

Abstract
Essentials.
Patients with liver diseases may acquire substantial changes in all components of hemostasis.
Hemostasis is in unstable balance due to simultaneous changes in pro‐ and antihemostatic systems.
Intrahepatic activation of hemostasis may contribute to disease progression.
Optimal strategies for prevention and treatment of bleeding and thrombosis are currently unknown.
Patients with liver diseases may develop alterations in all components of the hemostatic system. Thrombocytopenia, low levels of coagulation factors and inhibitors, low levels of fibrinolytic proteins, and increased levels of endothelial‐derived proteins such as von Willebrand factor are all part of the coagulopathy of liver disease. Due to concomitant changes in pro‐ and antihemostatic drivers, the net effects of these complex hemostatic changes have long been unclear. According to current concepts, the hemostatic system of patients with liver disease is in an unstable balance, which explains the occurrence of both bleeding and thrombotic complications. This review will discuss etiology and management of bleeding and thrombosis in liver disease and will outline unsolved clinical questions. In addition, we will discuss the role of intrahepatic activation of coagulation for progression of liver disease, a novel paradigm with potential consequences for the general management of patients with liver disease.
Keywords: acute liver failure, bleeding, cirrhosis, hypercoagulability, thrombosis
1. INTRODUCTION
The liver is a central organ in the homeostasis of the hemostatic system. The liver synthesizes the majority of plasma proteins involved in hemostasis including pro‐ and anticoagulant factors, pro‐ and antifibrinolytic factors, and thrombopoietin. In patients with advanced liver diseases complex alterations in the hemostatic system arise which are summarized in Table 1.1 Although it is commonly assumed many of these changes are related to decreased hepatic synthesis, a consumptive coagulopathy of systemic or intrahepatic origin may also contribute.2, 3
Table 1.
Alterations in the hemostatic system in patients with liver disease that impair (left) or promote (right) hemostasis
| Changes that impair hemostasis | Changes that promote hemostasis |
|---|---|
| Thrombocytopenia | Elevated levels of von Willebrand Factor (VWF) |
| Platelet function defects | Decreased levels of ADAMTS‐13 |
| Enhanced production of nitric oxide and prostacyclin | Elevated levels of factor VIII |
| Low levels of factors II, V, VII, IX, X, and XI | Decreased levels of protein C, protein S, antithrombin, α2‐macroglobulin, and heparin cofactor II |
| Vitamin K deficiency | Low levels of plasminogen |
| Dysfibrinogenemia | |
| Low levels of α2‐antiplasmin, factor XIII, and TAFI | |
| Elevated t‐PA levels |
Source: Modified from the European Association for the Study of the Liver from Lisman et al.1 with permission.
The net effects of the complex hemostatic changes in liver diseases have long been unclear. In the next sections, we will provide arguments for a “reset” balance in the hemostatic system in most patients with cirrhosis and acute liver failure.4, 5 Interestingly, any of the components of the hemostatic system may be simultaneously altered in patients with liver diseases. The fact that the hemostatic system can deal relatively well with such extensive changes teaches us about the resilience of the hemostatic system in general. In this review we will summarize laboratory and clinical features of the altered hemostatic system in patients with liver diseases.
2. WHY LIVER DISEASES WERE PREVIOUSLY CONSIDERED AS BLEEDING DISORDERS
Routine tests of hemostasis such as the prothrombin time (PT), activated partial thromboplastin time (APTT), and platelet count are frequently abnormal in these patients and these test results all indicate a hypocoagulable status. In patients with acute liver failure, abnormal routine hemostasis tests are present per definition since an international normalized ratio (INR) >1.5 is part of the definition of the syndrome. Although the use of the INR in this context seems peculiar since the INR was developed and validated only for monitoring of vitamin K antagonists, the hepatology community misuses this test extensively. It is not only part of the definition of acute liver failure, but also part of the prognostic score that are used to prioritize patients with cirrhosis on the waiting list for a liver transplant. Ironically, there is immense laboratory‐to‐laboratory variation in the INR in plasma samples from patients with liver disease.6, 7 This way, transplant candidates in centers with reagents yielding lower INR results may be at increased risk for dying on the waiting list.
Two clinical observations appear to agree with liver diseases as being a bleeding disorder. First, spontaneous bleeding complications in patients with cirrhosis are common. However, it has now been well established that the most common bleeding complication, ruptured esophageal varices, is unrelated to a defective hemostatic system.8 Rather, this bleeding event relates to local vascular abnormalities in combination with portal hypertension. Also in acute liver failure, bleeding was common in studies presented in the 1970s.9 At that time, around one‐third of patients with acute liver failure died with bleeding as the proximate cause of death. In a recent series, however, spontaneous and clinically significant bleeding is rare at around 5%, and bleeding very rarely results in death.10 The reasons for this substantial reduction in bleeding are unclear, but it has to be noted that the intensive care management of patients with acute liver failure has been revolutionized since the 1970s. Second, bleeding during invasive procedures was a substantial problem. When liver transplantation was introduced as a standard clinical procedure in the 1980s, bleeding complicated most, if not all, procedures.11, 12 Massive amounts of blood products (red cell concentrates, plasma, and platelet concentrates) were required in many liver transplant procedures. The explanation for the massive blood loss during surgery was believed to be the preoperative coagulopathy which further aggravated during the (lengthy) surgical procedure.13
During the last two decades, however, there has been a tremendous decline in transfusion requirements during liver transplantation. In fact, more and more centers report that in a proportion of patients liver transplant procedures can now be performed without the requirement for any blood products.14, 15, 16 Part of the decline in transfusion requirements may be related to improvements in surgical and anesthesiological techniques and improvements in donor organ quality and preservation. However, the key factor in the decrease in transfusion requirements has been the understanding that preoperative correction of the abnormal routine hemostasis test results is not required, and may even do more harm than good. Preoperative correction of thrombocytopenia and elevated PT and APTT test results inevitably requires administration of substantial amounts of volume. In the liver disease patient with portal hypertension, increased plasma volume, and disturbed cardiac function, administration of fluids results in a further increase in portal and central venous pressure. Thus, when platelet concentrates and plasma are administered with the aim to improve the hemostatic status, the increased portal and central venous pressure may in fact promote bleeding when surgical damage is inflicted.17
3. REBALANCED HEMOSTASIS IN LIVER DISEASES
The PT and APTT are only sensitive for procoagulant proteins, and are therefore unlikely to predict the hemostatic status of patients with complex hemostatic alterations. In patients with liver diseases, both pro‐ and anticoagulant factors may be present in decreased levels, and the hemostatic status of such patients can only be appreciated by using tests that take the balance between pro‐ and anticoagulant factors into account. Similarly, the status of the primary hemostatic system and the fibrinolytic system can only be assessed using global tests. Using modern thrombin generation tests, in which thrombomodulin was added to allow for activation of the protein C system, it was shown that thrombin generation in patient plasma was comparable to that of controls, despite substantially prolonged PT and APTT values in the patients.18 Thus, a concomitant decline in pro‐ and anticoagulant factors results in a reset balance in the coagulation system, and a similar argument applies for platelet function and fibrinolysis.19, 20, 21, 22 Some studies have even shown evidence for enhanced hemostatic capacity in patients with liver diseases, which is in sharp contrast to the old dogma of liver diseases as a bleeding disorder.23, 24, 25, 26, 27, 28, 29 Below we will separately address laboratory and clinical evidence for rebalanced hemostasis in cirrhosis and acute liver failure.
3.1. The hemostatic status in patients with cirrhosis
Mild to moderate thrombocytopenia is common in cirrhosis, and older studies have also indicated functional platelet defects.30, 31, 32 Thrombocytopenia in cirrhosis is multifactorial as reviewed previously.1, 33 An unexplored potential contributor to the thrombocytopenia of liver disease are circulating histones. Histones have been shown to induce thrombocytopenia in mice34 and have been implicated in thrombocytopenia in patients admitted to intensive care.35 As levels of circulating histones are slightly elevated in patients with cirrhosis, and substantially elevated in patients with acute liver failure,36 the role of histones in the thrombocytopenia of liver disease should be further explored. Next to their thrombocytopenia, patients with cirrhosis may have a prolonged skin bleeding time indicative of platelet defects.37 However, more recent data suggest that intrinsic platelet function in patients with cirrhosis is normal38 or even hyperactive,28 although net platelet function may be decreased due to thrombocytopenia and/or anemia.39 Technical issues complicate the interpretation of many published studies on platelet function in cirrhosis.40 Laboratory studies using flow‐based models have shown that the thrombocytopenia of cirrhosis may be balanced by highly elevated levels of von Willebrand factor (VWF).20, 38 In addition, plasma levels of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) are decreased in patients with liver disease.41 Decreased ADAMTS13 does not result in an increase in the multimeric size of VWF. In fact, a decrease of higher molecular weight multimers is present in plasma from patients with cirrhosis, which is presumably related to proteolysis by other VWF‐cleaving proteases such as plasmin.42, 43 Nevertheless, decreased ADAMTS13 in cirrhosis may promote primary hemostasis, as ADAMTS13 is also responsible for regulation of thrombus growth by proteolysis of VWF within a growing thrombus.44 Clinical evidence for an important compensatory role of VWF in the thrombocytopenia of cirrhosis may be deduced from studies in which thrombopoietin receptor agonists have been used to increase the platelet count in patients with cirrhosis. Elevation of the platelet count was associated with an increased risk of thrombosis, which may be attributable to the high VWF levels,45, 46 although alternative explanations for the thrombotic risk of thrombopoietin receptor agonists in these patients cannot be excluded. On the other hand, it has been suggested that thrombocytopenia (and not prolonged routine coagulation tests) increase the risk for procedure‐associated bleeding,47 although not all studies agree.48
Thrombin generation tests have shown normal‐to‐increased hemostatic potential in plasma from patients with cirrhosis when thrombin generation tests were performed in the presence of thrombomodulin.18, 23, 24, 25, 26, 27 Initially, it was concluded that the endogenous thrombin potential (ETP) in cirrhosis is normal18, but multiple subsequent studies have actually shown increased thrombin generation.23, 24, 25, 26, 27 Increasingly, the coagulation status of a patient with cirrhosis is not reported as the thrombin generating capacity, but rather as a ratio between thrombin generation tests performed in absence and presence of thrombomodulin, or as ratios between FVIII and protein C.49, 50, 51 Unfavorable ratios are referred to as “procoagulant imbalance,” which we feel is confusing terminology. We have argued against the use of ETP or FVIII/protein C ratios as estimates of hemostatic capacity as these ratios may be misleading, are difficult to interpret, and have no clear clinical correlation.52 Rather, we feel that ETP results obtained in the presence of thrombomodulin are the most adequate representation of coagulation status in patients with complex alterations in their hemostatic system. Nevertheless, these studies have demonstrated that the PT and APTT are inadequate to estimate the hemostatic status of patients with cirrhosis.
Besides preserved thrombin generating capacity, we have recently demonstrated procoagulant properties of the fibrinogen molecule. Despite reduced fibrinogen plasma levels, fibrin clot permeability, a measure of clot structure and quality, was decreased in patients with cirrhosis, which was attributable to oxidative modifications in the fibrinogen molecule.29
Cirrhosis has long been thought to be associated with a hyperfibrinolytic status related to increased levels of tissue‐type plasminogen activator which are insufficiently balanced by fibrinolytic inhibitors.53 However, using a global plasma‐based assay we demonstrated that the fibrinolytic system in cirrhosis was rebalanced due to a commensurate decline in pro‐ and antifibrinolytic factors.19 Importantly, a typical fibrinolytic bleeding rarely occurs in non‐surgical patients with cirrhosis, which supports the laboratory evidence of rebalanced fibrinolysis. Nevertheless, some studies using plasma‐based or whole blood assays indicating accelerated fibrinolysis in cirrhosis have questioned our findings.54, 55 Conversely, in large series of patients that were studied with thromboelastography, none had a fibrinolysis rate outside of the reference range.25, 56
Whole blood clot formation as tested by viscoelastic tests have been demonstrated to be normal in patients with cirrhosis,56 but also number of studies have demonstrated profound hypocoagulability.57, 58, 59, 60 Viscoelastic tests have (whole blood) clot formation as the endpoint, which is an obvious advantage to thrombin generation tests or routine diagnostic tests such as the PT, which have thrombin generation or the time point of fibrinogen to fibrin conversion in plasma as the endpoint. In addition, viscoelastic tests are sensitive for the activity of coagulation factor XIII.61 However, an obvious limitation of viscoelastic tests in patients with complex disorders of hemostasis is that it is not a true representation of hemostatic balance as it lacks activation of the anticoagulant protein C system, and is insensitive for von Willebrand factor. Given the profound changes in VWF and the protein C pathway in cirrhosis, it appears unlikely that thromboelastography forms a valid representation of overall hemostatic balance in these patients.
It has to be noted that most studies on the hemostatic balance in patients with cirrhosis have been performed in mixed cohorts of relatively well‐compensated patients. Although hemostatic changes in patients with cirrhosis from different etiologies are similar, they are certainly not identical. For example, patients with cholestatic cirrhosis appear in a more hypercoagulable state as compared to patients with noncholestatic cirrhosis,62 and patients with nonalcoholic fatty liver disease associated cirrhosis are somewhat more prothrombotic as compared to patients with alcohol‐induced cirrhosis.63 Also, it has been hypothesized that the hemostatic balance may be no longer maintained in patients with decompensated disease. However, in recent studies we demonstrated normo‐ to hypercoagulability in patients with acutely decompensated cirrhosis and acute‐on‐chronic liver failure using thrombin generation tests (unpublished data).
Although the hemostatic status of patients with cirrhosis appears in balance, there are clear hypo‐ and hypercoagulable features which may contribute to bleeding or thrombosis. Hypocoagulable features include hypofibrinogenemia,29 decreased clot formation and stability in some studies using viscoelastic testing,60 delayed fibrin polymerization,64 and hyperfibrinolysis.65 Hypercoagulable features include platelet hyperreactivity,28 enhanced thrombin generation,23, 24, 25, 26, 27 increased production of intravascular tissue factor,66 and prothrombotic properties of the fibrin clot.29
3.2. The hemostatic status of patients with acute liver failure
Thrombocytopenia is less common in patients with acute liver failure as compared to cirrhosis. The pathophysiology of thrombocytopenia in acute liver failure has not been extensively addressed, but recent studies indicate that platelet activation may contribute.67 Substantially elevated levels of highly procoagulant microparticles, mainly from platelet origin, have been demonstrated in plasma from patients with acute liver failure. These microparticles have been suggested to be the results of platelet fragmentation in a process resembling disseminated intravascular coagulation of sepsis. Indeed, intravascular (or intrahepatic) activation of hemostasis has been demonstrated in animal models of acute liver failure as will be outlined in the section on intrahepatic thrombosis as a contributor of disease progression. Whether the circulating procoagulant microparticles have a role in hemostasis has not been directly investigated.
Patients with acute liver failure, similar to patients with cirrhosis, have highly elevated levels of VWF and substantially decreased levels of ADAMTS13.22 Although ADAMTS13 levels are undetectable in a proportion of patients, a reduced proportion of high molecular weight multimers was detected, which is likely related to VWF proteolysis by proteases other than ADAMTS13.20 Interestingly, levels of ADAMTS13 measured on admission to the hospital have been related to outcome, which has been proposed to be a consequence of increased formation of intrahepatic platelet thrombi in those patients with low levels of ADAMTS13.
The alterations in coagulation proteins are more extensive in patients with acute liver failure compared to patients with cirrhosis. Levels of the liver‐derived factors are substantially decreased, and can become as low as 1–10% of normal.21, 68 Nevertheless, thrombomodulin‐modified thrombin generation in patients with acute liver failure was shown to be normal or increased relative to healthy controls.21, 68 In addition, thromboelastography test results are consistent with rebalanced hemostasis in acute liver failure.69
In contrast to cirrhosis, acute liver failure is characterized by a profound hypofibrinolytic status, which is likely related to substantially elevated plasma levels of PAI‐1 and low plasminogen levels.21
3.3. Clinical evidence of intact hemostatic capacity in cirrhosis and acute liver failure
What clinical evidence exists to support the theory of rebalanced hemostasis in liver disease and to refute the value of routine diagnostic tests such as the PT and platelet count in assessing hemostatic competence in liver diseases? First, more and more centers report transfusion‐free liver transplantation in a substantial proportion of patients.14, 15, 16 Although transfusion requirements still vary widely between centers,70 there is no doubt that this lengthy procedure with substantial surgical damage does not per se result in major blood loss. A recent study from a single center showed that almost 80% of a series of 700 consecutive patients were transplanted without any transfusion requirements.16 These data strongly argue against liver disease being associated with a bleeding diathesis. Secondly, although bleeding is common is cirrhosis, most bleeding episodes are unrelated to hemostatic dysfunction (eg, bleeding from ruptured esophageal varices). Bleeding problems such as bruising, purpura, epistaxis, gingival bleeding, menorrhagia, and bleeding associated with invasive procedures may be related to defective hemostasis. However, in some cases, elevated venous pressure may contribute to bleeding problems that at first sight appear as a consequence of deranged hemostasis. Interestingly, the extent of coagulopathy as measured by the PT or platelet count does not appear predictive of bleeding complications.71 In acute liver failure, clinically significant bleeding is rare, and in contrast to patients with cirrhosis bleeding from esophageal varices is virtually absent, which is explained largely by the usual absence of portal hypertension. Third, patients with liver disease are not protected from thrombotic events, and increasing clinical data refute the twentieth‐century concept of patients with liver diseases being “auto‐anticoagulated.” Cirrhosis has been identified as a risk factor for venous thrombosis,72 and in patients with acute liver failure, thrombotic complications are even more common than bleeding in patients with acute liver failure in recent series.69
3.4. The limited stability of the hemostatic balance in liver diseases
Although laboratory and clinical evidence supports the concept of rebalanced hemostasis in the “average” patient with liver disease, the new hemostatic balance appears much more fragile as compared to the hemostatic balance in patients with intact liver function (Figure 1). The limited balance of hemostasis in liver disease is in part explained by the low levels of pro‐ and antihemostatics, but may very well also be related to the distinct hypo‐ and hypercoagulable features of the patient with liver disease as discussed in the paragraph on “The hemostatic status in patients with cirrhosis.” As depicted in Figure 1, the patient with liver disease is constantly struggling to remain in hemostatic balance as a result on the low levels of hemostatic factors on each end of the hemostatic scale and by dynamic changes in pro‐ and antihemostatic processes. Therefore, it is likely difficult to predict using laboratory tests or clinical scores which patient is more likely to tip towards a bleeding diathesis and which one is more likely to develop thrombosis. The clinical reality is that patients may present with bleeding and thrombosis simultaneously, and obviously management of such patients is a particularly difficult clinical challenge.
Figure 1.
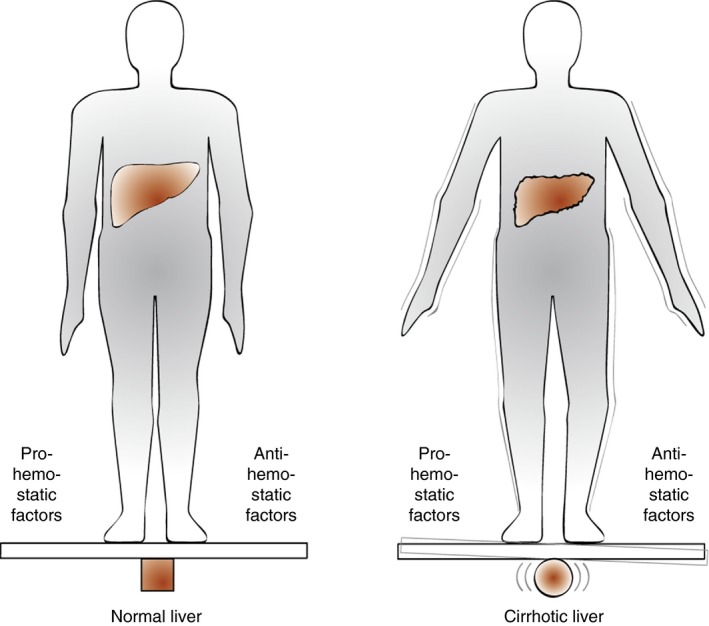
The hemostatic balance in patients with liver disease as compared to that of healthy individuals. This cartoon depicts the stable hemostatic balance in healthy individuals and shows that although the hemostatic system in patients with liver disease is (re)balanced, the balance is fragile and may easily tip to either a hypo‐ or a hypercoagulable status. Modified from the European Association for the Study of the Liver from Lisman et al.116 with permission
Furthermore, a number of disease‐related factors can actively tip the balance towards bleeding or thrombosis. Factors that promote bleeding include renal failure and infection, both of which are common in liver disease.73, 74 In addition, alterations in flow, endothelial activation, disruption of the endothelial glycocalix, and generation of procoagulant microparticles all potentially predispose to a thrombotic phenotype.
4. PREVENTION AND MANAGEMENT OF BLEEDING IN LIVER DISEASES
4.1. Bleeding in cirrhosis
We will limit our discussion to the management of “hemostatic” bleeding. Bleeding due to mechanical causes such as ruptured esophageal varices and true surgical bleeds are beyond the scope of this review. Prevention of bleeding during invasive procedures starts with a very restrictive fluid infusion policy the rationale for which is extensively outlined by us elsewhere.75, 76 As is evident from the liver transplant data, routine correction of abnormal routine test of hemostasis (platelet count, PT, APTT) by administration of platelet concentrates or plasma is not indicated and might even do harm.77 Undesired side‐effects of blood product administration include fluid overload and exacerbation of portal hypertension (which may paradoxically increase bleeding risk) in addition to general transfusion‐related side effects. In addition, complete correction of prolonged coagulation tests is almost never achieved.78 Although some investigators argue that a correction of the platelet count may be helpful in preventing bleeding in high risk procedures,71 clinical data substantiating this are lacking. It is even debated whether a low preprocedural platelet count increases procedural bleeding risk.47, 48 Nevertheless, preprocedural administration of platelet concentrates are still common, and thrombopoietin receptor agonists have been tested in clinical trials and are considered as potential alternatives for platelet transfusions,79 although they may be associated with an risk for thrombotic events.46 Based on our experience in liver transplantation, we believe it is best to wait with blood product administration until a hemostatic bleed actually occurs. Prophylactic administration of antifibrinolytics have been shown to substantially reduce blood product use during liver transplantation, and given the relative safety of such agents, they may be helpful in a prophylactic setting.80 Prophylactic correction of coagulation by low volume products such as prothrombin complex concentrates has theoretical advantages over administration of plasma and the effect of such agents on blood loss during liver transplantation is currently being investigated in a randomized controlled clinical trial.81 Other measures that are likely helpful in preventing bleeding is adequate control of infections and renal failure. Finally, a structured bleeding history should be part of the work‐up of patients with liver diseases for (elective) invasive procedures, although admittedly the value of this screening method for predicting bleeding in this population has not been thoroughly investigated.
When bleeding does occur, it is a challenge to decide on the most effective pro‐hemostatic therapy as bleeding may be caused by failure of any of the hemostatic systems (platelets, coagulation, fibrinolysis). Thromboelastography may be helpful in deciding which factor(s) require correction. Although thromboelastography‐based transfusion algorithms are rapidly gaining popularity,82, 83 convincing clinical evidence that such algorithms lead to an optimal restoration of hemostatic capacity is lacking. Pros and cons of various prohemostatic strategies in patients with liver diseases have been discussed by us elsewhere.84
4.2. Bleeding in acute liver failure
Prophylactic administration of blood products in patients with acute liver failure is not recommended.10 Not only is the risk of spontaneous or procedure‐related bleeding low, administration of blood products may cause worsening of intracranial hypertension. Moreover, the INR is an important prognostic indicator in acute liver failure, which is obscured by administration of plasma. Some authorities have recommended cautious prophylaxis with recombinant factor VIIa prior to high‐risk procedures, notably intracranial pressure monitors.85 However, the uncertain benefit and risk for thrombosis requires further evaluation of this strategy before broad use in this setting can be recommended.86
5. PREVENTION AND MANAGEMENT OF THROMBOSIS IN LIVER DISEASES
Thrombotic events, including venous thrombosis and portal vein thrombosis are frequent in patients with cirrhosis.72, 87, 88, 89, 90 In addition, anticoagulation for arterial events and atrial fibrillation may be required.
There are two major issues with antihemostatic treatment of patients with liver diseases. First, the liver and kidneys are involved in metabolic activation or clearance of a number of the drugs we use on a day‐to‐day basis in general thrombosis management. This results in unpredictable pharmacokinetics and for that reason only, some of these drugs are contraindicated for patients with liver diseases. However, despite these contraindications, the use of contraindicated drugs may be desired or deemed essential in selected cases. The unpredictable pharmacokinetics of anticoagulant drugs necessitates careful monitoring, which is a particular challenge in patients with liver diseases. Monitoring of vitamin K antagonists is difficult since the INR is already prolonged at baseline in patients with advanced disease. In such patients, the appropriate target INR has not been established. In addition, there is tremendous laboratory‐to‐laboratory variation in the INR in liver disease patients, which complicates studies on optimal target ranges.6 Recent studies have indicated that monitoring of heparins by standard anti‐Xa assays give a substantial underestimation of the true circulating heparin mass.91, 92 In clinical practice the use of anti‐Xa monitoring of heparins may therefore lead to incorrect and potentially dangerous dose escalations. Only anti‐Xa assays in which excess exogenous antithrombin is present in the reagent give accurate anti‐Xa levels in plasma from patients with cirrhosis.91 The second caveat of using antihemostatic drugs in patients with liver disease is altered drug potency. The complex hemostatic changes of liver disease were recently shown to impact drug potency (at least in vitro).92, 93, 94 Using in vitro thrombin generation tests it has been demonstrated that the extent by which thrombin generation is inhibited by various anticoagulant drugs may differ tremendously between patients and controls. The differential effects were proportional to the severity of disease indicating that the extent of alteration in the hemostatic system determines the deviation in potency. Confusingly, some drugs appear to have an increased anticoagulant potency, whereas others are less effective in plasma from patients with cirrhosis. Although it has not yet been established whether these in vitro alterations in potency are relevant in vivo, it may be that for optimal management of anticoagulant drugs in patients with liver diseases a monitoring test (or combination of tests) will be required that assesses both drug levels and drug potency.
As patients with cirrhosis are at risk for venous thrombosis, standard thromboprophylaxis should be applied upon immobilization, hospitalization, and post‐surgery, even in patients with a prolonged INR. Anticoagulant drugs (LMWH, vitamin K antagonists) have been used to treat established portal vein thrombosis, with resolution of the thrombus in a proportion of patients.95 Although the safety profile of LMWH in this context has been excellent and much better than that of vitamin K antagonists, prolonged treatment may be required which is a burden for the patient. For this reason, the use of direct oral anticoagulants is considered by an increasing number of centers,96, 97 despite the fact that there is virtually no clinical experience with these agents in patients with advanced liver disease, which were all excluded from the large randomized trials, and the theoretical issues with dosing and monitoring. One study has suggested that LMWH is also effective in prevention of portal vein thrombosis (PVT),98 but the results of that study await confirmation.
6. INTRAHEPATIC THROMBOSIS AS A CONTRIBUTOR TO DISEASE PROGRESSION
The most exciting advance in the field of thrombosis in liver diseases is the accumulating evidence of intrahepatic activation of coagulation as a contributor to disease progression. In the 1980s it was first recognized that microthrombi can be found in rodent models of liver disease,99, 100 and in the 1990s the first reports of microthrombi within human livers appeared.101 A functional role of these microthrombi has been suggested based on experimental animal models and clinical studies. In models of chronic liver disease, anticoagulant or antiplatelet drugs slow down the progression of disease.101, 102 Conversely, animals homozygous for factor V Leiden showed accelerated disease progression.102 Confusingly, in some models of cholestasis‐induced fibrosis, complete absence of platelets or fibrinogen paradoxically increases disease progression, suggesting that microthrombi may not always be harmful.103, 104 Fibrin and platelets have also been implicated in tissue repair mechanisms, which may partly explain the contradictory findings in some models.105, 106 Nevertheless, the results of human studies outlined in the subsequent paragraphs of this section suggest that in humans the harmful actions of intrahepatic coagulation activation are dominant.
In rodent models of acute liver failure, intrahepatic fibrin formation has also been demonstrated.107 Similar to result in rodent models of fibrosis, administration of anticoagulants reduced progression of disease in acute liver failure. Activation of coagulation in this model was demonstrated to rely on tissue factor expressed on hepatocytes.108 Whereas most tissue factor in a healthy liver is in an encrypted state, insults such as hepatocyte necrosis induced by acute liver failure appear to result in tissue factor decryption, resulting in coagulation activation.
Observational studies in humans have suggested a faster disease progression of patients with fibrosis who were also carriers of factor V Leiden, although not all studies agree.109, 110 In addition, patients with hemophilia and hepatitis‐related fibrosis appeared to have a slower disease progression compared to patients with hepatitis without hemophilia.111 The combined results of experimental animal studies and observational human studies have led to the proposal that anticoagulant drugs may be used as adjunct therapy in patients with early cirrhosis to prevent disease progression, decompensation, and delay or prevent liver transplantation or death.112 Indeed, a recent randomized clinical study showed that prolonged daily administration of low‐molecular‐weight heparin substantially delayed decompensation and death, without significant side effects.98 Although the results of this study require confirmation, these findings could potentially revolutionize the management of cirrhosis, possibly as part of a “polypill” program as proposed previously.112 Currently, two randomized controlled trials are assessing efficacy and safety of LMWH and rivaroxaban in patients with established cirrhosis with decompensation and survival as primary endpoints (clinicaltrials.gov identifiers NCT02643212 and NCT02271295). Whether antihemostatic treatment will also become part of the management of acute liver failure is less certain and clinical application may be limited by the perceived bleeding risk. Furthermore, a small study conducted in the 1970s found no evidence for a beneficial effect of heparin in patients with acute liver failure.113
Two hypotheses for the mechanism by which intrahepatic activation of coagulation leads to disease progression have been proposed. Initially, the theory of “parenchymal extinction” was proposed, in which physical blockade of the microcirculation by platelets, fibrin, or a platelet and fibrin‐containing thrombus is held responsible for the progression of disease.114 In this theory, tissue ischemia caused by these microthrombi is held responsible for the effects on disease progression. In recent years, a second hypothesis in which cellular activation by coagulation proteases is key in driving disease progression is gaining popularity. Thrombin (and factor Xa)‐mediated activation of protease activated receptors on stellate cells results in activation of these cells resulting in collagen production by the stellate cells.115 Alternatively, the mechanism may involve thrombin activation of protease activated receptors on platelets resulting in the release of profibrogenic molecules such as platelet‐derived growth factor.3 In models of acute liver failure activation of protease‐activated receptors also contribute to disease progression, although it has not been established activation of which cell types are responsible for the effect.107 The potential mechanisms explaining the pathogenic role of coagulation activation for progression of liver disease are summarized in Figure 2. Clearly, the different pathways are not mutually exclusive.
Figure 2.
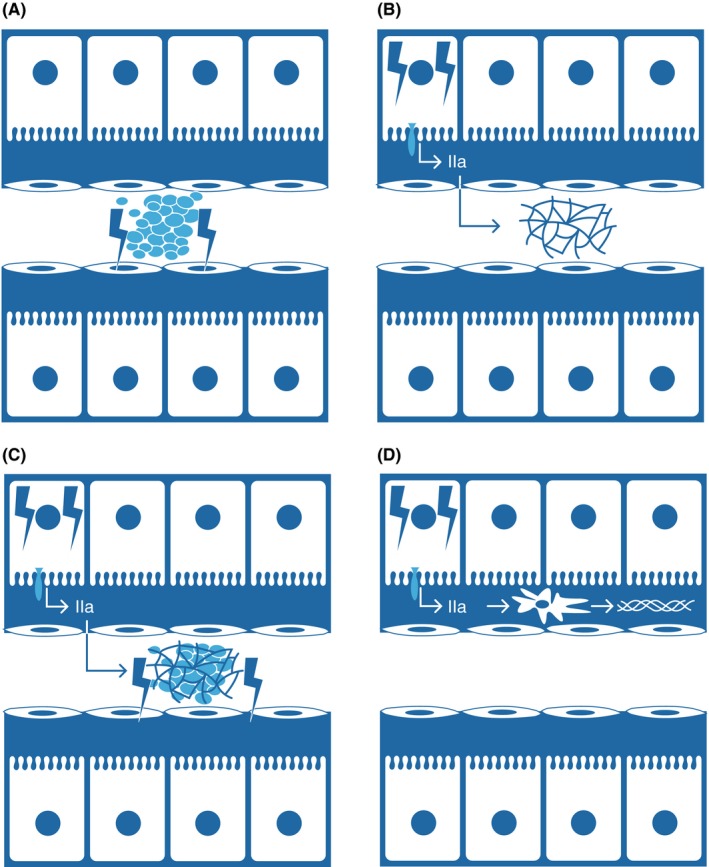
Potential mechanisms involved in progression of liver disease by intrahepatic activation of hemostasis. (A) Intrahepatic activation of endothelial cells results in the formation of platelet microthrombi in the sinusoid. Such microthrombi result in disease progression via the results of microischemia of the downstream tissue. (B) Hepatocellular injury results in decryption of hepatocyte tissue factor, the generation of thrombin (IIa), and eventually fibrin deposition in the sinusoid. (C) Concomitant activation of hepatocytes and hepatic endothelium results in the formation of platelet and fibrin‐containing microthrombi in the sinusoid. (D) Thrombin generated via decryption of hepatocyte tissue factor activates hepatic stellate cells to synthesize collagen
7. CONCLUSION
The complex hemostatic changes that frequently occur in patients with liver diseases results in a “rebalanced” hemostatic system. This reset hemostatic balance, however, has a limited stability which explains the occurrence of both bleeding and thrombotic complications in these patients. Prediction, prevention management, and monitoring of bleeding or thrombosis in patients with liver diseases are complicated as a result of the extensive baseline changes in the hemostatic system. As such, it is very difficult to provide guidelines for management. Some management advice has been published previously,71, 84, 116, 117, 118, 119, 120 but we would like to stress that most advice is based on expert opinion only. Table 2 lists unsolved clinical questions that require additional study. Importantly, carefully designed clinical studies on the efficacy and safety of pro‐ and antihemostatic strategies need to be performed. An exciting area of research regards the use of antihemostatic agents to prevent progression of disease. Whether such strategies are truly effective and sufficiently safe awaits confirmation in larger clinical trials.
Table 2.
Unsolved clinical questions on hemostatic management in patients with liver diseases
| Prediction | Prevention | Treatment | Monitoring | Dosing | |
|---|---|---|---|---|---|
| Management of bleeding |
|
|
|
|
|
| Management of thrombosis |
|
|
|
|
|
VKA, vitamin K antagonist; DOAC, direct oral anticoagulant.
ADDENDUM
T. Lisman and R.J. Porte contributed equally to the development of the concepts outlined in the paper. T. Lisman wrote the initial version of the manuscript and R.J. Porte revised the paper. No funding for writing this review was obtained.
RELATIONSHIP DISCLOSURE
We have no conflicts of interest to report
Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost. 2017;1:150–161. 10.1002/rth2.12028
REFERENCES
- 1. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol. 2002;37:280–7. [DOI] [PubMed] [Google Scholar]
- 2. Bakker CM, Knot EA, Stibbe J, Wilson JH. Disseminated intravascular coagulation in liver cirrhosis. J Hepatol. 1992;15:330–5. [DOI] [PubMed] [Google Scholar]
- 3. Anstee QM, Wright M, Goldin R, Thursz MR. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis. 2009;13:117–26. [DOI] [PubMed] [Google Scholar]
- 4. Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood. 2010;116:878–85. [DOI] [PubMed] [Google Scholar]
- 5. Lisman T, Stravitz RT. Rebalanced hemostasis in patients with acute liver failure. Semin Thromb Hemost. 2015;41:468–73. [DOI] [PubMed] [Google Scholar]
- 6. Trotter JF, Olson J, Lefkowitz J, Smith AD, Arjal R, Kenison J. Changes in international normalized ratio (INR) and model for endstage liver disease (MELD) based on selection of clinical laboratory. Am J Transplant. 2007;7:1624–8. [DOI] [PubMed] [Google Scholar]
- 7. Tripodi A, Baglin T, Robert A, Kitchen S, Lisman T, Trotter JF. Subcommittee on Control of Anticoagulation of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Reporting prothrombin time results as international normalized ratios for patients with chronic liver disease. J Thromb Haemost. 2010;8:1410–2. [DOI] [PubMed] [Google Scholar]
- 8. Garcia‐Tsao G, Bosch J. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med. 2010;362:823–32. [DOI] [PubMed] [Google Scholar]
- 9. Gazzard BG, Portmann B, Murray‐Lyon IM, Williams R. Causes of death in fulminant hepatic failure and relationship to quantitative histological assessment of parenchymal damage. Q J Med. 1975;44:615–26. [PubMed] [Google Scholar]
- 10. Munoz SJ, Stravitz RT, Gabriel DA. Coagulopathy of acute liver failure. Clin Liver Dis. 2009;13:95–107. [DOI] [PubMed] [Google Scholar]
- 11. Lewis JH, Bontempo FA, Cornell F, et al. Blood use in liver transplantation. Transfusion. 1987;27:222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farrar RP, Hanto DW, Flye MW, Chaplin H. Blood component use in orthotopic liver transplantation. Transfusion. 1988;28:474–8. [DOI] [PubMed] [Google Scholar]
- 13. Porte RJ, Knot EA, Bontempo FA. Hemostasis in liver transplantation. Gastroenterology. 1989;97:488–501. [DOI] [PubMed] [Google Scholar]
- 14. Ramos HC, Todo S, Kang Y, Felekouras E, Doyle HR, Starzl TE. Liver transplantation without the use of blood products. Arch Surg. 1994;129:528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Boer MT, Molenaar IQ, Hendriks HG, Slooff MJ, Porte RJ. Minimizing blood loss in liver transplantation: Progress through research and evolution of techniques. Dig Surg. 2005;22:265–75. [DOI] [PubMed] [Google Scholar]
- 16. Massicotte L, Thibeault L, Roy A. Classical notions of coagulation revisited in relation with blood losses, transfusion rate for 700 consecutive liver transplantations. Semin Thromb Hemost. 2015;41:538–46. [DOI] [PubMed] [Google Scholar]
- 17. Massicotte L, Lenis S, Thibeault L, Sassine MP, Seal RF, Roy A. Effect of low central venous pressure and phlebotomy on blood product transfusion requirements during liver transplantations. Liver Transpl. 2006;12:117–23. [DOI] [PubMed] [Google Scholar]
- 18. Tripodi A, Salerno F, Chantarangkul V, et al. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology. 2005;41:553–8. [DOI] [PubMed] [Google Scholar]
- 19. Lisman T, Leebeek FW, Mosnier LO, et al. Thrombin‐activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology. 2001;121:131–9. [DOI] [PubMed] [Google Scholar]
- 20. Lisman T, Bongers TN, Adelmeijer J, et al. Elevated levels of von Willebrand factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44:53–61. [DOI] [PubMed] [Google Scholar]
- 21. Lisman T, Bakhtiari K, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT. Intact thrombin generation and decreased fibrinolytic capacity in patients with acute liver injury or acute liver failure. J Thromb Haemost. 2012;10:1312–9. [DOI] [PubMed] [Google Scholar]
- 22. Hugenholtz GC, Adelmeijer J, Meijers JC, Porte RJ, Stravitz RT, Lisman T. An unbalance between von willebrand factor and ADAMTS13 in acute liver failure: implications for hemostasis and clinical outcome. Hepatology. 2013;58:752–61. [DOI] [PubMed] [Google Scholar]
- 23. Gatt A, Riddell A, Calvaruso V, Tuddenham EG, Makris M, Burroughs AK. Enhanced thrombin generation in patients with cirrhosis‐induced coagulopathy. J Thromb Haemost. 2010;8:1994–2000. [DOI] [PubMed] [Google Scholar]
- 24. Youngwon N, Kim JE, Lim HS, Han KS, Kim HK. Coagulation proteins influencing global coagulation assays in cirrhosis: hypercoagulability in cirrhosis assessed by thrombomodulin‐induced thrombin generation assay. Biomed Res Int. 2013;2013:856754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kleinegris MC, Habets CA, van de Sande AJ, et al. Liver cirrhosis is associated with hypercoagulability, decreased clot strength and normal fibrinolysis. J Thromb Haemost. 2013;11:49–50. [Google Scholar]
- 26. Groeneveld D, Porte RJ, Lisman T. Thrombomodulin‐modified thrombin generation testing detects a hypercoagulable state in patients with cirrhosis regardless of the exact experimental conditions. Thromb Res. 2014;134:753–6. [DOI] [PubMed] [Google Scholar]
- 27. Lebreton A, Sinegre T, Pereira B, Lamblin G, Duron C, Abergel A. Plasma hypercoagulability in the presence of thrombomodulin but not of activated protein C in patients with cirrhosis. J Gastroenterol Hepatol. 2017;32:916–24. [DOI] [PubMed] [Google Scholar]
- 28. Raparelli V, Basili S, Carnevale R, et al. Low‐grade endotoxemia and platelet activation in cirrhosis. Hepatology. 2017;65:571–81. [DOI] [PubMed] [Google Scholar]
- 29. Hugenholtz GC, Mccrae FL, Adelmeijer J, et al. Procoagulant changes in fibrin clot structure in patients with cirrhosis are associated with oxidative modifications of fibrinogen. J Thromb Haemost. 2016;15:1054–66. [DOI] [PubMed] [Google Scholar]
- 30. Laffi G, Cominelli F, Ruggiero M, et al. Altered platelet function in cirrhosis of the liver: Impairment of inositol lipid and arachidonic acid metabolism in response to agonists. Hepatology. 1988;8:1620–6. [DOI] [PubMed] [Google Scholar]
- 31. Laffi G, Marra F, Gresele P, et al. Evidence for a storage pool defect in platelets from cirrhotic patients with defective aggregation. Gastroenterology. 1992;103:641–6. [DOI] [PubMed] [Google Scholar]
- 32. Laffi G, Marra F, Failli P, et al. Defective signal transduction in platelets from cirrhotics is associated with increased cyclic nucleotides. Gastroenterology. 1993;105:148–56. [DOI] [PubMed] [Google Scholar]
- 33. Violi F, Basili S, Raparelli V, Chowdary P, Gatt A, Burroughs AK. Patients with liver cirrhosis suffer from primary haemostatic defects? Fact or fiction? J Hepatol. 2011;55:1415–27. [DOI] [PubMed] [Google Scholar]
- 34. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alhamdi Y, Abrams ST, Lane S, Wang G, Toh CH. Histone‐associated thrombocytopenia in patients who are critically ill. JAMA. 2016;315:817–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wen Z, Lei Z, Yao L, et al. Circulating histones are major mediators of systemic inflammation and cellular injury in patients with acute liver failure. Cell Death Dis. 2016;7:e2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mannucci PM, Vicente V, Vianello L, et al. Controlled trial of desmopressin in liver cirrhosis and other conditions associated with a prolonged bleeding time. Blood. 1986;67:1148–53. [PubMed] [Google Scholar]
- 38. Lisman T, Adelmeijer J, de Groot PG, Janssen HL, Leebeek FW. No evidence for an intrinsic platelet defect in patients with liver cirrhosis–studies under flow conditions. J Thromb Haemost. 2006;4:2070–2. [DOI] [PubMed] [Google Scholar]
- 39. Escolar G, Cases A, Vinas M, et al. Evaluation of acquired platelet dysfunctions in uremic and cirrhotic patients using the platelet function analyzer (PFA‐100): influence of hematocrit elevation. Haematologica. 1999;84:614–9. [PubMed] [Google Scholar]
- 40. Caldwell S, Lisman T. The cirrhotic platelet: Shedding light on an enigma. Hepatology. 2017;65:407–10. [DOI] [PubMed] [Google Scholar]
- 41. Uemura M, Fujimura Y, Matsumoto M, et al. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Thromb Haemost. 2008;99:1019–29. [DOI] [PubMed] [Google Scholar]
- 42. Federici AB, Berkowitz SD, Lattuada A, Mannucci PM. Degradation of von Willebrand factor in patients with acquired clinical conditions in which there is heightened proteolysis. Blood. 1993;81:720–5. [PubMed] [Google Scholar]
- 43. Tersteeg C, de Maat S, De Meyer SF, et al. Plasmin cleavage of von Willebrand factor as an emergency bypass for ADAMTS13 deficiency in thrombotic microangiopathy. Circulation. 2014;129:1320–31. [DOI] [PubMed] [Google Scholar]
- 44. Shida Y, Nishio K, Sugimoto M, et al. Functional imaging of shear‐dependent activity of ADAMTS13 in regulating mural thrombus growth under whole blood flow conditions. Blood. 2008;111:1295–8. [DOI] [PubMed] [Google Scholar]
- 45. Afdhal NH, Giannini EG, Tayyab G, et al. ELEVATE Study Group . Eltrombopag before procedures in patients with cirrhosis and thrombocytopenia. N Engl J Med. 2012;367:716–24. [DOI] [PubMed] [Google Scholar]
- 46. Lisman T, Porte RJ. Eltrombopag before procedures in patients with cirrhosis and thrombocytopenia. N Engl J Med. 2012;367:2055–6. [DOI] [PubMed] [Google Scholar]
- 47. Giannini EG, Greco A, Marenco S, Andorno E, Valente U, Savarino V. Incidence of bleeding following invasive procedures in patients with thrombocytopenia and advanced liver disease. Clin Gastroenterol Hepatol. 2010;8:899–902. [DOI] [PubMed] [Google Scholar]
- 48. Napolitano G, Iacobellis A, Merla A, et al. Bleeding after invasive procedures is rare and unpredicted by platelet counts in cirrhotic patients with thrombocytopenia. Eur J Intern Med. 2017;38:79–82. [DOI] [PubMed] [Google Scholar]
- 49. Tripodi A, Primignani M, Chantarangkul V, et al. An imbalance of pro‐ vs anti‐coagulation factors in plasma from patients with cirrhosis. Gastroenterology. 2009;137:2105–11. [DOI] [PubMed] [Google Scholar]
- 50. Tripodi A, Chantarangkul V, Primignani M, et al. Thrombin generation in plasma from patients with cirrhosis supplemented with normal plasma: considerations on the efficacy of treatment with fresh‐frozen plasma. Intern Emerg Med. 2012;7:139–44. [DOI] [PubMed] [Google Scholar]
- 51. Kalambokis GN, Oikonomou A, Christou L, et al. Von Willebrand factor and procoagulant imbalance predict outcome in patients with cirrhosis and thrombocytopenia. J Hepatol. 2016;65:921–8. [DOI] [PubMed] [Google Scholar]
- 52. Potze W, Sanyal AJ, Lisman T. Reply to: “Procoagulant imbalance in patients with non‐alcoholic fatty liver disease”. J Hepatol. 2017;66:250–1. [DOI] [PubMed] [Google Scholar]
- 53. Leebeek FW, Kluft C, Knot EA, de Maat MP, Wilson JH. A shift in balance between profibrinolytic and antifibrinolytic factors causes enhanced fibrinolysis in cirrhosis. Gastroenterology. 1991;101:1382–90. [DOI] [PubMed] [Google Scholar]
- 54. Colucci M, Binetti BM, Branca MG, et al. Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology. 2003;38:230–7. [DOI] [PubMed] [Google Scholar]
- 55. Rijken DC, Kock EL, Guimaraes AH, et al. Evidence for an enhanced fibrinolytic capacity in cirrhosis as measured with two different global fibrinolysis tests. J Thromb Haemost. 2012;10:2116–22. [DOI] [PubMed] [Google Scholar]
- 56. Stravitz RT. Potential applications of thromboelastography in patients with acute and chronic liver disease. Gastroenterol Hepatol (N Y). 2012;8:513–20. [PMC free article] [PubMed] [Google Scholar]
- 57. Tripodi A, Primignani M, Chantarangkul V, et al. The coagulopathy of cirrhosis assessed by thromboelastometry and its correlation with conventional coagulation parameters. Thromb Res. 2009;124:132–6. [DOI] [PubMed] [Google Scholar]
- 58. Lentschener C, Flaujac C, Ibrahim F, et al. Assessment of haemostasis in patients with cirrhosis: Relevance of the ROTEM tests?: A prospective, cross‐sectional study. Eur J Anaesthesiol. 2016;33:126–33. [DOI] [PubMed] [Google Scholar]
- 59. De Pietri L, Bianchini M, Rompianesi G, Bertellini E, Begliomini B. Thromboelastographic reference ranges for a cirrhotic patient population undergoing liver transplantation. World J Transplant. 2016;6:583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kleinegris MC, Bos MH, Roest M, et al. Cirrhosis patients have a coagulopathy that is associated with decreased clot formation capacity. J Thromb Haemost. 2014;12:1647–57. [DOI] [PubMed] [Google Scholar]
- 61. Bedreli S, Sowa JP, Malek S, et al. Rotational thromboelastometry can detect factor XIII deficiency and bleeding diathesis in patients with cirrhosis. Liver Int. 2017;37:562–8. [DOI] [PubMed] [Google Scholar]
- 62. Ben‐Ari Z, Panagou M, Patch D, et al. Hypercoagulability in patients with primary biliary cirrhosis and primary sclerosing cholangitis evaluated by thrombelastography. J Hepatol. 1997;26:554–9. [DOI] [PubMed] [Google Scholar]
- 63. Potze W, Siddiqui MS, Boyett SL, et al. Preserved hemostatic status in patients with non‐alcoholic fatty liver disease. J Hepatol. 2016;65:980–7. [DOI] [PubMed] [Google Scholar]
- 64. Martinez J, Palascak JE, Kwasniak D. Abnormal sialic acid content of the dysfibrinogenemia associated with liver disease. J Clin Invest. 1978;61:535–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Violi F, Ferro D, Basili S, et al. Hyperfibrinolysis resulting from clotting activation in patients with different degrees of cirrhosis. the CALC group. Coagulation abnormalities in liver cirrhosis. Hepatology. 1993;17:78–83. [PubMed] [Google Scholar]
- 66. Saliola M, Lorenzet R, Ferro D, et al. Enhanced expression of monocyte tissue factor in patients with liver cirrhosis. Gut. 1998;43:428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stravitz RT, Bowling R, Bradford RL, et al. Role of procoagulant microparticles in mediating complications and outcome of acute liver injury/acute liver failure. Hepatology. 2013;58:304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Habib M, Roberts LN, Patel RK, Wendon J, Bernal W, Arya R. Evidence of rebalanced coagulation in acute liver injury and acute liver failure as measured by thrombin generation. Liver Int. 2014;34:672–8. [DOI] [PubMed] [Google Scholar]
- 69. Stravitz RT, Lisman T, Luketic VA, et al. Minimal effects of acute liver injury/acute liver failure on hemostasis as assessed by thromboelastography. J Hepatol. 2012;56:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ozier Y, Pessione F, Samain E, Courtois F; French Study Group on Blood Transfusion in Liver Transplantation . Institutional variability in transfusion practice for liver transplantation. Anesth Analg. 2003;97:671–9. [DOI] [PubMed] [Google Scholar]
- 71. Northup PG, Caldwell SH. Coagulation in liver disease: A guide for the clinician. Clin Gastroenterol Hepatol. 2013;11:1064–74. [DOI] [PubMed] [Google Scholar]
- 72. Ambrosino P, Tarantino L, Di Minno G, et al. The risk of venous thromboembolism in patients with cirrhosis. A systematic review and meta‐analysis. Thromb Haemost. 2017;117:139–48. [DOI] [PubMed] [Google Scholar]
- 73. Noris M, Remuzzi G. Uremic bleeding: Closing the circle after 30 years of controversies? Blood. 1999;94:2569–74. [PubMed] [Google Scholar]
- 74. Goulis J, Armonis A, Patch D, Sabin C, Greenslade L, Burroughs AK. Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gastrointestinal hemorrhage. Hepatology. 1998;27:1207–12. [DOI] [PubMed] [Google Scholar]
- 75. Westerkamp AC, Lisman T, Porte RJ. How to minimize blood loss during liver surgery in patients with cirrhosis. HPB (Oxford). 2009;11:453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Alkozai EM, Lisman T, Porte RJ. Bleeding in liver surgery: prevention and treatment. Clin Liver Dis. 2009;13:145–54. [DOI] [PubMed] [Google Scholar]
- 77. de Boer MT, Christensen MC, Asmussen M, et al. The impact of intraoperative transfusion of platelets and red blood cells on survival after liver transplantation. Anesth Analg. 2008;106:32–44. [DOI] [PubMed] [Google Scholar]
- 78. Youssef WI, Salazar F, Dasarathy S, Beddow T, Mullen KD. Role of fresh frozen plasma infusion in correction of coagulopathy of chronic liver disease: a dual phase study. Am J Gastroenterol. 2003;98:1391–4. [DOI] [PubMed] [Google Scholar]
- 79. Qureshi K, Patel S, Meillier A. The use of thrombopoietin receptor agonists for correction of thrombocytopenia prior to elective procedures in chronic liver diseases: review of current evidence. Int J Hepatol. 2016;2016:1802932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Porte RJ, Molenaar IQ, Begliomini B, et al. Aprotinin and transfusion requirements in orthotopic liver transplantation: a multicentre randomised double‐blind study. EMSALT study group. Lancet. 2000;355:1303–9. [DOI] [PubMed] [Google Scholar]
- 81. Arshad F, Ickx B, van Beem RT, et al. Prothrombin complex concentrate in the reduction of blood loss during orthotopic liver transplantation: PROTON‐trial. BMC Surg 2013;13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang SC, Shieh JF, Chang KY, et al. Thromboelastography‐guided transfusion decreases intraoperative blood transfusion during orthotopic liver transplantation: Randomized clinical trial. Transplant Proc. 2010;42:2590–3. [DOI] [PubMed] [Google Scholar]
- 83. Saner FH, Gieseler RK, Akiz H, Canbay A, Gorlinger K. Delicate balance of bleeding and thrombosis in end‐stage liver disease and liver transplantation. Digestion. 2013;88:135–44. [DOI] [PubMed] [Google Scholar]
- 84. Lisman T, Caldwell SH, Burroughs AK, et al. Coagulation in Liver Disease Study Group . Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol. 2010;53:362–71. [DOI] [PubMed] [Google Scholar]
- 85. Shami VM, Caldwell SH, Hespenheide EE, Arseneau KO, Bickston SJ, Macik BG. Recombinant activated factor VII for coagulopathy in fulminant hepatic failure compared with conventional therapy. Liver Transpl. 2003;9:138–43. [DOI] [PubMed] [Google Scholar]
- 86. Pavese P, Bonadona A, Beaubien J, et al. FVIIa corrects the coagulopathy of fulminant hepatic failure but may be associated with thrombosis: a report of four cases. Can J Anaesth. 2005;52:26–9. [DOI] [PubMed] [Google Scholar]
- 87. Northup PG, McMahon MM, Ruhl AP, et al. Coagulopathy does not fully protect hospitalized cirrhosis patients from peripheral venous thromboembolism. Am J Gastroenterol 2006;101:1524‐8; quiz 1680. [DOI] [PubMed] [Google Scholar]
- 88. Sogaard KK, Horvath‐Puho E, Gronbaek H, Jepsen P, Vilstrup H, Sorensen HT. Risk of venous thromboembolism in patients with liver disease: a nationwide population‐based case‐control study. Am J Gastroenterol. 2009;104:96–101. [DOI] [PubMed] [Google Scholar]
- 89. Gulley D, Teal E, Suvannasankha A, Chalasani N, Liangpunsakul S. Deep vein thrombosis and pulmonary embolism in cirrhosis patients. Dig Dis Sci. 2008;53:3012–7. [DOI] [PubMed] [Google Scholar]
- 90. Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther. 2010;31:366–74. [DOI] [PubMed] [Google Scholar]
- 91. Potze W, Arshad F, Adelmeijer J, et al. Routine coagulation assays underestimate levels of antithrombin‐dependent drugs but not of direct anticoagulant drugs in plasma from patients with cirrhosis. Br J Haematol. 2013;163:666–73. [DOI] [PubMed] [Google Scholar]
- 92. Senzolo M, Rodriguez‐Castro KI, Rossetto V, et al. Increased anticoagulant response to low‐molecular‐weight heparin in plasma from patients with advanced cirrhosis. J Thromb Haemost. 2012;10:1823–9. [DOI] [PubMed] [Google Scholar]
- 93. Potze W, Arshad F, Adelmeijer J, et al. Differential in vitro inhibition of thrombin generation by anticoagulant drugs in plasma from patients with cirrhosis. PLoS ONE. 2014;9:e88390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Potze W, Adelmeijer J, Lisman T. Decreased in vitro anticoagulant potency of rivaroxaban and apixaban in plasma from patients with cirrhosis. Hepatology. 2015;61:1435–6. [DOI] [PubMed] [Google Scholar]
- 95. Rodriguez‐Castro KI, Simioni P, Burra P, Senzolo M. Anticoagulation for the treatment of thrombotic complications in patients with cirrhosis. Liver Int. 2012;32:1465–76. [DOI] [PubMed] [Google Scholar]
- 96. Intagliata NM, Henry ZH, Maitland H, et al. Direct oral anticoagulants in cirrhosis patients pose similar risks of bleeding when compared to traditional anticoagulation. Dig Dis Sci. 2016;61:1721–7. [DOI] [PubMed] [Google Scholar]
- 97. De Gottardi A, Trebicka J, Klinger C, et al. VALDIG Investigators . Antithrombotic treatment with direct‐acting oral anticoagulants in patients with splanchnic vein thrombosis and cirrhosis. Liver Int. 2017;37:694–9. [DOI] [PubMed] [Google Scholar]
- 98. Villa E, Camma C, Marietta M, et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology. 2012;143:1253–60. [DOI] [PubMed] [Google Scholar]
- 99. MacPhee PJ, Dindzans VJ, Fung LS, Levy GA. Acute and chronic changes in the microcirculation of the liver in inbred strains of mice following infection with mouse hepatitis virus type 3. Hepatology. 1985;5:649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wanless IR, Belgiorno J, Huet PM. Hepatic sinusoidal fibrosis induced by cholesterol and stilbestrol in the rabbit: 1. morphology and inhibition of fibrogenesis by dipyridamole. Hepatology. 1996;24:855–64. [DOI] [PubMed] [Google Scholar]
- 101. Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: Possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21:1238–47. [PubMed] [Google Scholar]
- 102. Anstee QM, Goldin RD, Wright M, Martinelli A, Cox R, Thursz MR. Coagulation status modulates murine hepatic fibrogenesis: Implications for the development of novel therapies. J Thromb Haemost. 2008;6:1336–43. [DOI] [PubMed] [Google Scholar]
- 103. Kodama T, Takehara T, Hikita H, et al. Thrombocytopenia exacerbates cholestasis‐induced liver fibrosis in mice. Gastroenterology. 2010;138:2487–98. [DOI] [PubMed] [Google Scholar]
- 104. Luyendyk JP, Kassel KM, Allen K, et al. Fibrinogen deficiency increases liver injury and early growth response‐1 (egr‐1) expression in a model of chronic xenobiotic‐induced cholestasis. Am J Pathol. 2011;178:1117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Drew AF, Liu H, Davidson JM, Daugherty CC, Degen JL. Wound‐healing defects in mice lacking fibrinogen. Blood. 2001;97:3691–8. [DOI] [PubMed] [Google Scholar]
- 106. Lesurtel M, Graf R, Aleil B, et al. Platelet‐derived serotonin mediates liver regeneration. Science. 2006;312:104–7. [DOI] [PubMed] [Google Scholar]
- 107. Ganey PE, Luyendyk JP, Newport SW, et al. Role of the coagulation system in acetaminophen‐induced hepatotoxicity in mice. Hepatology. 2007;46:1177–86. [DOI] [PubMed] [Google Scholar]
- 108. Sullivan BP, Kopec AK, Joshi N, et al. Hepatocyte tissue factor activates the coagulation cascade in mice. Blood. 2013;121:1868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wright M, Goldin R, Hellier S, et al. Factor V leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut. 2003;52:1206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Goulding C, O'Brien C, Egan H, et al. The impact of inherited prothrombotic risk factors on individuals chronically infected with hepatitis C virus from a single source. J Viral Hepat. 2007;14:255–9. [DOI] [PubMed] [Google Scholar]
- 111. Assy N, Pettigrew N, Lee SS, Chaudhary RK, Johnston J, Minuk GY. Are chronic hepatitis C viral infections more benign in patients with hemophilia? Am J Gastroenterol. 2007;102:1672–6. [DOI] [PubMed] [Google Scholar]
- 112. Tsochatzis EA, Bosch J, Burroughs AK. New therapeutic paradigm for patients with cirrhosis. Hepatology. 2012;56:1983–92. [DOI] [PubMed] [Google Scholar]
- 113. Gazzard BG, Clark R, Borirakchanyavat V, Williams R. A controlled trial of heparin therapy in the coagulation defect of paracetamol‐induced hepatic necrosis. Gut. 1974;15:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21:1238–47. [PubMed] [Google Scholar]
- 115. Fiorucci S, Antonelli E, Distrutti E, et al. PAR1 antagonism protects against experimental liver fibrosis. role of proteinase receptors in stellate cell activation. Hepatology. 2004;39:365–75. [DOI] [PubMed] [Google Scholar]
- 116. Shah NL, Intagliata NM, Northup PG, Argo CK, Caldwell SH. Procoagulant therapeutics in liver disease: a critique and clinical rationale. Nat Rev Gastroenterol Hepatol. 2014;11:675–82. [DOI] [PubMed] [Google Scholar]
- 117. Lisman T, Kamphuisen PW, Northup PG, Porte RJ. Established and new‐generation antithrombotic drugs in patients with cirrhosis ‐ possibilities and caveats. J Hepatol. 2013;59:358–66. [DOI] [PubMed] [Google Scholar]
- 118. Weeder PD, Porte RJ, Lisman T. Hemostasis in liver disease: Implications of new concepts for perioperative management. Transfus Med Rev. 2014;28:107–13. [DOI] [PubMed] [Google Scholar]
- 119. Donohue CI, Mallett SV. Reducing transfusion requirements in liver transplantation. World J Transplant. 2015;5:165–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Valla D. Splanchnic vein thrombosis. Semin Thromb Hemost. 2015;41:494–502. [DOI] [PubMed] [Google Scholar]