGF21 + rIL17A and single administration of PsTag600‐FGF21 significantly reduced liver vacuolization and intrahepatic lipid without marked difference in efficacy (Figure 3A, B, G). However, NASH mice treated with rIL17A alone had relatively exacerbating NASH, which was not significantly different from those of vehicle‐treated NASH mice (Figure 3). Thus, we suggest that the anti‐inflammatory effects of PsTag600‐FGF21 were dependent on decreases in IL17A in NASH mice.
Figure 6.
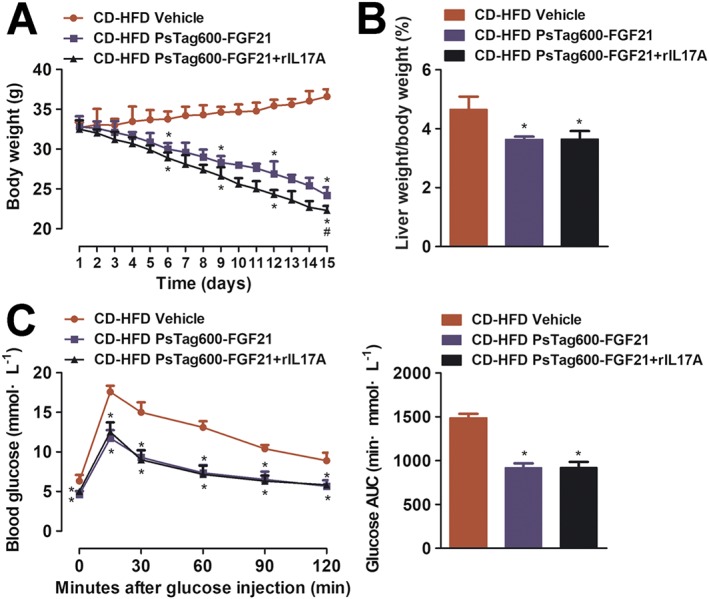
The effect of recombinant IL17A on body weight, liver weight and glucose tolerance in NASH mice. (A) In a 15 day pharmacological study, change in body weight was monitored every day. NASH mice were injected i.p. with 3.7 mg·kg−1 PsTag600‐FGF21 every 3 days and 25 μg·kg−1 rIL17A twice a day. (B) Liver weight/body weight (%) of mice after 15 days of treatment. (C) For OGTT, blood glucose and the glucose AUC from 0 to 120 min were measured. Data shown are means ± SD (n = 10 per group). *P < 0.05, significantly different from vehicle‐treated NASH mice; # P < 0.05, significantly different from PsTag600‐FGF21‐treated NASH mice.
PsTag600‐FGF21 indirectly suppressed Th17 cell differentiation and IL17A expression
The effects of PsTag600‐FGF21 on down‐regulation Th17‐IL17A axis in vivo raised the question of the mechanism(s) underlying these effects. We first investigated the influence of PsTag600‐FGF21 on Th17 cell differentiation in vitro. However, PsTag600‐FGF21 which was present during differentiation had no effect on IL17A production in Th17 cells (Figure 7A). We next tested whether a β‐klotho exists in naïve CD4+ T cells. As shown in Figure 7B, Western blot analysis demonstrated that a β‐klotho protein was expressed in differentiated 3T3‐L1 adipocytes, but not in naïve CD4+ T cells. These results indicated that the effects of PsTag600‐FGF21 on Th17 cell differentiation were indirect.
Figure 7.
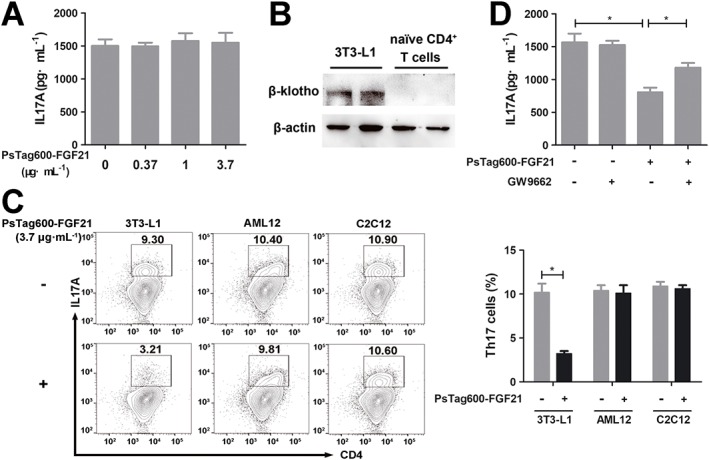
PsTag600‐FGF21 indirectly suppressed Th17 cell differentiation and IL17A expression. (A) Naïve CD4+ T cells were treated with indicated concentrations of PsTag600‐FGF21 during differentiation to Th17 cells. The supernatant IL17A levels were assayed after differentiation. (B) Naïve CD4+ T cells were analysed for the expression of FGF21 receptor β‐klotho. Differentiated 3T3‐L1 adipocytes were used as a positive control. (C) Coculture of naïve CD4+ T cells and differentiated 3T3‐L1 adipocytes /AML12 cells/C2C12 myoblasts with/without PsTag600‐FGF21 (3.7 μg·mL−1) during differentiation to Th17 cells. Flow cytometry analysis and quantification of IL17A expression on CD4+ T cells after differentiation. (D) Co‐culture of differentiated 3T3‐L1 adipocytes and naïve CD4+ T cells were pretreated with GW9662 (10.0 μmol·L−1) for 1 h, followed by incubation with PsTag600‐FGF21 (3.7 μg·mL−1) during differentiation to Th17 cells. The supernatant IL17A levels were assayed after differentiation. Data shown are means ± SD (n = 6 per group). *P < 0.05, significantly different as indicated.
Data presented in earlier research suggested that the major functions of FGF21 were mediated through β‐klotho/http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1808&familyId=323&familyType=CATALYTICRECEPTOR pathway (Kharitonenkov and Dimarchi, 2015; Fisher and Maratos‐Flier, 2016). The receptor FGFR1c was expressed broadly, but β‐klotho expression was more restricted to tissues such as the liver, pancreas, adipose tissue, skeletal muscle and brain (Gimeno and Moller, 2014; Zhang and Li, 2014). Thus, we investigated whether PsTag600‐FGF21 regulated the production of IL17A by Th17 cells through direct effects on FGF21 target tissues. Naïve CD4+ T cells were cocultured with differentiated 3T3‐L1 adipocytes, AML12 cells and C2C12 myoblasts respectively. As shown in Figure 7C, a significant decrease in Th17 cells only resulted from differentiated 3T3‐L1 adipocytes that were PsTag600‐FGF21 treated. Meanwhile, differentiated 3T3‐L1 adipocytes without PsTag600‐FGF21 had no influence on Th17 cell differentiation (Supporting Information Figure S4). As native FGF21 had been shown to activate http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595 in adipocytes, we next investigated the role of this transcription factor in PsTag600‐FGF21‐suppressd Th17 cell differentiation (Dutchak et al., 2012; Lin et al., 2013). Pretreatment of differentiated 3T3‐L1 adipocytes with the PPARγ antagonist GW9662 partly blocked the effects of PsTag600‐FGF21 in Th17 cell differentiation and IL17A expression (Figure 7D). Thus, these results revealed that adipocytes and PPARγ were involved in the indirect effects of PsTag600‐FGF21 on Th17 cell differentiation.
PsTag600‐FGF21‐treated adipocytes suppressed Th17 cell differentiation via adiponectin
Administration of PsTag600‐FGF21 (0.37 and 3.7 mg·kg−1) resulted in a marked increase in plasma levels of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3726 in NASH mice (Figure 2I). Serum adiponectin was significantly increased at 3 days after treatment with the high dose of PsTag600‐FGF21 (3.7 mg·kg−1) (Supporting Information Figure S5). Meanwhile, adiponectin production in differentiated 3T3‐L1 adipocytes was induced by PsTag600‐FGF21 treatment (Figure 8A, B), consistent with previous research (Holland et al., 2013). Furthermore, previous studies found that exogenous adiponectin inhibited Th17 cell differentiation from naïve CD4+ T cells (Shibata et al., 2015; Zhang et al., 2016). These results prompted us to examine whether adiponectin released from differentiated 3T3‐L1 adipocytes was elevated by PsTag600‐FGF21 treatment and suppressed Th17 cell differentiation. First of all, we examined the in vitro effects of adiponectin on Th17 cell differentiation. As shown in Figure 8C, IL17A production in Th17 cells were significantly reduced by exposure to adiponectin. Next, the expression of adiponectin receptors, Adipo1 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=650&familyId=106&familyType=OTHER, were assessed in naïve CD4+ T cells. The results of Western blots showed that only Adipo1 receptors were expressed in naïve CD4+ T cells (Figure 8D). Furthermore, to determine whether adiponectin regulated IL17A expression through Adipo1 receptors, naïve CD4+ T cells were transfected with siRNA against this receptor, leading to a reduction of about 74% in its mRNA (Figure 8E). siRNA against Adipo1 receptors also down‐regulated protein levels of these receptors in naïve CD4+ T cells (Figure 8E). Specific knockdown of AdipoR1 attenuated suppression of adiponectin‐induced IL17A expression in Th17 cells (Figure 8F). Finally, to clarify whether PsTag600‐FGF21‐treated adipocytes can modulate Th17 cell differentiation through an adiponectin‐dependent pathway, differentiated 3T3‐L1 adipocytes were transfected with siRNA against adiponectin and naïve CD4+ T cells were transfected with siRNA against Adipo1 receptors respectively. Control experiments had showed that siRNA against adiponectin could regulate mRNA and protein levels of adiponectin in adipocytes (Figure 8G). As shown in Figure 8H and Supporting Information Figure S6, IL17A expression was significantly increased when adiponectin in adipocytes or Adipo1 receptors in naïve CD4+ T cells were specifically knocked‐down.
Figure 8.
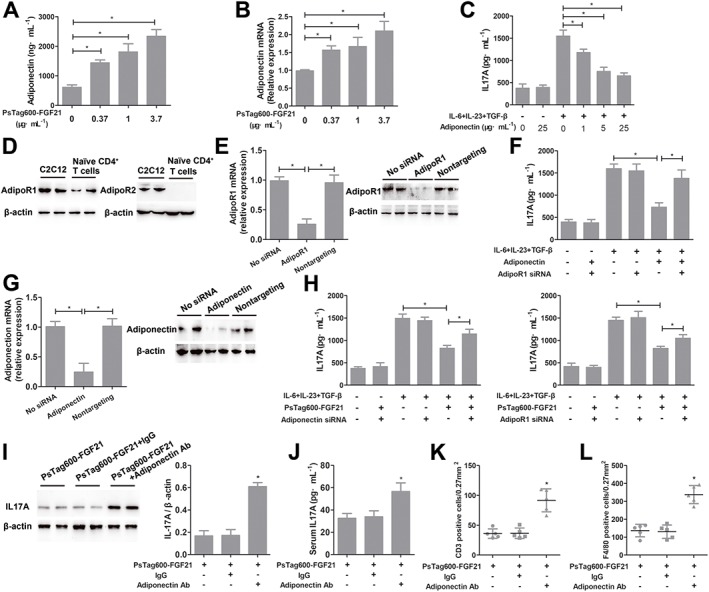
PsTag600‐FGF21 suppressed Th17 cell differentiation via adiponectin (n = 5 per group). (A, B) Differentiated 3T3‐L1 adipocytes were incubated with indicated concentrations of PsTag600‐FGF21 for 24 h. Adiponectin concentration in the conditioned medium and relative adiponectin mRNA abundance were analysed. *P < 0.05, significantly different as indicated. (C) Naïve CD4+ T cells were treated with indicated concentrations of adiponectin during differentiation to Th17 cells. The levels of IL17A in the supernatants were assayed after differentiation. *P < 0.05, significantly different as indicated. (D) Naïve CD4+ T cells were analysed for the expression of the adiponectin receptors, Adipo1 and Adipo2. C2C12 muscle cells were used as a positive control. (E) Naïve CD4+ T cells were transfected with Adipo1 receptor siRNA or non‐targeting control siRNA. Relative abundance of Adipo1 receptor mRNA and protein levels were analysed. *P < 0.05, significantly different as indicated. (F) Naïve CD4+ T cells were transfected with Adipo1 receptor siRNA and were treated with adiponectin (5 μg·mL−1) during differentiation to Th17 cells. The levels of IL17A in the supernatants were assayed after differentiation. *P < 0.05, significantly different as indicated. (G) Differentiated 3T3‐L1 adipocytes were transfected with adiponectin siRNA or nontargeting control siRNA. Relative adiponectin mRNA abundance and protein levels were analysed. *P < 0.05, significantly different as indicated. (H) Differentiated 3T3‐L1 adipocytes were transfected with siRNA against adiponectin, or naïve CD4+ T cells were transfected with siRNA against Adipo1 receptors respectively. Differentiated 3T3‐L1 adipocytes and naïve CD4+ T cells were cocultured with PsTag600‐FGF21 (3.7 μg·mL−1) during differentiation to Th17 cells. The levels of IL17A in the supernatants were assayed after differentiation. *P < 0.05, significantly different as indicated. (I) Western blot analysis of IL17A protein levels in the livers was measured in PsTag600‐FGF21‐treated NASH mice with/without an anti‐adiponectin antibody. Quantitation of results of Western blot using Quantity One® (Bio‐Rad) *P < 0.05, significantly different from PsTag600‐FGF21‐treated NASH mice. (J) Serum IL17A in mice described in (I) (*P < 0.05 vs. PsTag600‐FGF21‐treated NASH mice). (K, L) Quantification of CD3 and F4/80 from Supporting Information Figure S7. Data shown are means ± SD. *P < 0.05, significantly different from PsTag600‐FGF21‐treated NASH mice.
We further blocked adiponectin signalling in PsTag600‐FGF21‐treated NASH mice using an antibody against adiponectin. Blocking adiponectin raised the levels of IL17A protein in liver and blood of PsTag600‐FGF21‐treated NASH mice (Figure 8I, J). This also significantly increased intrahepatic CD3+ T cells and F4/80+ macrophages in PsTag600‐FGF21‐treated NASH mice, compared with the single administration of PsTag600‐FGF21 (Figure 8K, L and Supporting Information Figure S7). Taken together, these data suggested that PsTag600‐FGF21 depended on adiponectin to suppress Th17 cell differentiation and IL17A expression.
Discussion
Therapeutic development of native FGF21 has presented problems, such as its inherently short plasma half‐life and poor drug‐like properties (Owen et al., 2014; Kharitonenkov and Dimarchi, 2015). The recombinant PsTag polypeptide technology showed potential for extending t1/2 that could replace the traditional chemical polymer, PEG, in order to design biological drugs with tailored pharmacological properties to improve studies of, and therapies for, various diseases (Podust et al., 2015; Yin et al., 2016). This recombinant PsTag polypeptide consists of five amino acids (Pro, Ser, Thr, Ala and Gly), is an uncharged, hydrophilic and unstructured biological polymer and has been shown to be safe and poorly immunogenic (Yin et al., 2016). In the present study, we explored the efficacy of a new, long‐acting FGF21 attached to a PsTag600 polypeptide, in a CD‐HFD‐induced NASH mouse model and revealed some of the mechanisms responsible for this activity. Our results suggested that PsTag600‐FGF21 may be a treatment option for patients with NASH.
Previous studies had already demonstrated that treatment with FGF21 reduced circulating glucose and lipid levels, leading to body weight loss in obese animals (Gimeno and Moller, 2014; Fisher and Maratos‐Flier, 2016). However, these studies did not assess the effects of FGF21 on hepatic inflammation and fibrosis. Only two experimental studies of native FGF21 or FGF21 analogues had implicated FGF21 in the treatment of MCD diet‐induced NASH (Fisher et al., 2014; Ju et al., 2016). The MCD diet‐fed mouse model has a number of disadvantages, including decreased bodyweight, serum triglycerides and glucose levels (Kubota et al., 2013). By using CD‐HFD to induce a type of NASH associated with obesity and a human NASH‐like pathology, we could focus on the effects of long‐acting FGF21 on weight loss, as PsTag600‐FGF21 treatment potently induced weight loss in DIO mice (Yin et al., 2016). In the present study, chronic administration of PsTag600‐FGF21 also reduced the body weight and improved the insulin sensitivity, as expected. Therefore, we suggest that PsTag600‐FGF21 may be useful in the treatment of NASH associated with obesity.
Inflammation is one of the many defining pathological characteristics of NASH (Wolf et al., 2014; Heymann and Tacke, 2016; Wang et al., 2017). Although disordered lipid metabolism and fat accumulation in the liver are the common characteristics of both simple steatosis and NASH, steatosis might not be required for the progression of NASH in some cases (Gomes et al., 2016; Liu et al., 2016a). Many studies have shown that inflammation aggravated the metabolic disorders of NASH and suggested that inflammation determined the long‐term prognosis of NASH (Yilmaz, 2012; Gomes et al., 2016). Therefore, therapeutic drugs for NASH must also reverse the inflammation in the liver. The mechanism by which FGF21 regulated hepatic metabolism had been relatively clear (Fisher et al., 2014; Fisher and Maratos‐Flier, 2016). PsTag600‐FGF21 enhanced hepatocyte fatty acid oxidation by up‐regulating ACSL/FATP expression and promoting mitochondrial β‐oxidation of fatty acids, consistent with the role of native FGF21 in hepatic metabolism. Thus, we must also study the anti‐inflammatory effect of PsTag600‐FGF21 resulting in the reversal of NASH.
Inflammation in NASH is frequently triggered by mobilized Th17 cells and increased IL17A levels (Harley et al., 2014; Gomes et al., 2016). In addition, recombinant IL17A injection in chow‐fed C57BL/6 mice mimicked NASH (Gomes et al., 2016). The most interesting finding of the present study was that the anti‐inflammatory activity of PsTag600‐FGF21 in NASH was related to its down‐regulation of IL17A production. Adiponectin receptors were known to be expressed on T cells and adiponectin could directly inhibit Th17 cells via the SIRT1/PPARγ/http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=600t pathway (Shibata et al., 2015; Zhang et al., 2016). Additionally, adiponectin and FGF21 regulated glucose and lipid metabolism in similar ways and were functionally linked (Kharitonenkov and Adams, 2013). Several recent studies demonstrated that FGF21 was a positive regulator of adiponectin and that FGF21 depended on adiponectin to exert a systemic effect on energy metabolism and insulin sensitivity (Holland et al., 2013; Lin et al., 2013). The known action of FGF21 as a potent regulator of adiponectin secretion hinted that PsTag600‐FGF21 may exhibit anti‐inflammatory activity attributable to adiponectin. Our data revealed that PsTag600‐FGF21 depended on adiponectin to suppress Th17 cell differentiation and IL17A expression. There is good evidence for Th17 cells and its effector cytokine IL17A playing major roles in inflammation and autoimmunity (Bettelli et al., 2008; Korn et al., 2009). Modulating Th17 cell‐mediated inflammatory response may be one of the mechanisms for FGF21 benefiting autoimmune diseases. PsTag600‐FGF21, a long‐acting FGF21, was implicated in the control of the expression and release of many proteins and lipids from adipose tissue, not just adiponectin. Further studies directed at identifying these proteins and lipids could not only offer new insights into the anti‐inflammatory actions of PsTag600‐FGF21 but also unveil novel actions of PsTag600‐FGF21.
Our study showed that the therapeutic effects of PsTag600‐FGF21 on NASH were to reverse hepatic steatosis and fibrosis and reduce inflammation. Furthermore, PsTag600‐FGF21 down‐regulated IL17A production through the FGF21‐adiponectin‐IL17A axis, which could be a mechanism by which PsTag600‐FGF21 exerts anti‐inflammatory effects in the NASH model. Taken together, the results showed that PsTag600‐FGF21 exerted a number of pharmacological effects in NASH mice and could therefore be an effective drug candidate.
Author contributions
J.Y., L.B., X.G. and W.B. designed the research study; L.B. and J.Y. performed most of the experiments and analysed the data; W.G. and Q.W. performed some part of the research; L.B. and J.Y. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The circulating levels of FGF21 were measured every day for 7 days. Mice were injected intraperitoneally with a single dose on day 0 (n = 6/group). (Data are means ±SD. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S2 Liver cholesterol and triglycerides were detected after 7 days (n = 6/group). (Data are means ±SD. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S3 Relative mRNA levels of key genes associated with hepatic metabolism in livers. (n = 6/group). (Data are means ±SD. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S4 Coculture of naïve CD4+ T cells and differentiated 3 T3‐L1 adipocytes during differentiation to Th17 cells. Flow cytometry analysis and quantification of IL17A expression on CD4+ T cells after differentiation. (Data are means ±SD).
Figure S5 Effects of chronic administration with PsTag600‐FGF21 (3.7 mg·kg−1) on serum levels of adiponectin. (Data are means ±SD; n = 10/group. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S6 Differentiated 3 T3‐L1 adipocytes were transfected with nontargeting siRNA or naïve CD4+ T cells were transfected with nontargeting siRNA respectively. Differentiated 3 T3‐L1 adipocytes and naïve CD4+ T cells were cocultured with PsTag600‐FGF21 (3.7 μg/mL) during differentiation to Th17 cells. The supernatant IL17A levels were detected after differentiation. (Data are means ±SD; n = 6/group. *P < 0.05).
Figure S7 Tissue immunohistochemical analyses. Representative IHC on liver sections after the treatment of PsTag600‐FGF21, PsTag600‐FGF21 + control IgG, PsTag600‐FGF21 + Adiponectin Ab in the CD‐HFD induced NASH mice (200 × magnification). (n = 5/group).
Table S1 PCR Primer Sequences.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (81430082, 81703402 and 81473216), the National Science and Technology Major Project (2018ZX09201001‐003‐002), the project funded by China Postdoctoral Science Foundation (2017M611957), the Fundamental Research Funds for the Central Universities (2015XPT02), a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the ‘111 Project’ from the Ministry of Education of China and the State Administration of Foreign Expert Affairs of China (111‐2‐07).
Bao, L. , Yin, J. , Gao, W. , Wang, Q. , Yao, W. , and Gao, X. (2018) A long‐acting FGF21 alleviates hepatic steatosis and inflammation in a mouse model of non‐alcoholic steatohepatitis partly through an FGF21‐adiponectin‐IL17A pathway. British Journal of Pharmacology, 175: 3379–3393. 10.1111/bph.14383.
Contributor Information
Wenbing Yao, Email: wbyao@cpu.edu.cn.
Xiangdong Gao, Email: xdgao@cpu.edu.cn.
References
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiardes S, Shapiro H, Rozin S, Shibolet O, Elinav E (2016). Non‐alcoholic fatty liver and the gut microbiota. Mol Metab 5: 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Oukka M, Kuchroo VK (2008). Induction and effector functions of T (H)17 cells. Nature 453: 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy S, Syed BA (2016). Nonalcoholic steatohepatitis (NASH) drugs market. Nat Rev Drug Discov 15: 745–746. [DOI] [PubMed] [Google Scholar]
- Chackelevicius CM, Gambaro SE, Tiribelli C, Rosso N (2016). Th17 involvement in nonalcoholic fatty liver disease progression to non‐alcoholic steatohepatitis. World J Gastroenterol 22: 9096–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MZ, Chang JC, Zavala‐Solorio J, Kates L, Thai M, Ogasawara A et al (2017). FGF21 mimetic antibody stimulates UCP1‐independent brown fat thermogenesis via FGFR1/βKlotho complex in non‐adipocytes. Mol Metab 6: 1454–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ et al (2012). Fibroblast growth factor‐21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 148: 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JM, Frahm JL, Li LO, Coleman RA (2010). Acyl‐coenzyme A synthetases in metabolic control. Curr Opin Lipidol 21: 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T et al (2014). Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline‐deficient diets. Gastroenterology 147: 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Maratos‐Flier E (2016). Understanding the physiology of FGF21. Annu Rev Physiol 78: 223–241. [DOI] [PubMed] [Google Scholar]
- Gimeno RE, Moller DE (2014). FGF21‐based pharmacotherapy–potential utility for metabolic disorders. Trends Endocrinol Metab 25: 303–311. [DOI] [PubMed] [Google Scholar]
- Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A et al (2016). Metabolic inflammation‐associated IL‐17A causes non‐alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30: 161–175. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley ITW, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M et al (2014). IL‐17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 59: 1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann F, Tacke F (2016). Immunology in the liver – from homeostasis to disease. Nat Rev Gastroenterol Hepatol 13: 88–110. [DOI] [PubMed] [Google Scholar]
- Holland W, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM et al (2013). An FGF21‐adiponectin‐ceramide axis controls energy expenditure and insulin action in mice. Cell Metab 17: 790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T (2015). Research perspectives on the regulation and physiological functions of FGF21 and its association with NAFLD. Front Endocrinol 6: 1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju HL, Kang YE, Chang JY, Park KC, Kim HW, Kim JT et al (2016). An engineered FGF21 variant, LY2405319, can prevent non‐alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res 8: 4750–4763. [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, Adams AC (2013). Inventing new medicines: the FGF21 story. Mol Metab 3: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, Dimarchi R (2017). Fibroblast growth factor 21 night watch: advances and uncertainties in the field. J Intern Med 281: 233–246. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Dimarchi RD (2015). FGF21 revolutions: recent advances illuminating FGF21 biology and medicinal properties. Trends Endocrinol Metab 26: 608–617. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med 12: 561–563. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta ML, Behling C, Contos MJ, Cummings OW et al (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK (2009). IL‐17 and Th17 Cells. Annu Rev Immunol 27: 485–517. [DOI] [PubMed] [Google Scholar]
- Kubota N, Kado S, Kano M, Masuoka N, Nagata Y, Kobayashi T et al (2013). A high‐fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non‐alcoholic steatohepatitis in mice. Clin Exp Pharmacol Physiol 40: 422–430. [DOI] [PubMed] [Google Scholar]
- Li LO, Klett EL, Coleman RA (2010). Acyl‐CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta 1801: 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Tian H, Lam KSL, Lin S, Hoo RL, Konishi M et al (2013). Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab 17: 779–789. [DOI] [PubMed] [Google Scholar]
- Liu J, Xu Y, Hu Y, Wang G (2015). The role of fibroblast growth factor 21 in the pathogenesis of non‐alcoholic fatty liver disease and implications for therapy. Metab Clin Exp 64: 380–390. [DOI] [PubMed] [Google Scholar]
- Liu W, Baker RD, Bhatia T, Zhu L, Baker SS (2016a). Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci Cmls 73: 1969–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Huang S, Shi K, Zhao C, Chen L, Braddock M et al (2014). The role of fibroblast growth factor 21 in the pathogenesis of liver disease: a novel predictor and therapeutic target. Expert Opin Ther Targets 18: 1305–1313. [DOI] [PubMed] [Google Scholar]
- Liu W, Struik D, Nies VJM, Jurdzinski A, Harkema L, De Bruin A et al (2016b). Effective treatment of steatosis and steatohepatitis by fibroblast growth factor 1 in mouse models of nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 113: 2288–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Kesarwala AH, Eggert T, Medinaecheverz J, Kleiner DE, Jin P et al (2016). NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531 (7593): 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado MV, Diehl AM (2016). Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150: 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti GA, Machado MV, Diehl AM (2013). NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 10: 656–665. [DOI] [PubMed] [Google Scholar]
- Musso G, Cassader M, Gambino R (2016). Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 15: 249–274. [DOI] [PubMed] [Google Scholar]
- Nakagawa H (2015). Recent advances in mouse models of obesity‐ and nonalcoholic steatohepatitis‐associated hepatocarcinogenesis. World J Hepatol 7: 2110–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Xunshan D, Morgan DA, Katie Colbert C, Bookout AL, Kamal R et al (2014). FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab 20: 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podust VN, Balan S, Sim BC, Coyle MP, Ernst U, Peters RT et al (2015). Extension of in vivo half‐life of biologically active molecules by XTEN protein polymers. J Control Release 240: 52–66. [DOI] [PubMed] [Google Scholar]
- Reitman ML (2013). FGF21 mimetic shows therapeutic promise. Cell Metab 18: 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Tada Y, Hau CS, Mitsui A, Kamata M, Asano Y et al (2015). Adiponectin regulates psoriasiform skin inflammation by suppressing IL‐17 production from γδ‐T cells. Nat Commun 6: 7687. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Soejima Y, Fukusato T (2012). Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 18: 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V et al (2016). A long‐acting FGF21 molecule, PF‐05231023, decreases body weight and improves lipid profile in non‐human primates and type 2 diabetic subjects. Cell Metab 23: 427–440. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Matsubara T, Krausz KW, Patterson AD, Gonzalez FJ (2012). Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 56: 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yun G, Song J, Chao T, Man W, Que L et al (2017). The TIR/BB‐loop mimetic AS‐1 prevents non‐alcoholic steatohepatitis and hepatic insulin resistance by inhibiting NLRP3‐ASC inflammasome activation. Br J Pharmacol 174: 1841–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K et al (2014). Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross‐talk with hepatocytes. Cancer Cell 26: 549–564. [DOI] [PubMed] [Google Scholar]
- Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE (2013). From NAFLD to NASH to cirrhosis‐new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 10: 627–636. [DOI] [PubMed] [Google Scholar]
- Yilmaz Y (2012). Review article: is non‐alcoholic fatty liver disease a spectrum, or are steatosis and non‐alcoholic steatohepatitis distinct conditions? Aliment Pharmacol Ther 36: 815–823. [DOI] [PubMed] [Google Scholar]
- Yin J, Bao L, Hong T, Wang Q, Gao X, Yao W (2016). Genetic fusion of human FGF21 to a synthetic polypeptide improves pharmacokinetics and pharmacodynamics in a mouse model of obesity. Br J Pharmacol 173: 2208–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li Y (2014). Fibroblast growth factor 21, the endocrine FGF pathway and novel treatments for metabolic syndrome. Drug Discov Today 19: 579–589. [DOI] [PubMed] [Google Scholar]
- Zhang K, Guo Y, Ge Z, Zhang Z, Da Y, Li W et al (2016). Adiponectin suppresses T Helper 17 cell differentiation and limits autoimmune CNS inflammation via the SIRT1/PPARγ/RORγt pathway. Mol Neurobiol 54: 4908–4920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The circulating levels of FGF21 were measured every day for 7 days. Mice were injected intraperitoneally with a single dose on day 0 (n = 6/group). (Data are means ±SD. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S2 Liver cholesterol and triglycerides were detected after 7 days (n = 6/group). (Data are means ±SD. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S3 Relative mRNA levels of key genes associated with hepatic metabolism in livers. (n = 6/group). (Data are means ±SD. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S4 Coculture of naïve CD4+ T cells and differentiated 3 T3‐L1 adipocytes during differentiation to Th17 cells. Flow cytometry analysis and quantification of IL17A expression on CD4+ T cells after differentiation. (Data are means ±SD).
Figure S5 Effects of chronic administration with PsTag600‐FGF21 (3.7 mg·kg−1) on serum levels of adiponectin. (Data are means ±SD; n = 10/group. *P < 0.05 vs vehicle‐treated NASH mice).
Figure S6 Differentiated 3 T3‐L1 adipocytes were transfected with nontargeting siRNA or naïve CD4+ T cells were transfected with nontargeting siRNA respectively. Differentiated 3 T3‐L1 adipocytes and naïve CD4+ T cells were cocultured with PsTag600‐FGF21 (3.7 μg/mL) during differentiation to Th17 cells. The supernatant IL17A levels were detected after differentiation. (Data are means ±SD; n = 6/group. *P < 0.05).
Figure S7 Tissue immunohistochemical analyses. Representative IHC on liver sections after the treatment of PsTag600‐FGF21, PsTag600‐FGF21 + control IgG, PsTag600‐FGF21 + Adiponectin Ab in the CD‐HFD induced NASH mice (200 × magnification). (n = 5/group).
Table S1 PCR Primer Sequences.