

Abstract
Essentials.
Platelet function defects may cause atypical bleeding symptoms in immune thrombocytopenia (ITP).
An isolated platelet defect of collagen‐induced aggregation was explored in a patient with ITP.
ITP mediated by antibodies to glycoprotein (GP) VI curtail receptor function.
Inclusion of GPVI in diagnostic antibody detection assays may improve their diagnostic utility.
Idiopathic immune thrombocytopenia (ITP) is an autoimmune disorder characterized by relapsing/ remitting thrombocytopenia. Bleeding complications are infrequent with platelet counts above 30×109/L, and this level is commonly used as a threshold for treatment. The question of another/ co‐existent diagnosis or an alternate mechanism of platelet destruction arises when bleeding is experienced with platelet counts above this threshold. We report a case of anti‐GPVI mediated ITP that was diagnosed following investigations performed to address this key clinical question. A patient with ITP experienced exaggerated bruising symptoms despite a platelet count of 91×109/L. Platelet functional testing showed an isolated platelet defect of collagen‐induced aggregation. Next generation sequencing excluded a pathogenic variant of GP6, and anti‐GPVI antibodies that curtailed GPVI function were confirmed by extended platelet phenotyping. We propose that anti‐GPVI mediated ITP may be under‐recognized, and that inclusion of GPVI in antibody detection assays may improve their diagnostic utility and in turn, facilitate a better understanding of ITP pathophysiology and aid individualized treatment approaches.
Keywords: collagen, immune, platelet membrane glycoproteins, purpura, thrombocytopenia
1. CASE REPORT
A 41‐year‐old female presented to our haematology clinic for the investigation of a platelet function defect characterized by an isolated reduction of collagen induced aggregation that had been detected by light transmission aggregometry (LTA) in the setting of a long‐standing history of primary immune thrombocytopenia (ITP). The cause of the platelet function defect (PFD) was postulated to be secondary to an acquired autoantibody in the setting of ITP or to an inherited glycoprotein (GP) VI mutation. The undefined nature and clinical significance of the platelet defect had delayed surgery in the past and hence diagnosis was important to inform future treatment, and predict bleeding risk.
Immune thrombocytopenia had been diagnosed 19 years prior following mucocutaneous bleeding associated with a platelet count of 10×109/L. Subsequent full blood count examinations, bone marrow biopsy and response to immunosuppression were consistent with this diagnosis. Six years before her referral she received pulsed high dose corticosteroid, as well as intravenous immunoglobulin followed by dapsone for a relapse of severe thrombocytopenia (platelet count 2×109/L). This approach had achieved a complete response and dapsone was later ceased electively. No other treatment for ITP had been required following that period. Other medications included low dose amitryptiline (12.5 mg nocte) for chronic pain relating to osteonecrosis of the hip following steroid treatment, and alprazolam (0.5 mg) for insomnia.
Platelet function studies were requested prior to elective laparoscopic pelvic surgery, due to bruising symptoms that were inconsistent with the degree of thrombocytopenia (platelet count 91×109/L). Results of the LTA confirmed a PFD with absent platelet responses to collagen (1.25 μg/mL), with only slow and reduced response to this agonist at a higher concentration (6 μg/mL). Normal aggregation was noted with adenosine diphosphate (2.5 μmol/L) and arachidonic acid (1.6 mmol/L).
At the time of our initial consultation, the patient's full blood count demonstrated mild thrombocytopenia with no other abnormalities evident on the peripheral blood film (Figure 1A). ITP screening blood tests1 were repeated and excluded an underlying inflammatory condition or secondary acquired cause for thrombocytopenia. An isolated platelet defect in response to collagen was confirmed by LTA (Figure 1B). Platelet flow cytometry demonstrated normal surface expression of GPIb‐IX, integrin αIIbβ3, GPIV, and α2β1 with reduced expression of the collagen receptor GPVI (Figure 1C) and Western blots of patient platelet lysates using an anti‐GPVI cytoplasmic tail antibody confirmed the absence of both intact (60‐kDa band) and cleaved GPVI (~10‐kDa band) respectively (Figure 1D). Next generation sequencing was performed, and excluded a mutation in the coding sequence of GPVI.
Figure 1.
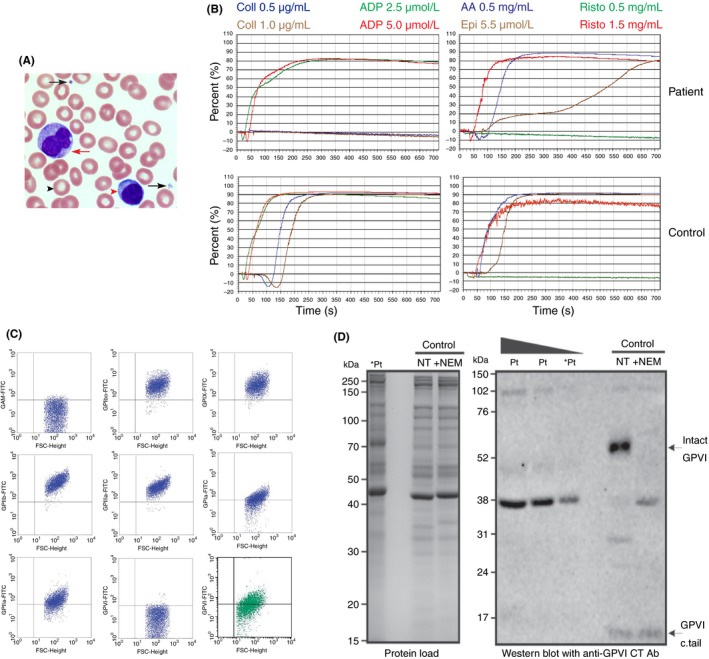
Patient platelet morphology and function. (A) Representative image of a blood film (100× magnification) demonstrating normal platelet (black arrows), red cell (black arrow head), monocyte (red arrow), and lymphocyte (red arrow head) morphology. (B) Platelet responses to agonists, collagen (Coll) (0.5 and 1.0 μg/mL), adenosine diphosphate (ADP) (2.5 and 5 μmol/L), arachidonic acid (AA) (0.5 mg/mL), epinephrine (Epi) (5.5 mol/L) and ristocetin (Risto) (0.5 and 1.5 mg/mL) as measured by LTA are shown for the patient (top) and healthy control (bottom). (C) Flow cytometry of patient platelets (blue dot plots) and a healthy donor (green dot plot) demonstrating reduced GPVI platelet surface expression on patient platelets. (D) Intact GPVI was detectable by western blot of non‐treated (NT) washed platelet lysates from a healthy donor (control) but not at three different loadings of platelet lysates from the patient (Pt; lanes labelled *Pt were loaded with equivalent volumes of lysate). Treatment with 5 mmol/L NEM for 15 minutes (+NEM) to cause shedding of GPVI enabled detection of a ~10 kDa band (GPVI c.tail)
To determine the presence of anti‐GPVI antibodies and elucidate their mechanism of action additional investigations were performed. Aggregation of donor platelets by patient serum was demonstrated by mixing 1:1 volume/volume with washed donor platelets. Rapid aggregation was noted which was blocked by pre‐incubation of donor platelets with either 10 μg/mL IV.3 (function blocking monoclonal antibody against FcγRIIa) or by pre‐treatment of donor platelets with 1 mmol/L NEM (a non‐specific treatment of platelets which induces metalloproteolytic shedding of GPVI). NEM‐treated platelets remained responsive to ristocetin demonstrating GPIb‐IX‐V and associated signalling pathways were functional, and indicating that the aggregating property of patient serum required functional FcγRIIa and intact GPVI (Figure 2A). These data are consistent with patient serum containing an anti‐GPVI autoantibody that caused platelet activation requiring an intact GPVI/ FcγRIIa complex.
Figure 2.
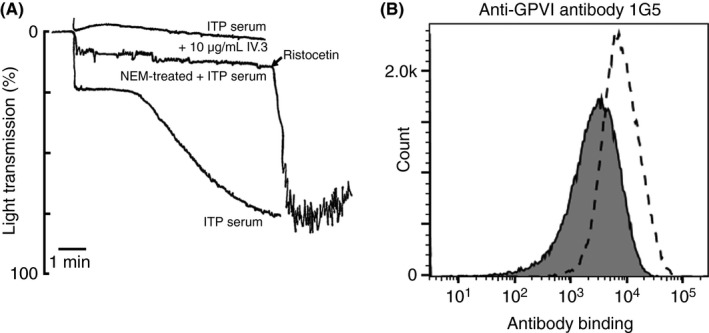
Patient serum aggregates donor washed platelets in an FcγRIIa‐ and GPVI‐dependent manner. Washed platelets were prepared as described previously18 and light transmission aggregometry was performed. (A) Patient serum was mixed with an equal volume of donor platelets or donor platelets pre‐treated with 10 μg/mL FcγRIIa‐blocking antibody IV.3 or 1 mmol/L NEM to remove GPVI. NEM‐treated platelets did not aggregate in the presence of patient serum but aggregated upon addition of 1.5 mg/mL ristocetin. (B) Flow cytometry of donor platelets using anti‐GPVI monoclonal antibody 1G5 to detect GPVI after 3 hours’ incubation at 37°C with equal volume of patient serum (filled histogram) or control serum (open histogram)
Flow cytometry of donor platelets incubated for 3 hours with control or patient serum was performed to evaluate whether patient serum could induce loss of platelet surface receptors (GPVI and others) from donor platelets. Expression levels of platelet surface receptors on donor platelets were monitored by flow cytometry. Incubation with patient but not control serum resulted in significant generation of microparticles, and so all measured platelet receptors (αIIbβ3, GPIbα, CD9, and GPVI) were reduced, however GPVI was clearly highly susceptible to loss by this treatment with 79% receptor clearance occurring during the experimental time frame (Figure 2B). Taken together, this data confirmed a rare occurrence of an anti‐GPVI autoantibody that engages both GPVI and FcγRIIa leading to platelet activation and aggregation. The observed loss of GPVI is probably via immunoreceptor tyrosine‐based activation motif (ITAM)‐mediated metalloproteolytic shedding as described in other cases.2, 3
2. DISCUSSION
Primary ITP is an acquired thrombocytopenia that is not associated with a systemic trigger including viral infection, connective tissue disorder, drug therapy, or lymphoproliferative neoplasm).1 Thrombocytopenia ensues following platelet destruction that is mediated by the production of platelet specific immunoglobulin‐G (most commonly targeting platelet αIIbβ3 and GPIb‐IX) by B‐lymphocytes, as well as, reduced platelet production secondary to T‐lymphocyte mediated platelet, and megakaryocyte cytotoxicity.4
GPVI is an integral platelet surface receptor that is expressed in a non‐covalent complex with the platelet Fc receptor γ chain (FcRγ).5 Surface expression and function of GPVI is dependent on its association with FcRγ.6 Binding of GPVI by agonists that include collagen, collagen‐related peptide, and snake toxins7, 8 induce structural changes of both GPVI and FcRγ leading to phosphorylation of Src and Syk tyrosine kinases.8 These in turn initiate an intracellular signaling pathway causing eventual shape change and activation of integrin αIIbβ3 with resultant platelet aggregation mediated by αIIbβ3 binding of von Willebrand factor or fibrinogen.8
Autoantibodies to GPVI have been reported in ITP9, 10, 11, 12, 13 and as in our case, activate platelets in a GPVI/ FcRγIIa complex dependent manner.13 Subsequent loss of platelet surface GPVI causing reduced responsiveness to collagen is likely mediated through autoantibody engagement of GPVI and FcRγIIa that results in metalloproteolytic shedding of GPVI by ADAM10 on platelets.3, 13
The diagnosis of ITP is made following exclusion of secondary causes of thrombocytopenia, as there is no single diagnostic assay.1, 4 Routine use of antiplatelet antibody assays, such as the monoclonal antibody immobilization of platelet antigens assay (MAIPA), that measures antibodies bound to specific platelet glycoproteins for the diagnosis ITP is not recommended, due to lack of reported sensitivity (50%‐80%) and specificity (80%) of this platform in the setting of ITP.1, 14 The standard MAIPA platform measures antibodies bound to platelet antigens, αIIbβ3 and GPIb‐IX, and does not include antibodies to α2β1 and GPVI, although the latter have both been described in cases of ITP.9, 10, 11, 12, 13, 15 It is feasible that the sensitivity and specificity of this assay may be improved by the addition of these additional antigenic targets into standard MAIPA platforms, thereby validating its use for diagnostic purposes. The identification of anti‐platelet antibodies at diagnosis has been reported to be associated with a higher likelihood of disease chronicity and bleeding symptoms.16 Therefore, the utility of these assays prospectively could also potentially impact treatment and monitoring strategies.
Major bleeding including intracerebral haemorrhage is rare in ITP and occurs predominantly in patients with platelet counts below 10×109/L.17 A platelet count of less than 30×109/L is generally regarded as a threshold for treating ITP.1 Thrombopoietin (TPO) regulates thrombopoiesis through activation of TPO receptors on the megakaryocyte cell surface, resulting in increased platelet production. Two second generation TPO agonists, Eltrombopag and Romiplostim, are approved for the treatment of ITP.1 Romiplostim treatment may directly upregulate the expression of platelet GPVI through receptor‐mediated demethylation of the GP6 promoter.18 Moreover, Gardiner and colleagues19 reported normalized GPVI levels and platelets responses to collagen by LTA in a patient with anti‐GPVI mediated ITP following 6 months of treatment with Romiplostim. This case illustrates that anti‐GPVI antibodies can impair receptor function contributing to increased mucocutaneous bleeding symptoms at platelet counts above the threshold of 30×109/L. Taken together therefore, identification of anti‐GPVI antibodies at diagnosis may aid treating clinicians to elect to pursue closer surveillance schedules, or even embark on “disease”‐specific therapies with TPO agonists at platelet counts higher than the accepted threshold.
Finally, this case demonstrates the utility of genetic based testing in uncharacterized platelet disorders. In our report, the absence of a GP6 mutation suggested that an acquired rather than inherited PFD was responsible for the loss of GPVI expression. Moreover, it provided reassurance to the patient who had concerns regarding transmission of a possible clinically significant variant to offspring.
In summary, we describe a rare case of anti‐GPVI mediated ITP and showed that these antibodies not only cause initial platelet activation, but result in loss of platelet surface GPVI causing clinically appreciable platelet dysfunction that is characterized by mild bleeding symptoms at platelet counts not usually associated with bleeding in ITP. In the case described here, elective surgery was delayed due to concerns of the etiology and significance of the patient's platelet defect. Ultimately an alternate non‐surgical treatment strategy was pursued which facilitated investigation and identification of the patient's platelet dysfunction. Future peri‐operative care will be guided by recommendations for the management of mild bleeding disorders.20
We propose that anti‐GPVI mediated ITP may be underappreciated, as LTA is generally not performed for bleeding symptoms associated with ITP, and probably more significantly, GPVI is not currently included in standard MAIPA assays. Inclusion of GPVI as a protein target, together with other known antigenic markers (α2β1), may improve diagnostic utility of this assay, provide prognostic information and may facilitate individualized treatment approaches.
AUTHOR CONTRIBUTIONS
D. J. Rabbolini, S. Dunkley and C. M. Ward provided biological samples and analyzed clinical data. W. S Stevenson, M‐C. Morel‐Kopp and D. J. Rabbolini performed and analyzed next‐generation sequencing data. E. E. Gardiner, A. Jahangiri, C. Lee, D. J. Rabbolini and M.‐C. Morel‐Kopp performed and analyzed laboratory investigations. D. J. Rabbolini, E. E. Gardiner, W. S. Stevenson and C. M. Ward wrote the manuscript.
RELATIONSHIP DISCLOSURES
The authors declare no competing financial interests.
ACKNOWLEDGEMENTS
The authors acknowledge excellent laboratory assistance from Ms. Christine Lee and Ms. Anila Jahangiri.
Rabbolini DJ, Gardiner EE, Morel‐Kopp M‐C, et al. Anti‐glycoprotein VI mediated immune thrombocytopenia: An under‐recognized and significant entity? Res Pract Thromb Haemost. 2017;1:291–295. 10.1002/rth2.12033
REFERENCES
- 1. Neunert C, Lim W, Crowther M, Cohen A, Solberg L Jr, Crowther MA; American Society of Hematology . The ASH 2011 evidence‐based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190–207. [DOI] [PubMed] [Google Scholar]
- 2. Gardiner EE, Karunakaran D, Arthur JF, et al. Dual ITAM‐mediated proteolytic pathways for irreversible inactivation of platelet receptors: de‐ITAM‐izing FcγRIIa. Blood. 2008;111:165–74. [DOI] [PubMed] [Google Scholar]
- 3. Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb‐IX‐V by ADAM family metalloproteinases. J Thromb Haemost. 2007;5:1530–7. [DOI] [PubMed] [Google Scholar]
- 4. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood. 2017;129:2829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcαR and the natural killer receptors. J Biol Chem. 1999;274:29019–24. [DOI] [PubMed] [Google Scholar]
- 6. Nieswandt B, Bergmeier W, Schulte V, Rackebrandt K, Gessner JE, Zirngibl H. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRγ chain. J Biol Chem. 2000;275:23998–4002. [DOI] [PubMed] [Google Scholar]
- 7. Andrews RK, Kamiguti AS, Berlanga O, Leduc M, Theakston RD, Watson SP. The use of snake venom toxins as tools to study platelet receptors for collagen and von Willebrand factor. Haemostasis. 2001;31:155–72. [DOI] [PubMed] [Google Scholar]
- 8. Nieswandt B, Watson SP. Platelet‐collagen interaction: is GPVI the central receptor? Blood. 2003;102:449–61. [DOI] [PubMed] [Google Scholar]
- 9. Sugiyama T, Okuma M, Ushikubi F, Sensaki S, Kanaji K, Uchino H. A novel platelet aggregating factor found in a patient with defective collagen‐induced platelet aggregation and autoimmune thrombocytopenia. Blood. 1987;69:1712–20. [PubMed] [Google Scholar]
- 10. Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen‐induced aggregation and adhesion. J Clin Invest. 1989;84:1440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arai M, Yamamoto N, Moroi M, Akamatsu N, Fukutake K, Tanoue K. Platelets with 10% of the normal amount of glycoprotein VI have an impaired response to collagen that results in a mild bleeding tendency. Br J Haematol. 1995;89:124–30. [DOI] [PubMed] [Google Scholar]
- 12. Boylan B, Chen H, Rathore V, et al. Anti‐GPVI‐associated ITP: an acquired platelet disorder caused by autoantibody‐mediated clearance of the GPVI/FcRgamma‐chain complex from the human platelet surface. Blood. 2004;104:1350–5. [DOI] [PubMed] [Google Scholar]
- 13. Gardiner EE, Al‐Tamimi M, Mu FT, et al. Compromised ITAM‐based platelet receptor function in a patient with immune thrombocytopenic purpura. J Thromb Haemost. 2008;6:1175–82. [DOI] [PubMed] [Google Scholar]
- 14. Brighton TA, Evans S, Castaldi PA, Chesterman CN, Chong BH. Prospective evaluation of the clinical usefulness of an antigen‐specific assay (MAIPA) in idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Blood. 1996;88:194–201. [PubMed] [Google Scholar]
- 15. Warner MN, Moore JC, Warkentin TE, Santos AV, Kelton JG. A prospective study of protein‐specific assays used to investigate idiopathic thrombocytopenic purpura. Br J Haematol. 1999;104:442–7. [DOI] [PubMed] [Google Scholar]
- 16. Grimaldi D, Canoui‐Poitrine F, Croisille L, et al. Antiplatelet antibodies detected by the MAIPA assay in newly diagnosed immune thrombocytopenia are associated with chronic outcome and higher risk of bleeding. Ann Hematol. 2014;93:309–15. [DOI] [PubMed] [Google Scholar]
- 17. Lee MS, Kim WC. Intracranial hemorrhage associated with idiopathic thrombocytopenic purpura: report of seven patients and a meta‐analysis. Neurology. 1998;50:1160–3. [DOI] [PubMed] [Google Scholar]
- 18. Kanaji S, Kanaji T, Jacquelin B, et al. Thrombopoietin initiates demethylation‐based transcription of GP6 during megakaryocyte differentiation. Blood. 2005;105:3888–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gardiner EE, Thom JY, Al‐Tamimi M, et al. Restored platelet function after romiplostim treatment in a patient with immune thrombocytopenic purpura. Br J Haematol. 2010;149:625–8. [DOI] [PubMed] [Google Scholar]
- 20. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost. 2007;5:164–74. [DOI] [PubMed] [Google Scholar]