

Abstract
Essentials.
The optimal dosing strategy of rivaroxaban for patients at the extremes of body weight is not known.
A pharmacokinetic study was conducted based in real‐world patients in a London teaching hospital.
In the cohort of patients studied, weight on its own did not impact significantly on rivaroxaban pharmacokinetics.
A larger study with patients in the weight categories of interest from the real‐world population is required to further clarify the situation.
Background
There is concern amongst clinicians that the fixed dosing strategy of rivaroxaban for the treatment of venous thromboembolism (VTE) might not be optimal in those patients under or overweight.
Objective
To develop a pharmacokinetic model for rivaroxaban, based on real‐world patients, specifically focusing on the impact of patients’ body weight on rivaroxaban pharmacokinetics.
Patients/methods
One hundred and one patients prescribed rivaroxaban prophylactic or treatment doses for the prevention or treatment of VTE were recruited at a London teaching hospital. Subjects had up to 3 rivaroxaban concentrations measured during a single dosing period (trough, 1 and 3 hours post dose). Population pharmacokinetic analyses was conducted to develop a rivaroxaban model, which was subsequently evaluated.
Results
A one‐compartment model with between‐subject variability on rivaroxaban clearance and volume of distribution, with a combined (additive and proportional) error model, best fitted the data. Following a full covariate analysis, creatinine clearance on rivaroxaban clearance was found to be the significant covariate impacting on the pharmacokinetic profile of rivaroxaban in the dataset.
Conclusions
Our results suggest that the most important covariate impacting on rivaroxaban pharmacokinetics is creatinine clearance and the weight alone has little effect. These findings are in line with previous studies for rivaroxaban. Larger datasets, from real‐world patients who are followed longitudinally, should be conducted to provide front‐line clinicians with further reassurance when prescribing rivaroxaban for the acute management of VTE.
Keywords: anticoagulation, obesity, pharmacokinetics, rivaroxaban, weight
1. INTRODUCTION
The direct Xa‐inhibitors are attractive anticoagulants relative to traditional anticoagulants.1 Post‐regulatory studies, along with real‐world studies, suggest that these direct oral anticoagulants (DOACs) are safe and effective,2, 3, 4 however, since their availability in clinical practice there has been some debate about whether the one‐size‐fits‐all approach with these agents is the best way to use them.5, 6 A particular cohort of patients where uncertainty exists with this dosing strategy are those patients at the extremes of weight. In their summary of product characteristics, the manufacturers of rivaroxaban state that in patients who are at the extremes of weight (<50 kg or >120 kg), only a small influence of weight on patients’ rivaroxaban plasma concentrations (<25%) is observed and no dose adjustment is necessary.7 This conclusion is derived from a small pharmacokinetic study in 36 healthy volunteers, administered 10 mg of rivaroxaban8; how well the results from such a study translate to clinical outcomes is difficult to determine. The lack of robust data has left clinicians wondering if the fixed dosing strategy truly is sufficient for those in obese category class II or III and perhaps too much for patients <50 kg.
With the prevalence of obesity increasing substantially in recent years, obese patients are no longer exceptional in clinical practice. This issue is particularly relevant in the acute management of venous thromboembolism (VTE), where it's important to efficiently manage the thrombosis and prevent further morbidity and mortality. Equally, more frail patients, and/or underweight patients are presenting to clinic with acute VTE and one questions the wisdom in prescribing the same dose that would be given to patients of no frailty, often larger individuals. The DOACs dosing strategy is very different to that employed by low molecular weight heparin (LMWH), where dosing is based on a mg/kg or IU/kg basis, despite the LMWHs having predictable pharmacokinetic profiles.9 Weight is also a well‐recognized covariant, impacting on the response to warfarin therapy.10
Whilst pharmacokinetic studies and pharmacokinetic models already exist for rivaroxaban,11, 12, 13, 14 they do not currently provide sufficient evidence for clinicians prescribing for patients at the extremes of weight, in determining whether the doses are safe and effective. In recent years, both a sub‐analysis of the Einstein Deep Vein Thrombosis/Pulmonary Embolism (DVT/PE) studies15 and a UK study16 have begun to assess the effect of extremes of body weight and clinical outcomes with standard‐dose rivaroxaban in the treatment of VTE, with both reporting that the fixed‐dose rivaroxaban strategy did not impact on clinical outcomes in those at the extremes of weight through different methods employed in these individual studies. Current Scientific and Standardisation Committee (SSC) guidance of the International Society of Thrombosis and Haemostasis (ISTH) on the use of DOACs in obese patients makes three recommendations17: (i) to use appropriate standard dosing of DOACs in patients up to 40 kg/m2 and weight 120 kg for VTE prevention and treatment and prevention of ischemic stroke and systemic arterial embolism in NVAF, (ii) not to use DOACs in patients with a BMI >40 kg/m2 or weight >120 kg due to limited data and available Pharmacokinetic/Pharmacodynamic (PK/PD) evidence suggesting decreased drug exposure, peak concentration, and shorter elimination half‐lives with increasing weight, and (iii) if DOACs are used in patients with a BMI >40 kg/m2 or weight >120 kg, then to check drug‐specific peak and trough level. If the drug level is reported back within the expected range, continuation of the DOAC seems reasonable. The guideline authors suggest if the drug level is found to be below the expected range, then the guidance committee suggest changing to a Vitamin K Antagonist (VKA) rather than adjusting the dose of the DOAC. No guidance is provided by the SSC on what to do in patients with a weight <50 kg. The ISTH guidance is interesting, as it implies that all DOACs have a similar pharmacokinetic profile. Whilst it's acknowledged that all of the DOACs have a more predictable pharmacokinetic profile relative to VKAs, each DOAC has its own distinct pharmacokinetic profile worthy of individual consideration when considering patients at the extremes of weight.
Clinicians need more robust data before they can be sure of the optimal dosing strategy of each individual DOAC in this particular cohort of patients. In our hospital, rivaroxaban is the first line agent for the management of VTE and we wanted to begin to better understand the relationship between body weight and rivaroxaban exposure, particularly for the management of acute VTE.
2. OBJECTIVES
To develop a pharmacokinetic model for rivaroxaban, based on real‐world patients, specifically focusing on the impact of patients’ body weight on rivaroxaban pharmacokinetics.
3. METHODS
3.1. Study setting and recruitment
This study saw collaboration between the departments of haematology and orthopaedic surgery at King's College Hospital (KCH). KCH is a 1000‐bedded London teaching hospital based in South East London, providing tertiary care for cardiology, neurology, haematology, and liver specialities. The orthopaedic surgery department at KCH conducts ~10 elective knee or hip replacements per week. Following surgery, patients are routinely prescribed VTE thromboprophylaxis with enoxaparin (a LMWH), administered by subcutaneous injection for the duration of their inpatient stay and then rivaroxaban 10 mg daily following discharge from hospital. Rivaroxaban is continued for 2 weeks for elective knee replacement patients and 4 weeks for elective hip replacements.
The thrombosis service at KCH provides acute and chronic management of all patients who have suffered an acute DVT/PE and for patients managed with long‐term anticoagulation for the prevention of recurrent VTE. At the time of the study (2013), first‐line treatment with LMWH and warfarin was gradually phased out and replaced with rivaroxaban at a dose of 15 mg twice daily for 3 weeks, followed by 20 mg daily thereafter, following rivaroxaban's license and NICE approval in the UK in the summer of 2012.18
Patients were eligible for the study if they were 18 years of age or older and were prescribed rivaroxaban for the aforementioned indications in these departments. Patients were excluded from the study if they had a documented allergy to rivaroxaban, if they had significant renal function impairment (CrCl <30 mL/min–using the Cockcroft Gault method of calculation19), if they were prescribed the following concomitant drugs which are known to significantly impact on rivaroxaban concentrations: clarithromycin, telithromycin, HIV protease inhibitors, ketaconazole, itraconazole, voriconazole, and posaconazole, or if they had significant liver disease or deranged baseline coagulation.
For the orthopaedic population, eligible patients were identified soon after their procedure on the orthopaedic ward. For the VTE population, eligible patients were identified following a positive diagnosis through either the outpatient DVT service or thrombosis clinic in the haematology department.
All patients provided informed written consent prior to participation. At the time of consent, all participants were given a book to record the exact times they took their rivaroxaban tablets each day until they were seen by SB for the index study visit for blood sampling. Where doses were missed (if any), patients were asked to record these in the book provided by SB.
3.2. Blood sampling
Subjects were asked to take their rivaroxaban tablets at a specific time of day in relation to their follow‐up visit to the hospital as part of routine clinical care. On the day of their follow‐up visit to the hospital (index study visit), patients had up to three blood samples drawn (at different times), to assess how the concentration of rivaroxaban was changing with time. The number of samples the patients had drawn was dictated by the patients themselves (minimum of 1 and maximum of 3). On the day of their visit, patients arrived to the hospital not having taken their rivaroxaban. SB then took a blood sample (trough) and asked the patient to take their rivaroxaban. One hour later, a further sample was taken and a third sample was taken 3‐4 hours post dose.
3.3. Sampling handling and analysis
Anti‐Xa activity was used to characterize the rivaroxaban concentration. For determination of anti‐Xa activity, 2.7 mL blood (9 vol) sample was collected in 0.109 M (3.2% trisodium citrate) Becton‐Dickinson Vacutainer. Following collection, the sample was centrifuged in a Rotina 420 R centrifuge (Hettich Zentrifugen) and double spun for 7 minutes at 2500 g and frozen within 1 hour of sample collection. The samples were stored at −40°C until analyzed. Samples were thawed and analyzed in weekly batches using the STA anti‐Xa assay (Diagnostica Stago, Asnieres‐sur‐Seine, France), with appropriate rivaroxaban calibrators and controls, on the STA‐R evolution analyzer (Diagnostica Stago) in the laboratory at King's College Hospital. Results were reported as ng/mL. The lower and upper limits of quantification for this assay are <20 ng/mL and >500 ng/mL, respectively. This functional assay has been shown to correlate well with analysis conducted using turbulent flow liquid chromatography with high‐resolution mass spectrometry.20
3.4. Analysis and PK modelling
Pharmacokinetic (PK) analysis was conducted using the method of non‐linear mixed effects modelling. Population PK modelling fits mathematical models to describe pharmacokinetic data that arises from more than one individual.21 The method does not require each subject to provide sufficient data to characterize their own PK profile, as PK information is shared between individuals to develop the population PK profile.21 The method therefore allows the use of sparse sampling study designs, having an obvious advantage when applied in the clinic setting. A typical population PK model integrates both a covariate and a statistical model.22, 23 The covariate model describes relationships between PK parameters and patient characteristics. The statistical model describes the variance in PK between and within individuals as well as residual variance due to biological variability, measurement errors, and errors in the fit of the model to the data.
3.5. Developing the rivaroxaban model
Initially, several structural base models were developed, eg, a one‐ and two‐compartment model and the model which best fit the data was selected. Goodness‐of‐fit plots, a statistical improvement in the fit of the model to the data using the objective function (minus twice the log‐likelihood of the data), assessment of the precision of the parameter estimates, and residual variability were the criterion used to evaluate and choose the base model to take forward for full covariate analysis. The specific covariates evaluated as part of this analysis were those which had a mechanistic meaning: age, weight, BMI, lean body weight, creatinine, creatinine clearance, ethnicity, and gender. The covariate analysis involved a classical graphical approach, plotting individual estimates of the pharmacokinetic parameters random effects (ETAs) against each of the covariates being considered for inclusion in the model. Each selected covariate was then tested by univariate addition into the base model to confirm its relevance. A decrease in objective function value of at least 6.64 (P < .01), was required to retain the covariate in the intermediate model. All significant covariates were then simultaneously added to the base model and their continued relevance was evaluated using a stepwise backward elimination method, where each covariate was removed singularly from the model. An increase in the objective function value of greater than 10.82 (P < .001) was required to retain the covariate in the final model.
Both the base and the final models were evaluated using a non‐parametric bootstrap procedure (1000 replicates).24 The final model was additionally evaluated using a visual predictive check,25 where the 5th, 50th, and 95th prediction intervals, simulated from the posterior distribution of the final model parameter estimates were overlaid with the 5th, 50th, and 95th percentiles from the observed data. A well‐performing model would see the observed percentiles and simulated prediction intervals superimposed.
Pharmacokinetic analysis was conducted using NONMEM (ICON plc, Dublin, Ireland) version 7.2.1326 and graphical analysis associated with the PK modelling and simulation was conducted using “R” version 2.14.1.40.
3.6. Ethical approval
The study was approved by the London Harrow Ethics Committee; REC reference number: 12/LO/1951 and the local Research and Development Committee at King's College Hospital.
4. RESULTS
During the recruitment period (June 1, 2013‐April 30, 2014), 101 patients consented to take part in the study. Demographic information on these patients is presented in Table 1. Ninty‐seven patients were either being managed for acute VTE or the secondary prevention of VTE and 4 patients were prescribed prophylactic rivaroxaban in the context of elective orthopaedic surgery prophylaxis. The 101 subjects provided 193 samples for PK modelling purposes, 24 patients (24%) providing 3 samples at the index study visit, 45 patients providing 2 samples (44%), and 32 subjects (32%) providing 1 sample.
Table 1.
Demographic information on the patients recruited
| Patient demographics (n = 101) | |
|---|---|
| Age, years (mean range) | 52 (20‐86) |
| Male/female (%) | 58/42 |
| Body weight, kg (mean ± SD) | 88.0 (23.4) |
| <50 kg (%) | 2 |
| 50‐100 kg (%) | 81 |
| >100 kg (%) | 17 |
| Lean body weight, kg (mean ± SD) | 57.0 (11.3) |
| BMI, kg/m2 (%) | |
| 16‐18.49 | 1 |
| 18.5‐24.9 | 27 |
| 25‐29.9 | 32 |
| 30‐34.9 | 21 |
| 35‐39.9 | 14 |
| ≥40 | 6 |
| Creatinine clearance, (%) | |
| >80 mL/min | 67 |
| 50‐79 mL/min | 25 |
| 30‐49 mL/min | 7.8 |
| <30 mL/min | 0.2 |
| Indication for anticoagulation (%) | |
| Acute VTE first event | 58 |
| Acute recurrent event | 26 |
| Secondary prevention of VTE | 12 |
| Elective orthopaedic VTE prophylaxis | 4 |
| Dose of rivaroxaban (%) | |
| 20 mg once a day | 38 |
| 15 mg twice a day | 57 |
| 10 mg once a day | 4 |
| 15 mg once a day | 1 |
| Proportion of samples provided at each time point (%) | |
| Trough only | 32 |
| Trough and 1 hour post dose | 44 |
| Trough, 1 and 3 hours post dose | 24 |
| Ethnicity (%) | |
| White | 74 |
| Afro‐Caribbean | 21 |
| Other | 5 |
BMI, body mass index; SD, standard deviation; VTE, venous thromboembolism.
Figure 1 illustrates the breadth of samples generated in this study, in relation to body weight, and Figure 2 illustrates the breadth of concentrations in relation to time after dose of the samples generated by this study. Of these, 14 samples were below or above the level of quantification (BLQ or ALQ) of the assay being utilized. ALQ or BLQ were handled using a mixed‐method approach. For BLQ <20 ng/mL, where results were obtained from the analyzer, these were entered into the model as reported, otherwise half BLQ (0.10 ng/mL) were entered into the dataset. ALQ values (>500 ng/mL) were entered into the dataset, as 500 ng/mL. The total BLQ or ABL for the dataset comprised <10% of the total dataset, and research suggests that when this is the case, that no one single approach is superior for handling BLQ/ABL for PK modelling purposes.27
Figure 1.
Breadth of rivaroxaban samples obtained with respect to bodyweight, in relation to time after dose
Figure 2.

Breadth of rivaroxaban concentrations obtained with respect to time after dose
4.1. Rivaroxaban base model development
Several base models were initially explored (ie, one‐ and two‐compartment models). A one‐compartment model with between‐subject variability on rivaroxaban clearance and volume of distribution with a combined (additive and proportional) error model best fitted the data (Table 2, with associated 1000 bootstrap).
Table 2.
Base model PK estimates with associated bootstrap (1000 replicates)
| Parameter | Base model estimates (%SE) | 1000 Bootstrap median (2.5th‐97.5th) |
|---|---|---|
| CL (L/h) | 8.59 (7) | 8.35 (7.34‐9.55) |
| V d (L) | 104 (13) | 99.10 (79.2‐130.4) |
| K a (/h) | 1.32 (24) | 1.28 (0.78‐2.28) |
| ωCL (%CV) | 56 (25) | 73.8 (61.6‐81.8) |
| ωVd (%CV) | 66 (52) | 82.6 (56.6‐94.3) |
| Proportional error (%) | 28.6 (84) | 51.3 (5.6‐65.6) |
| Additive error (ng/mL) | 0.018 (38) | 0.018 (0.008‐0.027) |
| Objective function | −853.710 | – |
CL, clearance; K a, absorption rate constant; SE, standard error; V d, volume of distribution; ωCL, between subject variability on rivaroxaban clearance; ωVd, between subject variability on rivaroxaban volume of distribution.
A full covariate analysis was then conducted evaluating the following selected covariates: weight, lean body weight, creatinine, creatinine clearance, ethnicity, gender, and BMI (see Table 3). Only creatinine clearance on clearance met the criteria for inclusion to the final model.
Table 3.
Univariate covariate addition to the base model
| Model | Covariate | ∆OBV |
|---|---|---|
| 4 | CL × (CrCl/79)0.434 | −14.65 |
| 5 | CL × (Wt/87)0.0403 | −0.05 |
| 6 | CL × (Age/51)−0.335 | −6.91 |
| 7 | CL × (Cre/81)−0.44 | −5.46 |
| 8 | CL × (LBW/58)0.275 | −1.45 |
| 9 | CL × (BMI/30)−0.126 | −0.55 |
| 10 | CL × (ETHN)−0.038 | −0.09 |
| 11 | V d × (ETHN)0.139 | −0.64 |
| 12 | V d × (BMI/30)−0.027 | −0.01 |
| 13 | V d × (LBW/58)0.264 | −0.69 |
| 15 | V d × (Cre/81)0.454 | −0.30 |
| 16 | V d × (Age/51)0.0539 | −0.07 |
| 17 | V d × (Wt/87)0.0657 | −0.07 |
| 18 | V d × (CrCl/79)−0.137 | −0.65 |
∆OBV represents the change in the base model objective function with the addition of the covariate. The base line OBV was −853.710. All models minimised successfully. Gender was modelled as a “what‐if” statement and was not found to impact significantly on CL or V d (∆OBV 0.07 for CL and ∆OBV 0.09 for V d).
BMI, body mass index; CL, clearance; CrCl, creatinine clearance; Cre, serum creatinine; ETHN, ethnicity; LBW, lean body weight; V d, volume of distribution; Wt, actual body weight.
The final rivaroxaban PK estimates for typical values of CL and V d thus can be represented mathematically as:
Table 4 provides the estimates for the PK parameters for the final model developed.
Table 4.
The typical value of clearance and volume of distribution from the final model developed with the associated bootstrap results
| Parameter | Final covariate model (%SE) | 1000 Bootstrap median (2.5th‐97.5th) |
|---|---|---|
| CL (L/h) | 8.86 (7) | 8.57 (7.58‐9.79) |
| V d (L) | 101 (12) | 95 (75.3‐125) |
| K a (/h) | 1.21 (34) | 1.21 (0.72‐2.13) |
| FACCrCl | 0.434 (30) | 0.416 (0.215‐0.619) |
| ωCL (%CV) | 48 (99) | 64.5 (53.8‐78.2) |
| ωVd (%CV) | 60 (247) | 82.1 (38.7‐96.1) |
| Proportional error (%) | 31 (215) | 52.7 (5.62‐67.2) |
| Additive error (ng/mL) | 0.016 (112) | 0.016 (0.006‐0.025) |
| Objective function | −868.355 | – |
CL, clearance; FACCrCL is the exponent on creatinine clearance as a covariate on clearance; K a, absorption rate constant; SE, standard error; V d, volume of distribution; ωCL, between subject variability on rivaroxaban clearance; ωVd, between subject variability on rivaroxaban volume of distribution.
Finally, a visual predictive check was conducted (Figure 3), demonstrating a well‐performing rivraoxaban PK model.
Figure 3.
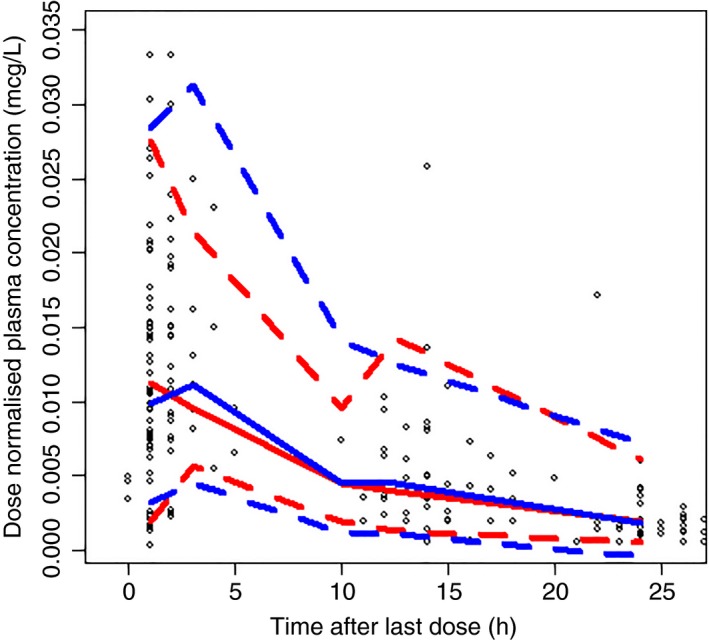
Visual predictive check from final model. Red lines represent the observed anti‐Xa activities and the blue lines represent the simulated anti‐Xa activities. Fifth (dashed), median, and 95th (dashed) percentiles are presented for both sets of concentrations
5. DISCUSSION
This study aimed to develop a PK model for rivaroxaban derived from real‐world patients, with a particular focus on body weight. From the dataset generated, a relatively well‐performing PK model for rivaroxaban was developed (Table 5 in comparison to PK models published by the manufacturers of rivaroxaban) and the data suggests that the single best co‐variant predicting rivaroxaban exposure is creatinine clearance.
Table 5.
Comparison of previous PK models of rivaroxaban and the model developed in this study
| PK parameter | Mueck et al.11 | Mueck et al.12 | Mueck et al.13 | Xu et al.14 | Present study |
|---|---|---|---|---|---|
| Compartment model | One | One | One | One | One |
| CL (L/h), (%SE) | 7.51 (4.1) | 7.3 (4) | 5.67 (3.70) | 6.48 (2.21) | 8.86 (7) |
| V d (L), (%SE) | 58.2 (4.9) | 49.1 (4.3) | 54.4 (3.80) | 57.9 (1.16) | 101 (12) |
| K a (/h), (%SE) | 1.49 (10.0) | 1.81 (8.3) | 1.23 (5.0) | 1.24 (3.28) | 1.21 (34) |
| ωCL (%CV) | 38.2 (10.0) | 38.6 (8.3) | 39.9 (7.60) | 31.3 (4.72) | 48 (99) |
| ωVd (%CV) | 32.4 (23.0) | – | 28.8 (11.4) | 10.0 (3.66) | 60 (247) |
| ωKa (%CV) | – | – | – | 139 (0.30) | – |
| Proportional residual error (%) (%SE) | 52.6 (3.0) | 37.1 (4.0) | 40.7 (3.20) | – | 31.0 (215) |
| Additive error (ng/mL) (%SE) | – | – | – | 0.352 (1.09) | 0.016 (112) |
CL, clearance; K a, absorption rate constant; SE, standard error; V d, volume of distribution; ωCL, between subject variability on rivaroxaban clearance; ωVd, between subject variability on rivaroxaban volume of distribution.
Elimination of rivaroxaban is via a dual pathway: renal excretion and metabolic degradation. Approximately 1/3 of rivaroxaban is eliminated as unchanged active drug in the urine, with active renal secretion accounting for 30% and glomerular filtration for 6%. The specific transporters involved with active renal secretion of rivaroxaban are P‐glycoprotein (P‐gp) and breast cancer resistance protein (BCRP). The remaining two‐thirds of rivaroxaban is metabolized by several cytochrome P450 enzymes (CYP 3A4/5, CYP2J2) and CYP‐independent mechanisms.28 Given this, and the fact that active renal secretion predominates renal clearance, perhaps it's not surprising that renal function is the significant predictor of rivaroxaban exposure. It's also important to remember that this clearance mechanism is unique to rivaroxaban and what is reported here, may not be the case for the other currently available DOACs. We were surprised that weight did not feature more significantly as a covariant in our study. There are three possible reasons for this: (i) weight is not an important determinant of rivaroxaban pharmacokinetics and the current manufacturers recommendations are correct, (ii) we did not have enough patients represented in the weight categories of interest in our study to pick this out correctly, or (iii) the CrCl equation utilized in this study and commonly used in clinical practice, already has weight accounted for, as follows:
where weight in the present study was computed using a LBW equation,29 and F = 1.04 for females and 1.23 for males.
Research suggests that both renal and liver activity increases in the obese,30 therefore one might predict that clearance also increases in this population. Han and colleagues31 stipulate that (i) obese patients exhibit a higher absolute drug clearance compared to their normally weighted counterparts, (ii) clearance of drugs does not increase linearly with total body weight, however (iii) clearance and lean body weight (as calculated through a mechanistically derived lean body weight equation) are linearly correlated. Perhaps with more patients in the specific weight categories of interest, weight would come through as important covariant on rivaroxaban pharmacokinetics and further data is clearly required to clarify this.
On closer examination of the model developed here, the estimates for V d are greater than that described in the PK models previously published by the manufacturers of rivaroxaban. This illustrates how the typical pharmacokinetic estimates will vary in the real‐world population in comparison to those volunteers who took part in the earlier studies and confirms the need for further PK studies similar to the present study, with larger datasets in the future, from real‐world patients.
Some data from other studies is becoming to emerge. Di Nisio and colleagues15 completed a sub‐analysis of the EINSTEIN DVT/PE studies, with the aim of determining the incidence of major bleeding in patients with low body weight and recurrent VTE in patients with high body weight. Using a Cox proportional hazards model, no association was found between bodyweight and BMI and risk of recurrent VTE, major bleeding or clinically relevant bleeding. The authors of this sub‐analysis conclude that body weight is not associated with an increased risk of major bleeding or recurrent VTE in patients with either a low or high bodyweight. However, the data from this study is derived from the clinical trials of rivaroxaban, and its questionable how well represented the extremes of weight were in these studies. Archillage and colleagues16 assessed 167 acute VTE patients, stratified into three groups based on weight (<50 kg, 50‐120 kg, >120 kg), with patients having rivaroxaban concentration measured 2‐4 hours post dose (peak). Patients were followed for a median of 14 months. The authors report that peak rivaroxaban plasma concentrations were significantly higher in patients with a lower body weight (<50 kg). Those patients with a weight >120 kg had comparable rivaroxaban peak concentrations to those of standard body weight. The authors also report that weight did not appear to impact on clinical outcomes. Although the results from this small study are encouraging, it's important to note that peak sampling is not considered the best time‐point, when evaluating the clearance of a drug, which would be the case when wanting to evaluate the impact of weight. Trough sampling would have been more informative. More recently a study in the setting of bariatric surgery, where prophylactic doses of rivaroxaban were given pre‐ and post‐bariatric surgery, found single doses of rivaroxaban resulted in similar systematic drug exposures prior to and after bariatric surgery.32 The median weights of patients in this study having sleeve gastrectomy was 137 kg (range 112‐153) and 101.5 kg (range 96‐120) for Roux‐en‐Y gastric bypass, ie, not excessively obese.
The findings from these afore‐mentioned studies are reassuring, and in our study, we also found patients’ rivaroxaban concentrations to be comparable when comparing the standard body weight patients and those in the obese category.
Our findings suggest that rivaroxaban certainly behaves differently from other traditional anticoagulants, in the extremes of weight population, ie, that the dose of anticoagulant does not need to increase in line with weight. The findings we report here are specific to rivaroxaban. Whilst the general principal of what we found might translate to other DOACs, due to their unique pharmacokinetic profiles, specific studies with each DOAC should be conducted to assess if the same is true for each one.
The results of our study, should be considered in the context of their limitations. We conducted this study, at a time when experience with rivaroxaban was emerging and the agent had newly become available; the number of patients prescribed the drug in the extremes of weight category was less at the time of recruitment compared to our current practice now and that described in ISTH guidance.17 Therefore, the number of patients in the extremes of weight categories in the present study, could have been greater. Despite this, a well performing rivaroxaban model was developed and with further pharmacokinetic data coupled with outcome data, further direction to offer clinicians should be possible.
6. CONCLUSION
Our study developed a pharmacokinetic model rivaroxaban model focusing on the impact body weight had on rivaroxaban exposure across a wide weight range, derived from a real‐world population. Our study suggests that weight on its own is not a good predictor of rivaroxaban exposure, and found renal function computed by the Cockcroft–Gault equation to be a significant covariant explaining rivaroxaban exposure. Further studies with larger data‐sets from the extremes of weight cohort of patients will confirm/refute the findings from our work.
RELATIONSHIP DISCLOSURE
SB was supported through an unrestricted grant from Bayer plc. Bayer had no role in the design, data collection, analysis or writing of the report for this study.
AUTHOR CONTRIBUTIONS
SB collected and helped with analysis of the data. JPP, LNR, RKP, VK and RA designed the study. JPP and BG developed the PK model described. JPP drafted the manuscript, which was critically reviewed by all authors.
ACKNOWLEDGMENTS
The authors would like to thank the patients who agreed to participate in this study.
Barsam SJ, Patel JP, Roberts LN, et al. The impact of body weight on rivaroxaban pharmacokinetics. Res Pract Thromb Haemost. 2017;1:180–187. 10.1002/rth2.12039
Contributor Information
Jignesh P. Patel, Email: jig.patel@kcl.ac.uk.
Roopen Arya, https://twitter.com/aryaroopen.
REFERENCES
- 1. Sterne JA, Bodalia PN, Bryden PA, et al. Oral anticoagulants for primary prevention, treatment and secondary prevention of venous thromboembolic disease, and for prevention of stroke in atrial fibrillation: systematic review, network meta‐analysis and cost‐effectiveness analysis. Health Technol Assess. 2017;21:1–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beyer‐Westendorf J, Förster K, Pannach S, et al. Rates, management and outcome of bleeding complications during rivaroxaban therapy in daily care: results from the Dresden NOAC registry. Blood. 2014;124:955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ageno W, Mantovani LG, Haas S, et al. Safety and effectiveness of oral rivaroxaban versus standard anticoagulation for the treatment of symptomatic deep‐vein thrombosis (XALIA): an international, prospective, non‐interventional study. Lancet Haematol. 2016;3:e12–21. [DOI] [PubMed] [Google Scholar]
- 4. Camm AJ, Amarenco P, Haas S, et al.; on behalf of the Xantus Investigators . XANTUS: a real‐world, prospective, observational study of patients treated with rivaroxaban for stroke prevention in atrial fibrillation. Eur Heart J. 2016;37:1145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel JP, Couchman L, Chitongo PB, Flanagan RJ, Arya R. New anticoagulants: dosing and monitoring. BMJ. 2015;350:h1585. [DOI] [PubMed] [Google Scholar]
- 6. Powell JR. Are new oral anticoagulant dosing recommendations optimal for all patients? JAMA. 2015;313:1013–4. [DOI] [PubMed] [Google Scholar]
- 7. Bayer plc . Summary of product characteristics of rivaroxaban. 2016. http://www.medicines.org.uk/emc/medicine/25586 (accessed March 11, 2017).
- 8. Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of rivaroxaban (BAY 59‐7939) in healthy subjects. J Clin Pharmacol. 2007;47:218–26. [DOI] [PubMed] [Google Scholar]
- 9. Patel JP, Roberts LN, Arya R. Anticoagulating obese patients in the modern era. Br J Haematol. 2011;155:137–49. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi H, Wilkinson GR, Nutescu EA, et al. Different contributions of polymorphisms in VKORC1 and CYP2C9 to intra‐ and inter‐population differences in maintenance dose of warfarin in Japanese, Caucasians and African‐Americans. Pharmocogen Genom. 2006;16:101–10. [DOI] [PubMed] [Google Scholar]
- 11. Mueck W, Borris LC, Dahl OE, et al. Population pharmacokinetics and pharmacodynamics of once‐ and twice‐daily rivaroxaban for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Haemost. 2008;100:453–61. [PubMed] [Google Scholar]
- 12. Mueck W, Eriksson BI, Bauer KA, et al. Population pharmacokinetics and pharmacodyanamics of rivaroxaban – an oral, direct factor Xa inhibitor in patients undergoing major orthopaedic surgery. Clin Pharmacokinet. 2008;47:203–16. [DOI] [PubMed] [Google Scholar]
- 13. Mueck W, Lensing AWA, Agnelli G, Decousus H, Prandoni P, Misselwitz F. Population pharmacokinetic analyses in patients treated for acute deep‐vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50:675–86. [DOI] [PubMed] [Google Scholar]
- 14. Xu S, Moore K, Burton P, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban in patients with acute coronary syndromes. Br J Clin Pharmacol. 2012;74:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Nisio M, Vedovati MC, Riera‐Mestre A, et al. Treatment of venous thromboembolism with rivaroxaban in relation to body weight – a sub‐analysis of the Einstein DVT/PE studies. Thromb Haemost. 2016;116:739–46. [DOI] [PubMed] [Google Scholar]
- 16. Arachillage DRJ, Reynolds R, Devey T, Maclean R, Kitchen S, van Veen JJ. Effects of extremes of body weight on drug level in patient treated with standard dose of rivaroxaban for venous thromboembolism. Thromb Res. 2016;147:32–5. [DOI] [PubMed] [Google Scholar]
- 17. Martin K, Beyer‐Westendorf J, Davidson BL, Huisman MV, Sandset PM, Moll S. Use of the direct oral anticoagulants in obese patients: guidance from the SSC of the ISTH. J Thromb Haemost. 2016;14:1308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. National Institute for Clinical Excellence . TA 261 – Rivaroxaban for the treatment of DVT and prevention of recurrent DVT and PE, July 2012 [cited 2017 July 7]. Available from http://www.nice.org.uk/guidance/ta261.
- 19. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. [DOI] [PubMed] [Google Scholar]
- 20. Gous T, Couchman L, Patel JP, Paradzai C, Arya R, Flanagan RJ. Measurement of the direct oral anticoagulants apixaban, dabigatran, edoxaban, and rivaroxaban in human plasma using turbulent flow liquid chromatography with high resolution mass spectrometry. Ther Drug Monit. 2014;36:597–605. [DOI] [PubMed] [Google Scholar]
- 21. Duffull SB, Wright DFB, Winter HR. Interpreting population pharmacokinetic‐pharmacodynamic analyses – a clinical viewpoint. Br J Clin Pharmacol. 2011;71:807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheiner LB. The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab Rev. 1984;15:153–71. [DOI] [PubMed] [Google Scholar]
- 23. Aarons L. Population pharmacokinetics: theory and practice. Br J Clin Pharmacol. 1991;32:669–70. [PMC free article] [PubMed] [Google Scholar]
- 24. Davison AC, Hinkley DV. Bootstrap methods and their application. New York: Cambridge University Press; 1997. [Google Scholar]
- 25. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82:17–20. [DOI] [PubMed] [Google Scholar]
- 26. Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user's guides (1989–2009). Ellicott City, MD: Icon Development Solutions; 2009. [Google Scholar]
- 27. Keizer RJ, Jansen RS, Rosing H, Beijnen JH, Schellens JHM, Huitema ADR. Incorporation of extrapolated concentration data below the limit of quantification in population PK analyses. Presented as a poster at the Population Approach Group in Europe Annual Meeting, 2010. [cited 2017 July 2]. Available from http://www.page-meeting.org/?abstract=1722
- 28. Mueck W, Stampfuss J, Kubitza D, Becka M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet. 2014;53:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44:1051–65. [DOI] [PubMed] [Google Scholar]
- 30. Chagnac A, Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol Ren Physiol. 2000;278:F817–22. [DOI] [PubMed] [Google Scholar]
- 31. Han PY, Duffull SB, Kirkpatrick CMJ, Green B. Dosing in obesity: a simple solution to a big problem. Clin Pharmacol Ther. 2007;82:505–8. [DOI] [PubMed] [Google Scholar]
- 32. Kroll D, Stirnimann G, Vogt A, et al. Pharmacokinetics and pharmacodynamics of single doses of rivaroxaban in obese patients prior to and after bariatric surgery. Br J Clin Pharmacol. 2017;83:1466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]