

Abstract
Essentials.
Idarucizumab, a monoclonal antibody fragment, binds dabigatran with high affinity and specificity.
In this phase 1 trial, healthy Japanese males received idarucizumab alone or with dabigatran.
Idarucizumab achieved immediate, complete and sustained reversal of dabigatran anticoagulation.
Idarucizumab was well tolerated and demonstrated no pro‐coagulant effects.
Background
Idarucizumab is a humanized monoclonal antibody fragment that specifically binds with high affinity to dabigatran.
Objectives
This study investigated the safety, tolerability and pharmacokinetics of idarucizumab alone and with dabigatran at steady state, and the effects of idarucizumab on dabigatran‐induced anticoagulation.
Patients/Methods
This was a two‐part, phase I, randomized, placebo‐controlled, double‐blind, rising‐dose trial in healthy Japanese males. Part 1: 32 subjects (males) received single idarucizumab doses (1, 2, 4 or 8 g [n=6/dose group]) or placebo (n=2/dose group). Part 2: 48 males received dabigatran (220 mg bid) followed by idarucizumab (n=9/dose group) 1, 2, 4 or 5 g (2×2.5 g), or placebo (n=3/dose group). Anti‐idarucizumab antibodies (ADAs) and idarucizumab effect on anticoagulation parameters (diluted thrombin time [dTT], ecarin clotting time [ECT], activated partial thromboplastin time [aPTT] and thrombin time [TT]) were assessed.
Results
No adverse events were reported in subjects receiving idarucizumab. After single doses of idarucizumab (alone or at steady state of dabigatran), maximum plasma concentration was achieved around the end of each infusion. Mean all anticoagulation parameters fell below the upper limit of normal immediately after idarucizumab infusion in all dose groups; the effect was sustained at 4 and 2×2.5 g over the entire measurement period until 72 h. At 1‐ and 2‐g doses, partial return of the anticoagulant effect occurred. Idarucizumab alone had no effect on coagulation parameters. Treatment‐emergent ADAs occurred in 6/60 males receiving idarucizumab.
Conclusions
Idarucizumab infusion achieved immediate, complete and sustained reversal of dabigatran‐induced anticoagulation in Japanese volunteers. Idarucizumab was well tolerated with no procoagulant effects. Trial registration number: ClinicalTrials.gov NCT02028780 (completed)
Keywords: dabigatran etexilate, idarucizumab, Japanese, reversal agent, safety
1. INTRODUCTION
The direct oral anticoagulant (DOAC) dabigatran is a direct thrombin inhibitor. Dabigatran etexilate (DE), the prodrug of dabigatran, is approved for prevention of stroke in patients with atrial fibrillation (AF) and for the treatment and secondary prevention of venous thromboembolism. In clinical trials and real‐world observational studies, dabigatran was at least as effective as warfarin, with a favorable bleeding profile.1, 2, 3, 4, 5
A sub‐analysis in Japanese patients from the Randomized Evaluation of Long‐term anticoagulant therapY (RE‐LY) trial indicated similar safety and efficacy for DE vs warfarin and higher bleeding rates for warfarin in Asians vs non‐Asians6, 7; this was attributed to postulated genetic differences in blood coagulation between Asian and non‐Asian subjects. The reductions in hemorrhagic stroke and major bleeding rates were comparable between Asians vs non‐Asian patients treated with DE.8
Nevertheless, all anticoagulants are associated with risk of severe bleeding.9, 10, 11, 12 Owing to the relatively short half‐life of dabigatran (12‐14 hour),13 cessation of treatment and standard supportive care (eg, volume replacement) can be used to manage bleeding in many patients with adequate renal function, but this may be inadequate in emergency situations.13, 14, 15, 16
Consequently, there has been a demand for rapid, specific reversal agents for use in cases of rare severe bleeding or when urgent surgery or interventions are needed. One such reversal agent is idarucizumab, a humanized monoclonal antibody fragment, which binds dabigatran with high affinity and specificity.17
A phase I study of idarucizumab in healthy, predominantly Caucasian, male volunteers demonstrated that peak plasma exposure of idarucizumab was achieved at, or shortly after a 5‐minute intravenous infusion; this was followed by rapid elimination within 24 hour.18 Idarucizumab treatment in Caucasian volunteers receiving dabigatran led to immediate, complete, and sustained reversal of anticoagulation and was well tolerated.19 In an interim analysis of a phase III trial in dabigatran‐treated patients with severe bleeding or in need of an urgent surgery/procedure, idarucizumab completely reversed the anticoagulant effect of dabigatran within minutes.17 Safety and efficacy data in Asians are limited.
The present phase I study in healthy Japanese male volunteers was designed to confirm the safety, tolerability, and pharmacokinetics of idarucizumab alone and with dabigatran at steady state, and to explore the effect of ascending doses of idarucizumab on the pharmacokinetics of dabigatran and the reversal of dabigatran anticoagulant activity (pharmacodynamics).
2. MATERIALS AND METHODS
2.1. Study design
This randomized, double‐blind (within dose groups), placebo‐controlled, single‐center trial was conducted at Medical Co. LTA Sumida Hospital, Tokyo, Japan (January‐August 2014). It comprised two parts; in both parts, male subjects were randomized in a 3:1 ratio, idarucizumab to placebo. (ClinicalTrials.gov registration no. NCT02028780).
Part 1 was a consecutive rising‐dose investigation of the safety, tolerability, and pharmacokinetics of a range of intravenous doses of idarucizumab alone (Figure 1A).
Figure 1.
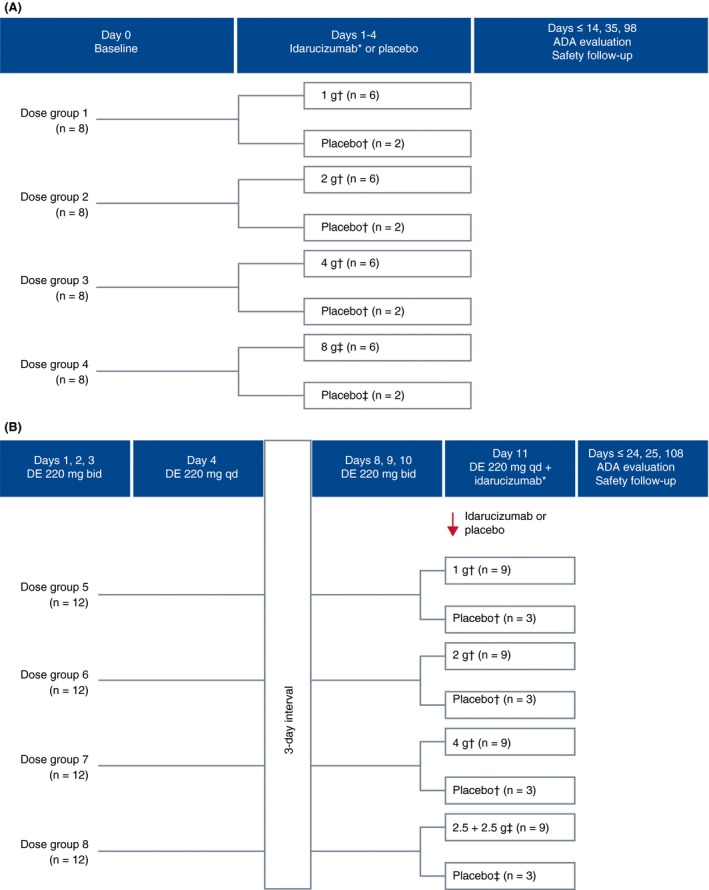
Study design. (A) Part 1. Subjects received either placebo (n=2/dose group) or single intravenous doses of idarucizumab, 1, 2, or 4 g as a 5‐minute infusion or 8 g as a 1‐hour infusion (n=6/group). ADA=antidrug antibody. *21‐hour interval between dose levels. †Dose as a 5‐minute infusion. ‡Dose as a 1‐hour infusion. (B) Part 2. Subjects received oral DE for 4 days (220 mg bid: days 1, 2, and 3; 220 mg: the morning of day 4) followed by a 3‐day wash‐out period. DE was readministered on days 8, 9, and 10 at 220 mg bid, with a morning 220‐mg dose on day 11. On day 11, idarucizumab doses of 1, 2, or 4 g were administered as 5‐minute intravenous infusions ~2 hour after the last dabigatran dose corresponding to the timing around the maximum plasma concentration (C max) of dabigatran; 5 g was administered as two 5‐minute infusions of 2.5 g separated by a 15‐minute interval. In each 12‐person dose group, 3 subjects were randomized to placebo and 9 to idarucizumab. ADA, antidrug antibody; bid, twice daily; DE, dabigatran etexilate; qd, once daily. *21‐hour interval between dose levels. †Dose as a 5‐minute infusion. ‡Dose as two 5‐minute infusions separated by 15 minute
Part 2 investigated the safety, tolerability, and pharmacokinetics of idarucizumab in the presence of dabigatran. It also explored the pharmacokinetics of dabigatran after administration of idarucizumab and the ability of different doses of idarucizumab to reverse the anticoagulant activity of dabigatran through measurement of pharmacodynamic parameters (Figure 1B).
Idarucizumab doses were selected based on the phase I trial of healthy Caucasian male volunteers.18, 19 In both parts of the present study, idarucizumab was started at a dose of 1 g as a safety precaution. The next dose level was administered after evaluation of the safety data covering at least a 21‐hour period. Subjects (males) remained on site for at least 72 hour following drug administration at each dose level. To allow for monitoring of the effects of the highest dose of idarucizumab (8 g), the active drug, or its placebo, was administered first in two subjects, then in a further two, followed by the remaining four. Both the subject and investigator were blinded to treatment with the study drug or placebo, although the current dose level was known.
A dosing schedule of DE 220 mg bid for the first 3 days and once daily on day 4 in a fasting state was selected to investigate safety. The same dosing schedule for DE was repeated on day 8 and ended on day 11 to examine the efficacy of idarucizumab to reverse the anticoagulant activity of DE. This dose of DE was selected to cover peak plasma concentration levels in the RE‐LY trial where plasma concentration around 2‐hour post‐dose achieved was 184 ng/mL20 and corresponded with the dosing schedule of the phase I trial in Caucasian subjects (males)19 (NCT identifier, NCT01688830). At this dosage, the C max of dabigatran at steady state covers the C max of dabigatran after administration of 150 mg bid to the patient population in the phase III global study conducted in patients with nonvalvular atrial fibrillation including Japanese individuals (unpublished data).
2.2. Study subjects and conduct
This two‐part study randomized 80 healthy male Japanese volunteers aged ≥20 and ≤45 years (body mass index ≥18.5 and <25.0 kg/m2). Exclusion criteria included any abnormal findings at the screening examination; clinically relevant comorbidities including gastrointestinal, hepatic, renal, cardiovascular, metabolic, central nervous system, immunological or hormonal disorder; history or evidence of a blood disorder; and subjects unable to refrain from smoking during hospitalization (or a current smoker >10 cigarettes/day or equivalent).
The study protocol was approved by an institutional review board. The trial was reported in line with CONSORT guidelines and conducted according to the Declaration of Helsinki, the International Conference on Harmonization good clinical practice (GCP), and relevant regulatory and GCP requirements. All subjects provided written informed consent.
The randomization list was generated using a validated pseudo‐random number generator and a supplied seed number to create reproducible and nonpredictable allocation.
2.3. Antidrug antibody analysis
For the analysis of anti‐idarucizumab antibodies (ADAs), samples were collected at baseline, end of study (EOS), and 4‐week and 3‐month follow‐ups. ADAs were detected in human plasma samples by a validated bridging electrochemiluminescence method (Covance Laboratories Inc., Chantilly, VA, USA). Positive samples were subjected to a confirmatory assay and additional analyses. ADAs were classified as pre‐existing (present in the baseline sample before study drug administration) or treatment‐emergent (present/raised after idarucizumab administration).
ADA titer was determined as the highest dilution factor at which the mean signal for the sample remains above or at the assay cut‐point level. The titer is expressed as the dilution factor. For example, for a sample that was positive with a 40‐fold dilution but negative with an 80‐fold dilution, the assigned titer would be 40. The assay did not have an initial minimum required dilution. Therefore, a titer of 1 is assigned to a sample that was positive only when assayed undiluted.
2.4. Pharmacokinetic/pharmacodynamic analyses
Pharmacokinetic and pharmacodynamic analyses were performed using validated assay methods.21, 22 The detailed methods of analyses are provided in the supplementary material.
2.5. Outcomes
2.5.1. Safety outcomes
The primary safety endpoint was the number (%) of subjects with adverse events (AEs) assessed by the investigator to be drug‐related. Safety outcomes included AEs, laboratory tests (including hematology, clinical chemistry, urine analysis, coagulation parameters and antidrug antibodies), electrocardiogram (ECG), vital signs and local tolerability.
2.5.2. Pharmacokinetic parameters
Pharmacokinetic parameters in part 1 were: idarucizumab C max, area under the curve (AUC0‐∞), and excretion after 72 hour (Ae0‐72; Ae0‐73 for the 8‐g group). In part 2, these were: idarucizumab C max, AUC0‐∞, Ae0‐72, total dabigatran Ae0‐74 and unbound dabigatran AUC2‐12 on days 4 and 11.
2.5.3. Pharmacodynamic parameters
The secondary pharmacodynamic endpoint in study part 2 was: area under the effect curve between 2 and 12 hour on days 4 and 11 for dTT. Additional clotting tests (ECT, TT, aPTT) were included.
Upper limit of normal (ULN) reference values for the coagulation parameters were determined based on mean values +2 standard deviation of all individual baseline measurements obtained before treatment with 4‐ or 8‐g idarucizumab without DE (part 1), and before the start of administration of DE in all study arms (part 2).
2.6. Statistical analyses
The planned sample size (n=80) was based on previous phase I trials of idarucizumab. A group size between 8 and 12 per dose level is commonly used in healthy subject trials and was considered sufficient for exploratory investigation of single/multiple dose safety and pharmacokinetics.23 Descriptive statistics for safety, pharmacokinetic, and pharmacodynamic endpoints were calculated. Dose proportionality of idarucizumab was assessed using a regression model with 95% confidence interval for the slope.
All parameters are presented using descriptive statistics; no formal statistical tests were undertaken.
3. RESULTS
3.1. Subjects
In total, 81 males were enrolled in the study; 80 were randomized to part 1 (n=32) or part 2 (n=48) (Figure 2). All subjects in part 1 and all randomized subjects from part 2 (one subject was withdrawn before randomization because of AEs [bruising of the leg, hemorrhage and swelling] after receiving dabigatran) completed the study (Figure 2). Demographic characteristics were similar between idarucizumab and placebo groups (Table 1).
Figure 2.
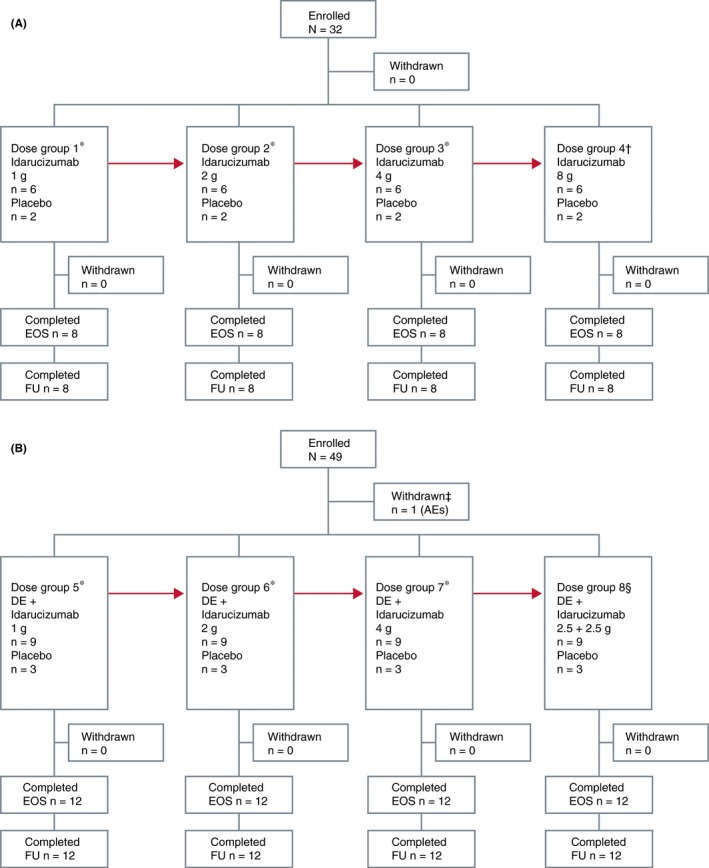
CONSORT diagram: flow of patients through study. (A) Part 1 and (B) part 2. AEs, adverse events; DE, dabigatran etexilate; EOS, end of study; FU, follow‐up. *Infusion time=5 minute. †Infusion time=1 hour. ‡In “dabigatran period.” §Infusion time=5+5 minute with an interval of 15 minute
Table 1.
Demographics and baseline characteristics
| Part 1 | |||||||
|---|---|---|---|---|---|---|---|
| Treatment | Placebo | Idarucizumab | Total | ||||
| Idarucizumab dose and infusion time | 5 minute | 1 hour | 1 g 5 minute | 2 g 5 minute | 4 g 5 minute | 8 g 1 hour | |
| Number of subjects, n | 6 | 2 | 6 | 6 | 6 | 6 | 32 |
| Age, mean, years (±SD) | 28.7 (10.5) | 32.5 (4.9) | 31.5 (10.9) | 27.8 (7.7) | 31.5 (8.3) | 32.2 (8.1) | 30.5 (8.5) |
| Weight, mean, kg (±SD) | 68.7 (5.5) | 65.5 (3.5) | 66.0 (9.8) | 62.3 (6.0) | 67.2 (6.6) | 61.5 (6.7) | 65.2 (7.0) |
| BMI, mean, kg/m2 (±SD) | 22.9 (1.3) | 21.6 (1.1) | 21.8 (1.9) | 22.0 (2.3) | 22.8 (1.6) | 21.1 (2.3) | 22.1 (1.9) |
| Part 2 | |||||||
|---|---|---|---|---|---|---|---|
| Treatment | DEa+placebo | DEa+placebo | DEa+idarucizumab | Total | |||
| Idarucizumab dose and infusion time | 5 minute | 5 minute+5 minute | 1 g 5 minute | 2 g 5 minute | 4 g 5 minute | 2.5 + 2.5 g 5 minute+5 minute | |
| Number of subjects, n | 9 | 3 | 9 | 9 | 9 | 9 | 48 |
| Age, mean, years (±SD) | 24.7 (3.3) | 29.7 (5.0) | 23.8 (3.1) | 25.7 (3.1) | 25.4 (2.7) | 24.4 (1.4) | 25.1 (3.1) |
| Weight, mean, kg (±SD) | 61.4 (6.2) | 60.7 (5.9) | 57.0 (4.5) | 61.0 (8.2) | 65.0 (4.3) | 62.8 (7.6) | 61.4 (6.5) |
| BMI, mean, kg/m2 (±SD) | 21.4 (1.8) | 20.2 (1.6) | 20.6 (1.3) | 21.3 (1.7) | 21.5 (2.0) | 21.2 (2.0) | 21.2 (1.7) |
BMI, body mass index; DE, dabigatran etexilate; SD, standard deviation.
DE 220 mg twice daily.
3.1.1. Safety outcomes
AEs
In part 1, one of six (16.7%) subjects receiving placebo (5‐minute infusion) had an elevated blood creatine phosphokinase level, which was mild and assessed as drug‐related by the investigator; no AEs were reported (drug‐related or otherwise) in remaining subjects. There were no AEs reported in part 2. Thus, in both parts of the study, there were no severe or serious AEs in randomized patients. Additionally, no clinically significant changes were seen in other laboratory and safety parameters, including 12‐lead ECG, vital signs and local infusion tolerability.
ADA pooled analysis
Of the 80 subjects in both study parts, 14 (17.5%) were positive for pre‐existing ADA, with positive titers ranging from 1 to 20. For 12 of these subjects (four placebo, eight idarucizumab), titers of pre‐existing ADA were relatively constant throughout the study.
Six of 60 (10%) idarucizumab‐treated subjects developed treatment‐emergent ADAs with positive titers ranging from 1 to 40, the latter value being reported in one subject. Two subjects had pre‐existing ADAs, and their ADA titers increased at follow‐up visits at 4 weeks and 3 months. Four subjects had no detectable ADA at baseline, but positive ADA titers were recorded at follow‐up visits. Of these six males, two had transient responses (one observed at the EOS and 4‐week time‐points but not at the 3‐month follow‐up; one observed only at EOS); four had possibly persistent responses, including the two males with pre‐existing antibodies who displayed increased titers during treatment. All of these responses were classified as possibly persistent because they were detected at 3‐month follow‐up and there was no later sampling point to determine if there was any subsequent decline in antibody titer. No relationship was found between dose of idarucizumab and the titer of treatment‐emergent antibodies. Among samples testing positive for ADA (pre‐existing and treatment‐emergent including follow‐up periods) the distribution of titer values was: titer value 1, n=4; titer value 2, n=16; titer value 4, n=2; titer value 8, n=2; titer value 10, n=1; titer value 16, n=1; titer value 20, n=3; titer value 40, n=1.
Idarucizumab pharmacokinetics
Plasma concentration–time profiles of idarucizumab in the presence and absence of dabigatran were similar but concentrations were slightly lower in the presence of dabigatran compared with those of the same dose without dabigatran (Figure 3). Pharmacokinetic parameters for idarucizumab were comparable whether administered alone or at dabigatran steady state (Table 2), except for urinary excretion and renal clearance (CLR).
Figure 3.
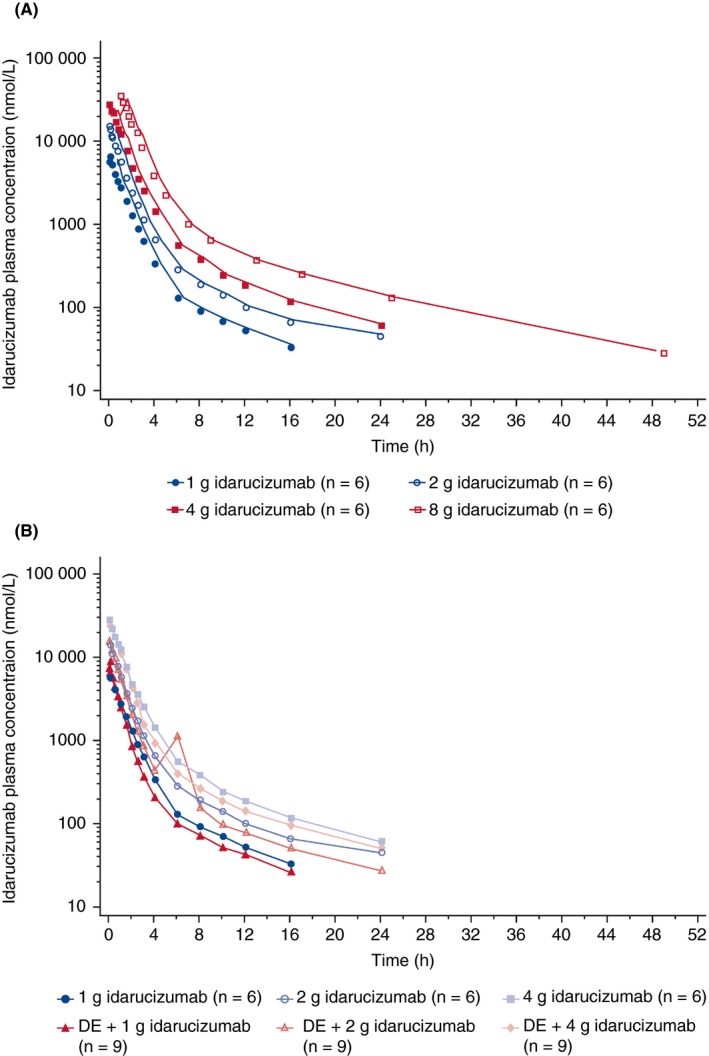
Mean plasma concentration–time profiles of idarucizumab. (A) Idarucizumab alone 1‐4 g (5‐minute infusion) or 8 g (1‐hour infusion) (semi‐log scale). (B) Idarucizumab alone 1‐4 g (5‐minute infusion) or idarucizumab + dabigatran at steady state (semi‐log scale). DE, dabigatran etexilate
Table 2.
Pharmacokinetic parameters of idarucizumab alone and with dabigatran etexilate
| Part 1 | Idarucizumab | |||||||
|---|---|---|---|---|---|---|---|---|
| Idarucizumab dose | 1 g (n=6) | 2 g (n=6) | 4 g (n=6) | 8 g (n=6) | ||||
| Infusion time, min | 5 | 5 | 5 | 60 | ||||
| gMean | gCV, % | gMean | gCV, % | gMean | gCV, % | gMean | gCV, % | |
| AUC0‐∞, nmol·h/L | 9150 | 15.0 | 19 500 | 17.8 | 37 600 | 14.4 | 76 800 | 14.8 |
| AUC0‐∞ norm, nmol·h/L/mg | 9.15 | 15.0 | 9.76 | 17.8 | 9.39 | 14.4 | 9.60 | 14.8 |
| C max, nmol/L | 6810 | 10.1 | 15 700 | 10.9 | 28 100 | 14.3 | 37 600 | 6.88 |
| C max, norm, nmol/L/mg | 6.81 | 10.1 | 7.86 | 10.9 | 7.03 | 14.3 | 4.70 | 6.88 |
| t max, ha | 0.142 | 0.117‐0.167 | 0.117 | 0.0830‐0.167 | 0.0830 | 0.0830‐0.117 | 1.06 | 1.00‐1.08 |
| t1/2, h | 6.38 | 15.4 | 10.3 | 31.4 | 7.62 | 6.46 | 9.70 | 9.98 |
| t1/2, 2, ha | 0.629 | 22.3 | 0.645 | 32.4 | 0.707 | 25.4 | 1.04 | 53.2 |
| Fe0‐72, %d | 7.38 | 54.9 | 14.6 | 165 | 24.5 | 52.4 | 41.3 | 25.0 |
| CL, mL/min | 38.1 | 15.0 | 35.7 | 17.8 | 37.1 | 14.4 | 36.3 | 14.8 |
| CLR,0‐4, mL/mind | 3.32 | 45.3 | 6.23 | 180 | 10.7 | 48.1 | 16.7 | 32.4 |
| VSS, L | 6.21 | 14.1 | 7.45 | 26.4 | 6.17 | 17.7 | 6.85 | 14.9 |
| Part 2 | Dabigatran etexilate 220 mg+idarucizumab | |||||||
|---|---|---|---|---|---|---|---|---|
| Idarucizumab dose | 1 g (n=9) | 2 g (n=9) | 4 g (n=9) | 2.5+2.5 g (n=9) | ||||
| Infusion time, min | 5 | 5 | 5 | 5+5 | ||||
| gMean | gCV, % | gMean | gCV, % | gMean | gCV, % | gMean | gCV, % | |
| AUC0‐∞, nmol·h/L | 8590 | 14.2 | 19 200 | 18.5 | 34 500c | 16.6c | 43 300 | 8.25 |
| AUC0‐∞ norm, nmol·h/L/mg | 8.59 | 14.2 | 9.59 | 18.5 | 8.63c | 16.6c | 8.67 | 8.25 |
| C max, nmol/L | 9510 | 33.8 | 17 600 | 16.8 | 30 200 | 17.7 | 30 100 | 11.5 |
| C max, norm, nmol/L/mg | 9.51 | 33.8 | 8.78 | 16.8 | 7.54 | 17.7 | 6.03 | 11.5 |
| t max, ha | 0.167 | 0.0830‐0.167 | 0.117 | 0.0830‐0.167 | 0.117 | 0.0830‐0.333 | 0.417 | 0.417‐0.583 |
| t 1/2, h | 6.01 | 33.3 | 7.31 | 13.5 | 9.06c | 24.1a | 7.91 | 9.33 |
| t 1/2, 2, ha | 0.520 | 59.6 | 0.674 | 26.5 | 0.668 | 34.3 | 0.758 | 13.0 |
| Fe0‐72, %d | 20.1 | 35.1 | 33.0 | 12.5 | 50.9 | 16.4 | 49.3 | 18.1 |
| CL, mL/min | 40.6 | 14.2 | 36.3 | 18.5 | 40.4 | 16.6 | 40.2 | 8.25 |
| CLR,0‐4, mL/mind | 9.34 | 28.5 | 14.5 | 19.1 | 23.6 | 21.6 | 22.9 | 21.5 |
| VSS, L | 5.57 | 28.0 | 5.45 | 19.7 | 6.38 | 27.5 | 6.53 | 10.2 |
AUC, area under the curve; CL, total clearance; CLR,0‐4, renal clearance between 0 and 4 hour; C max, maximum plasma concentration; Fe0‐72, fraction excreted in urine between 0 and 72 hour; gCV, geometric coefficient of variation; gMean, geometric mean; t max, time to maximum plasma concentration; t1/2, terminal half‐life; t1/2, 2, initial half‐life; fe, fraction excreted in urine; VSS, volume of distribution at steady state.
Median (min–max).
Initial half‐life.
n=8.
fe0‐73[%] and CLR,0‐7 for 8 g.
After administration of a single dose of idarucizumab 1‐8 g alone or at dabigatran steady state, C max was achieved directly at the end of each infusion, followed by a biphasic decline in plasma concentrations with a rapid initial phase and a longer terminal phase. After administration of two idarucizumab doses 15 minute apart, C max was achieved around the end of the second infusion.
AUC, as a measure of exposure, increased dose‐proportionally with or without dabigatran in the range of idarucizumab 1‐8 g. C max increased proportionally in the absence of dabigatran; however, it increased sub‐proportionally in the presence of dabigatran in the range of idarucizumab 1‐4 g. For C max, the 8‐g dose group was excluded due to the different infusion time. Inter‐individual variability was low.
The geometric mean (gMean) initial half‐life (t 1/2, 2) and gMean terminal elimination half‐life of idarucizumab, administered with or without dabigatran, were 0.52‐1.04 hour and 6.0‐10.3 hour, respectively, and were independent of dose. Volume of distribution of idarucizumab, administered with or without dabigatran, was ~5.5‐7.5 L, and total plasma clearance was ~36‐41 mL/min, both being relatively constant across the dose range. Urinary excretion and CLR of idarucizumab at 1‐4‐g doses in the presence of dabigatran at steady state were approximately two to three times higher than without dabigatran, but were comparable at 5‐ and 8‐g doses.
Dabigatran pharmacokinetics
The plasma concentration–time profile of active dabigatran alone was as expected13 (Figure 4A). Plasma concentrations of unbound dabigatran (Figure 4B) and active dabigatran decreased to below the limit of quantification (BLQ) or close to BLQ immediately post‐idarucizumab. After idarucizumab infusion, plasma concentrations of total dabigatran rapidly increased by approximately five‐ to six‐fold in all dose groups, reaching a maximum in ~0.5 hour (Figure 4A), which reflects the rapid redistribution and binding of unbound dabigatran from the peripheral compartment as soon as the central portion is bound by idarucizumab.
Figure 4.
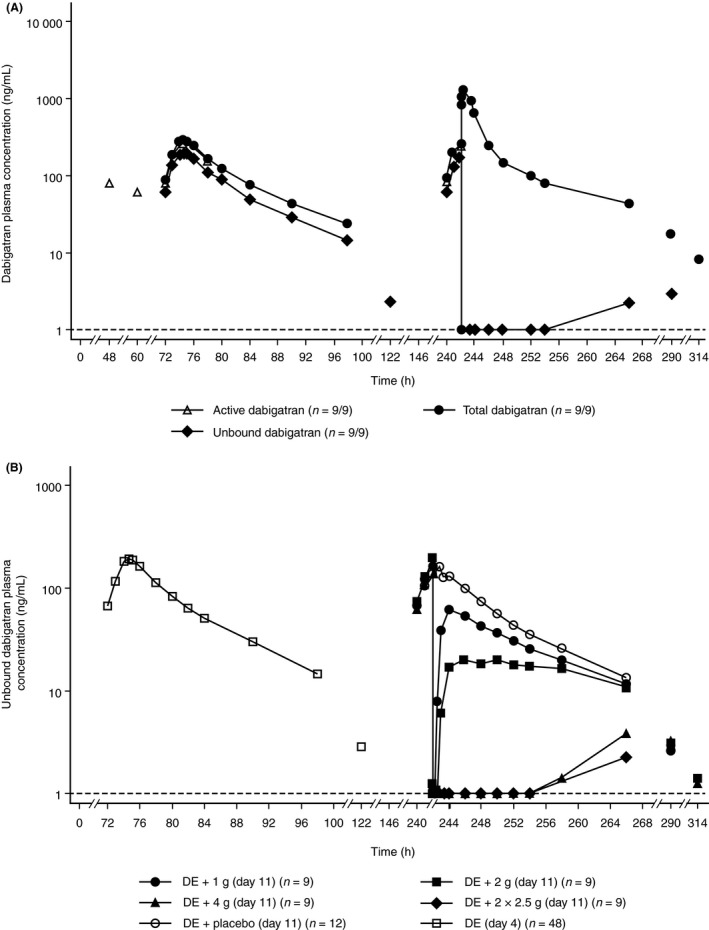
Arithmetic mean plasma concentration–time profiles. (A) Total dabigatran, unbound dabigatran, and active dabigatran in the absence (day 4) or presence (day 11) of idarucizumab 2.5+2.5 g. (B) Unbound dabigatran in the absence (day 4) or presence (day 11) of idarucizumab 1, 2, 4, and 5 g at dabigatran steady state (semi‐log‐scale). DE, dabigatran etexilate
At lower doses of 1 and 2 g of idarucizumab, measurable levels of unbound dabigatran were found up to 48 and 72 hour after administration, respectively. At the 4‐g dose of idarucizumab, unbound dabigatran was measurable only from 24 to 72 hour and 24 to 48 hour at low levels below 4 ng/mL. In the 2×2.5‐g idarucizumab dose group, measurable levels of unbound dabigatran were present only after 48 hour, much later than the timeframes at the lower doses (Figure 4B).
3.1.2. Pharmacodynamic effects on coagulation parameters
Idarucizumab alone
In subjects receiving idarucizumab alone, no differences were observed in coagulation parameters (dTT, ECT, TT, aPTT) between pre‐ and post‐dose measurements, regardless of idarucizumab dose. Idarucizumab had no apparent effect on endogenous thrombin formation, as measured by CAT.
Idarucizumab reversal of dabigatran‐induced anticoagulation
On day 4, administration of DE resulted in prolongation of clotting times, as expected (data not shown). Idarucizumab 1, 2, 4, or 2.5+2.5 g resulted in immediate and complete reversal of anticoagulant effect, shown by mean dTT falling to below the ULN; the reversal effect was not observed after placebo (Figure 5A and B). Mean dTT remained below the ULN during the observed period for up to 72 hour after administration of idarucizumab 4 g (Figure 5A) and 2.5+2.5 g (Figure 5B). At lower doses, mean coagulation times showed a partial return of the anticoagulant effect of dabigatran after 1‐2 hour following administration of 1‐ or 2‐g idarucizumab (Figure 5A). ECT (Figure 5C and D), aPTT (Figure 5E and F), and TT (Figure 5G and H) showed similar profiles to dTT post‐idarucizumab.
Figure 5.
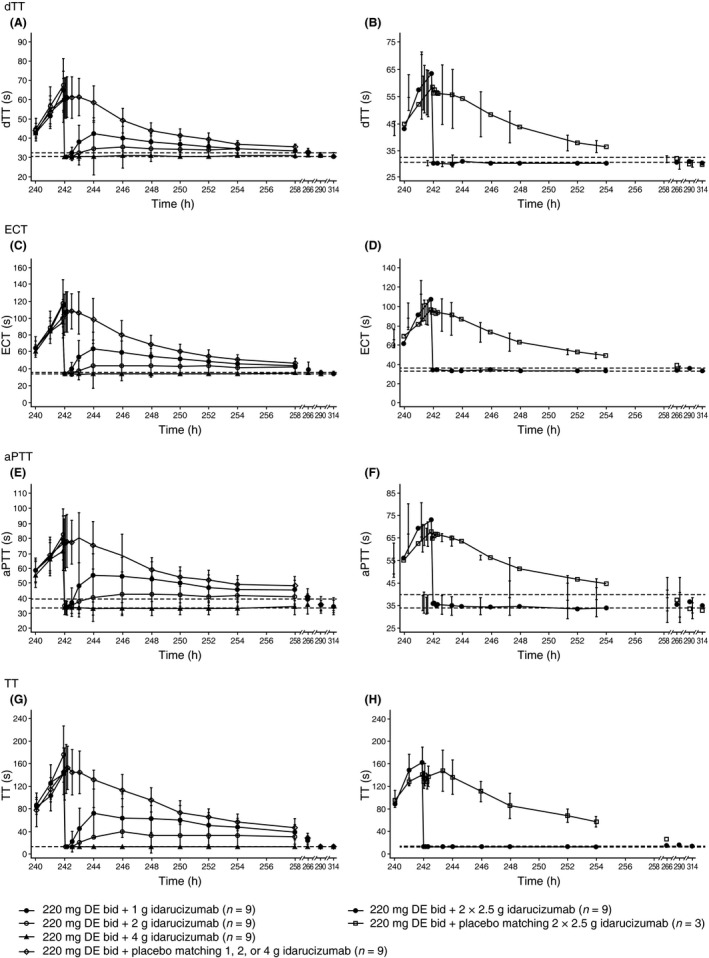
Effect of idarucizumab or placebo infusion on day 11 of coagulation variables, by dose group. Effect of idarucizumab (1, 2, 4, 2.5+2.5 g) and placebo on (A, B) diluted thrombin time (dTT), (C, D) ecarin clotting time (ECT), (E, F) activated partial thromboplastin time (aPTT), and (G, H) thrombin time (TT). bid, twice daily; DE, dabigatran etexilate
As expected, thrombin generation and derived parameters were raised after the last dose of dabigatran. In particular, the thrombin lag time ratio and thrombin time‐to‐peak ratio increased approximately 5‐ and 2.5‐fold, respectively. The ratio of baseline values to post‐idarucizumab thrombin generation parameter values demonstrated an increase relative to baseline; post‐idarucizumab (2‐5 g) and were close to 1. Changes in thrombin lag time ratio and thrombin time to peak ratio were most pronounced (Table 3).
Table 3.
Ratios of thrombin generation parameters relative to baseline, 2.5 h after the last dose of dabigatran etexilate (0.5 h after idarucizumab or placebo, if administered)
| Day 4 | Day 11 | |||||
|---|---|---|---|---|---|---|
| ER2.5,ss | DE 220 mg (n=48) | DE 220 mg+placebo (n=12) | DE 220 mg+1 g (n=9) | DE 220 mg+2 g (n=9) | DE 220 mg+4 g (n=9) | DE 220 mg+2.5+2.5 g (n=9) |
| Thrombin lag time ratio | ||||||
| Mean | 5.20 | 4.78 | 1.36 | 1.08 | 1.00 | 0.991 |
| CV, % | 34.9 | 32.2 | 44.9 | 7.70 | 9.48 | 16.0 |
| Thrombin AUC ratio | ||||||
| Mean | 0.580 | 0.638 | 1.05 | 0.986 | 0.970 | 0.967 |
| CV, % | 35.6 | 26.1 | 12.1 | 8.89 | 14.7 | 12.1 |
| Thrombin peak ratio | ||||||
| Mean | 0.755 | 0.847 | 1.14 | 1.02 | 1.08 | 1.11 |
| CV, % | 35.6 | 24.1 | 11.8 | 16.1 | 18.2 | 36.9 |
| Thrombin time to peak ratio | ||||||
| Mean | 2.62 | 2.45 | 1.06 | 1.03 | 0.941 | 0.955 |
| CV, % | 30.8 | 27.1 | 19.1 | 7.24 | 6.20 | 19.6 |
AUC, area under the curve; CV, coefficient of variation; DE, dabigatran etexilate; ER2.5,ss, thrombin time to peak 2.5 hour.
4. DISCUSSION
In this phase I study the safety and pharmacokinetics of idarucizumab were investigated in healthy Japanese males for the first time. Administration of idarucizumab at doses of 4 and 2.5+2.5 g led to immediate, complete, and sustained reversal of dabigatran‐induced anticoagulation for the duration of the observation period (up to 72 hour); at 1 and 2 g doses of idarucizumab, a partial return of the anticoagulant effect of dabigatran was observed (1‐2 hour after idarucizumab administration); idarucizumab was safe and well tolerated at all doses. These findings support results obtained in a largely Caucasian population.18, 19
4.1. Safety profile
There were no unexpected or clinically relevant safety concerns reported and no AEs considered by the investigators to be related to idarucizumab.
An increase in prothrombotic effect is a potential risk following reversal of anticoagulation; a meta‐analysis of trials in vitamin K antagonist–treated patients receiving prothrombin complex concentrates for anticoagulation reversal suggested a low but quantifiable risk of thromboembolism.15 It could not be determined whether this represents underlying risk or is a prothrombotic effect. Despite being structurally similar to thrombin, idarucizumab has not exhibited specific thrombin‐like enzymatic activity24; in in vitro studies, idarucizumab showed no binding to known thrombin substrates and had no activity in coagulation or platelet aggregation tests.24, 25
Idarucizumab demonstrated no prothrombotic effects, alone or during reversal of dabigatran anticoagulation, as assessed by endogenous thrombin generation parameters. These parameters were not affected by idarucizumab alone and returned to baseline values (but not beyond) during reversal. Idarucizumab administered alone at doses up to 8 g demonstrated no effect on coagulation (assessed by dTT, ECT, aPTT, or TT) in these healthy Japanese volunteers. These findings are consistent with preclinical and clinical studies showing no prothrombotic effect resulting from idarucizumab use.19, 20, 21
As a specific humanized Fab given as a single intravenous dose, idarucizumab has low potential for immunogenic reactions. Treatment‐emergent ADAs were found in 6 of 60 subjects who received idarucizumab (in 2 of these males, ADAs were pre‐existing but increased after treatment), 4 of which were possibly persistent and 2 transient. These primary, treatment‐emergent ADA responses do not occur until after idarucizumab has been completely cleared. Any effect of treatment‐emergent ADA on idarucizumab PK/PD could only occur if high titer ADA are present at the time of a subsequent dose of idarucizumab. It has been estimated that at the highest titer observed in the present study, a titer of 40, the amount of ADA circulating is equivalent (on a molar basis) to <0.1% of a 5 g dose of idarucizumab.26 It is likely that the ADA titer would need to be many times higher than 40 to have an effect on PK/PD of a subsequent dose of idarucizumab. In a pooled analysis of phase I studies, no AEs were indicative of immunogenic reactions, and the pharmacokinetics and pharmacodynamics of idarucizumab were unaffected.26
The safety profile of idarucizumab in Japanese male subjects was comparable to that in phase I studies in which the majority of volunteers were healthy Caucasians; idarucizumab was well tolerated. The proportion of subjects with pre‐existing antibodies that were cross‐reactive with idarucizumab was also similar.18, 19
4.1.1. Pharmacokinetic and pharmacodynamic profiles
The pharmacokinetic profile of idarucizumab displayed plasma concentrations reaching a peak at the end of the idarucizumab infusion, ensuring immediate availability of the reversal agent for dabigatran binding followed by rapid elimination (t 1/2, 2=0.63‐1.04 hour). This rapid elimination of unbound idarucizumab may suggest that effective dabigatran anticoagulation could be re‐established by dabigatran treatment as early as 24 hour post‐idarucizumab administration, although this timeframe may vary depending upon the demographic profile of the patients. This has been demonstrated in a phase Ib study in elderly, renally impaired male and female volunteers.27
The urinary excretion and CLR of idarucizumab with dabigatran were approximately two to three times higher than with idarucizumab alone. This was not due to adsorption to container, because the assay remained accurate after several dilutions, proven by the bioanalytical validation study.
The reversal of anticoagulant effects verified by the disappearance of unbound dabigatran was sustained at doses of 4 and 2.5+2.5 g and was consistent with the total body load of dabigatran and the 1:1 stoichiometry of the binding properties of idarucizumab to dabigatran.24 At the 220 mg bid dose the median steady state C max on day 4 was 286 ng/mL for total dabigatran, with 10‐90th percentiles of 136‐432 ng/mL. The steady state C max at the 90th percentile was comparable to the dabigatran exposure at 2 hour post‐dose steady state plasma concentration at the 90th percentile (383 ng/mL, 150 mg bid) as observed in the RE‐LY study.20 The increase in plasma concentrations of total dabigatran observed immediately after idarucizumab administration reflected redistribution of dabigatran from the peripheral compartment and bound to idarucizumab in the central compartment. This bound dabigatran was inactive dabigatran; there was no measurable anticoagulant activity. Idarucizumab binds to and inactivates dabigatran in the central compartment, which approximated the central compartment plus interstitial space. This binding reduces the unbound dabigatran centrally and, in a dynamic equilibrium, dabigatran in the peripheral tissues moves into the central compartment. If unbound idarucizumab remains, this redistributed dabigatran is inactivated by idarucizumab.22 If there is no free idarucizumab remaining, the coagulation parameters re‐elevate. The sustained effect corresponds to the almost irreversible binding shown in preclinical studies.24 The slow off‐rate of idarucizumab binding to dabigatran24 means that dabigatran remains bound to idarucizumab until the complex is eliminated from the body through a combination of renal catabolism and, for doses ≥1 g, urinary excretion.19 Lower doses of idarucizumab (1 and 2 g) were associated with a partial return of anticoagulant effect, reflecting redistribution of unbound dabigatran that was not bound by idarucizumab, since there was no remaining free idarucizumab.19
The pharmacokinetic profile of idarucizumab in Japanese male subjects was comparable to results from phase I studies with predominantly healthy Caucasian males; differences in exposure were negligible after weight normalization, since mean body weight was lower in Japanese subjects. Mean body weight values in Caucasian subjects were ~80 kg for both study parts, and in Japanese male subjects were ~65 kg in part 1 and 61 kg in part 2. Since exposure to dabigatran is higher in Japanese subjects (AUC2‐12,ss 1480 ng/mL, n=48, geometric coefficient of variation [gCV]=44.1%) than in Caucasians (1090 ng/mL, n=35, gCV=56.4%), a 4‐g idarucizumab dose was needed for sustained reversal, compared with the 2‐g idarucizumab dose that was sufficient for sustained reversal in Caucasians.18
4.2. Study strengths and limitations
Although double‐blind conditions were maintained within each dose group, investigators and subjects knew the current dose level due to the risk‐minimizing sequential dose escalation scheme. Measurement of objective parameters of coagulation limited the potential for bias this could have introduced. The present study did not include women. However, the pharmacokinetic profiles of idarucizumab were similar in males and females (data not shown).28 The safety profile of idarucizumab in healthy volunteers may differ from that in the target patient population, who are much older and likely to have life‐threatening underlying conditions and multiple comorbidities. This could result in different responses to treatment. Interim data from bleeding patients or patients requiring emergency surgery have not identified any safety concerns. The rapid, complete and sustained reversal of anticoagulant effects in patients with a 5‐g dose was similar to that seen in healthy subjects.29 The effect of idarucizumab on bleeding was not determined in this study, although reversal of anticoagulation was demonstrably effective through measurement of objective parameters. This is the only trial in healthy volunteers to include idarucizumab at the 2.5+2.5 g dose with 15‐minute intervals between infusions; this dose was investigated in the phase III Reversal of Dabigatran Anticoagulant Effect With Idarucizumab (RE‐VERSE AD) trial.29
5. CONCLUSIONS
In this first study of idarucizumab in a healthy Japanese male population, both regimens of idarucizumab administration (4 and 2.5+2.5 g) led to immediate, complete, and sustained reversal of dabigatran‐induced anticoagulation. Idarucizumab doses of 1 and 2 g were insufficient for sustained reversal of anticoagulant effects of dabigatran in this population. Idarucizumab was safe and well tolerated, and did not exhibit a prothrombotic effect or an effect on coagulation parameters. These pharmacokinetic and safety data could potentially inform dosing regimens for labeling and use in phase III trials, which include Japanese patients treated with dabigatran who have rare uncontrolled bleeding or who require urgent reversal of anticoagulation prior to an emergency procedure or surgery.
ADDENDUM
All authors had access to study data. Idarucizumab and matching placebo were provided by the Department of Pharmaceutical Development of Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach an der Riβ, Germany. M. Yasaka was the external expert of local clinical development for idarucizumab in Japan. I. Ikushima was the principal investigator for this trial and was responsible for the safe and ethical conduct of the trial. A. Harada was the pharmacokineticist for this trial, and was responsible for pharmacokinetic/pharmacodynamic analysis and interpretation. S. Imazu was the trial clinical monitor, responsible for coordinating the activities required to manage the trial in accordance with applicable regulations and internal standard operating procedures. A. Taniguchi was the statistician for this trial and was responsible for statistical analyses. S. Norris was the trial bioanalyst responsible for quantitation of idarucizumab plasma concentrations and for assessment of anti‐idarucizumab antibody responses. D. Gansser was the bioanalyst for the trial with responsibility for the quantitation of total and unbound dabigatran. J. Stangier and M. Schmohl were involved in the trial as biomarker analysts. P. Reilly was the internal medical expert and was responsible for coordinating international clinical development for idarucizumab. The authors were fully responsible for all content and editorial decisions were involved at all stages of manuscript development and have approved the final version.
RELATIONSHIP DISCLOSURE
AH, SI, AT, SN, DG, JS, MS, and PR are employees of Boehringer Ingelheim. MY has received lecture fees (over 1 million Yen) from Nippon Boehringer Ingelheim, Bayer Yakuhin, Bristol‐Myers Squibb and Daiichi‐Sankyo, and research funding (over 2 million Yen) from Sanofi. II has no conflicts of interest to declare.
Supporting information
ACKNOWLEDGMENTS
Funding: The study was funded by Boehringer Ingelheim Pharma GmbH & Co. KG. Medical writing assistance, supported by Boehringer Ingelheim Pharma GmbH & Co. KG, was provided by Sarah Mohamad, MPharm, MRPharmS and Anil Dandu, MPharm, PhD of PAREXEL.
Yasaka M, Ikushima I, Harada A, et al. Safety, pharmacokinetics and pharmacodynamics of idarucizumab, a specific dabigatran reversal agent in healthy Japanese volunteers: a randomized study. Res Pract Thromb Haemost. 2017;1:202–215. 10.1002/rth2.12029
Funding Information
This study was funded by Boehringer Ingelheim.
REFERENCES
- 1. Connolly SJ, Ezekowitz MD, Yusuf S, Reilly PA, Wallentin L. Newly identified events in the RE‐LY trial. N Engl J Med. 2010;363:1875–6. [DOI] [PubMed] [Google Scholar]
- 2. Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361:2342–52. [DOI] [PubMed] [Google Scholar]
- 3. Schulman S, Kakkar AK, Goldhaber SZ, et al. Treatment of acute venous thromboembolism with dabigatran or warfarin and pooled analysis. Circulation. 2014;129:764–72. [DOI] [PubMed] [Google Scholar]
- 4. Schulman S, Kearon C, Kakkar AK, et al. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med. 2013;368:709–18. [DOI] [PubMed] [Google Scholar]
- 5. Majeed A, Hwang HG, Connolly SJ, et al. Management and outcomes of major bleeding during treatment with dabigatran or warfarin. Circulation. 2013;128:2325–32. [DOI] [PubMed] [Google Scholar]
- 6. Hori M, Connolly SJ, Ezekowitz MD, Reilly PA, Yusuf S, Wallentin L. Efficacy and safety of dabigatran vs. warfarin in patients with atrial fibrillation–sub‐analysis in Japanese population in RE‐LY trial. Circulation. 2011;75:800–5. [DOI] [PubMed] [Google Scholar]
- 7. Hori M, Connolly SJ, Zhu J, et al. Dabigatran versus warfarin: effects on ischemic and hemorrhagic strokes and bleeding in Asians and non‐Asians with atrial fibrillation. Stroke. 2013;44:1891–6. [DOI] [PubMed] [Google Scholar]
- 8. Hori M, Fukaya T, Kleine E, Reilly PA, Ezekowitz MD, Connolly SJ. Efficacy and safety of dabigatran etexilate vs. warfarin in Asian RE‐LY patients according to baseline renal function or CHADS2 score. Circulation. 2015;79:2138–47. [DOI] [PubMed] [Google Scholar]
- 9. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361:1139–51. [DOI] [PubMed] [Google Scholar]
- 10. Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883–91. [DOI] [PubMed] [Google Scholar]
- 11. Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981–92. [DOI] [PubMed] [Google Scholar]
- 12. Giugliano RP, Ruff CT, Braunwald E, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093–104. [DOI] [PubMed] [Google Scholar]
- 13. van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate–a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103:1116–27. [DOI] [PubMed] [Google Scholar]
- 14. Weitz JI, Pollack CV Jr. Practical management of bleeding in patients receiving non‐vitamin K antagonist oral anticoagulants. Thromb Haemost. 2015;114:1113–26. [DOI] [PubMed] [Google Scholar]
- 15. Dentali F, Marchesi C, Giorgi PM, et al. Safety of prothrombin complex concentrates for rapid anticoagulation reversal of vitamin K antagonists. A meta‐analysis. Thromb Haemost. 2011;106:429–38. [DOI] [PubMed] [Google Scholar]
- 16. Heidbuchel H, Verhamme P, Alings M, et al. EHRA practical guide on the use of new oral anticoagulants in patients with non‐valvular atrial fibrillation: executive summary. Eur Heart J. 2013;34:2094–106. [DOI] [PubMed] [Google Scholar]
- 17. Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373:511–20. [DOI] [PubMed] [Google Scholar]
- 18. Glund S, Moschetti V, Norris S, et al. A randomised study in healthy volunteers to investigate the safety, tolerability and pharmacokinetics of idarucizumab, a specific antidote to dabigatran. Thromb Haemost. 2015;113:943–51. [DOI] [PubMed] [Google Scholar]
- 19. Glund S, Stangier J, Schmohl M, et al. Safety, tolerability, and efficacy of idarucizumab for the reversal of the anticoagulant effect of dabigatran in healthy male volunteers: a randomised, placebo‐controlled, double‐blind phase 1 trial. Lancet. 2015;386:680–90. [DOI] [PubMed] [Google Scholar]
- 20. Reilly PA, Lehr T, Haertter S, et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE‐LY Trial (Randomized Evaluation of Long‐Term Anticoagulation Therapy). J Am Coll Cardiol. 2014;63:321–8. [DOI] [PubMed] [Google Scholar]
- 21. Stangier J. Clinical pharmacokinetics and pharmacodynamics of the oral direct thrombin inhibitor dabigatran etexilate. Clin Pharmacokinet. 2008;47:285–95. [DOI] [PubMed] [Google Scholar]
- 22. Stangier J, Feuring M. Using the HEMOCLOT direct thrombin inhibitor assay to determine plasma concentrations of dabigatran. Blood Coagul Fibrinolysis. 2012;23:138–43. [DOI] [PubMed] [Google Scholar]
- 23. Shamoo AE. The myth of equipoise in phase 1 clinical trials. Medscape J Med. 2008;10:254. [PMC free article] [PubMed] [Google Scholar]
- 24. Schiele F, van Ryn J, Canada K, et al. A specific antidote for dabigatran: functional and structural characterization. Blood. 2013;121:3554–62. [DOI] [PubMed] [Google Scholar]
- 25. van Ryn J, Schurer J, Kink‐Eiband M, Clemens A. Reversal of dabigatran‐induced bleeding by coagulation factor concentrates in a rat‐tail bleeding model and lack of effect on assays of coagulation. Anesthesiology. 2014;120:1429–40. [DOI] [PubMed] [Google Scholar]
- 26. Norris S, Ramael S, Ikushima I, et al. Evaluation of the immunogenicity of the dabigatran reversal agent idarucizumab during phase I studies. Br J Clin Pharmacol. 2017;83:1815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glund S, Stangier J, Schmohl M, et al. Idarucizumab, a specific antidote for dabigatran: Immediate, complete and sustained reversal of dabigatran induced anticoagulation in elderly and renally impaired subjects. Blood. 2014;124:344.24914142 [Google Scholar]
- 28. Glund S, Stangier J, van Ryn J, et al. Effect of age and renal function on idarucizumab pharmacokinetics and idarucizumab‐mediated reversal of dabigatran anticoagulant activity in a randomized, double‐blind, crossover phase Ib study. Clin Pharmacokinet. 2017;56:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pollack CV Jr, Reilly PA, Bernstein R, et al. Design and rationale for RE‐VERSE AD: A phase 3 study of idarucizumab, a specific reversal agent for dabigatran. Thromb Haemost. 2015;114:198–205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials