

Summary
Essentials.
Increasing evidences supports a role for splicing defects in multiple disorders.
For antithrombin (AT) deficiency only 7% of mutations disturb intronic splicing sequences.
Our study of 141 unrelated cases with AT deficiency found higher rate of splicing defects (>13%).
A wide range of gene defects cause different types of AT deficiency through aberrant splicing.
Background
There is increasing evidence supporting the relevance of aberrant splicing in multiple disorders. In antithrombin deficiency only 22 intronic mutations affecting splicing sites (7% of SERPINC1 mutations) are considered as splicing mutations.
Methods
SERPINC1 was analyzed by Sanger sequencing and MLPA in 141 unrelated cases with antithrombin deficiency. Plasma antithrombin was studied by functional and western blot assays, purified by FPLC and characterized by proteomic analysis. In silico predictions on splicing was done with the Human Splicing Finder software.
Results
We detected 89 different SERPINC1 defects, 13 with potential effect on splicing. Ten cases presented 9 mutations disturbing splicing sites, 5 new. Three gross or small gene defects also disturbed a correct splicing. Interestingly, the first duplication of a single exon ever described (c.1154‐13_1218+115dup), caused mild deficiency (75%). A deeper intronic mutation (c.1154‐14G>A), identified in three unrelated patients with traces of disulphide dimers of antithrombin in plasma, created a cryptic splicing site that might generate a variant with 4 additional in frame residues according to in silico predictions. This aberrant splicing was confirmed by proteomic analysis of the dimer purified from plasma.
Conclusions
A high proportion of cases with antithrombin deficiency (up to 13%) may be explained by an aberrant splicing. Up to 15% of mutations in SERPINC1: splicing site variations, gross gene defects and deep intronic mutations, may affect a correct splicing with three potential consequences type I, type II, and even moderate antithrombin deficiency.
Keywords: antithrombin deficiency, intron, mutations, splicing, thrombosis
1. INTRODUCTION
Antithrombin is a serpin (serin‐protease inhibitor) considered as the main endogenous anticoagulant in humans. It exerts its anticoagulant function by inhibiting crucial serine proteases of the clotting cascade, mainly thrombin and factor Xa.1 Thus, inherited antithrombin deficiency significantly increases the risk of thrombosis (20‐ to 40‐fold). Congenital antithrombin deficiency is a heterogeneous disorder that shows an autosomal dominant pattern of inheritance. Deficiency is estimated to have a prevalence of about 0.02% in the general population, while accounting for 1% of cases with confirmed deep vein thrombosis.2 Two deficiency states are described: type I deficiency characterized by a parallel reduction in both functional and antigenic levels of antithrombin in plasma; and type II deficiency distinguished by the production and secretion of a variant protein to the plasma circulation with null or impaired inhibitor activity.3
The gene encoding antithrombin, SERPINC1, is located on the long arm of chromosome 1 (q23‐25), and comprises 7 exons and 6 introns spanning 13 574 bp of genomic DNA (ENSG00000117601). Non‐coding regions consist of 11 980 bp (88% from the whole gene).4, 5
Up to 80% of cases with suspicion of inherited antithrombin deficiency are caused by SERPINC1 gene defects6, although other gene defects may also cause antithrombin deficiency.7 According to the Human Gene Mutation Database (HGMD), missense or nonsense mutations constitute more than 50% to the genetic defects identified in SERPINC1, followed by small deletions (19%), gross deletions, and small insertions (8.5% and 8.1%, respectively). Only 22 mutations affecting introns that might theoretically disrupt the natural mRNA processing have been described (7% of mutations identified in SERPINC1), all associated with type I deficiency. As different evidences are supporting the increased role of splicing defects on different human genetic diseases8, 9, the aim of this study was to identify new SERPINC1 genetic variations underlying antithrombin deficiency through an incorrect splicing in one of the largest cohorts of unrelated patients with this strong thrombophilia.
2. MATERIALS AND METHODS
2.1. Patients
During the last 20 years, our center recruited 141 unrelated Caucasian cases from different hospitals with suspicion of antithrombin deficiency (plasma anti‐FXa activity <80% of a reference plasma generated with 100 healthy blood donors) presenting 89 different SERPINC1 gene defects.
The study was approved by the Ethics Committees of the Hospital General Universitario Reina Sofia in Murcia, Spain. All patients or their relatives, and controls (blood donors for the reference plasma) provided written informed consent.
2.2. Blood collection
Blood samples were collected without venous stasis from an antecubital vein in citrate‐anticoagulated tubes (109 mmol L−1) and processed within 24 hour after extraction. The tubes were centrifuged for 15 minute at 2000 g at room temperature, and the separated plasma was aliquoted and stored at −70°C until analysis. Genomic DNA was purified by the salting out following Puregen Blood Core KitB (Qiagen, Madrid, Spain) procedure and stored at −20°C.
The first sample was obtained at least 6 months after the thrombotic event, when the patient was not under any anticoagulant therapy. No patient was evaluated under heparin treatment.
2.3. Antithrombin evaluation
2.3.1. Functional and antigenic assays
Antithrombin deficiency was identified by using different commercial functional methods available in each hospital10, and confirmed in the sample delivered to our laboratory by evaluating plasma anti‐FXa activity using a chromogenic method in the presence of heparin (HemosIL Liquid Antithrombin, Instrumentation Laboratory, Munich, Germany) in a plate reader (Synergy HT, Biotek).
Antigen levels were measured by immunodiffusion (Laurell) or home‐made ELISA using rabbit polyclonal anti‐human antithrombin (A9522, Sigma‐Aldrich), mouse monoclonal anti‐human antithrombin (A5816, Sigma‐Aldrich) and anti‐mouse ECL IgG horseradish peroxidase (NA931 Amersham Biosciences, Piscataway, NJ, USA).
The normal range of antithrombin measurements (anti‐FXa and antigen levels) was 80‐120% with coefficient of variation of 5‐7%.
2.3.2. Western blot analysis
Plasma antithrombin was evaluated by Western blot using different electrophoretic conditions described elsewhere.11 The blotting was performed by using rabbit anti‐human antithrombin polyclonal antibody (A9522, Sigma‐Aldrich) followed by donkey anti‐rabbit IgG‐horseradish peroxidase conjugate (NA9340, GE Healthcare, Barcelona, Spain) as secondary antibody.
Detection was carried out with an ECL kit (Amersham Biosciences, Piscataway, NJ, USA) in the ImageQuant LAS4000mini equipment (Exon Biotec, GE Healthcare).
2.3.3. Protein purification
Antithrombin disulphide‐linked dimers from plasma of patients with c.1154‐14G>A or the c.334C>T mutations were purified as described before.12 Briefly, citrated plasma of patients were subjected to heparin affinity chromatography on HiTrap Heparin columns (GE Healthcare), followed by ion exchange chromatography on HiTrap Q columns (GE Healthcare) and a gel filtration. All procedures were done in an ÄKTA Purifier (GE Healthcare). Finally, proteins eluted were desalted through a dialysis tubing (Sigma‐Aldrich) and stored at −70°C, prior to analysis. After electrophoretic analysis (SDS‐PAGE), bands corresponding to dimers purified from patients with c.1154‐14G>A mutation and wild type antithrombin, were subjected to in‐gel digestion with 200 ng of sequencing grade modified Trypsin (Promega, Madrid, Spain) in 50 mmol L−1 ammonium bicarbonate for 16 hour at 37°C, after a denaturation step with DTT (10 mmol L−1, 30 minute, 40°C) and an alkylation step with Iodoacetamide (25 mmol L−1, 30 minute, room temperature). The resulting peptides were extracted with 1% formic acid, 50% acetonitrile and evaporated to dryness prior to LC‐MSMS analysis.
2.3.4. LC−ESI−MS/MS analysis
Microcapillary reverse‐phase LC was performed with a nanoAcquity system (Waters, Saint‐Quentin, France). Reversed phase separation of trypsin digests was performed with an Atlantis, C18, 3 μm, 75 μm×10 cm Nano Ease fused silica capillary column (Waters) buffered in 5% acetonitrile with 0.2% formic acid. After injection of sample, the column was washed for 5 minute with the same buffer as above, and the peptides were eluted using a linear gradient of 5‐50% acetonitrile in 30 minute at a constant flow rate of 0.2 μL min−1. The column was coupled online with a Q‐TOF Micro (Waters) using a PicoTipnanospray ionization source (Waters). The heated capillary temperature was 80°C and the spray voltage was 1.8‐2.2 kV. MS/MS data were collected in an automated, data‐dependent mode. The three most intense ions in each survey scan were sequentially fragmented by collision induced dissociation (CID) using an isolation width of 2.5 and a relative collision energy of 35%. Data processing was performed with MassLynx 4.0. Database searching was done with MASCOT Daemon v2.3.0 (MatrixScience, London, UK) against UniprotKB/Swiss‐Prot database, and against an in‐house database including Antithrombin sequence plus c.1154‐14G>A variant. Mass error tolerance was 50 ppm for parent and 0.1 Da for fragments; there were no restrictions on molecular weight and isoelectric point; the fixed modification was carbamidomethylation of cysteine; the variable modification was oxidation of methionine.
2.3.5. Genetic analysis
The exons and flanking regions, as well as 1500 bp of the promoter region of SERPINC1, were sequenced in all patients with antithrombin deficiency.13 Gross deletions were analyzed by multiplex ligation‐dependent probe amplification (MLPA) using the SALSAMLPA Kit P227 SerpinC1 (MRC Holland, Amsterdam, The Netherlands).
2.3.6. In silico analysis of potential splicing signals
To analyze the potential effect of mutations on sequences potentially involved in splicing, we used the Human Splicing Finder software (HSF, version 3.014). We consider potentially relevant those motifs (acceptor or donor sites) that are disrupted or created as a consequence of a specific variant with a score variation (difference between the reference wild type and mutant motifas strengths) >20%, and in case of creating a new motif when it also reaches a score >80%.
3. RESULTS
3.1. Classical splicing mutations
Ten patients with severe thrombotic events (P1‐P10) and type I antithrombin deficiency (mean of 48% anti‐FXa activity and 51% of antigen levels) had 9 different heterozygous splice site mutations (Table 1). Remarkably, P2 (with c.1218 +1G>A mutation) had lower antigen and anti‐FXa activity than expected (Table 1), which may be explained by additional modulating factors. Five of these splicing site mutations were not previously described in available mutation databases (Table 1). All these mutations severely disturbed natural acceptor (N=7) or donor splicing sites (N=2). Three of them potentially affected the splicing of exon 6, two affecting exons 2 and 5, and single ones affecting the splicing of exons 1 and 3 (Table 1). It is remarkable that no mutation affecting the splicing of exon 4 has been described either in our cohort or in the HGMD.
Table 1.
Demographic and clinical data of patients with antithrombin deficiency caused by a potential aberrant splicing
| Patient | Age (years) | Variant ID | Mutation | Pathogenicitya | Other risk factors | Thrombosis | Anti‐FXa (%) | Ag (%) | Type of AT deficiency |
|---|---|---|---|---|---|---|---|---|---|
| P1 | 45 | CS013816 | c.1218+1G>A | Pathogenic | DVT | 50 | 60 | I | |
| P2 | 50 | CS013816 | c.1218+1 G>A | Pathogenic | DVT | 29 | 42 | I | |
| P3 | 40 | CS952107 | c.1218+1G>T | Pathogenic | DVT | 40 | 45 | I | |
| P4 | 29 | CS011025 | c.409‐1G>C | Pathogenic | CVT | 52 | 51 | I | |
| P5 | 27 | New | c.863‐2A>C | Pathogenic | PE | 55 | 50 | I | |
| P6 | 35 | New | c.1219‐3C>A | Pathogenic | FVL+/− | PE | 51 | 45 | I |
| P7 | 21 | New | c.42‐2A>C | Pathogenic | Fatal PE | 60 | 69 | I | |
| P8 | 31 | CS132987 | c.409‐2A>T | Pathogenic | AT Camb +/− | PE | 40 | 48 | I |
| P9 | 40 | New | c.1154‐2A>T | Pathogenic | DVT | 46 | 50 | I | |
| P10 | 15 | New | c.625‐2A>G | Pathogenic | 3 RecurrentDVT | 58 | 52 | I | |
| P11 | 29 | New | c.1219‐1_1248del | Pathogenic | Mesenteric& portal thrombosis | 47 | 50 | I | |
| P12 | 26 | New | Del exon 4 | Pathogenic | DVT | 50 | 55 | I | |
| P13 | 42 | New | c.1154‐13_1218+115dup | Pathogenic | DVT | 75 | 79 | I moderate | |
| P14 | 25 | CS941423 | c.1154‐14G>A | Pathogenic | PE | 52 | 46 | II | |
| P15 | 56 | CS941423 | c.1154‐14G>A | Pathogenic | DVT | 38 | 44 | II | |
| P16 | 32 | CS941423 | c.1154‐14G>A | Pathogenic | No (Severefamilyhistory) | 39 | 47 | II | |
| P17 | 28 | CM070261 | c.470A>G p.[Lys157Arg] | Pathogenic | FVL+/−& PT+/− | DVT&PE | 50 | 60 | I |
AG, Antigen levels; AT Cam, Antithrombin Cambridge II (p.Ala416Ser); CVT, Cerebral vein thrombosis; DVT, Deep venous thrombosis; FVL, Factor V Leiden; PE, Pulmonary embolism; PT, Prothrombin c.*97G>A; +/−, Heterozygous.
Pathogenicity according to American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines.29
3.2. Other variants causing antithrombin deficiency through an aberrant splicing
3.2.1. Small and gross gene defects
P11, a 29‐year‐old female with type I antithrombin deficiency (47% of anti‐FXa activity and 50% of antigen levels) (Figure 1) who developed portal, mesenteric, and splenic vein thrombosis during pregnancy, had a new 31 bp deletion in heterozygosis (c.1219‐1_1248del) affecting the last nucleotide of intron 6 and 30 bp of exon 7 (Table 1). According to HSF in silico predictions, this deletion disrupted the exon 7 wild type acceptor splicing site (from 90% to 61% of splicing probability).
Figure 1.
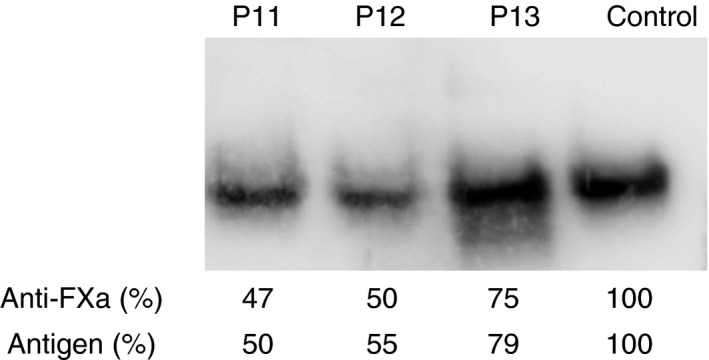
Plasma antithrombin in P11, P12, and P13 patients detected by Western Blot after electrophoretic separation in native‐PAGE. A healthy blood donor was used as a negative control. Anti‐FXa activity and antigen levels are also indicated for each case.
P12 and P13 (26‐ and 42‐year‐old patients with early deep venous thrombosis) presented gross gene rearrangements.
MLPA analysis of P12 revealed a profile suggesting a possible heterozygous deletion involving exon 4. Exon 4 must be absent from affected transcripts in P12. Accordingly, the processing of introns of the mutant transcript would be different from that followed by the wild type transcript, with altered reading frame resulting in a premature stop codon (Figure 2). This mechanism likely explains the type I antithrombin deficiency identified in P12 (Table 1 and Figure 1).
Figure 2.
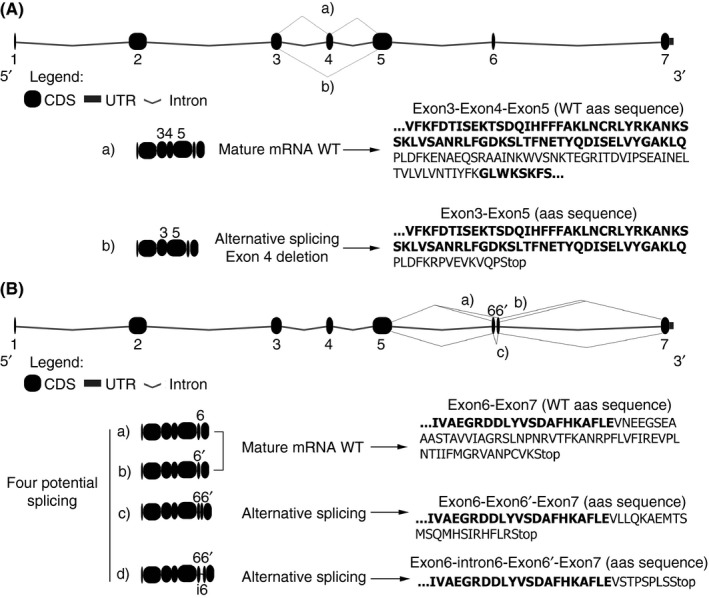
mRNA of SERPINC1 potentially generated by the gross deletion of exon 4 (A) and the 193 bp duplication involving exon 6 (B) with potential alternative splicing, two of them rendering normal RNA. Bold text is used to indicate each exon
In P13, PCR amplification and sequencing with primers flanking exon 6 revealed an insertion of 193 bp covering exon 6 (c.1154‐13_1218+115dup) (Table 1). Surprisingly, P13 with duplication of exon 6, had a moderate type I deficiency of antithrombin with anti‐FXa and antigen levels of around 75% (Table 1 and Figure 1). The 193 bp duplication involving exon 6 may generate different potential alternative splicing, two of them able to render normal mRNA processing of SERPINC1, which may explain the moderate antithrombin deficiency detected in this patient (Figure 2).
3.3. Deep intronic mutations
Although the design of primers used in this study does not allow detecting very deep intronic mutations, we identified one intronic variation not directly disturbing donor or acceptor sequences, c.1154‐14G>A, that might cause antithrombin deficiency. This mutation was detected in three unrelated patients from Portugal and two different regions of Spain: P14, P15 and P16 (Table 1). This mutation was previously described in a Dutch patient with type I antithrombin deficiency, and authors suggested this mutation to cause a splicing defect (CS941423).15 Patients with this mutation from our cohort also showed an apparent type I deficiency, with a mean of around 50% of functional activity and antigen levels (Table 1). However, and in agreement with a previous report16 our patients with this mutation presented traces of disulphide‐linked dimers in plasma, which had slightly slower electrophoretic mobility than those identified in patients with conformational missense mutations (c.334C>T; p.Pro112Ser)11 (Figure 3).
Figure 3.
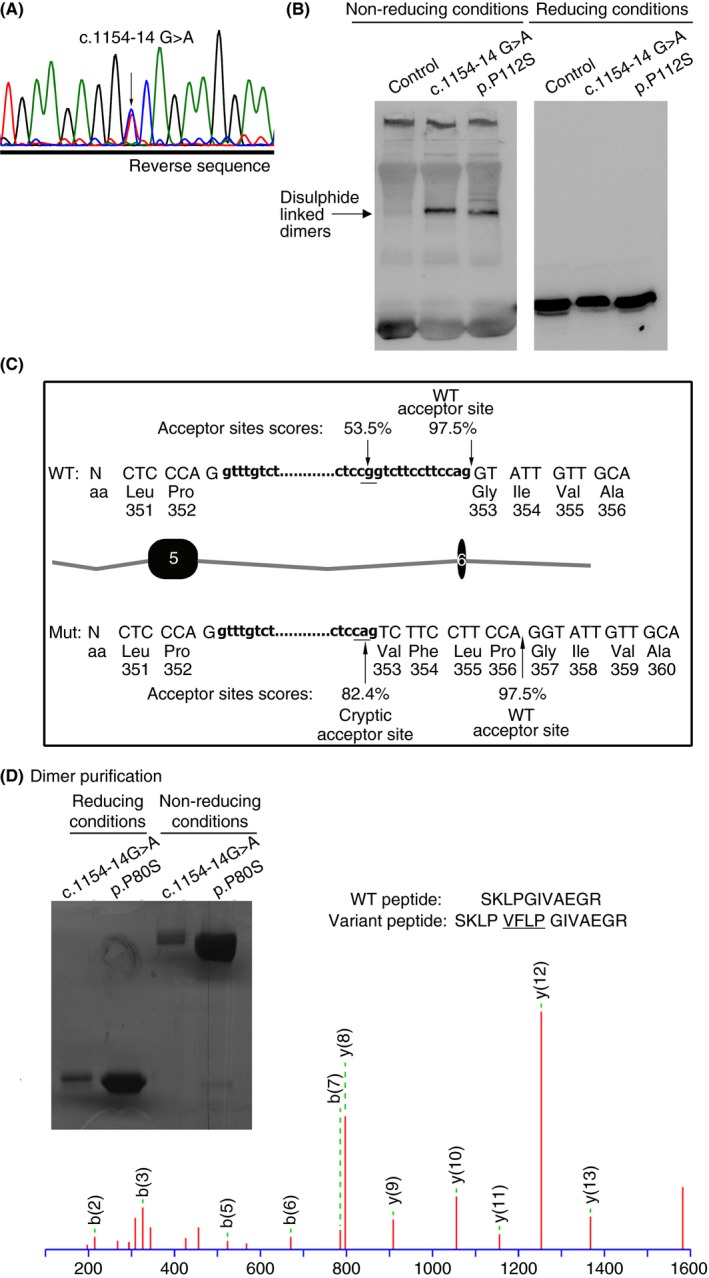
Identification and characterization of the c.1154‐14G>A mutation identified in three unrelated patients from our series (P14, P15 and P16). (A) Electropherogram showing the point mutation. (B) Electrophoretic analysis (SDS‐PAGE) of plasma antithrombin under reducing and not‐reducing conditions. Disulphide‐linked dimers are highlighted by an arrow. As control of a missense mutation also generating disulphide‐linked dimers, plasma from a patient with p.Pro112Ser mutation was also included. (C) Splicing predictions according to the results obtained with the Human Splicing Finder Software. (D) Purification of disulphide‐linked dimers from plasma of patients with antithrombin deficiency heterozygous for c.1154‐14G>A and p.Pro112Ser mutations and proteomic analysis of the protein obtained from the patient with c.1154‐14G>A mutation. N, nucleotides; aa, aminoacids.
In silico prediction suggested that c.1154‐14G>A creates a new cryptic acceptor sequence, which may potentially generate a variant protein with the in‐frame insertion of 4 residues at position 384 (Figure 3). Purification of disulphide‐linked dimers from plasma of patients with this mutation and further proteomic analysis confirmed the predicted splicing in the variant protein (Figure 3).
4. DISCUSSION
Point mutations are the most common cause of hereditary disease. Most of them (84% in the HGMD) have been considered to change a single aminoacid (missense) or generate a premature stop codon (nonsense).17, 18 However, in most cases mutation analyses are exclusively performed at the genomic DNA level, and the effect of a mutation on the encoded mRNA and protein is predicted from the primary sequence alone. Only few studies have evaluated experimentally the consequences of the mutation identified in a patient, and interestingly, about 50% of the mutations result in aberrant splicing.19, 20 Actually, there is increasing evidence that many human disease genes are caused by mutations (also located at exons) that affect pre‐mRNA splicing.8, 9 Additionally, it is important to remark that the deleterious consequences of a mutation causing an aberrant splicing will potentially be higher than those from a missense mutation. Mutations affecting splicing sites, donor or acceptor, have obvious consequences on splicing. However, also nonsense, missense, and even synonymous mutations can inactivate genes by inducing the splicing machinery to skip the mutant exons. In fact, it has been estimated that 1.6% of disease‐causing missense mutations can affect splicing, and recent predictions suggest that approximately 7% of exonic variants in the general population may disrupt splicing, which includes cryptic splicing.21, 22 Similarly, coding‐region single‐nucleotide polymorphisms might cause phenotypic variability by influencing splicing accuracy or efficiency. Finally, the relevance of deep intronic mutations in disease has not been established, as most studies are restricted to exons and flanking regions.
On SERPINC1 only 22 mutations (7% of the total mutations identified) are accepted to affect a correct splicing. Most of them affecting splicing sites and all described in section 2.1 with type I antithrombin deficiency, in accordance to the severe consequences that these types of mutations have in this structurally and functionally sensitive serpin. The results obtained in our cohort of 141 unrelated and consecutive patients with antithrombin deficiency are consistent with available data from mutation databases: 10 cases with 9 different mutations affecting splicing sites, 5 of them new, all with type I deficiency. These mutations represent 7% of our cohort and 10% of all different mutations identified in our study, which is similar to the rates described in other reports of large series.6, 23 However, we also identified 4 additional gene defects in 6 patients with particular features and unexpected consequences that also caused antithrombin deficiency by inducing an aberrant splicing. Thus, gross gene defects such as deletions or duplications of exons (the last one firstly described worldwide in SERPINC1) may cause antithrombin deficiency by significantly affecting the correct splicing. Actually, the only way to explain the mild antithrombin deficiency observed in the patient with exon 6 duplication (75% of anti‐FXa activity and 79% of antigen levels) is by alternative splicing (Figure 2). Additionally, deep intronic mutations may also cause antithrombin deficiency. In this framework, we would like to point out the effect of the point mutation affecting intron 5, 14 bp upstream exon 6. The cryptic acceptor splicing site generated by the nucleotide change G>A in intron 5 results in an altered mRNA with 12 bp added in frame that would code for 4 extra amino acids in the polypeptide chain.15 However, in contrast to original proposition that the mutant allele is not expressed at the protein level15 our study demonstrates that this variant is expressed and forms disulphide dimers by disturbing the correct folding of this serpin, like other missense mutations.11 This is the first evidence that an aberrant splicing may also cause a type II antithrombin deficiency.
In conclusion, our study shows new evidences supporting that splicing mutations may play a more important role than previously thought in human hereditary disease. Aberrant splicing underlies a high proportion of cases with antithrombin deficiency, at least twice than the numbers proposed nowadays. Our study also confirms that the distortion of the correct splicing might be the mechanism involved in antithrombin deficiency caused by different types of gene defects. In addition to classical splicing site variations, deep intronic mutations and other gene variations including gross gene defects may cause antithrombin deficiency through an aberrant splicing. Missense and nonsense mutations in SERPINC1 might also disturb a correct splicing, as suggested by a recent study.9 This large in silico study revealed that 21 out of 22 nonsense and 2 missense SERPINC1 mutations described in databases have high probability to affect the splicing.9 Interestingly, one of these mutations, CM070261 p.(Lys157Arg) was identified in a patient from our cohort (P17) who had a type I deficiency (Table 1) despite this mutation affects a residue involved in the binding of heparin.24 Therefore, it is possible that the number of mutations associated with aberrant splicing could be even higher than that suggested in this study, and it may increase if considering other regulatory elements involved in the splicing process. However, it is important to point out the requirement of further studies to verify the aberrant splicing suggested for mutations described in SERPINC1, which may include recombinant models, analysis of SERPINC1 transcripts and minigene models. Finally, our study shows that the consequences of an aberrant splicing are not restricted to the absence of protein encoded from the affected allele. As demonstrated in this study, an aberrant splicing in SERPINC1 may cause type I, type II or even moderate deficiency. As a final consideration; the elucidation of the pathological mechanism associated to one mutation is not only important from a basic point of view. It is also relevant for establishing the most appropriate and personalized therapeutic strategy at the molecular level, particularly with the promising results obtained to rescue mutations causing aberrant splicing.25, 26, 27, 28
AUTHOR CONTRIBUTION
MEM‐B and JC designed research, performed research, analyzed data and wrote the paper. RL‐G, and IM‐M performed research and reviewed the paper. SA, TSS, MFL, EW, LE provided patients. VV designed and reviewed the paper.
RELATIONSHIP DISCLOSURE
None of the authors have any disclosures relevant to this paper.
de la Morena‐Barrio ME, López‐Gálvez R, Martínez‐Martínez I, et al. Defects of splicing in antithrombin deficiency. Res Pract Thromb Haemost. 2017;1:216–222. 10.1002/rth2.12025
Funding information
This work was supported by PI15/00079 and CB15/00055 from ISCIII & FEDER; Fundación Española de Trombosis y Hemostasia (FETH), and GATRA Grifols Award. MEM‐B holds a fellowship from (FETH). IMM holds a “Miguel Servet” contract from ISCIII.
REFERENCES
- 1. Bjork I, Olson ST. Antithrombin. A bloody important serpin. Adv Exp Med Biol. 1997;425:17–33. [PubMed] [Google Scholar]
- 2. Martinelli I, De Stefano V, Mannucci PM. Inherited risk factors for venous thromboembolism. Nat Rev Cardiol. 2014;11:140–56. [DOI] [PubMed] [Google Scholar]
- 3. Lane DA, Bayston T, Olds RJ, et al. Antithrombin mutation database: 2nd (1997) update. For the Plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1997;77:197–211. [PubMed] [Google Scholar]
- 4. Bock SC, Harris JF, Balazs I, Trent JM. Assignment of the human antithrombin III structural gene to chromosome 1q23‐25. Cytogenet Cell Genet. 1985;39:67–9. [DOI] [PubMed] [Google Scholar]
- 5. Olds RJ, Lane DA, Chowdhury V, De Stefano V, Leone G, Thein SL. Complete nucleotide sequence of the antithrombin gene: evidence for homologous recombination causing thrombophilia. Biochemistry. 1993;32:4216–24. [DOI] [PubMed] [Google Scholar]
- 6. Luxembourg B, Delev D, Geisen C, et al. Molecular basis of antithrombin deficiency. Thromb Haemost. 2011;105:635–46. [DOI] [PubMed] [Google Scholar]
- 7. de la Morena‐Barrio ME, Martinez‐Martinez I, de Cos C, et al. Hypoglycosylation is a common finding in antithrombin deficiency in the absenceof a SERPINC1 gene defect. J Thromb Haemost. 2016;14:1549–60. [DOI] [PubMed] [Google Scholar]
- 8. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–98. [DOI] [PubMed] [Google Scholar]
- 9. Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de la Morena‐Barrio ME, Buil A, Anton AI, et al. Identification of antithrombin‐modulating genes. Role of LARGE, a gene encoding a bifunctional glycosyltransferase, in the secretion of proteins? PLoS ONE. 2013;8:e64998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corral J, Huntington JA, Gonzalez‐Conejero R, et al. Mutations in the shutter region of antithrombin result in formation of disulfide‐linked dimers and severe venous thrombosis. J Thromb Haemost. 2004;2:931–9. [DOI] [PubMed] [Google Scholar]
- 12. Martinez‐Martinez I, Johnson DJ, Yamasaki M, et al. Type II antithrombin deficiency caused by a large in‐frame insertion. Structural, functional and pathological relevance. J Thromb Haemost. 2012;10:1859–66. [DOI] [PubMed] [Google Scholar]
- 13. de la Morena‐Barrio ME, Sevivas TS, Martinez‐Martinez I, et al. Congenital disorder of glycosylation (PMM2‐CDG) in a patient with antithrombin deficiency and severe thrombophilia. J Thromb Haemost. 2012;10:2625–7. [DOI] [PubMed] [Google Scholar]
- 14. Desmet FO, Hamroun D, Lalande M, Collod‐Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jochmans K, Lissens W, Yin T, et al. Molecular basis for type 1 antithrombin deficiency: identification of two novel point mutations and evidence for a de novo splice site mutation. Blood. 1994;84:3742–8. [PubMed] [Google Scholar]
- 16. Emmerich J, Vidaud D, Alhenc‐Gelas M, et al. Three novel mutations of antithrombin inducing high‐molecular‐mass compounds. Arterioscler Thromb. 1994;14:1958–65. [DOI] [PubMed] [Google Scholar]
- 17. Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base‐pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. [DOI] [PubMed] [Google Scholar]
- 18. Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–81. [DOI] [PubMed] [Google Scholar]
- 19. Ars E, Kruyer H, Morell M, et al. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet. 2003;40:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teraoka SN, Telatar M, Becker‐Catania S, et al. Splicing defects in the ataxia‐telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64:1617–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krawczak M, Thomas NS, Hundrieser B, et al. Single base‐pair substitutions in exon‐intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–8. [DOI] [PubMed] [Google Scholar]
- 22. Mort M, Sterne‐Weiler T, Li B, et al. MutPred Splice: machine learning‐based prediction of exonic variants that disrupt splicing. Genome Biol. 2014;15:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caspers M, Pavlova A, Driesen J, et al. Deficiencies of antithrombin, protein C and protein S ‐ Practical experience in genetic analysis of a large patient cohort. Thromb Haemost. 2012;108:247–57. [DOI] [PubMed] [Google Scholar]
- 24. Ordonez A, de CC, Minano A, et al. Coexistence of three genetic risk factors in a Spanish thrombophilic family: Factor V Leiden, prothrombin 20210 and a new type I antithrombin deficiency. Thromb Haemost. 2007;97:153–5. [PubMed] [Google Scholar]
- 25. Hammond SM, Wood MJ. Genetic therapies for RNA mis‐splicing diseases. Trends Genet. 2011;27:196–205. [DOI] [PubMed] [Google Scholar]
- 26. Havens MA, Hastings ML. Splice‐switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016;44:6549–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nuzzo F, Radu C, Baralle M, et al. Antisense‐based RNA therapy of factor V deficiency: in vitro and ex vivo rescue of a F5 deep‐intronic splicing mutation. Blood. 2013;122:3825–31. [DOI] [PubMed] [Google Scholar]
- 28. Tajnik M, Rogalska ME, Bussani E, et al. Molecular basis and therapeutic strategies to rescue factor IX variants that affect splicing and protein function. PLoS Genet. 2016;12:e1006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation of sequence variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. J Mol Diagn. 2015;19:4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]