

Abstract
The PTEN gene encodes a master regulator protein that exerts essential functions both in the cytoplasm and in the nucleus. PTEN is mutated in the germline of both patients with heterogeneous tumor syndromic diseases, categorized as PTEN hamartoma tumor syndrome (PHTS), and a group affected with autism spectrum disorders (ASD). Previous studies have unveiled the functional heterogeneity of PTEN variants found in both patient cohorts, making functional studies necessary to provide mechanistic insights related to their pathogenicity. Here, we have functionally characterized a PTEN missense variant [c.49C>G; p.(Gln17Glu); Q17E] associated to both PHTS and ASD patients. The PTEN Q17E variant displayed partially reduced PIP3-catalytic activity and normal stability in cells, as shown using S. cerevisiae and mammalian cell experimental models. Remarkably, PTEN Q17E accumulated in the nucleus, in a process involving the PTEN N-terminal nuclear localization sequence. The analysis of additional germline-associated PTEN N-terminal variants illustrated the existence of a PTEN N-terminal region whose targeting in disease causes PTEN nuclear accumulation, in parallel with defects in PIP3-catalytic activity in cells. Our findings highlight the frequent occurrence of PTEN gene mutations targeting PTEN N-terminus whose pathogenicity may be related, at least in part, with the retention of PTEN in the nucleus. This could be important for the implementation of precision therapies for patients with alterations in the PTEN pathway.
Introduction
The PTEN gene encodes a phosphatase whose major activity is the dephosphorylation of phosphatidylinositol 3,4,5-trisphosphate (PIP3) to render phosphatidylinositol 4,5-bisphosphate (PIP2). This enzymatic activity counteracts the action of class I phosphatidylinositol 3-kinases (PI3K), thus negatively regulating the PI3K/AKT/mTOR pro-survival signaling pathway [1–3]. PTEN also dephosphorylates a wide array of proteins, some of which are related with the regulation of cell motility and migration [4]. In addition, PTEN possesses catalysis-independent activities, mainly mediated by protein–protein interactions [5]. Both, catalysis-dependent and -independent activities are associated with specific PTEN subcellular compartmentation. In particular, cytoplasmic localization and transient binding of PTEN to membranes is related with the dephosphorylation of PIP3, whereas PTEN nuclear localization is mostly associated with transcriptional regulation and DNA damage control mediated by protein–protein interactions [6–9]. The regulated intracellular shuttling of PTEN involves several molecular determinants in the PTEN protein, which include, among others, a cluster of phosphorylatable amino acids at PTEN C-terminal tail and a set of overlapping subcellular localization motifs at PTEN N terminus (NLMs, N-terminal localization motifs) [10–12]. Conformational changes in the PTEN protein that expose these motifs to targeting or anchoring moieties regulate PTEN catalytic activity and subcellular distribution in cells [8, 13–16].
The PTEN gene is the second tumor suppressor, after TP53, in terms of somatic mutation frequency in human tumors. PTEN is also mutated in the germline of patients with syndromic predisposition to several types of cancer, grouped as PHTS (PTEN hamartoma tumor syndrome) [17]. In addition, the PTEN gene is mutated in up to 15% of autism spectrum disroders (ASD) patients with macrocephaly, and other genes directly related with the PI3K/AKT/mTOR pathway, including the PIK3CA, PIK3R1, AKT1, and AKT3 protooncogenes, have also been found mutated in the germline of patients with tumor predisposition or with megalencephaly-related disorders [18–22]. This is in line with an important contribution for the effectors of the PI3K/AKT/mTOR pathway not only in tissue oncogenesis, but also in brain growth and development [23, 24]. In ASD patients, PTEN variants generated by missense mutations are prevalent, and although their functional impact in terms of PTEN catalytic activity is variable, many of these variants do not show severe intrinsic impaired catalytic activity but display diminished protein stability [25–27]. Intriguingly, several missense germline mutations at PTEN have been found on both PHTS and ASD patients, indicating a heterogeneous penetrance for these mutations, which could be due in part to PTEN functional alterations which go beyond its catalytic activity. In this study, we have functionally characterized a PTEN variant found on both PHTS and ASD patients [c.49C>G; p.(Gln17Glu); Q17E]. The PTEN Q17E variant retained most of PTEN stability and catalytic activity, but displayed abnormal subcellular localization, being enriched in the cell nucleus. Further analysis unveiled that disease-associated variants displaying amino acid substitutions at PTEN N-terminus frequently cause both PTEN nuclear accumulation and defects in PTEN catalytic activity in cells. Our findings suggest that misregulated PTEN nuclear accumulation could be related, at least in part, with the pathogenicity of a group of disease-associated PTEN variants.
Materials and methods
Cell culture, transfections, plasmids, and mutagenesis
Simian kidney COS-7 cells were grown in DMEM containing high glucose supplemented with 5% heat-inactivated fetal bovine serum (FBS), 1 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Human glioblastoma U87MG cells were grown in DMEM containing high glucose supplemented with 10% heat-inactivated FBS, 1 mM l-glutamine, 1 mM sodium pyruvate, 1% non-essential amino acids, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Cells were grown at 37 °C, 5% CO2. Cells were transfected by the DEAE-dextran method (COS-7 cells) or using Lipofectamine (ThermoFisher Scientific, USA) (U87MG cells), and processed after 48 h. For experiments of protein stability, cells were treated with 800 μg/ml cycloheximide (Sigma-Aldrich) for 6 h before lysis. The Saccharomyces cerevisiae strain YPH499 (MATa ade2-101 trp1-63 leu2-1 ura3-52 his3-∆200 lys2-801) was used for heterologous expression of mammalian proteins. YPH499 yeast cells were grown in synthetic complete (SC) medium, containing 0.17% yeast nitrogen base without amino acids, 0.5% ammonium sulfate supplemented with appropriate amino acids and nucleic acid bases, and added 2% glucose (SD), galactose (SG), or raffinose (SR), as required. Yeasts were transformed by standard procedures, and drop growth assays and GFP-AKT1 distribution analyses were performed as described [12, 28, 29]. pRK5 PTEN, pSG5 AKT1, pYES2 PTEN, and YCpLG myc-p110α-CAAX plasmids have been previously described [10, 28]. The pRK5 PTEN-GFP (C-terminal tagging) plasmid was made by PCR and subcloning into pRK5. The PTEN and PTEN-GFP amino acid substitution variants were made by PCR oligonucleotide site-directed mutagenesis as described [30], and mutations were confirmed by DNA sequencing. Nucleotide and amino acid numbering for PTEN variants correspond to reference sequences from accession numbers NM_000314.4 and NP_000305.3, respectively. Nomenclature of variants is according to ref. [31]. Variants and phenotypes data have been deposited in the LOVD gene variant database (http://www.lovd.nl/3.0/home) [32], with the following accession IDs: https://databases.lovd.nl/shared/variants/0000351723, 0000352569-0000352584, 0000352586-0000352590, and 0000353018.
Immunofluorescence and microscopy techniques
PTEN subcellular location in COS-7 cells immunofluorescence was determined by immunofluorescence as previously described, using mouse monoclonal anti-PTEN 425A and fluorescein-conjugated anti-mouse antibody [12, 33]. PTEN-GFP (green fluorescence protein) subcellular location in COS-7 cells was directly visualized by standard fluorescence microscopy. For quantitation of PTEN subcellular distribution, at least 50 positive cells were scored for each experiment. Cells were rated as showing nuclear staining (N), cytoplasmic staining (C), or staining within both the nucleus and the cytoplasm (N/C). Nuclei were identified by DAPI (Sigma-Aldrich, USA) staining. All pictures were taken under a ×20 magnification. Measurement of GFP-AKT1 plasma membrane localization in yeast, as an indirect indicator of cellular PIP3 levels, was performed by fluorescence microscopy, as described [12, 28]. More than 100 cells were examined and scored for each condition or experiment for either cytoplasmic or membrane-associated localization.
Immunoblotting
Whole-cell protein extracts from COS-7 and U87MG cells overexpressing ectopic PTEN variants were prepared by cell lysis in ice-cold M-PERTM lysis buffer (ThermoFisher Scientific) supplemented with PhosSTOP phosphatase inhibitor and cOmplete protease inhibitor cocktails (Roche, Switzerland), followed by centrifugation at 15,200 × g for 10 min and collection of the supernatant. Proteins (50–100 μg) were resolved in 10% SDS-PAGE under reducing conditions and transferred to PVDF membranes. Immunoblotting was performed using anti-phospho-Ser473-AKT and anti-AKT (Cell Signaling Technologies, USA), anti-PTEN 6H2.1 (Merck Millipore, USA), or anti-GAPDH (Santa Cruz Biotechnology, USA) antibodies, followed by horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse (Calbiochem, USA) antibodies. For determination of phospho-AKT content and PTEN protein stability, bands were quantified using an Image studioTM software with Odyssey® CLx Imaging System (LI-COR, USA).
Results and discussion
The PTEN Q17E variant targets the PTEN N-terminal localization motifs
The PTEN Q17E mutational variant [c.49C>G; p.(Gln17Glu)] has been reported both in PHTS and ASD patients [34, 35], suggesting a relevant functional impact on PTEN function. The PTEN residue Gln17 (Q17) is located within the PTEN N-terminal localization motifs (NLMs) of the PTEN PTP domain, which include a PIP2-binding motif (PBM, residues 6–15) [36], a nuclear localization signal (NLS, residues 8–32) [10], and a cytoplasmic localization signal (CLS, residues 19–25) [37]. As with other amino acids in this region, Q17 is well conserved among species, although in the fly D. melanogaster a Lys (K) is present in the equivalent position (Fig. 1a). This suggests that Q17 is important, but not essential, for PTEN function. The PTEN NLMs are surface-exposed, which facilitates their interaction with targeting or regulatory moieties, and they show the same topological orientation with the positively charged TI-loop at the PTEN PTP domain, as well as the CBR3- and Cα2-membrane-binding loops at the C2 domain (Fig. 1b) [8, 38, 39]. Together, these observations suggest a role for Q17 in regulation of the function and the subcellular partitioning of PTEN.
Fig. 1.
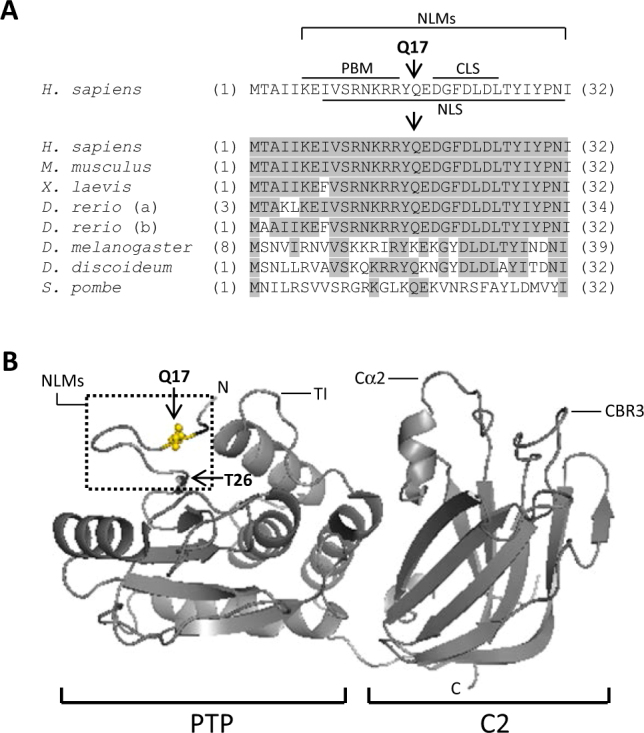
Localization and species conservation of Gln17 (Q17) residue in PTEN protein. a At the top, the human PTEN amino acid sequence (single-letter amino acid code) (accession number NP_000305.3) from residues 1–32 is shown, indicating the PTEN N-terminal localization motifs (NLMs): nuclear localization signal (NLS), PIP2-binding motif (PBM), and cytoplasmic localization signal (CLS). Residue Q17 is indicated with an arrow. At the bottom, an alignment of PTEN N-terminal amino acid sequence from different species is shown. Conserved residues are shadowed. Note that D. rerio possesses two PTEN-like genes. Numbers in brackets correspond to amino acid numbering. b Depiction of localization of Q17 residue in the PTEN 3D structure (accesion ID5R). Residues Q17 and Thr26 (T26) are indicated with an arrow. The localization of the TI-, Cα2-, and CBR3-membrane-binding loops is also indicated. PTP, protein tyrosine phosphatase domain; C2, C2-membrane-binding domain
The PTEN Q17E variant displays slightly reduced PIP3-phosphatase activity and is stable in cells
We have previously shown that a PTEN Q17A experimental variant [c.(49C>G; 50A>C); p.(Gln17Ala)] slightly affects the PIP3-phosphatase activity of PTEN in a heterologous yeast system without affecting PTEN nuclear/cytoplasmic distribution [12]. Here, we have analyzed, both in yeast and in mammalian cells transfected with the appropriate cDNAs, the functionality of the PTEN Q17E variant, in comparison with variants Q17A and Q17P [c.50A>C; p.(Gln17Pro)] (associated to a glioma tumor sample; cancer.sanger.ac.uk/cosmic). The PTEN Q17E variant displayed lower PIP3-phosphatase activity when compared with wild-type PTEN, both in yeast (Fig. 2a, b) and in mammalian cells (Fig. 2c), although in the case of mammalian cells the difference was not statistically significant. A similar effect was observed upon transfection of COS-7 PTEN-positive cells and U87MG PTEN-negative cells. Contrarily, PTEN Q17P behaved as a full loss-of-function PTEN variant, like the catalytically inactive PTEN C124S variant [c.371G>C; p.(Cys124Ser)], in both experimental settings (Fig. 2). Thus, we conclude that the PTEN Q17E variant is only partially affected in its in vivo catalytic activity. These results are in agreement with those reported by Nguyen et al., showing a similar in vitro PIP3-phosphatase activity for PTEN Q17E and PTEN wild type [40].
Fig. 2.

Functional activity of PTEN Q17E variant in cells. a, b Functional activity of PTEN Q17E variant in a heterologous yeast S. cerevisiae system. In a, a yeast growth drop assay is shown of cells transformed with different combinations of plasmids encoding a hyperactive form of the mammalian PI3K p110α catalytic subunit (p110α-CAAX) and PTEN variants, under glucose growth conditions (control, no induction of heterologous proteins) or galactose growth conditions (induction of heterologous proteins). The growth of yeast cells is inhibited by p110α-CAAX (p110α-CAAX + Vector), which converts essential pools of PIP2 into PIP3. This can be prevented by the expression of active PTEN [p110-CAAXα + PTEN wild type (WT)] but not catalytically inactive PTEN mutation (p110α-CAAX + PTEN C124S). PTEN Q17E partially rescued the effect of PI3K. In b, the activity of PTEN variants was quantified co-transforming the yeast with plasmids encoding a GFP-AKT1 reporter, which binds to PIP3 at the plasma membrane. Monitored microscopically, removal of the GFP-AKT1 reporter form the plasma membrane is a read-out of PTEN activity on PI3K-generated PIP3 substrate. Data are the average of three experiments on three different clones (n > 100 cells per clone). Bars correspond to the mean ± SD. Student’s t test P values are shown. c Functional activity of PTEN Q17E variant in mammalian cells. COS-7 or U87MG cells were co-transfected with different combinations of plasmids encoding AKT1 and PTEN variants (WT, PTEN wild type), and the phosphorylation of AKT (as an indirect measurement of PTEN catalytic activity) was monitored by immunoblotting using anti-pAKT (Ser473) antibody. The expression of total AKT, PTEN, and GAPDH (as a loading control) was also monitored using specific antibodies. In the top panels, representative experiments are shown. In the bottom panels, the pAKT/AKT ratio from each condition is shown, after quantification of the bands from at least three independent experiments. Student’s t test P values are shown. Note the slight decrease (statistically non-significant) in PTEN Q17E activity when compared with PTEN wild type
The ASD-associated PTEN variants frequently cause destabilization and degradation of the PTEN protein [25, 27]. Next, we analyzed the protein stability of the PTEN Q17E variant in transfected mammalian COS-7 and U87MG cells by measuring protein degradation in the presence of the protein synthesis inhibitor cycloheximide, in comparison with the unstable PTEN D252G variant [c.755A>G; p.(Asp252Gly)] (also found in ASD and PTHS patients) [27]. As shown, PTEN Q17E protein stability was similar to that of PTEN wild type, and higher to that displayed by PTEN D252G (Fig. 3). Thus, different to other PTEN variants associated to disease (such as Q17P or D252G), that strongly affect PTEN catalysis or protein stability, the PTEN Q17E variant displayed slightly diminished PIP3-phosphatase activity and normal protein stability in cells. Interestingly, the Q17E variant has not been reported up to date in cancer somatic mutation databases, such as COSMIC or cBioPortal (Supplementary Table 1, and data not shown).
Fig. 3.

Stability of PTEN Q17E in cells. COS-7 or U87MG cells were transfected with pRK5 plasmids encoding the indicated PTEN variants (WT, wild type), and cells were incubated in the presence of cycloheximide (CHX) for 6 h to monitor protein degradation. PTEN protein levels were measured by immunoblotting using anti-PTEN antibody. GAPDH is shown as loading control. In the top panels, representative experiments are shown. In the bottom panels, the PTEN protein levels after cycloheximide cell treatment are shown as the ratio 6 h/0 h ± SD, after quantification of the bands from at least three independent experiments. Student’s t test P values are shown. PTEN Q17E displayed similar stability than PTEN wild type
The PTEN Q17E variant accumulates in the nucleus
Next, we assessed the subcellular distribution of PTEN Q17E, in comparison with PTEN wild type, PTEN Q17A, and PTEN Q17P. Immunofluorescence analysis in transfected COS-7 cells revealed that PTEN Q17E, but not PTEN wild type or Q17A, accumulated in the nucleus (Fig. 4a). Remarkably, the glioma-associated PTEN Q17P variant also displayed increased nuclear accumulation, unveiling a crucial role for the Q17 residue in the control of PTEN nucleus–cytoplasm shuttling. As shown in Fig. 4a, a PTEN K13A experimental variant [c.(37A>G; 38A>C); p.(Lys13Ala)] with a non-functional N-terminal NLS [10, 12], did not show these changes, demonstrating the involvement of the N-terminal NLS in the nuclear accumulation of the PTEN Q17E and Q17P variants. When a PTEN-GFP protein was used to monitor PTEN subcellular localization in transfected COS-7 cells, the nuclear accumulation of PTEN Q17E and Q17P was also observed, and was dependent on the integrity on the N-terminal PTEN NLS as well (Fig. 4b). This is in agreement with the observation that a PTEN-GFP Q17E variant is modestly accumulated in the nucleus of D. discoideum cells [40]. Mutation of the PTEN catalytic Cys (PTEN C124S variant) had no effect on the subcellular distribution of PTEN Q17E or Q17P, indicating that the nuclear accumulation of these PTEN variants is independent of PTEN catalysis (Fig. 4b).
Fig. 4.

Subcellular localization of PTEN Q17E. a COS-7 cells were transfected with pRK5 plasmids encoding the indicated PTEN variants, and cells were analyzed by immunofluorescence using anti-PTEN antibody. In the top panels, representative images are shown. In the bottom panels, quantification of percentages of cells with nuclear (N), cytoplasmic (C), or nuclear/cytoplasmic (N/C) localization is shown. b COS-7 cells were transfected with plasmids encoding the indicated PTEN-GFP variants (C-terminal tagging) (WT, wild type) and analyzed by fluorescence microscopy. In the top panels, representative images are shown. In the bottom panels, quantification of percentages of cells with nuclear (N), cytoplasmic (C), or nuclear/cytoplasmic (N/C) localization is shown
Germline-associated variants targeting the PTEN N terminus cause PTEN nuclear accumulation
Next, we analyzed the subcellular localization of the complete set of N-terminal PTEN variants, expanding residues 12–32, found in the germline of PHTS or ASD patients, using PTEN-GFP as the reporter (Supplementary Table 1). As shown, nuclear accumulation was prominent on disease-associated variants targeting residues 17–26 (Fig. 5a and Supplementary Table 1). This is in agreement with the findings by Denning et al., which proposes the existence of a CLS at this PTEN region [37]. Alternatively, the 17–26 PTEN region could behave as a nuclear exclusion motif, as proposed for regions at the TI-, CBR3-, and Cα2-loops [8, 10], which hampers the exposure of the overlapping atypical NLS motif at residues 8–32. Interestingly, the PTEN PBM overlaps with the NLS (see Fig. 1a), and it has been proposed a dynamic and cooperative role for the PBM and TI-loop at the PTP domain, as well as the CBR3- and Cα2-loops at the C2 domain, for PTEN plasma membrane binding and activation [38, 41, 42]. Thus, the exposed loop at the N-terminal NLMs PTEN region (Fig. 1b) could work as a PTEN conformational sensor regulating both PTEN subcellular localization and lipid phosphatase activity. In our experiments, the PIP3 in vivo functional analysis showed a partial or total loss-of-function phenotype for most variants at the 17–26 region (Fig. 5b; Supplementary Table 1), suggesting a link between a defective catalytic activity in cells and aberrant nuclear accumulation. Our results argue that mutations affecting PTEN function and targeting the PTEN N-terminal region frequently favor PTEN nuclear accumulation in parallel with decreasing PTEN PIP3-phosphatase activity in cells. This suggests that PTEN nuclear accumulation could be a surrogate marker for loss of PIP3-phosphatase activity in some patients carrying PTEN N-terminal variants. In addition, N-terminal mutated nuclear PTEN could also manifest a dysfunctional role independently of its catalysis, in line with the nuclear retention of PTEN found in some tumors or in cells from some PHTS patients [43, 44]. In this regard, some PHTS-associated PTEN variants that target PTEN at other regions display nuclear accumulation in association with p53 downregulation and increased DNA damage [45, 46]. Further work is required to elucidate the nuclear mechanisms by which N-terminal mutated nuclear PTEN may be causative of PTEN-associated disease. This could shed some light on the variable qualitative penetrance of specific PTEN gene mutations found on both PHTS and ASD patients.
Fig. 5.

Functional analysis of PTEN N-terminal germline mutations. a COS-7 cells were transfected with plasmids encoding the indicated PTEN mutations (WT, wild type), in the background of PTEN-GFP, and cells were analyzed by fluorescence microscopy. Quantification of percentages of cells with nuclear (N), cytoplasmic (C), or nuclear/cytoplasmic (N/C) localization is shown. b In vivo PTEN activity in the yeast heterologous model. Yeast cells were co-transformed with plasmids encoding p110α-CAAX, GFP-AKT1, and the indicated PTEN variants (WT, wild type), and PIP3-phosphatase activity was assessed as in Fig. 2. The percentage of cells with GFP-AKT1 in the cytoplasm (C), indicative of PTEN PIP3-phosphatase activity, or in the plasma membrane (PM), within the population of transformant clones co-expressing p110α-CAAX, GFP-AKT1 and the respective PTEN variants, is represented. Data are the average of three experiments on three different clones (n ≥ 100 cells per clone)
Electronic supplementary material
Acknowledgements
This work was supported in part by grants SAF2013-48812-R, SAF2016-79847-R, BIO2013-44112-P, and BIO2016-75030-P from Ministerio de Economía y Competitividad (Spain), and grant 2013111011 from Gobierno Vasco, Departamento de Salud (Basque Country, Spain). JM was supported by a predoctoral grant from Gobierno Vasco (Programa de Formación de Personal Investigador no doctor, Departamento de Educación, Política Lingüística y Cultura del Gobierno Vasco). SL has been the recipient of a fellowship from Bilbao Bizkaia Kutxa (BBK) (Spain). We thank Prof. Johan T. den Dunnen for providing database submission support.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1038/s41431-018-0155-x) contains supplementary material, which is available to authorized users.
Contributor Information
Víctor J. Cid, Email: vicjcid@ucm.es
Rafael Pulido, Email: rpulidomurillo@gmail.com, Email: rafael.pulidomurillo@osakidetza.eus.
References
- 1.Pulido R. PTEN: a yin-yang master regulator protein in health and disease. Methods. 2015;77-78:3–10. doi: 10.1016/j.ymeth.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 3.Worby CA, Dixon JE. Pten. Annu Rev Biochem. 2014;83:641–69. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 4.Leslie NR, Maccario H, Spinelli L, Davidson L. The significance of PTEN’s protein phosphatase activity. Adv Enzym Regul. 2009;49:190–6. doi: 10.1016/j.advenzreg.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Papa AP. Protein Tyrosine Phosphatases in Cancer, pp 247–60 (Springer, New York, 2016).
- 6.Bassi C, Stambolic V. PTEN, here, there, everywhere. Cell Death Differ. 2013;20:1595–6. doi: 10.1038/cdd.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bononi A, Pinton P. Study of PTEN subcellular localization. Methods. 2015;77-78:92–103. doi: 10.1016/j.ymeth.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gil A, Andrés-Pons A, Pulido R. Nuclear PTEN: a tale of many tails. Cell Death Differ. 2007;14:395–9. doi: 10.1038/sj.cdd.4402073. [DOI] [PubMed] [Google Scholar]
- 9.Kreis P, Leondaritis G, Lieberam I, Eickholt BJ. Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Front Mol Neurosci. 2014;7:23. doi: 10.3389/fnmol.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gil A, Andrés-Pons A, Fernández E, et al. Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: Involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell. 2006;17:4002–13. doi: 10.1091/mbc.e06-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gil A, Lopez JI, Pulido R. Assessing PTEN subcellular localization. Methods Mol Biol. 2016;1388:169–86. doi: 10.1007/978-1-4939-3299-3_12. [DOI] [PubMed] [Google Scholar]
- 12.Gil A, Rodriguez-Escudero I, Stumpf M, Molina M, Cid VJ, Pulido R. A functional dissection of PTEN N-terminus: implications in PTEN subcellular targeting and tumor suppressor activity. PLoS ONE. 2015;10:e0119287. doi: 10.1371/journal.pone.0119287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrés-Pons A, Gil A, Oliver MD, Sotelo NS, Pulido R. Cytoplasmic p27Kip1 counteracts the pro-apoptotic function of the open conformation of PTEN by retention and destabilization of PTEN outside of the nucleus. Cell Signal. 2012;24:577–87. doi: 10.1016/j.cellsig.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282:23306–15. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- 15.Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA. 2009;106:480–5. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vazquez F, Devreotes P. Regulation of PTEN function as a PIP3 gatekeeper through membrane interaction. Cell Cycle. 2006;5:1523–7. doi: 10.4161/cc.5.14.3005. [DOI] [PubMed] [Google Scholar]
- 17.Mester JE, C PTEN hamartoma tumor syndrome. Handb Clin Neurol. 2015;132:129–37. doi: 10.1016/B978-0-444-62702-5.00009-3. [DOI] [PubMed] [Google Scholar]
- 18.Kelleher RJ, 3rd, Geigenmuller U, Hovhannisyan H, et al. High-throughput sequencing of mGluR signaling pathway genes reveals enrichment of rare variants in autism. PLoS ONE. 2012;7:e35003. doi: 10.1371/journal.pone.0035003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orloff MS, He X, Peterson C, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet. 2013;92:76–80. doi: 10.1016/j.ajhg.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riviere JB, Mirzaa GM, O’Roak BJ, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–40. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tilot AK, Frazier TW, 2nd, Eng C. Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurotherapeutics. 2015;12:609–19. doi: 10.1007/s13311-015-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J, Parada LF. PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol. 2012;22:873–9. doi: 10.1016/j.conb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Takei N, Nawa H. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci. 2014;7:28. doi: 10.3389/fnmol.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei H, Alberts I, Li X. The apoptotic perspective of autism. Int J Dev Neurosci. 2014;36:13–18. doi: 10.1016/j.ijdevneu.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 25.Johnston SB, Raines RT. Conformational stability and catalytic activity of PTEN variants linked to cancers and autism spectrum disorders. Biochemistry. 2015;54:1576–82. doi: 10.1021/acs.biochem.5b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodríguez-Escudero I, Oliver MD, Andrés-Pons A, Molina M, Cid VJ, Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet. 2011;20:4132–42. doi: 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- 27.Spinelli L, Black FM, Berg JN, Eickholt BJ, Leslie NR. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet. 2015;52:128–34. doi: 10.1136/jmedgenet-2014-102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrés-Pons A, Rodríguez-Escudero I, Gil A, et al. In vivo functional analysis of the counterbalance of hyperactive phosphatidylinositol 3-kinase p110 catalytic oncoproteins by the tumor suppressor PTEN. Cancer Res. 2007;67:9731–9. doi: 10.1158/0008-5472.CAN-07-1278. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Escudero I, Fernandez-Acero T, Bravo I, et al. Yeast-based methods to assess PTEN phosphoinositide phosphatase activity in vivo. Methods. 2015;77-78:172–9. doi: 10.1016/j.ymeth.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Mingo J, Erramuzpe A, Luna S, et al. One-tube-only standardized site-directed mutagenesis: an alternative approach to generate amino acid substitution collections. PLoS ONE. 2016;11:e0160972. doi: 10.1371/journal.pone.0160972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–63. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- 33.Andrés-Pons A, Valiente M, Torres J, et al. Functional definition of relevant epitopes on the tumor suppressor PTEN protein. Cancer Lett. 2005;223:303–12. doi: 10.1016/j.canlet.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 34.Nizialek EA, Mester JL, Dhiman VK, Smiraglia DJ, Eng C. KLLN epigenotype-phenotype associations in Cowden syndrome. Eur J Human Genet. 2015;23:1538–43. doi: 10.1038/ejhg.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanderver A, Tonduti D, Kahn I, et al. Characteristic brain magnetic resonance imaging pattern in patients with macrocephaly and PTEN mutations. Am J Med Genet A. 2014;164A:627–33. doi: 10.1002/ajmg.a.36309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–79. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- 37.Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene. 2007;26:3930–40. doi: 10.1038/sj.onc.1210175. [DOI] [PubMed] [Google Scholar]
- 38.Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci USA. 2003;100:7491–6. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JO, Yang H, Georgescu MM, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–34. doi: 10.1016/S0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen HN, Yang JM, Afkari Y, et al. Engineering ePTEN, an enhanced PTEN with increased tumor suppressor activities. Proc Natl Acad Sci USA. 2014;111:E2684–2693. doi: 10.1073/pnas.1409433111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lumb CN, Sansom MS. Defining the membrane-associated state of the PTEN tumor suppressor protein. Biophys J. 2013;104:613–21. doi: 10.1016/j.bpj.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shenoy S, Shekhar P, Heinrich F, et al. Membrane association of the PTEN tumor suppressor: molecular details of the protein-membrane complex from SPR binding studies and neutron reflection. PLoS ONE. 2012;7:e32591. doi: 10.1371/journal.pone.0032591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakarakos P, Theohari I, Nomikos A, et al. Immunohistochemical study of PTEN and phosphorylated mTOR proteins in familial and sporadic invasive breast carcinomas. Histopathology. 2010;56:876–82. doi: 10.1111/j.1365-2559.2010.03570.x. [DOI] [PubMed] [Google Scholar]
- 44.Yu W, He X, Ni Y, Ngeow J, Eng C. Cowden syndrome-associated germline SDHD variants alter PTEN nuclear translocation through SRC-induced PTEN oxidation. Hum Mol Genet. 2015;24:142–53. doi: 10.1093/hmg/ddu425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He X, Ni Y, Wang Y, Romigh T, Eng C. Naturally occurring germline and tumor-associated mutations within the ATP-binding motifs of PTEN lead to oxidative damage of DNA associated with decreased nuclear p53. Hum Mol Genet. 2011;20:80–89. doi: 10.1093/hmg/ddq434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lobo GP, Waite KA, Planchon SM, Romigh T, Nassif NT, Eng C. Germline and somatic cancer-associated mutations in the ATP-binding motifs of PTEN influence its subcellular localization and tumor suppressive function. Hum Mol Genet. 2009;18:2851–62. doi: 10.1093/hmg/ddp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.