

Key Points
TNP-470 skews DC differentiation to a phenotype with Th1-stimulatory features.
TNP-470–treated DC vaccine protects mice from tumors by tumor-specific immunogenicity induction in prophylactic and therapeutic settings.
Abstract
Fumagillin is an antiangiogenic and antineoplastic fungal natural product, and TNP-470 is one of its most potent analogs. Decades of studies revealed that TNP-470 has potent anticancer activities via destruction of neovasculature. In stark contrast, TNP-470 has been reported to suppress lymphocyte proliferation, thereby limiting its clinical potentials. In an attempt to investigate whether the similar or opposite immunomodulatory effect of TNP-470 could act on myeloid cells, we found that TNP-470 potentiates the immunogenicity of dendritic cells (DCs) toward a phenotype with T helper cell type 1 (Th1)–stimulatory features. Using DC vaccine on a murine melanoma cancer model, the TNP-470–treated DC vaccine could significantly induce tumor-specific immunogenicity and substantially enhance tumor eradication when compared with vehicle-treated DC vaccine in a prophylactic setting. Enhanced tumor-specific immunogenicity and delayed tumor progression were observed in a therapeutic setting upon the TNP-470–treated DC vaccine. Our data showed that TNP-470 potentiates Toll-like receptor signaling, including NF-κB activation, in DCs to transcriptionally activate interleukin-12 production, thus inducing a Th1-immune response. Our current study uncovers a novel immune function of TNP-470 in DCs and redefines its role as a novel class of small molecule immune adjuvant in DC-based cancer vaccine given potentiation of DC immunogenicity is a major roadblock in DC vaccine development. Our study not only provides a novel adjuvant for ex vivo–cultured patient-specific DC vaccines for cancer treatment but also discovers the distinct immunostimulatory function of TNP-470 in DCs of myeloid lineage that differs from its immunosuppressive function in lymphoid cells.
Visual Abstract
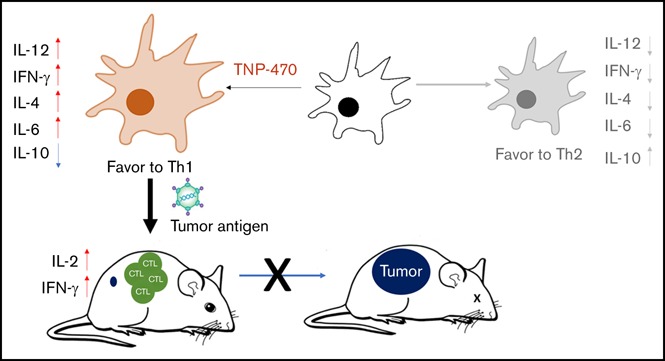
Introduction
Dendritic cells (DCs) are the master antigen-presenting cells (APCs) in the immunological synapse, where upon priming with APCs, naive T cells are activated and primary T-cell–mediated immune response is initiated.1 DCs are classified into 2 different statuses (immature and mature status) according to different phenotypic patterns and functions. In the immature stage, DCs reside in the periphery with high endocytic activity and are capable of engulfing exogenous antigens. Triggered by external stimulus such as different Toll-like receptor (TLR) agonists, DCs become mature and immunogenic. Mature DCs exhibit high expression of costimulatory molecules and secrete cascades of immunostimulatory and proinflammatory cytokines, therefore promoting T-cell activation and inducing T helper cell type 1 (Th1) immune response.2 The unique role of DCs in adaptive immunity makes DC therapy a promising immunotherapeutic approach in anticancer treatment.3 Different from a traditional chemotherapeutic approach in which severe side effects might occur due to the nonselective cytotoxicity, an immunotherapeutic approach uses the host’s immune system to elicit specific antitumor activity. As one of the personalized immunotherapeutic approaches, DC therapy exploits the ex vivo–generated patient-specific DCs, pulsed with specific tumor antigen, to trigger tumor-specific cytotoxicity and durable tumor rejection through induction of tumor-specific immunogenicity.4 The DC vaccine has been adopted in phase 1 and 2 trails for over 2 decades and has been reported to be well tolerated. However, the therapeutic efficacy of the DC vaccine is limited because the induced tumor-specific immunogenicity is dampened in the immunosuppressive tumor microenvironment created by tumors with aggressive features and/or late-stage tumors.
Tumor can escape from immune surveillance through different mechanisms. Coinhibitory receptors, such as CTLA-4 and PD-1,5 can be induced in tumor-infiltrated immune cells, therefore hampering the T-cell–mediated immune response. Regulatory T cells and myeloid-derived suppressor cells (MDSCs) can be infiltrated and accumulated around tumor tissue, therefore attenuating the antitumor immunity of cytotoxic T lymphocytes and blunting the antigen-presenting capacities of tumor-specific DCs and tumor-associated macrophages.6 Taken together, maintenance and potentiation of DC immunogenicity are the major challenges in developing an effective DC vaccine in anticancer treatment. DCVax has recently been approved by the US Food and Drug Administration (FDA) as an immunotherapeutic agent; its promising clinical outcome has been reported in phase 3 trials for treatment of glioblastoma multiforme.7 Provenge (sipuleucel-T), the other FDA-approved DC vaccine, has been adopted in treatment of progressive prostate cancer resulting in prolonged survival.8 To improve the therapeutic efficacy of the DC vaccine, current research has been focused on discovering different novel DC adjuvants targeting TLR-mediated signaling pathways.9-13
Fumagillin is a natural product extracted from fungal metabolites with pronounced antiangiogenic activities and has long been adopted in anticancer and antiarthritis treatments. Biochemical studies identified that methionine aminopeptidase 2 (MetAP2) was one of the molecular targets of fumagillin.14 High-throughput screening of synthetic fumagillin analogs in human endothelial cells revealed that the antiangiogenic activities of these analogs were highly correlated with their inhibitory potencies of MetAP2 enzymatic activities.15 TNP-470 is one of the most potent fumagillin analogs and has been shown to exhibit a wide spectrum of tumor-suppressive activities in different cancer models.16-18 Preclinical data showed that the polymeric form of TNP-470 could significantly inhibit tumorigenesis in vivo.19,20 In stark contrast to its antiangiogenic activity, TNP-470 was reported to inhibit lymphocyte proliferation upon alloantigen-induced stimulation.15 Apart from the suppressive effect on lymphocytes, study found that fumagillin could affect hematopoietic stem cell (HSC) initiation and expansion as MetAP2 was the one of molecular switches during these processes.21 In the current study, we investigated the immunomodulatory potential of TNP-470 in myeloid lineage. Our results showed that TNP-470 could potentiate the immunogenicity of DCs by transcriptional induction of interleukin-12 (IL-12), the Th1 cytokine, through activating the NF-κB pathway and potentiation of the p38 and JNK/MAPK signaling cascades. Furthermore, administration of TNP-470–treated antigen-primed DCs could promote in vitro and in vivo antigen-specific Th1 polarization. To investigate the immunological effect and therapeutic potentials of TNP-470 as a DC-specific adjuvant, TNP-470–treated DC vaccine was applied on the murine melanoma model, and the therapeutic efficacies (such as degrees of tumor growth, levels of tumor-specific cytotoxicity, and tumor-specific immunogenicity) were tested and compared with the vehicle-treated DC vaccine group. We observed enhanced tumor-specific immunogenicity and delayed tumor development in mice upon treatment with the TNP-470–treated DC vaccine, thereby suggesting TNP-470 could be a promising immune adjuvant in developing effective DC vaccine. In our current study, we identified that the fumagillin analog TNP-470 skews DC’s differentiation to the Th1-stimulatory phenotype, and this novel immunomodulatory function of TNP-470 makes it a novel class of immune-adjuvant DC vaccine for anticancer therapy.
Materials and methods
Mice and cell lines
All animal experiments were conducted complying with the ethics regulations of the Department of Health of Hong Kong government, the Committee on the Use of Live Animals in Teaching and Research, University of Hong Kong and the Committee on the Use of Human & Animal Subjects in Teaching and Research, Hong Kong Baptist University. Male C57BL/6N and Balb/c mice at 6 to 8 weeks of age were obtained from the Laboratory Animal Unit, University of Hong Kong and Laboratory Animal Service Centre, Chinese University of Hong Kong. Human monocytic cell line THP-1 was maintained in RPMI 1640 (Gibco) and murine melanoma B16-F10 was maintained in Dulbecco modified Eagle medium (Gibco).
Compound, antibodies, and reagents
TNP-470 was purchased from Sigma. Cytokines granulocyte macrophage colony-stimulating factor (GM-CSF) and IL-4 for bone marrow–derived DC (BMDC) generation was purchased from Peprotech. Fluorochrome-conjugated antibodies for flow cytometry were purchased from eBioscience and Biolegend: CD11c-allophycocyanin, CD80-phycoerythrin (PE), CD83–fluorescein isothiocyanate (FITC), CD86-FITC, T-bet–FITC, interferon γ (IFN-γ)–allophycocyanin. Antibodies for chromatin immunoprecipitation (ChIP) assay and western blot analysis were purchased from Cell Signaling Technology: polyclonal phospho-p38, polyclonal p38, monoclonal phospho-JNK, polyclonal JNK, monoclonal p65, monoclonal histone H3, anti-mouse c-rel polyclonal antibody (pAb), and anti-mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (mAb). Normal rabbit immunoglobulin G (IgG) for ChIP assay was purchased from Santa Cruz Biotechnology. Hamster anti-mouse CD3e antibody for T-cell receptor (TCR)–mediated stimulation was purchased from BD Pharmingen. Phorbol myristate acetate (PMA) and ionomycin for mitogen stimulation were purchased from Sigma. Lipopolysaccharide (LPS) for BMDC and human DC maturation was purchased from Invivogen. TLR agonists were purchased from Invivogen. Inhibitors (PD98059, SB203580, CAPE) were purchased from Tocris. Antigens for DC-pulsing were purchased from Sigma (ovalbumin [OVA]) and synthesized by GenScript (OVA peptide257-264, TRP-2180-188, glycoprotein 100 [gp100]25-33). Cross-linker disuccinimidyl glutarate for ChIP assay was purchased from Sigma. Radioimmunoprecipitation assay (RIPA) for ChIP assay and whole-cell lysate preparation were purchased from Thermo Fisher.
BMDC generation, lymphocytes, and CD4 cell collection and MLR
BMDCs were generated as follows. Bone marrow progenitors were collected from femur and tibia of mice, followed by red blood cell lysis. Bone marrow cells were then cultured in RPMI 1640 medium in the presence of GM-CSF (20 ng/mL) and IL-4 (4 ng/mL) with TNP-470 or vehicle control (dimethyl sulfoxide [DMSO]) for 6 days. On day 3, cells were replenished with fresh RPMI 1640 medium with GM-CSF/IL-4 and TNP-470/vehicle control. On day 6, nonadherent and loosely attached cells were harvested as immature DCs. To promote DC maturation, DCs were further stimulated with LPS (200 ng/mL) for different time durations according to experimental purposes. For some experiments required for antigen pulsing, OVA (50 µg/mL), tumor lysate (200 ng/mL), OVA 257-264 peptide SIINFEKL (10 µM), and TRP-2 180-188 peptide (10 µM) were added to immature DCs on day 6, followed by 16 hours of LPS stimulation.
To collect lymphocytes from mice, spleens and lymph nodes were collected, minced into small pieces, and passed through a 0.45-μm cell strainer. Single-cell suspension was then incubated with ACK lysis buffer to remove red blood cells and ready for experiment. To collect CD4 cells, lymphocytes collected as described were further incubated with CD4 magnetic microbeads (Miltenyi) at 4°C for 30 minutes. CD4 cells were then harvested by positive selection of microbead-bound cells according to the manufacturer’s instructions.
For syngeneic and allogenic mixed lymphocyte reaction (MLR), BMDCs from C57BL mice were cultured with lymphocytes from C57BL mice (syngeneic culture) or Balb/c mice (allogenic culture) at different DC-to-lymphocyte ratios (1:5, 1:10, and 1:20) for 72 hours (syngeneic culture) and 48 hours (allogenic culture) in a 96-well round plate; 0.5 μCi 3H-labeled thymidine was added in the last 16 hours of culture. Cell proliferation was evaluated by measuring the amount of incorporated thymidine. 3H-Thymidine radioactivity was measured using a 1450 MicroB counter. Culture supernatant from syngeneic and allogenic MLR coculture was collected to measure IL-2 secretion by enzyme-linked immunosorbent assay (ELISA).
DC/CD4 cells priming; in vitro and in vivo OVA polarization assays
To examine polarization effects of DCs, syngeneic DCs and CD4 cells were cultured for 7 days as priming process. Primed CD4 cells were treated with PMA as mitogenic stimulation or anti-CD3e as TCR-mediated stimulation. Polarization effects were evaluated by quantification of IFN-γ+ CD4 cells and T-bet+ CD4 cells upon different stimulations under flow cytometry analysis. For in vitro OVA polarization assay, OVA-specific immune responses were induced by footpad immunization of C57BL mice with OVA (10 µg), which was emulsified in complete Freud adjuvant.22,23 OVA-specific lymphocytes were collected from mice 7 days post-OVA immunization. To determine in vitro OVA-specific immunogenicity, OVA-pulsed DCs were cultured with OVA-specific lymphocytes, and culture supernatant from 24, 48, and 72 hours of culture was harvested to measure cytokine secretion by ELISA.
For in vivo OVA polarization assay, mice were immunized with TNP-470/vehicle-treated and OVA-pulsed BMDCs (1 × 106 BMDCs per mouse) on days 0 and 7. Lymphocytes were harvested on day 14 and stimulated with OVA (50 µg/mL) ex vivo. Culture supernatant from 24, 48, and 72 hours of culture was collected to measure cytokine secretion by ELISA. Th1 polarization effects were evaluated by quantifying the percentages of OVA-specific IFNγ+ CD4 and CD8 cells 3 days post ex vivo coculture with OVA. To determine OVA-specific cytotoxic T lymphocyte (CTL) responses, mice were immunized with TNP-470/vehicle-treated, OVA or SIINFEKL peptide-pulsed DCs on days 0, 7, and 14. Lymphocytes were harvested on day 21 and stimulated with SIINFEKL (1 µM) ex vivo. Culture supernatant from 24, 48, and 72 hours of culture was collected to measure IFN-γ secretion by ELISA.
ChIP assay and western blotting
Immature TNP-470/vehicle-treated BMDCs were stimulated with LPS for 0 and 2 hours, then proceed to ChIP assay as previously described.24 BMDCs were fixed with disuccinimidyl glutarate at room temperature for 45 minutes before formaldehyde cross-linking (room temperature for 15 minutes). Equivalent amounts of input DNA were immunoprecipitated. Chromatin was subjected to ChIP using anti-cRel (Cell Signaling Technology) and control rabbit IgG (Santa Cruz Biotechnology). For real-time polymerase chain reaction (PCR) of ChIP samples, the fold enrichment values were normalized to the control IgG using pairs of specific primer (supplemental Table 1).
To detect protein expression in whole-cell extract and nucleus, whole-cell lysate was prepared by cell lysis of BMDCs with RIPA supplemented with protease inhibitor cocktail (Roche). Nuclear fractions from BMDCs were prepared by incubating BMDCs with buffer A (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES]-KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride [PMSF]) at 4°C for 10 minutes, then cell pellets were further lysed with buffer C (20 mM HEPES-KOH pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol) at 4°C for 20 minutes. Protein concentrations were quantified by Bradford protein assay (Bio-Rad). Protein samples were subjected to electrophoresis in 8% to 12% sodium dodecyl sulfate (SDS)–polyacrylamide gels. Proteins were then transferred to polyvinylidene difluoride (PVDF) membrane. Membrane was blocked with 5% milk in Tris-buffered saline (TBS) with Tween 20 (TBST), incubated with primary antibodies, followed by corresponding secondary antibodies linked to horseradish peroxidase (HRP). Detection of chemiluminescence was revealed using WesternBright ECL (Advasta), developed and monitored by Las 4000 (Fuji).
NF-κB translocation assay
To detect NF-κB translocation, BMDCs on day 6 upon LPS stimulation were washed with ice-cold phosphate-buffered saline (PBS), then fixed with 4% paraformaldehyde at room temperature, followed by cell lysis in 0.5% Triton X-100. Primary antibody was NF-κB p65 and secondary antibody was Texas-Red anti-mouse IgG. The slides were mounted in VectaShield mounting medium with 4,6-diamidino-2-phenylindole (Vector Laboratories). Translocation of p65 was visualized under a Zeiss fluorescent microscope.
Prophylactic and therapeutic settings of B16 murine melanoma vaccination model
To generate DC vaccine against B16 murine melanoma for vaccination model, BMDCs were pulsed with tumor lysate, which was prepared by 3 rounds of freeze-and-thaw cycle against B16-F10 melanoma.25 For prophylactic setting, mice were immunized with 3 rounds of DC vaccine on 21, 14, and 7 days prior to tumor challenge. For therapeutic setting, mice were primarily challenged with B16 melanoma. When the size of the solid tumor reached ∼150 mm3 on day 7, mice were immunized with DC vaccine on day 7 and day 11, respectively. Tumor development and survival rate were monitored every other day. To evaluate tumor-specific immune response, lymphocytes from mice in different groups were collected and cultured with tumor lysate ex vivo for 24, 48, and 72 hours. Culture supernatant was collected to measure cytokine secretion (IL-2 and IFN-γ) by ELISA. Numbers of tumor-specific CTLs were determined by quantifying IFN-γ–producing CD8 cells upon ex vivo stimulation with corresponding tumor lysate for 72 hours. Tumor-specific cytotoxicity was evaluated by lactate dehydrogenase (LDH) assay (Roche). Briefly, lymphocytes from immunized mice in different groups were primarily cultured with tumor lysate ex vivo for 72 hours and served as effector cells, then these effector cells were cultured with B16 melanoma cells (as target cells) at effector-to-target ratios at 100:1 for 6 hours. Culture supernatant was collected and cytotoxicity against B16 was measured by LDH assay (Roche) as per the manufacturer’s protocol.
Real-time PCR, cytokine ELISA, and flow cytometry analysis
To measure gene expression, TNP-470/vehicle-treated BMDCs were stimulated with LPS for 4 hours, total RNA was then extracted using Trizol method. Complementary DNA (cDNA) was prepared by SuperScript III (Invitrogen). Expression levels of different genes (supplemental Table 1) were evaluated using ABI ViiA7 fast real-time PCR system and Sybr Green PCR master mix (Applied Biosystems). Reverse transcription–quantitative PCR (RT-qPCR) results were normalized against GAPDH. To measure secretion of different cytokines (IL-12, IL-10, IL-2, and IFN-γ), culture supernatant harvested at different time points were measured by cytokine ELISA kits (e-Bioscience and Biogend) as per the manufacturer’s protocol. For cell surface staining, cells were stained with fluorochrome-conjugated antibodies in 2% fetal bovine serum (FBS; in PBS) at 4°C, then washed with PBS and resuspend in 2% FBS (in PBS) for flow cytometric analysis using BD FACSCanto II. For intracellular cytokine IFN-γ staining, cells were fixed in fixation buffer at room temperature, then permeabilized in permeabilization buffer. Permeabilized cells were stained with IFN-γ–allophycocyanin at room temperature, then washed with permeabilization/wash buffer and resuspended in PBS. Intracellular cytokine secretion was quantified using BD FACSCanto II. To detect T-bet expression in CD4 cells by flow cytometry analysis, CD4 cells were fixed with fixation/permeabilization buffer at room temperature. Permeabilized cells were stained with anti-Tbet-PE at room temperature, then washed with PBS and resuspend in 2% FBS (in PBS). T-bet expression was quantified using BD FACSCanto II.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (La Jolla, CA) with the 2-tailed unpaired nonparametric Student t test. In some cases, grouped data were analyzed with 1-way analysis of variance (ANOVA) followed by the Bonferroni post hoc multiple comparison test. Results were considered as significant when P < .05.
Results
TNP-470 potentiates DC immunogenicity in response to TLR stimulation in vitro
Fumagillin affects HSCs initiation and expansion via binding to MetAP2, one of the key molecular switches involved in these processes. Apart from the suppressive effect on the HSC differentiation, fumagillin analog TNP-470 has been reported to inhibit lymphocyte proliferation in vitro.15 The fact that TNP-470’s inhibition on T-cell immunogenicity presents a great disadvantage as an antiangiogenic drug. This prompts us to ask whether TNP-470 has an immunomodulatory effect on the myeloid branch of HSCs. We first validated the effect of TNP-470 on T-cell proliferation by performing thymidine incorporation assay and measuring IL-2 secretion. Similar to previous studies showing the in vitro inhibitory effect on T-cell proliferation, TNP-470 could significantly inhibit CD3/CD28-mediated T-cell proliferation, the half maximal inhibitory concentration (IC50) of which is 2.87 nM (Figure 1A left), accompanied by substantial IL-2 reduction (Figure 1A right). We next asked whether TNP-470 could inhibit T-cell proliferation in vivo, we administrated mice with a high dosage of TNP-470 for 2 weeks (20 mg/kg, intraperitoneal injection every 2 days), a dosage that is effective for its antiangiogenic activity.26,27 Percentages of CD4 and CD8 cells in spleen and lymph node were then evaluated by flow cytometry analysis (supplemental Figure 1). We found that there was no significant change in the percentages of CD4 and CD8 cells between TNP-470 and solvent treatment groups implying that TNP-470 has no effect on T-cell homeostasis in vivo. We then evaluated the phenotypic patterns and functions of bone marrow–derived murine DCs in the presence of TNP-470 or vehicle control DMSO. Flow cytometry analysis showed that there was no significant change in the expression of costimulatory molecules between these 2 groups of DCs (supplemental Figure 2A), but TNP-470–treated DCs showed enhanced antigen-binding capacity (supplemental Figure 2B). Furthermore, TNP-470–treated DCs displayed a different cytokine secretion profile upon LPS stimulation (Figure 1B). These results suggest that TNP-470 could cause functional change(s) of DCs, especially the polarization effect(s) on T cells, which is characterized by the shift of IL-12/IL-10 balance.
Figure 1.

TNP-470 potentiates DC immunogenicity upon LPS stimulation in vitro. (A) In vitro inhibitory effects of TNP-470 on T lymphocytes. Lymphocytes were and CD28 with different concentrations of TNP-470 for 72 hours. 3H-Thymidine was added 16 hours prior to sample harvest. Proliferation of T lymphocytes was evaluated by the level of incorporated 3H-thymidine. Representative data from 3 independent studies are shown (left). Data presented as mean ± standard error of the mean (SEM; n = 3). IL-2 secretion from lymphocytes upon CD3/CD28 stimulation (right). Lymphocytes were stimulated with anti-mouse CD3e and CD28 in the presence of vehicle/TNP-470 (0.5 and 1 µM) for 24 hours. Culture supernatant was harvested and detected for IL-2 secretion. Data presented as mean ± SEM (n = 3) from 2 independent studies. ***P < .001 compared with vehicle. ###P < .001 TNP-470 (0.5 µM) vs TNP-470 (1 µM). BMDCs upon vehicle and TNP-470 treatment (5 nM) were stimulated with LPS (200 ng/mL) for 4 hours to evaluate cytokine mRNA expression and for 16 hours to evaluate cytokine secretion. (B) Cytokine secretion profile of TNP-470/vehicle-treated DCs upon LPS stimulation. **P < .01 and ***P < .001 TNP-470–treated DCs (TNP-DC) vs vehicle-treated DCs (CTR-DC). Data presented as mean ± SEM from 2 independent experiment. (C) Fold-of-change in expression of IL-12 and IL-10 from BMDCs, which were normalized with GAPDH. Data presented as mean ± SEM from 3 independent experiment. ***P < .001 TNP-470 DC vs CTR-DC. (D) Cytokine IL-12 and IL-10 secretion from culture supernatant. Data presented as mean ± SEM from 5 independent experiment. ***P < .001 TNP-DC vs CTR-DC; ###P < .001 imDC vs CTR-DC. (E) BMDCs was stimulated with LPS for 4 hours to evaluate gene expression levels (left) and 8 hours to examine protein levels (right) of SOCS1 and SOCS3. ***P < .001 TNP-DC vs CTR-DC. Data presented as mean ± SEM from 2 independent experiment.
The shift of IL-12/IL-10 balance was further validated at both gene expression level and cytokine secretion level (Figure 1C-D). IL-12 messenger RNA (mRNA) level was increased approximately eightfold in the presence of TNP-470 while the IL-10 mRNA level was reduced by >70%. IL-12 is the signature cytokine produced by APCs to skew Th1 polarization28 while IL-10, an immunosuppressive cytokine, triggers T-cell unresponsiveness and is involved in regulatory T-cell generation.29 Furthermore, suppressors of cytokine signaling (SOCS) family proteins, especially SOCS1 and SOCS3, are the negative regulators in TLR-signaling pathway, through which cascades of stimulatory and proinflammatory cytokines are produced. Gene expression and protein levels of SOCS1 and SOCS3 were significantly reduced in TNP-470–treated DCs compared with vehicle-treated DCs (Figure 1E). Reduction of SOCS1 and SOCS3 in TNP-470–treated DCs might lead to their cytokine-stimulatory properties.
We next examined whether TNP-470–treated DCs could trigger T-cell activation by coculturing these DCs with lymphocytes. In either syngeneic or allogenic DC/lymphocytes coculture, TNP-470–treated DCs significantly enhanced lymphocyte proliferation at all DC-to-lymphocyte ratios compared with vehicle-treated DCs (Figure 2A left and middle). Furthermore, results also showed that T-cell activation was enhanced by TNP-470–treated DCs, which was reflected by substantial IL-2 induction in lymphocytes upon coculture with TNP-470–treated DCs (Figure 2A right). All of these data imply that TNP-470 could shift the balance of IL-12/IL-10 to potentiate the immunogenicity of DCs and trigger the subsequent T-cell activation in the immunological synapse. We then asked whether the shift of IL-12/IL-10 balance by TNP-470 could happen in human DC cells. Human monocytic cell line THP-1 was differentiated into DCs with appropriate cytokine cocktail in the presence of TNP-470 or solvent control. We found induction of IL-12 mRNA expression and simultaneous reduction of IL-10 mRNA expression upon LPS stimulation in TNP-470–treated, differentiated THP-1 cells (supplemental Figure 3). The similar shift of IL-12/IL-10 balance in TNP-470–treated DCs between human and mouse suggested that TNP-470 would trigger similar modulatory effects in human T cells as those in murine counterparts.
Figure 2.

TNP-470–treated DCs induce T-cell activation in vitro and in vivo. (A) BMDCs were cultured with syngeneic (left) and allogenic (middle) lymphocytes at different ratios (1:5, 1:10, 1:20) for 72 hours (syngeneic culture) and 48 hours (allogenic culture). 3H-labeled thymidine (0.5 μCi/well) was added in the last 16 hours of culture and cell proliferation was measured by incorporated 3H-labeled thymidine. Data presented as mean ± SEM from 3 independent experiment. ***P < .001 TNP-DC coculture vs CTR-DC coculture. Culture supernatant from 72-hour syngeneic culture and 48-hour allogenic culture (DC:T-cell ratio = 1:5) were harvested and detected for IL-2 secretion (right). ***P < .001 TNP-DC coculture vs CTR-DC coculture. Data presented as mean ± SEM from 3 independent experiment. (B) Activation of in vitro OVA-specific CD4 T cells. CD4 cells from OVA-immunized mice were collected and cultured with TNP-470/vehicle- treated and OVA-pulsed DCs for 24 hours. Culture supernatant was detected for IL-2 secretion (left). ***P < .001 OVA-pulsed CTR-DC coculture vs OVA-pulsed TNP-DC coculture. Data presented as mean ± SEM from 3 independent experiment. Activation of OVA-specific lymphocytes in vivo. C57BL mice were immunized with OVA-pulsed DCs (TNP-470/vehicle-treated). Lymphocytes from mice in different groups were harvested and stimulated with OVA ex vivo. Culture supernatant from 24-, 48-, and 72-hour culture was detected for IL-2 secretion (right). *P < .05 lymphocytes from mice immunized with OVA-pulsed TNP-DC vs OVA-pulsed CTR-DC counterpart. Data presented as mean ± standard deviation (SD) from 3 independent experiment.
TNP-470–treated DCs skew T cells toward Th1 polarization and induce strong CTL response
Shift of the IL-12/IL-10 balance prompted us to investigate whether TNP-470–treated DCs favor Th1 immune response. Syngeneic naive CD4 cells were first primed with TNP-470/vehicle-treated DCs followed by mitogenic stimulation or TCR-mediated stimulation. For those CD4 cells primed with TNP-470–treated DCs, a significantly higher percentage of IFN-γ–producing CD4 cells was detected when compared with control (Figure 3A top). Higher percentage of T-bet–expressing CD4 cells were also observed in those primed with TNP-470–treated than those primed with vehicle-treated DCs (Figure 3A bottom). Induction of Th1 cytokine IFN-γ and Th1-driving transcription factor T-bet demonstrate that TNP-470–treated DCs could skew Th1 polarization in vitro. We next tested whether TNP-470–treated DCs could promote antigen-specific Th1 polarization. Footpad administration of mice with antigen emulsified in complete Freund adjuvant is a common approach in examining cellular immune response. OVA is the foreign antigen that triggers strong antigen-specific immunity upon foodpad administration and is commonly adopted in the study of antigen-specific T-cell polarization.22,23 To examine the in vitro antigen-specific T-cell polarization, OVA-specific immune response was induced by footpad immunization of OVA on mice. OVA-specific CD4 T cells were then harvested and cultured with OVA-pulsed DCs. Substantially higher levels of Th1 cytokines IL-12, IFN-γ (Figure 3B), and IL-2 (Figure 2B left) were detected in the coculture with TNP-470–treated DCs than those with vehicle-treated DCs, whereas the level of IL-4 decreased below detection limit (data not shown).
Figure 3.

TNP-470–treated DCs promote in vitro and in vivo Th1 polarization and induce strong CTL responses. Naive CD4 cells were primed with TNP-470/vehicle-treated DCs for 7 days. Primed CD4 cells were further stimulated with PMA (50 ng/mL) for 4 hours as mitogenic stimulation and anti-CD3e for 6 hours as TCR-mediated stimulation. Representative data of IFN-γ–producing CD4 cells and T-bet expressing CD4 cells by intracellular staining were showed (A, left) and percentages of corresponding cell population were summarized (A, right). ***P < .001 CD4 cells primed with TNP-DC vs CTR-DC. Data presented as mean ± SEM from 3 independent experiment. (B) In vitro OVA polarization assay. TNP-470/vehicle- treated and OVA-pulsed DCs were cultured with OVA-specific CD4 cells for 24 hours. Culture supernatant were detected for IL-12 (left) and IFN-γ (right) secretion. ***P < .001 Coculture of TNP-DCs and OVA-specific CD4 cells vs that of CTR-DCs and OVA-specific CD4 cells. Data presented as mean ± SEM from 3 independent experiment. (C) In vivo OVA polarization assay. C57BL mice were immunized with OVA-pulsed DCs (TNP-470/vehicle-treated) on day 0 and day 7. Lymphocytes from mice in different groups were harvested on day 14 and stimulated with OVA ex vivo. Culture supernatant from 24-, 48-, and 72-hour culture was detected for IFN-γ secretion (left). Data presented as mean ± SD from 3 independent experiment. Percentages of OVA-specific CD4 (middle) and CD8 cells (right) were quantified by flow cytometric analysis of IFN-γ+CD4+ and CD8+ cells upon 72 hours of ex vivo stimulation of OVA. Data presented as mean ± SD from 3 independent experiment. (D) OVA-specific CTL response. C57BL mice were immunized with OVA or OVA peptide (SIIFEKL)–pulsed DCs (TNP-470/vehicle- treated) on day 0, 7, and 14. Lymphocytes harvested on day 21 were stimulated with OVA peptide ex vivo and culture supernatant from 24-, 48-, and 72-hour culture was collected to detect IFN-γ secretion. **P < .01 and ***P < .001 TNP-DC vs CTR-DC (OVA /SIIFEKL-pulsed). ##P < .01 OVA-pulsed TNP-DC vs SIIFEKL-pulsed TNP-DC. Data presented as mean ± SD (n = 3).
Induction of Th1 cytokines implies that TNP-470–treated DCs could promote OVA-specific Th1 polarization in vitro. To determine the in vivo effect of TNP-470–treated DCs on antigen-specific immunogenicity, mice were immunized with TNP-470 /vehicle–treated, OVA-pulsed DCs. Lymphocytes from these immunized mice were then harvested and stimulated with OVA ex vivo. Higher levels of IFN-γ (Figure 3C left) and IL-2 (Figure 2B right) were detected from lymphocytes harvested from mice immunized with TNP-470–treated, OVA-pulsed DCs than those immunized with vehicle-treated, OVA-pulsed DCs, which are similar to the in vitro results showing that Th1 cytokines were significantly induced when TNP-470–treated, OVA-pulsed DCs were cultured with OVA-specific lymphocytes. In addition, a higher percentage of IFN-γ–producing CD4 cells and CD8 cells was detected in mice immunized with TNP-470–treated, OVA-pulsed DCs (Figure 3C middle and right). All of these data demonstrate that TNP-470– treated DCs induces OVA-specific Th1 polarization in mice. We then examined whether the induction of an OVA-specific Th1 response could trigger a strong CTL response in vivo. Compared with OVA, class I MHC–restricted OVA257-264 peptide (SIINFEKL) is more efficient to be cross-presented by DCs and induce CTL response.30 Similar to previous study, we found that lymphocytes from mice immunized with peptide-pulsed DCs could produce higher amount of IFN-γ than those immunized with OVA-pulsed DCs regardless of vehicle-treated DCs or TNP-470–treated DCs (Figure 3D). More importantly, TNP-470–treated DCs could potentiate the CTL response in mice regardless of immunization with OVA- or peptide-pulsed DCs as indicated by substantial IFN-γ induction from the harvested lymphocytes upon ex vivo stimulation of class I–restricted epitope peptide (Figure 3D). Of all immunized groups, lymphocytes from mice immunized with TNP-470–treated, peptide-pulsed DCs produced the highest amount of IFN-γ indicating a strong induced CTL response. Taken together, TNP-470–treated DCs could skew antigen (OVA)-specific Th1 immune response both in vitro and in vivo and subsequently trigger strong CTL response.
TNP-470 activates TLR signaling to mediate Th1-stimulatory immunogenicity through IL-12 induction in DCs
TLR signaling is responsible for DC maturation, leading to the expression of MHC class I and II molecules and the secretion of proinflammatory cytokines. Therefore, we examined the effect of TNP-470 on TLR signaling. Besides TLR4 agonist LPS, of the 4 different TLR agonists tested, TNP-470 enhanced Pam3SK (TLR1/2 agonist) and HKLM (TLR2 agonist) mediated IL-12 production (Figure 4A). We then investigated how TNP-470 would affect TLR signaling to mediate DC immunogenicity and IL-12 induction. First, we explored the effect of TNP-470 on p38/JNK signaling. Western blotting showed that TNP-470 could further increase LPS-induced phosphorylation compared with control (Figure 4B). TNP-470–mediated IL-12 induction was blunted by p38 inhibitor SB203580 and MAPK inhibitor PD098059 at 10 µM (Figure 4C). We then examined the effect of TNP-470 on NF-κB pathway as NF-κB is the key molecule regulating the transcription of cluster of genes involved in inflammation and immune response. NF-κB is a heterogeneous dimer of NF-κB/Rel family proteins and its transcriptional activity is regulated by the activation domain of p65/relA and c-Rel.31 Among the NF-κB/Rel family proteins, p65 and c-Rel are the 2 selective transcription factors contributing to IL-12p40 gene regulation via binding to the corresponding gene promoter regions.32 Because NF-κB directly mediates IL-12 transcription during DC maturation, we investigated the effect of TNP-470 on NF-κB activation. First, NF-κB inhibitor CAPE at both 2 and 10 µM significantly blunted the TNP-470–mediated IL-12 production (Figure 4D). RT-qPCR showed that nearly all NF-κB subunits (p52, p65, c-Rel, and relB) were significantly induced by TNP-470 upon LPS stimulation when compared with control (Figure 4E). We next tested whether TNP-470 promotes nuclear translocation of p65 and c-Rel. Upon LPS stimulation, a higher abundance of c-Rel and p65 was detected in the nuclear extract of TNP-470–treated DCs than those in vehicle-treated DCs (Figure 4F). Furthermore, NF-κB translocation assay showed that more p65 subunit was translocated into nucleus of TNP-470–treated DCs (Figure 4G). ChIP analysis of IL-12p40 promoter showed higher occupancy of c-Rel, but not p65 (data not showed), in TNP-470–treated DCs compared with vehicle-treated DCs (Figurer 4H). Our data collectively showed that TNP-470 activates NF-κB to increase IL-12 transcription. Taken together, TNP-470 potentiates multiple TLR-signaling pathways to mediate IL-12 induction and the subsequent Th1-stimulatory immunogenicity in DCs.
Figure 4.

TNP-470–treated DCs trigger NF-κB transactivation of IL-12 and potentiate p38/JNK activation. (A) TNP-470/vehicle-treated DCs were stimulated with different TLR agonists (5 nM, 16hr). **P < .01 TNP-DC vs CTR-DC upon agonist stimulation. Data presented as mean ± SEM from 3 independent experiment. (B) TNP-470/vehicle-treated DCs were stimulated with LPS (1 µg/mL) for 0, 30, and 60 min. Total protein lysate was blotted with antibodies against p38, phosphor-p38, JNK, phosphor-JNK and GAPDH. Representative images from 2 independent experiment were showed. (C) TNP-470/vehicle-treated DCs were stimulated with LPS for 24 hours in the presence of 2 µM and 10 µM p38 inhibitor SB203580 and MAPK inhibitor PD98059. IL-12 secretion was detected by cytokine ELISA. ***P < .001 compared with TNP-470–treated DCs without inhibitor. Data presented as mean ± SEM from 2 independent experiment. (D) TNP-470/vehicle-treated DCs were stimulated with LPS (200 ng/mL) for 24 hours in the presence of 2 µM and 10 µM NF-κB inhibitor CAPE. IL-12 secretion was detected by cytokine ELISA. ***P < .001 compared with TNP-470–treated DCs without inhibitor. Data presented as mean ± SEM from 2 independent experiment. (E) Fold-of-change in expression of different NF-κB subunits, p52, p65, c-Rel and relB, which were normalized with GAPDH, in TNP-470/vehicle-treated DCs (200 ng/mL LPS for 4 hours). *P < .05, **P < .01 and ***P < .001 TNP-DC vs CTR-DCs. Data presented as mean ± SEM from 2 independent experiment. (F) TNP-470/vehicle-treated DCs was stimulated with LPS for 30 minutes. Expression of cRel, p65 and histone H3 (loading control) from nuclear fractions were detected. Representative images from 2 independent experiment were showed. (G) NF-κB p65 translocation assay. TNP-470/vehicle-treated DCs were stimulated with LPS for 30 minutes. The subcellular localization of p65 was visualized by immunofluorescence using anti-p65 antibody (red) and nuclei were stained by DAPI (blue). Representative images from 3 independent experiment were showed. Original magnification ×40. (H) Recruitment of NF-κB subunit c-Rel to IL-12 promoter. TNP-470/vehicle-treated DCs were stimulated with LPS for 0 and 2 hours. Cells were then cross-linked and lysed. DNA was immunoprecipitated with anti-cRel and control IgG. Degree of c-Rel recruitment to IL12 promoter was quantified by real time PCR. *** P < .001 TNP-DC vs CTR-DC. Representative data presented as mean ± SEM from 2 independent experiment.
TNP-470–treated DC vaccine induces tumor-specific CTLs and prevents solid tumor development in prophylactic setting
Given that TNP-470–treated DCs induce in vitro and in vivo antigen-specific immunogenicity through Th1 polarization and activation, we further examined the utility of TNP-470 as a kind of immune-adjuvant for DC vaccine. Murine B16 melanoma vaccination model was generated to test whether TNP-470–treated DC vaccine could elicit protective effect on tumor development in prophylactic setting (Figure 5A).25 Mice upon immunization with TNP-470–treated DC vaccine showed significant enhancement in immunogenicity than mice in other groups including vehicle DC vaccine group. Lymphocytes from these mice substantially produced immunostimulatory cytokine IL-2 and IFN-γ upon ex vivo tumor lysate stimulation at different time points (Figure 5B). To examine the induction of tumor-specific immune response in TNP-470–treated DC vaccine group, we quantified the numbers of tumor-specific CTLs, the IFN-γ–producing CD8 cells, upon tumor lysate stimulation by flow cytometry analysis. Numbers of CTLs in TNP-470–treated DC vaccine group were increased substantially when compared with those in DC alone without pulsing. More importantly, there was a significant increase of CTLs in TNP-470–treated DC vaccine group than those in vehicle DC vaccine group, indicating directly that TNP-470 could elicit the enhancement of tumor-specific immunogenicity (Figure 5C). To examine whether TNP-470 mediated DC immunogenicity could elicit antitumor activities, we performed LDH assay to detect the levels of specific lysis against B16 melanoma. Similar to the higher amount of CTLs, significantly higher level of tumor lysis was detected in TNP-470–treated DC vaccine group than all other groups indicating that the potent antitumor activity of those CTLs (Figure 5D). Taken together, TNP-470–treated DC vaccine could elicit tumor-specific immunogenicity and tumor-killing activities. To assess whether the observed immunogenicity can be translated into clinical outcome, number of tumor-free mice, tumor volume development and tumor weight at end point were monitored. There was a significant increase in the number of tumor-free mice in TNP-470–treated DC vaccine group (Figure 5E). Moreover, the solid-tumor weight from the mice in TNP-470–treated DC vaccine group was significantly reduced by 90% when compared with the 2 control groups (Figure 5F). Though mice upon immunization with DC vaccine could delay the solid tumor development (P < .05), the protective effect was more pronounced in TNP-470–treated DC vaccine (P < .01) (supplemental Figure 4). These data demonstrated that the enhanced DC immunogenicity and induction of tumor-killing CTLs by TNP-470 could translate into beneficial outcome, which was reflected by increased number of tumor-free mice, reduced solid-tumor weight and delayed tumor development.
Figure 5.

Enhanced tumor-specific immunogenicity and protective effect(s) of TNP-470–treated DC vaccine in prophylactic B16 melanoma vaccination model. (A) Schematic diagram of prophylactic setting of vaccination model. Mice were immunized with PBS (solvent control), DC without tumor lysate pulsing (DC control), tumor lysate-pulsed TNP-470/vehicle-treated DC vaccine. (B) Lymphocytes from immunized mice in different groups were collected and stimulated with tumor lysate (50 µg/mL) for 24, 48 and 72 hours ex vivo. Culture supernatant was detected for secretion of IL-2 (left) and IFN-γ (right). **P < .01 and *** P < .001 TNP-DC vaccine vs vehicle DC vaccine. (C) To quantify amount of tumor-specific CTLs from mice in different groups, lymphocytes were stimulated with tumor lysate (50 µg/mL) for 72 hours ex vivo, followed by re-stimulation of PMA/Ionomycin for 6 hours. Cells were then stained with CD3, CD8 and IFN-γ. Tumor-specific CTLs were determined by flow cytometric analysis of CD3+CD8+IFN-γ+ cells. *P < .05 vehicle DC vaccine vs PBS solvent control. **P < .01 TNP-DC vaccine vs DC control. ***P < .001 TNP-DC vaccine vs PBS solvent control. ##P < .01 TNP-DC vaccine vs vehicle DC vaccine. Data presented as mean ± SD (n = 6) from 2 independent studies. (D) Tumor-specific cytolytic activity. Lymphocytes from mice in different groups were collected and stimulated with tumor lysate (50 µg/mL) for 72 hours ex vivo, then harvested as effector cells (E), and B16-F10 cells were used as target cells (T). Specific lysis was detected by LDH assay at E:T ratio = 100:1. **P < .01 TNP-DC vaccine vs vehicle DC vaccine; ***P < .001 TNP-DC vaccine vs PBS solvent control and DC control. Data presented as mean ± SD (n = 6) from 2 independent studies. (E) Percentages of tumor-free mice in different vaccination groups (n = 6). *P < .05 TNP-DC vaccine vs PBS solvent control. #P < .05 TNP-DC vaccine vs DC control. (n = 6, Modified Kaplan-Meier Survival Analysis). (F) Weight of solid tumor developed on mice on day 26 (end point). *P < .05 TNP-DC vaccine vs PBS solvent control. **P < .01 TNP-DC vaccine vs DC control. Data presented as mean ± SD (n = 6) from 2 independent studies.
TNP-470–treated DC vaccine has added effect on the delayed tumor progression in therapeutic setting
Because TNP-470–treated DC vaccine elicited protective effect on melanoma development in prophylactic setting through induction of tumor-specific immunogenicity, we next examined whether similar effect(s) could be observed in a practical therapeutic setting. In therapeutic setting (Figure 6A), mice were primarily challenged with B16 melanoma, then mice were immunized on DC vaccine when the size of solid tumor reached about 150 mm3 on day 7. On day 11, mice were subjected to second round of DC vaccine immunization and the tumor-specific immunogenicity induced by TNP-470–treated DC vaccine was examined at the end point of experiment. Similar to the observation in prophylactic model, upon ex vivo stimulation of tumor lysate, substantial induction of immunostimulatory cytokine IL-2 and IFN-γ (Figure 6B) was detected in lymphocytes harvested from mice immunized with TNP-470–treated DC vaccine when compared with other groups. To further examine whether TNP-470–treated DC vaccine could elicit potent CTL response against B16 melanoma, we quantified the number of CTLs from lymphocytes upon ex vivo tumor lysate stimulation by flow cytometry analysis and tested the tumor-killing capacities of these CTLs by LDH assay. We found that the percentage of CTLs in TNP-470–treated DC vaccine group were significantly increased than those in vehicle DC vaccine group (Figure 6C). In testing the tumor-killing capacities, CTLs from both vehicle DC vaccine group and TNP-470–treated DC vaccine group elicited significantly higher level of tumor lysis against B16 melanoma than those from solvent control group, however, tumor-killing capacity of CTLs in TNP-470–treated DC vaccine group was higher than those in vehicle DC vaccine group with marginal significance (Figure 6D). We next examined whether the induction of tumor-specific immunogenicity by TNP-470–treated DC vaccine could translate into therapeutic benefit during tumor progression. The tumor progression was significantly delayed in mice upon immunization with TNP-470–treated DC vaccine while the delayed effect in mice upon vehicle DC vaccine was not (Figure 6E). Also, the delay of tumor progression was significantly better in TNP-470–treated DC vaccine group when compared with the vehicle DC vaccine group at multiple time points.
Figure 6.

Enhanced tumor-specific immunogenicity and delayed tumor progression by TNP-470–treated DC vaccine in therapeutic B16 melanoma vaccination model. (A) Schematic diagram of therapeutic setting of vaccination model. Solid tumor was developed on mice by subcutaneous injection of B16 melanoma. When size of solid tumor reached around 150 mm3 (day 7), mice were immunized with PBS (solvent control), DC without tumor lysate pulsing (DC control), tumor lysate-pulsed TNP-470/vehicle-treated DC vaccine on day 7 and day 11, respectively. (B) Lymphocytes from immunized mice in different groups were collected and stimulated with tumor lysate (50 µg/mL) for 24, 48, and 72 hours ex vivo. Culture supernatant was detected for secretion of IL-2 (top) and IFN-γ (bottom). **P < .01 and *** P < .001 TNP-DC vaccine vs PBS solvent control; ##P < .01 TNP-DC vaccine vs vehicle DC vaccine. (C) To quantify amount of tumor-specific CTLs from mice in different groups, lymphocytes were stimulated with tumor lysate (50 µg/mL) for 72 hours ex vivo, followed by re-stimulation of PMA/Ionomycin for 6 hours. Cells were then stained with CD3, CD8 and IFN-γ. Tumor-specific CTLs were determined by flow cytometric analysis of CD3+CD8+IFN-γ+ cells. ***P < .05 TNP-/vehicle DC vaccine vs PBS solvent control; ##P < .01 TNP-DC vaccine vs DC control/ vehicle DC vaccine. Data presented as mean ± SD (n = 5). (D) Tumor-specific cytolytic activity. Lymphocytes from mice in different groups were collected and stimulated with tumor lysate (50µg/mL) for 72 hours ex vivo, then harvested as effector cells (E), and B16-F10 cells were used as target cells (T). Specific lysis was detected by LDH assay at E:T ratio = 100:1. **P < .01 vehicle DC vaccine vs PBS solvent control; ***P < .001 TNP-DC vaccine vs PBS solvent control. Data presented as mean ± SD (n = 5). (E) Solid tumor development in therapeutic setting of B16 melanoma vaccination model. Mice with solid tumor (around 150 mm3 on day 7) were vaccinated with vehicle PBS, DC without tumor lysate pulsing, tumor lysate-pulsed vehicle DC vaccine, TNP-470–treated DC vaccine. Tumor volume of solid B16 melanoma developed on mice from day 7 to day 15. Data are presented as mean ±SD (n = 5) *P < .05, TNP-DC vaccine vs vehicle DC vaccine; #P < .05, ##P < .01 TNP-DC vaccine vs PBS solvent control.
As TNP-470–treated DC vaccine could elicit strong CTL response in either prophylactic or therapeutic setting, we extended our study by evaluating the effect of TNP-470–treated DC vaccine on the CTL response in mice upon immunization with either tumor lysate-pulsed or Trp2-pulsed DC vaccines. Trp2180-188 peptide is the class I MHC–restricted epitope recognized by CTLs to elicit antitumor response. Between mice immunized with either tumor lysate–pulsed or Trp2-pulsed DC vaccines, lymphocytes from TNP-470–treated DC vaccine groups significantly produced higher levels of IFN-γ upon ex vivo tumor lysate stimulation regardless of lysate or antigen pulsing (supplemental Figure 5A). Moreover, in terms of fold induction of IFN-γ production caused by TNP-470–treated DC vaccine when compared with vehicle DC vaccine, lymphocytes from Trp2-pulsed DC vaccine group exhibited a significantly higher fold induction (supplemental Figure 5B). These results indicated that specific antigen peptide Trp2-pulsed DCs might induce a stronger CTL response. Furthermore, similar to IFN-γ fold induction, LDH assay (supplemental Figure 5C) showed that immunization with TNP-470–treated DC vaccine in mice could elicit higher levels of tumor lysis when compared with vehicle DC vaccine group regardless of pulsing with lysate or peptide. Moreover, between the 2 TNP-470–treated DC vaccine, the tumor-killing capacity in lymphocytes from Trp2-pulsed DC vaccine group was higher than tumor lysate-pulsed DC vaccine group with marginal significance, thereby indicating stronger antitumor activity of these CTLs. All these data demonstrated that TNP-470–treated DC could induce tumor-specific immunogenicity, especially CTL response, to elicit tumor killing capacities in prophylactic and therapeutic vaccination models.
Collectively, our data revealed that TNP-470 could be adopted as an immune adjuvant for DC vaccine in cancer treatment given boosting DC immunogenicity is one of the biggest challenges in DC vaccine development.
Discussion
Fumagillin analog TNP-470, a drug under clinical trial for cancer treatment, warrants many studies due to its antiangiogenic effect. In this study, we reported that TNP-470 could potentiate DC immunogenicity and serve as an immune adjuvant in DC-based cancer vaccine. TNP-470 was reported to effectively suppress lymphocyte proliferation in vitro, therefore presenting a disadvantage for it to be a potent anticancer drug. As expected, we observed a potent in vitro inhibitory effect of TNP-470 on T-cell proliferation and IL-2 secretion. To the contrary, long term and direct treatment of TNP-470 in mice had no significant effect on the distribution of CD4 and CD8 T cells in spleen and lymph nodes, suggesting that the suppressive effects of TNP-470 on T cells in vivo is minimal when its antiangiogenic activity is potent. Similar observation was reported in the study of the less potent fumagillin showing that no significant change of T-cell proliferation in lung could be detected upon direct administration of fumagillin.33 Ma et al21 reported that application of fumagillin could affect HSC initiation and expansion elucidating the potential effect of fumagillin and its analogs on hematopoietic function. We found that TNP-470 at 5 nM had no obvious effect on colony formation of bone marrow progenitors (supplemental Figure 6A) and cell apoptosis of these progenitors (supplemental Figure 6B), thus implying TNP-470 might affect hematopoietic cell differentiation but not cell survival. Upon LPS stimulation, cytokine secretion profile in TNP-470–treated DCs altered and demonstrated an immunostimulatory pattern. Interestingly, Th1-polarizing cytokine IL-12 was substantially induced and immunosuppressive cytokine IL-10 was significantly reduced. Shift of IL-12/IL-10 balance suggested the Th1-polarized nature of the TNP-470–treated DCs. Our results, for the first time, demonstrated that TNP-470 modulates DC differentiation, making them as a kind of immunogenic APCs which favor to Th1 polarization. We also demonstrated that these “Th1 favorable DCs” upon TNP-470 treatment could enhance therapeutic potential of DC vaccine in cancer treatment. Given that DCVax and provenge (2 FDA-approved DC vaccines) has been adopted in clinical trials with promising outcomes,7,8 this is especially important when DC immunogenicity poses the greatest challenge for DC vaccine development.
DCs, the immune cells bridging the innate and adaptive immunity, represent a promising target in cancer therapy. However, the low therapeutic efficacy of DC vaccine limits its clinical use mainly due to the immunosuppressive nature of the tumor microenvironment.34-36 To overcome these challenges, different studies aim at boosting up DC immunogenicity to elicit strong Th1 immune response and effective CTL response.37 The in vivo OVA-specific Th1 and CTL responses induced by TNP-470–treated DCs prompted us to evaluate the therapeutic potential of these DCs in cancer immunotherapy. We found that DC vaccine could protect mice from melanoma development to some extent, but mice upon immunization with TNP-470–treated DC vaccine could have an additional protection from tumor development in both prophylactic and therapeutic settings. Accompanied with the additional tumor protection, these mice showed enhanced Th1 immune responses and elicited effective tumor-specific CTL responses. Compared with the prophylactic setting, in the therapeutic setting, the tumor protection induced by vehicle DC vaccine was much worse than TNP-470–treated DC vaccine, and it was probably due to less rounds of vaccinations and the aggressive nature of melanoma. Although potentiation of DC immunogenicity presents the major challenge for DC vaccine development, this observation highlights the exact utility and the requisite role of a DC adjuvant in improving the tumor protection of a DC vaccine. Our data revealed that TNP-470 could be exploited in DC vaccine as an immune adjuvant in cancer immunotherapy to boost up tumor-specific immunogenicity and cytolytic activity. Distinct from other studies in which the tumor-specific immunogenicity was enhanced by coadministration of DC vaccine and immune adjuvant, the current study showed that TNP-470 pretreatment could enhance the immunogenicity of DC vaccine and tumor-killing CTL responses. Furthermore, administration of this DC vaccine has no adverse effect on T cells in the host as TNP-470 has only been applied to the ex vivo DC culture. Our data also supported that TNP-470 enhances DC immunogenicity against not to a single type of antigen but to wide types of antigens indicating that TNP-470 helps DCs to present different antigens.
TLR signaling is responsible for IL-12 production and subsequent induction of DC immunogenicity. In DCs, we found that TNP-470 could promote NF-κB translocation in either resting or activation status. Our results also showed that TNP-470 enhanced recruitment of c-Rel, one of the 5 NF-κB subunits, to the IL-12p40 promoter and therefore upregulated IL-12 transcription. The unique role of c-Rel in IL-12 production was highlighted in a study showing that enhanced recruitment of c-Rel, but not other NF-κB subunits, to IL-12 promoter contributed to the abnormal IL-12 synthesis in IL-10–deficient DCs.38 Apart from the NF-κB activation, we found that phosphorylation of p38 and JNK were substantially induced in TNP-470–treated DCs and application of p38 inhibitor and JNK inhibitor could partially attenuate IL-12 induction mediated by TNP-470. Our study found that TNP-470 could potentiate TLR1/2/4 signaling through activation of NF-κB and MAPK pathways, therefore inducing IL-12 transcription and the subsequent DC immunogenicity.
We, for the first time, discovered the novel immunomodulatory effect of TNP-470 on DCs. TNP-470 could potentiate TLR1/2/4 pathways in DCs to induce IL-12 production and the subsequent Th1 immune response. The enhanced DC immunogenicity by TNP-470 could translate into clinical benefits as a novel adjuvant for DC vaccine. Previous study15 showed that the inhibitory effect on T lymphocyte proliferation and the antiangiogenic effect on endothelial cells by fumagillin analogs are correlated to their inhibitory potencies of MetAP2 activities. Although MetAP2 was identified as one of the primary binding targets of fumagillin and TNP-470, global expression analysis showed that MetAP2 expression was much lower in DCs than T lymphocytes. Furthermore, the distinct TLR-signaling pathways in DCs imply that the immunomodulatory effects of TNP-470 on DCs might be MetAP2-independent. It will be of interest to identify the DC-specific binding target(s) of TNP-470 in the future study. In the current study, we reported the distinct function of TNP-470 in DCs as a kind of immune adjuvant. Besides providing a repositioning perspective of TNP-470 in cancer therapy, TNP-470 also serves as an example of a small molecule with distinctive effects on DC immunogenicity and T-cell proliferation through our study.
In summary, our findings revealed TNP-470 skews DC differentiation toward Th1-stimulatory phenotype, and this novel biological function allows TNP-470 to be a potent immune adjuvant in ex vivo DC-based cancer vaccine.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Yan Chen, Jiao Peng, and Julia Tsang for technical assistance and critical discussion; Dan Yang for financial assistance; and Hong Lok Lung for critical reading of the manuscript. The authors are grateful to Chris Wong, Yu Zhiling, and Polly Leung for various reagents.
This work was supported by the Hong Kong Baptist University (HKBU) SDF 15-1012-P04, HKBU FRG2/16-17/061, RGC General Research Fund HKBU12143316.
Authorship
Contribution: D.H.-H.H. and R.H.-F.W. designed the research, analyzed results, made the figures, and wrote the paper/revision(s); and D.H.-H.H. performed experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for R.H.-F.W. is Anven AlzdX, Berkeley, CA.
Correspondence: Roger Hoi-Fung Wong, Department of Biology, Hong Kong Baptist University, T1014 Cha Chi-Ming Science Tower, Kowloon Tong, Hong Kong; e-mail: rogerwong@hkbu.edu.hk; roger_wong@berkeley.edu; rwong@anvenalzdx.com.
References
- 1.Strioga M, Schijns V, Powell DJ Jr, Pasukoniene V, Dobrovolskiene N, Michalek J. Dendritic cells and their role in tumor immunosurveillance. Innate Immun. 2013;19(1):98-111. [DOI] [PubMed] [Google Scholar]
- 2.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23(1):975-1028. [DOI] [PubMed] [Google Scholar]
- 3.Constantino J, Gomes C, Falcão A, Neves BM, Cruz MT. Dendritic cell-based immunotherapy: a basic review and recent advances. Immunol Res. 2017;65(4):798-810. [DOI] [PubMed] [Google Scholar]
- 4.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117(5):1195-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rolinski J, Hus I. Breaking immunotolerance of tumors: a new perspective for dendritic cell therapy. J Immunotoxicol. 2014;11(4):311-318. [DOI] [PubMed] [Google Scholar]
- 7.Hdeib A, Sloan AE. Dendritic cell immunotherapy for solid tumors: evaluation of the DCVax® platform in the treatment of glioblastoma multiforme. CNS Oncol. 2015;4(2):63-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholz M, Yep S, Chancey M, et al. . Phase I clinical trial of sipuleucel-T combined with escalating doses of ipilimumab in progressive metastatic castrate-resistant prostate cancer. ImmunoTargets Ther. 2017;6:11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin CC, Pan IH, Li YR, et al. . The adjuvant effects of high-molecule-weight polysaccharides purified from Antrodia cinnamomea on dendritic cell function and DNA vaccines. PLoS One. 2015;10(2):e0116191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang WT, Lai TH, Chyan YJ, et al. . Specific medicinal plant polysaccharides effectively enhance the potency of a DC-based vaccine against mouse mammary tumor metastasis. PLoS One. 2015;10(3):e0122374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Ma X, Su C, et al. . Uric acid enhances the antitumor immunity of dendritic cell-based vaccine. Sci Rep. 2015;5(1):16427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seth A, Heo MB, Sung MH, Lim YT. Infection-mimicking poly(γ-glutamic acid) as adjuvant material for effective anti-tumor immune response. Int J Biol Macromol. 2015;75:495-504. [DOI] [PubMed] [Google Scholar]
- 13.Kang TH, Kim YS, Kim S, et al. . Pancreatic adenocarcinoma upregulated factor serves as adjuvant by activating dendritic cells through stimulation of TLR4. Oncotarget. 2015;6(29):27751-27762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci USA. 1997;94(12):6099-6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turk BE, Su Z, Liu JO. Synthetic analogues of TNP-470 and ovalicin reveal a common molecular basis for inhibition of angiogenesis and immunosuppression. Bioorg Med Chem. 1998;6(8):1163-1169. [DOI] [PubMed] [Google Scholar]
- 16.Yamaoka M, Yamamoto T, Ikeyama S, Sudo K, Fujita T. Angiogenesis inhibitor TNP-470 (AGM-1470) potently inhibits the tumor growth of hormone-independent human breast and prostate carcinoma cell lines. Cancer Res. 1993;53(21):5233-5236. [PubMed] [Google Scholar]
- 17.Shusterman S, Grupp SA, Barr R, Carpentieri D, Zhao H, Maris JM. The angiogenesis inhibitor tnp-470 effectively inhibits human neuroblastoma xenograft growth, especially in the setting of subclinical disease. Clin Cancer Res. 2001;7(4):977-984. [PubMed] [Google Scholar]
- 18.Nahari D, Satchi-Fainaro R, Chen M, et al. . Tumor cytotoxicity and endothelial Rac inhibition induced by TNP-470 in anaplastic thyroid cancer. Mol Cancer Ther. 2007;6(4):1329-1337. [DOI] [PubMed] [Google Scholar]
- 19.Kruger EA, Figg WD. TNP-470: an angiogenesis inhibitor in clinical development for cancer. Expert Opin Investig Drugs. 2000;9(6):1383-1396. [DOI] [PubMed] [Google Scholar]
- 20.Benny O, Fainaru O, Adini A, et al. . An orally delivered small-molecule formulation with antiangiogenic and anticancer activity. Nat Biotechnol. 2008;26(7):799-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma AC, Fung TK, Lin RH, et al. . Methionine aminopeptidase 2 is required for HSC initiation and proliferation. Blood. 2011;118(20):5448-5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamala T. Hock immunization: a humane alternative to mouse footpad injections. J Immunol Methods. 2007;328(1-2):204-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maret A, Coudert JD, Garidou L, et al. . Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor alpha expression in hematopoietic cells. Eur J Immunol. 2003;33(2):512-521. [DOI] [PubMed] [Google Scholar]
- 24.Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136(6):1056-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fields RC, Shimizu K, Mulé JJ. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95(16):9482-9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohta Y, Watanabe Y, Tabata T, et al. . Inhibition of lymph node metastasis by an anti-angiogenic agent, TNP-470. Br J Cancer. 1997;75(4):512-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Frischer JS, New T, et al. . TNP-470 promotes initial vascular sprouting in xenograft tumors. Mol Cancer Ther. 2004;3(3):335-343. [PubMed] [Google Scholar]
- 28.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. 2004;172(1):61-69. [DOI] [PubMed] [Google Scholar]
- 29.Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006;117(4):433-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Met O, Buus S, Claesson MH. Peptide-loaded dendritic cells prime and activate MHC-class I-restricted T cells more efficiently than protein-loaded cross-presenting DC. Cell Immunol. 2003;222(2):126-133. [DOI] [PubMed] [Google Scholar]
- 31.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18(1):621-663. [DOI] [PubMed] [Google Scholar]
- 32.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci USA. 2000;97(23):12705-12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kass D, Bridges RS, Borczuk A, Greenberg S. Methionine aminopeptidase-2 as a selective target of myofibroblasts in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2007;37(2):193-201. [DOI] [PubMed] [Google Scholar]
- 34.Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22(8):1897-1906. [DOI] [PubMed] [Google Scholar]
- 35.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spitzer MH, Carmi Y, Reticker-Flynn NE, et al. . Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168(3):487-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong H, Shurin MR, Han B. Optimizing dendritic cell-based immunotherapy for cancer. Expert Rev Vaccines. 2007;6(3):333-345. [DOI] [PubMed] [Google Scholar]
- 38.Hoentjen F, Sartor RB, Ozaki M, Jobin C. STAT3 regulates NF-kappaB recruitment to the IL-12p40 promoter in dendritic cells. Blood. 2005;105(2):689-696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.