

Abstract
Whole cell membrane capacitance is an electrophysiological property of the plasma membrane that serves as a biomarker for stem cell fate potential. Neural stem and progenitor cells (NSPCs) that differ in ability to form neurons or astrocytes are distinguished by membrane capacitance measured by dielectrophoresis (DEP). Differences in membrane capacitance are sufficient to enable the enrichment of neuron- or astrocyte-forming cells by DEP, showing the separation of stem cells on the basis of fate potential by membrane capacitance. NSPCs sorted by DEP need not be labeled and do not experience toxic effects from the sorting procedure. Other stem cell populations also display shifts in membrane capacitance as cells differentiate to a particular fate, clarifying the value of sorting a variety of stem cell types by capacitance. Here, we describe methods developed by our lab for separating NSPCs on the basis of capacitance using several types of DEP microfluidic devices, providing basic information on the sorting procedure as well as specific advantages and disadvantages of each device.
Keywords: Neural stem cell, progenitor cell, dielectrophoresis, membrane capacitance, cell sorting, microfluidics
1. Introduction
1.1 Capacitance as a means to identify and sort NSPCs
Neural stem and progenitor cells (NSPCs) have therapeutic potential to treat neurological diseases and injuries [1] since they provide neuroprotection and differentiate into the three cell types of the central nervous system - neurons, astrocytes, and oligodendrocytes [2]. NSPCs expanded to generate sufficient numbers of cells for transplantation form a heterogeneous population of cells with varying ratios of progenitors linked to distinct fates, such as neuron progenitors and astrocyte progenitors [3,4]. Quantitative measures to assess or reduce the heterogeneity of transplanted cells are critical and current marker based approaches to identify progenitors are limited.
Whole cell membrane capacitance is an electrophysiological property of the plasma membrane that identifies and enriches cells at distinct stages of differentiation in multiple stem cell lineages, including NSPCs. Whole cell membrane capacitance can be measured using dielectrophoresis (DEP), which is a technique that uses electric fields to analyze or separate cells. NSPCs, neurons and astrocytes exhibit distinct behaviors in DEP and NSPCs can be prospectively sorted from neurons using DEP [5,6]. Undifferentiated mouse and human NSPC populations containing more cells destined to become neurons after differentiation can be distinguished from those with more astrocyte-forming cells by capacitance, and membrane capacitance dynamically reflects declining numbers of neurogenic cells in human NSPCs [3,5]. Differences in whole cell membrane capacitance are sufficient for enrichment of neurogenic and astrogenic progenitors from heterogeneous populations of mouse NSPCs [7,8]. Cell surface components play a role in measured membrane capacitance and initial studies suggest carbohydrates on NSPCs contribute to the behavior of these cells in DEP [7]. A variety of store-charging carbohydrates are added to cell surface proteins and lipids via N-linked glycosylation, and we hypothesize these are detectable with DEP due to polarization. Membrane capacitance identifies and enables the enrichment of undifferentiated cells from differentiated progeny in the hematopoietic stem cell (HSC) [9,10], mesenchymal/adipose-derived stem cell (MSC, ADSC) [11–16], muscle [17] and embryonic stem (ES) [18–20] cell lineages, indicating the usefulness of this biomarker in stem cell studies and advantages to sorting stem cells by capacitance using DEP.
This methods paper focuses on three unique DEP microfluidic devices and the two-step sorting scheme we developed for NSPCs, which can be extended to other cell systems. The initial step defines responses of cells to specific frequencies in DEP, and the subsequent step separates cells at a specific frequency in a DEP-based microfluidic device. For a more general review of microfluidic separation devices, see Hyun and Jung, Electrophoresis 2013, 34, 1028–1041 [21].
1.2 DEP phenomena relevant for cell sorting by capacitance
DEP can be used to characterize the cell biophysical properties capacitance, permittivity, and conductance. DEP is ideal for cell separations because it is a label free, rapid, straightforward method capable of separating desired cell subpopulations from heterogeneous mixtures without changing functionality. Characterizing NSPC biophysical properties with DEP can increase understanding of their diverse functions and provide a label free biomarker of cell phenotype. DEP utilizes non-uniform electric fields to polarize cells and induce movement based on the dielectric properties of the cell membrane, cytoplasm, and structurally dominant organelles [22]. Electric fields are delivered using electrodes supplied with alternating current (AC), in which current changes direction as defined by the frequency of the applied electric field, or direct current (DC), in which there is no frequency component and current flows in one direction. AC and DC electric fields provide controlled cell movements in DEP based on the shape of the electric field [23]. DEP in DC-based systems is induced by physical barriers to the current that create local constrictions in the electric field to generate non-uniform electric fields [24]. In our case, AC electric fields are preferred because of the selectivity provided with the frequency component.
Cells in electric fields have distinct dielectric dispersions that can be used to identify or separate cells in heterogeneous populations. Key properties extracted from cell behavior in electric fields are capacitance, a cell’s ability to store electrical energy [25], permittivity, a cell’s ability to resist an electric field [26], and conductance, a cell’s ability to conduct electric charge [27]. All of these properties can be determined from frequency dependent responses of cells and characterized by the β dispersion region. At radio frequencies (β-region), 100 kHz to 10 MHz, and in low conductivity mediums (~100 μS/cm) cell dielectric dispersions of spherical cells are dominated by the plasma membrane at lower frequencies, and higher frequencies penetrate the cell surface to probe the cytoplasm [28–30]. Since the DEP responses of NSPCs that differ in fate are in the low frequency β-region [5], we focus here on properties dominated by the plasma membrane.
Maxwell-Wagner interfacial polarizations govern cell responses in the β-region. Polarized cells will display either positive DEP (pDEP), in which cells move to high electric field gradient areas, or negative DEP (nDEP), in which cells repel from high electric field gradient areas (Fig.1) [22]. Pictorially, this means cells will appear along electrode edges for pDEP and not along electrode edges for nDEP. Cells experiencing nDEP may be above or in the same plane as the electrodes but far away from the electrode edges. In DEP, a cell is placed in a conductive medium and an electric field is applied. The field interacts with ions available in the medium causing them to move and align around the cell (interfacial polarization, Fig.1) [31]. The movement and alignment of ions on the outside of the cell are affected by the content and properties of the cell surface [26]. Thus, different cell surfaces will affect polarization in contrasting ways and cause cells to have distinct pDEP and nDEP responses.
Figure 1.
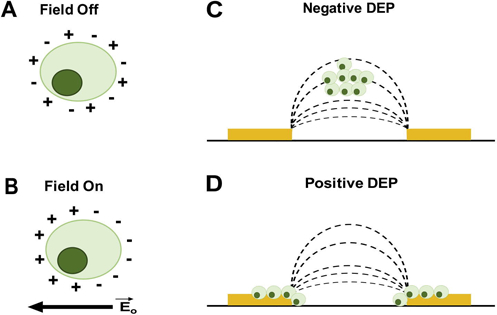
Cell polarization and DEP response. Light green circle represents a cell and the dark green circle is the nucleus. (A) When the electric field is off ions are randomly oriented around the cell (no polarization). (B) Turning on the electric field causes ions to redistribute and align around the cell (polarization). The nucleus is not expected to reorient in the cell with polarization. (C) After polarization, cell movement toward the low strength part of the AC electric field, away from electrodes, is negative DEP (nDEP). (D) In positive DEP (pDEP), cells move toward the high strength part of the electric field, attracted to electrode edges. Yellow rectangles in (C) and (D) represent electrodes and dashed lines are the electric field lines. (C) and (D) adapted with permission from [33].
The induced DEP force that causes cell movement is given by [22] where εmed is the medium permittivity (unitless), Re[fCM] is the real part of the Clausius-Mossotti factor (unitless) and describes cell motion in the electric field, R is the cell radius (µm), and is electric field. The induced DEP force () can be tuned by adjusting the frequency, magnitude and shape of the electric field. Cell size impacts the DEP response, therefore knowing NSPC size becomes important because larger cells have a different DEP response than smaller cells.
1.3 Membrane capacitance calculated from cell behavior in DEP
Cell DEP behavior is quantified experimentally by measuring pDEP and nDEP responses at specific frequencies. Cells have two characteristic crossover frequencies (fxo), at which there is no net movement in response to the electric field, influenced by cell external and internal structures. The low fxo is determined by cell size, shape, and plasma membrane with typical values between 10–100 kHz, but reported as high as ~4 MHz. The second, higher fxo is influenced by the cell cytoplasm and is typically above 10MHz in low conductivity media [32]. Using the low fxo along with other data points from the DEP response spectra, the dielectric properties of the membrane (permittivity, conductance, and capacitance) are estimated. Whole cell membrane capacitance, Cmem, is a function of fxo and given by Cmem = σmed /2πrfxo [22], where r is the cell radius (μm) and σmed is the medium conductivity (μS/cm). Further, membrane permittivity, εmem, is proportional to Cmem, εmem = Cmemd/4πr2ε0 [29], where d is the membrane thickness and ε0 = 8.85×10−12 is the vacuum permittivity. And membrane conductance (Gmem) is proportional to membrane conductivity, σmem, given by Gmem = σmem/d [27]. These equations for Cmem, εmem, and Gmem are valid for experiments conducted in low conductivity buffer solutions (~100 μS/cm) [27]. Membrane thickness is estimated as 7×10−9 m, which is related to the length of fatty acid chains in the membrane. As cell characterization techniques improve we will obtain better estimations for membrane thickness.
NSPCs that generate either more neurons or more astrocytes have distinct whole cell membrane capacitance, Cmem, values and thus can be sorted by DEP [3,7–8]. They also vary in the lower fxo, but do not differ in cell size [3]. Mouse NSPCs from earlier embryonic stages of cerebral cortical development generate more neurons (cultures contain more neuron progenitors) and have a Cmem value of 8.2 0.5 mF/m2. Mouse NSPCs from later developmental stages that generate more astrocytes (cultures contain more astrocyte progenitors) have a Cmem value of 10.7 0.6 mF/m2. DEP-based separation devices enrich NPs at high frequencies and APs at low frequencies from mouse NSPCs, showing that a difference in capacitance of approximately 2.5 mF/m2 is sufficient to enable differential enrichment of progenitors.
2. Separation of NSPCs with DEP devices
2.1 Ideal parameters for DEP-based sorting of NSPCs
An ideal DEP-based microfluidic device for NSPC separation will achieve high selectivity/purity and high cell throughput.2,3 For mouse NSPCs cultured from the embryonic day 12 cortex, maximum enrichment of neuron progenitors (NPs) is 3.3-fold and astrocyte progenitors (APs) is 5-fold to obtain populations at 100% purity (unsorted populations contain approximately 30% NPs and 20% APs). This assumes no other biological processes prevent the isolation of a population of 100% purity. Optimal throughput for DEP-based sorting devices would be on the order of 1.4×106 cells/hr) to avoid the need for expansion of cells post-sorting to generate large numbers of cells [8]. Transplantation of NSPCs into animal models requires a range of 75,000 to 1.5×106 cells per animal; with 10 animals per treatment group this means 106 – 107 cells are needed for a transplant experiment [34–36]. For clinical translation, approximately 4–12 patients are needed for a Phase I trial, and with each patient receiving 106 to 108 cells, depending on study design [37,38]. Thus, obtaining 107 to 109 cells post-sort is desirable. Therefore, high cell throughput here is defined as a sorting rate of 1.4×106 cells/hr; sorting for 4 hours allows 109 cells obtainable after 10 to 12 days of post-sorting cell expansion [3]. Other important qualities are label-free technology (no cell tagging with antibodies for detection), simple fabrication (assemble 10 devices/day), low cost ($20/microfluidic device), programmability (capable of automating steps), short electric field exposure (5 mins) and experimental (4 hrs or less) times, and user-friendly (trained technician can operate easily).
2.2 Cell considerations for sorting with DEP
The ability of DEP to effectively analyze and sort stem cell populations such as NSPCs is a fairly recent finding in the field, making it important to determine whether exposure of NSPCs to the buffer conditions or electric fields necessary for DEP affect cell behavior. Early studies showed that both mouse and human NSPCs maintained high levels of viability in osmotically balanced low conductivity buffer used for DEP (~100 μS/cm) for up to 4 hours, which is much longer than needed for many DEP characterization experiments [3,5]. Critical studies directly assessed the effects of DEP on NSPC function by testing whether exposure of cells to electric fields for varying times affected the survival, proliferation, or differentiation potential of human and mouse NSPCs [33]. The induced DEP movement of cells forms the basis for their analysis and sorting occurs rapidly, on the order of seconds. Therefore, NSPCs were exposed to electric fields for times ranging from 1 to 30 minutes in osmotically balanced low conductivity buffer to adequately cover the possible times needed for DEP-based experiments. Short-term DEP exposure (1 minute or less) at all frequencies had no effect on cell survival, proliferation, or differentiation [33]. Exposure at 5 minutes induced a slight effect on survival at 50 kHz but not at other frequencies and no effect on proliferation or differentiation [33]. NSPCs treated with electric fields at any frequency for up to 30 minutes showed no effects on either cell proliferation measured by DNA synthesis and cell cycle kinetics or differentiation [33]. However, exposure to DEP electric fields at frequencies near the crossover frequency (50 kHz or 100 kHz) for times ranging from 10 to 30 minutes decreased survival of NPSCs to a maximum of 30% cell loss after 30 minutes of exposure [33]. These findings inform the design of NSPC experiments utilizing DEP and define limits on the exposure time at frequencies near the crossover frequency. In sum, exposure to electric fields is not harmful to human or mouse NSPCs at the short times needed for DEP-based analysis or sorting in low conductivity buffer. If higher conductivity buffer is desired for DEP analysis then the buffer must be osmotically balanced to sustain membrane integrity and cell viability.
Many DEP-based devices are microscale, in part due to the nature of the electric field gradient necessary for DEP that puts limits on the dimensions of certain types of DEP devices. In some cases, this results in relatively low cell numbers post DEP sorting. Since NSPCs are proliferative, one option is to expand cells after sorting to increase cell numbers as long as post-sorting enrichment is maintained during cell expansion. Expansion of mouse NSPCs after DEP-based sorting was directly tested by sorting cells at a frequency that enriches for astrocyte progenitors and measuring cell expansion as well as astrocyte progenitor enrichment over 2 weeks [8]. The total number of cells generated at each passage was not different for DEP-sorted and control cells and yielded approximately 109 total cells after 2 weeks, thus generating large numbers of cells and confirming that DEP sorting did not alter cell proliferation. Fate potential analysis across the expansion period demonstrated no loss in enrichment of astrocyte progenitors. Hence, DEP-sorted NSPCs can be expanded to generate sufficient quantities of cells for techniques such as cell transplantation while retaining enrichment of the cell population of interest.
2.3 Preparation of cells for analysis or sorting
NSPCs are cultured in usual growth medium prior to DEP analysis or sorting. For example, in our experiments mouse NSPCs are grown as suspension cultures in NSPC proliferation media (DMEM supplemented with 1xB27, 1xN2, 1 mM sodium pyruvate, 2 mM glutamine, 1 mM N-acetylcysteine, 20 ng/mL EGF, 10 ng/ml FGF, and 2 μg/mL heparin) as neurospheres [5,33]. Good cell culturing techniques are essential for successfully analyzing and sorting NSPCs using DEP since healthy cells help to ensure reliable results [5,7]. Cells must be transferred to a low conductivity, osmotically balanced DEP buffer prior to DEP analysis or sorting [5,31]. DEP buffer is composed of 8.5% (w/v) sucrose, 0.3% (w/v) glucose, and deionized H2O. RPMI-1640 media is added to adjust buffer conductivity to ~100–110 μS/cm. To prepare mouse NSPCs for DEP, NSPC neurospheres are dissociated using NeuroCult Chemical Dissociation kit (Stem Cell Technologies) and cells are washed with DEP buffer 3 times prior to resuspension in DEP buffer at a final concentration of 1–3×106 cells/ml. Centrifugation during washes should use the lowest possible centrifugal force to pellet cells to avoid high shear forces that might damage the cells when in DEP buffer. NSPCs are sorted as undifferentiated cells, but they are differentiated post-sorting to determine their fate potential. For mouse NSPCs, differentiation is induced post-sorting by plating cells on laminin-coated coverslips in medium lacking EGF, FGF, and heparin.
Prior to a sort, NSPCs are analyzed in DEP and a characteristic trapping curve is generated to determine sorting frequencies. Two types of DEP devices have been used for trapping curve analyses, the DEP microwell device and the 3DEP analyzer. The DEP microwell device has planar electrodes and the transition from nDEP to pDEP is determined by counting the percentage of cells attracted to and trapped along the electrode edges. The 3DEP analyzer (LabTech, East Sussex, UK), a microdevice with 20 microwells containing 3-D electrodes surrounding the microwells, is also used to determine sorting frequencies by rapidly producing NSPCs’ DEP spectra. This system measures cell movement in DEP by plotting light intensity in the well, which shifts with cell motion at each frequency. Six to 8 DEP spectra are produced using the 3DEP analyzer and averaged per set of cells. A normalized DEP trapping curve is created by subtracting the minimum intensity from the average intensity and scaling by the difference in the maximum and minimum intensity values, INORMALIZED = Iavg-Imin/Imax-Imin. This estimates the percent of cells that trapped due to pDEP at specific frequencies ranging from 2 kHz to 20 MHz. For AP enrichment, the sorting frequency is determined as the frequency at which 30% of the NSPCs are in pDEP, as determined from the pre-sort trapping curve analysis. Cell controls include DEP buffer control and 1 MHz control to generate cells that have gone through the device but were not enriched or sorted.
2.4 DEP devices for sorting NSPCs
NSPCs can be sorted with DEP microfluidic devices. Our lab has implemented three unique DEP microfluidic device designs and sorting schemes to enrich NSPCs. Each device incorporates inlet ports to introduce cells and the DEP buffer solution into the device, gold electrodes designed to maximize the electric field strength and trap cells near electrode edges (pDEP force), and outlet port(s) to easily recover sorted cells. A function generator AFG320 (Tektronic, Beaverton, OR) is used to power the electrodes. Cells are introduced into the device via an open well or by using a fluidic pumping system: manually with syringes, automated with syringe pumps (Harvard Apparatus PicoPlus, Holliston, MA), or with a compressed nitrogen tank pressure regulator pump system (described further below under “Large Capacity Electrode Array (LCEA)” microfluidic device).
Attention to detail in device fabrication is critical for robust devices that can stand the rigors of use with complex biological samples [6,8]. Each device was fabricated using standard cleanroom techniques. Electrode arrays were created by coating glass substrates with 200 titanium and 1000 gold using electron beam evaporation (Temescal CV-8). The electrode features were patterned with AZ 4620 photoresist (AZ Electronic Materials, Branchburg, NJ, USA), and then carefully etched around the photoresist pattern leaving behind the desired electrode geometry. Our typical electrode dimensions are 50 μm electrode width and 50 μm gap between electrodes although other geometries are possible [33]. PDMS (polydimethylsiloxane) is used to create microfluidic channels or wells for the devices. The structure and dimensions of the PDMS are created with a PDMS mold, which is made by patterning SU-8 2025 photoresist (MicroChem Corp., Newton, MA, USA) to create the desired features. Uncured PDMS (184 silicone elastomer, Dow Corning Corp., Midland, MI, USA) is poured onto the mold, cured at >75 °C in an oven for at least 3 hours, and then cut to desired size (suitable for secure bonding to the glass substrate with the electrodes). For channels, the PDMS mold is set to a height of 30–50 μm and 200–1500 μm width and channel inlet holes are created with a 23G needle and outlet holes are punched using a 3 mm diameter biopsy punch. PDMS is cleaned with tape, treated with oxygen plasma (Plasma Cleaner/Sterilizer, Harrick, Ithaca, NY, USA), and then irreversibly bonded to the glass substrate containing the electrodes. Wires are attached to the electrodes using either conductive silver epoxy (MG Chemicals, Toronto, Ontario, Canada) or solder to create ground and positive connections. The final device is assembled and ready for sorting. Prior to loading cells, the device should be washed sequentially with 70% ethanol, milliQ H2O, 5% BSA-PBS solution, and the DEP buffer solution (all solutions except ethanol should be sterile filtered). Typical equipment and chemicals used for DEP experiments are listed in Appendix A, Table A.1 and A.2. Our lab has used a DEP microwell device sorter, DEP-assisted continuous sorter (DACS), and large capacity electrode array (LCEA) to sort NSPCs.
Table A.1.
Equipment lists for DEP experiments.
| Equipment list | Supplier |
|---|---|
| Function generator | Tektronix (model AFG320) |
| Microscope | Olympus (model BX41) |
| Camera (collect video of sorting) | Canon (model EOS Rebel T2i) |
| Syringe pump | Harvard Apparatus (model PicoPlus) |
| Pressure regulator | SMC Pneumatics (model ITV1011–21N1S4) |
| Electron Beam Evaporator | Temescal CV-8 **Researchers without access to this equipment can use pre-coated glass substrates commercially available through Sigma Aldrich, New Wave Thin film, and AMS Biotechnology, etc. |
| Spin coater | Laurell Photoresist Spinner |
| UV exposure lamp | Oriel UV Flood Exposure System |
| Plasma cleaner | Harrick |
| 3DEP Analyzer | LabTech |
| Tubing (to supply cells to device) | Tygon® (part # AAD04103) |
| Conductivity Meter | Thermo Scientific (model Orion 4 STAR) |
Table A.2.
Chemical list for DEP experiments.
| Chemical list | Supplier |
|---|---|
| Sucrose | Fisher BioReagents (cat # BP220–1) |
| Glucose | Fisher Scientific (cat # D16–1) |
| RPMI-1640 Medium | GE Life Sciences (cat # SH30255.01) |
| Ethanol | Rossville Gold Shield (Proof 200) |
| Bovine Serum Albumin (BSA) | Rocky Mountain Biologicals, Inc. (part # BSA-BSH-25G) |
| Polydimethylsiloxane (PDMS) | Dow Corning (Sylgard® 184 silicone elastomer kit) |
| Positive photoresist | Shipley 1827 |
| Developer | MF-319 developer |
| Gold etchant | Potassium iodide, KI:I2:H2O=4:1:40 |
| Titanium etchant | 4% Hydrofluoric acid |
| SU-8 photoresist | MicroChem Corp. (Series # 2025) |
| Conductive silver epoxy | MG Chemicals (cat # 8331–14G) |
2.4.1 DEP microwell device sorter [8]
The DEP microwell device has been used to sort NSPCs with relatively high throughput. This device has a simple design consisting of a 3 by 5 microwell array (15 microwells total) [33]. Microwells are created with PDMS and planar interdigitated gold electrodes are located at the bottom of each microwell (Fig. 2A). The electrodes are spaced 50 μm apart and are 50 μm width. A function generator is connected using gold pads at the top and bottom of the microwell array. This device has been used to successfully sort APs from a heterogeneous mouse NSPC population [8]. The open wells make this device simpler than other DEP microfluidic devices with inlet and outlet channels. A suspension of 3×106 cells/mL NSPCs in DEP buffer solution is placed in each microwell (40 μL per well) and allowed to settle for ~10 mins so cells are in close proximity to the electrodes on the bottom of the well prior to application of the electric field. The preselected sorting frequency from trapping curve analysis is applied at 3 Vpp for less than 5 minutes. Cells trap along the electrode edges and cells that do not experience pDEP remain in suspension above the electrodes. While the electric field is on, two 20 μL washes of DEP buffer are used to remove non-trapped cells. The field is turned off and the cells released from the electrode edges are collected and transferred to a collection vial containing NSPC growth medium. This process is repeated multiple times in the microwell array until sufficient numbers of cells are collected. Results show that AP enrichment (~1.4-fold) is possible with this simple DEP microwell device (Fig. 2B,C) and enrichment is maintained over 4 passages to generate 109 cells for further study [8].
Figure 2.
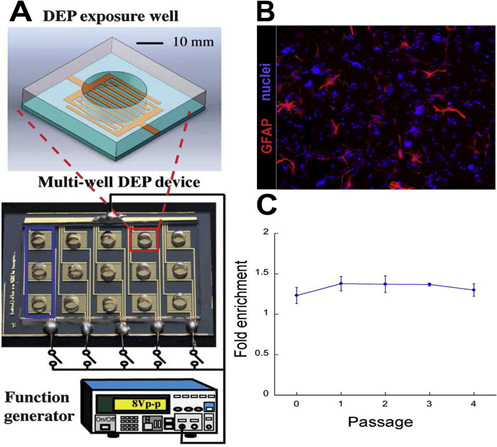
DEP microwell device separates NSPCs at a single frequency. (A) The DEP microwell device consists of a 3 by 5 microwell array. Each microwell consists of PDMS walls surrounding planar interdigitated electrodes at the bottom of the microwell to deliver the electric field (zoom-in box). A function generator is connected using gold pads on the top and bottom of the well array. (B) Mouse NSPCs were sorted at 100 kHz in the DEP microwell device then differentiated in the absence of growth factors to allow generation of glial fibrillary acidic protein (GFAP)-positive astrocytes from APs. All cell nuclei were stained with Hoechst (blue). (C) AP fold enrichment was quantified as the percentage of astrocytes generated from 100 kHz sorted cells relative to unsorted 1 MHz control Analysis over 4 passages reveals maintenance of enrichment. Reprinted with permission from [8,33].
The advantages of the DEP microwell device include its ease of use, the large electrode arrays that supply high throughput, and the easy visualization of cell behavior along the electrode arrays. However, the manual washing steps before cell collection adds a source for human error and can reduce the purity of the collected cells. This device design is primarily compatible with separation at a single frequency for each sort. Additionally, the device operation is not automated, making it labor intensive.
2.4.2 DEP Assisted Continuous Sorter (DACS) [6,7]
A second method for sorting NSPCs uses the DACS device (Fig. 3A). This sophisticated device has microfluidic channels with 3 castellated electrode regions for cell trapping and an additional electrode array to screen newly sorted cells. The main interdigitated stems of the castellated electrodes are 50 μm wide, 50 μm length, and 150 μm space between electrode stems. Each square protrusion is spaced such that a 45° angle is formed corner to corner. Microfluidic channels, dimensions 500 μm width and 40 μm height, include one main channel containing the cell trapping electrodes that branches off to two outlet ports and 3 perpendicular channels crossing the main channel for collection of isolated cells. Castellated interdigitated electrodes are used for cell trapping since fluid flow will give a shear force in one direction when cells are brought into the device for trapping and in the perpendicular direction when flow is switched to the collection outlets. Thus, DEP forces are optimized for both flow configurations. Fluid flow in the main and perpendicular collection channels is controlled with valves. For separation, NSPCs suspended in DEP buffer enter the device at 1–2×106 cells/mL through the inlet and flow along the main channel to the castellated electrode regions (Fig. 3C), where all cells are trapped at a higher frequency (f2) and 8 Vpp. Frequencies for sorting are determined using DEP trapping curves as described above (Fig. 3B). The unsorted mNSPC trapping curve displayed a gradual slope indicating the presence of cellular subpopulations; a steeper slope indicates a more homogeneous cell population while heterogeneous populations generate a more gradual curve, Appendix B Figure B.1. [5]. Once NSPCs are trapped, fluid flow is stopped by closing valves in the main channel (Fig. 3C2). The frequency is then reduced from f2 to f1 releasing a subset of cells from the electrodes (Fig. 3C3). Released cells are targeted to collection outlets by opening the valves in the three perpendicular channels while the electric field is maintained at f1. Screening electrodes, located in the middle perpendicular outlet channel, are used to assess the DEP trapping curve of the sorted cells by sequentially shifting the frequency applied to the screening electrodes and measuring the percentage of cells in pDEP at each frequency. Fluid flow in DACS is provided by a syringe pump connected to the outlet channels off the main channel operating at 1.0–1.5 μL/min.
Figure 3.
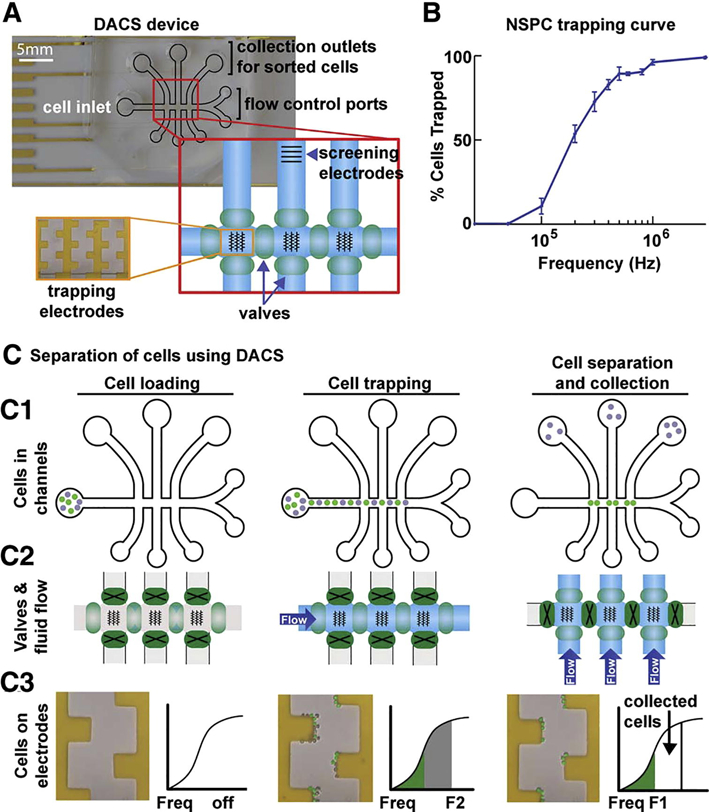
DEP-assisted continuous sorter (DACS) separates NSPCs in specific frequency bands. (A) DACS device consisting of microfluidic channels (black outline) with a main channel for loading cells, perpendicular channels for cell retrieval, and ports connected to syringe pumps for fluid flow control. Fluid flow is switched from main to perpendicular channels by opening and closing pneumatic valves (green ovals, red zoomed-in box). The castellated electrode arrays in the main channel are shown schematically by black lines in the red enlarged box and as viewed through the microscope in the orange enlarged box. Screening electrodes positioned in the perpendicular outlet channel allow post-sorting cell analysis (enlarged red box). (B) A trapping curve of E12 mouse NSPCs is used to select frequency ranges for separation. (C) DACS cell separation illustrated in three phases—cell loading, cell trapping, and cell separation and collection. C1 depicts cell position in channels, C2 shows valve operation and fluid flow in channels, C3 displays electrodes and separation of two cell populations. (C1) Cells are loaded into inlet, flow through the main channel and trap on electrodes using 8 Vpp , and are sorted with one population (gray cells) directed toward collection outlets while the other (green cells) remains in the main channel. (C2) Pneumatic valves (green ovals) control fluid flow; open valves (light green ovals) allow fluid flow and closed valves (dark green ovals with black X) stop flow. Flow is changed to the perpendicular channels for cell collection. (C3) Initially during cell loading the electric field is off and no cells are trapped on the electrodes. When the electric field is applied with f2 both cell populations are trapped (gray and green pseudo-colored cells); cell and trapping curve colors (gray and green) correspond. Frequency is reduced from f2 to f1 and the gray cells are released from the electrodes and flow to collection outlets. Reprinted with permission from [7].
Figure B.1.
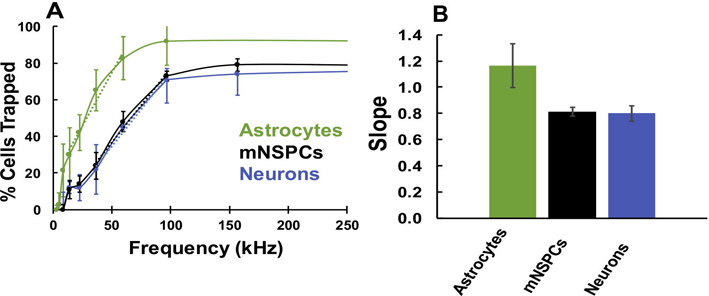
DEP trapping curves reflect population heterogeneity. (A) Normalized trapping curves of mouse NSPCs (mNSPCs), primary astrocytes and primary neurons generated with the 3DEP Analyzer. The colored dashed lines are best fit trend lines for each curve. (B) The trapping curve slopes were determined by adapting signal processing tools to fit a linear trend line [28]. n = 3 independent experiments. Error bars represent ± S.E.M. One-way ANOVA with Tukey post hoc test for multiple comparisons showed no significant difference between the slopes of the three cell types.
Sorting NSPCs with DACS yielded enriched populations of APs and neuron progenitors (NPs). Multiple frequency bins ranging from 0–400 kHz were selected to collect NPs and APs and sorted cells were plated, differentiated, and stained with either mouse anti-Map2 and rabbit anti-TuJ1 to detect neurons formed from NPs, or mouse anti-GFAP, to identify astrocytes generated from APs. NPs were enriched in higher frequency bands, 300–400 kHz and 200–300 kHz, with 1.7-fold and 1.5-fold enrichment, respectively (Fig. 4A,B). At 300–400 kHz, 52% of cells formed neurons, compared to 31% in buffer control samples. APs were enriched in lower frequency bands, with 1.5-fold enrichment in the 0–200 kHz band, (Fig. 4A,B) and 30% of cells generating astrocytes versus 19% in buffer control. DACS achieves better NP enrichment than fluorescence activated cell sorting (FACS) with PSA-NCAM (Fig. 4C). Therefore, it is advantageous to use this sophisticated DACS device for sorting NPs and APs.
Figure 4.
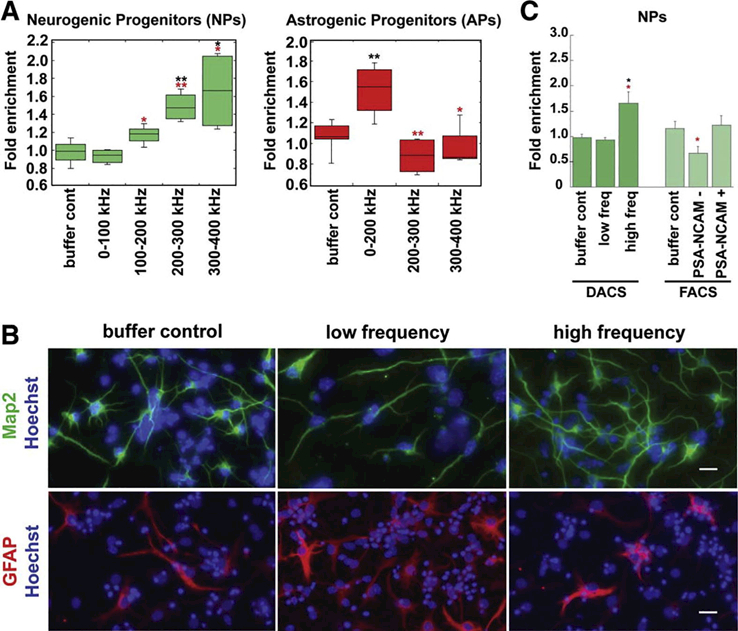
DACS device enriches APs and NPs from E12 mouse NSPCs. (A) NPs are enriched 1.7-fold in high frequency bands and APs are enriched 1.5-fold at low frequency bands using 8 Vpp; fold enrichment from immunostaining quantitation and relative to 1 MHz controls. (B) Buffer control, low frequency and high frequency sorted cells were differentiated and immunostained to detect neurons or astrocytes. Representative images demonstrate that more Map2-positive neurons were formed from NPs enriched at higher frequencies while greater GFAP-positive astrocytes differentiated from APs enriched at lower frequencies. Low frequency for Map2 is 0–100 kHz and 0–200 kHz for GFAP. High frequency is 300–400 kHz for both Map2 and GFAP. Cell nuclei are stained with Hoechst and appear blue. Scale bars = 20 μm. (C) NP fold enrichment is higher with DACS sorting than with FACS with PSA-NCAM antibody. Black asterisks denote significance compared to DEP buffer control and red asterisks denote significance of low frequency sorted cells compared to high frequency sorted cells or PSA-NCAM (−) compared to PSA-NCAM (+) cells (*p<0.05, **p<0.01). Reprinted with permission from [7].
Advantages of the DACS device include its automated design that helps to reduce human error in sorting. The ability to collect multiple frequency bands allows analysis of several samples from a single sort. One disadvantage of this device is the small number of cells collected after ~6 hrs. While it is possible to expand cells after sorting, more cells are desirable to facilitate supplementary post-sorting characterization and other studies. The sophisticated but complicated design of the DACS device makes it more difficult to use.
2.4.3 Large Capacity Electrode Array (LCEA) sorter [8]
The LCEA device includes planar interdigitated electrodes, 50 μm wide with 50 μm spacing, and a microfluidic channel, 1500 μm wide and 30 μm height, with two inlets and one outlet (Fig. 5A). Cells in DEP buffer and DEP wash buffer enter the device from separate sample tubes connected to a pressure source to drive fluid flow through two inlet channels controlled with manual valves (Fig. 5B). When cells enter the device the DEP wash buffer inlet is closed and when the DEP buffer is needed to wash cells away the cell inlet is closed. Once cells for sorting have reached the electrode region, the electric field is turned on at high frequency such that all viable cells experience pDEP (7 Vpp and 1 MHz). This initial cell trapping is repeated with low fluid flow until a large number of cells are visible on the interdigitated electrodes (cell loading). After the desired number of cells are loaded (~1000 cells) the cell inlet is closed and the DEP wash buffer inlet is opened to wash away any loosely trapped cells. Cells are released by reducing the frequency in 100 kHz increments to create frequency bins and cells are collected for each bin at the outlet port. This trap and release sorting scheme can create multiple bins of sorted cells, as shown schematically (Fig. 5C-H). Similar to the previously described devices, APs were enriched 1.9-fold in low frequency bins (Fig. 5I,J).
Figure 5.
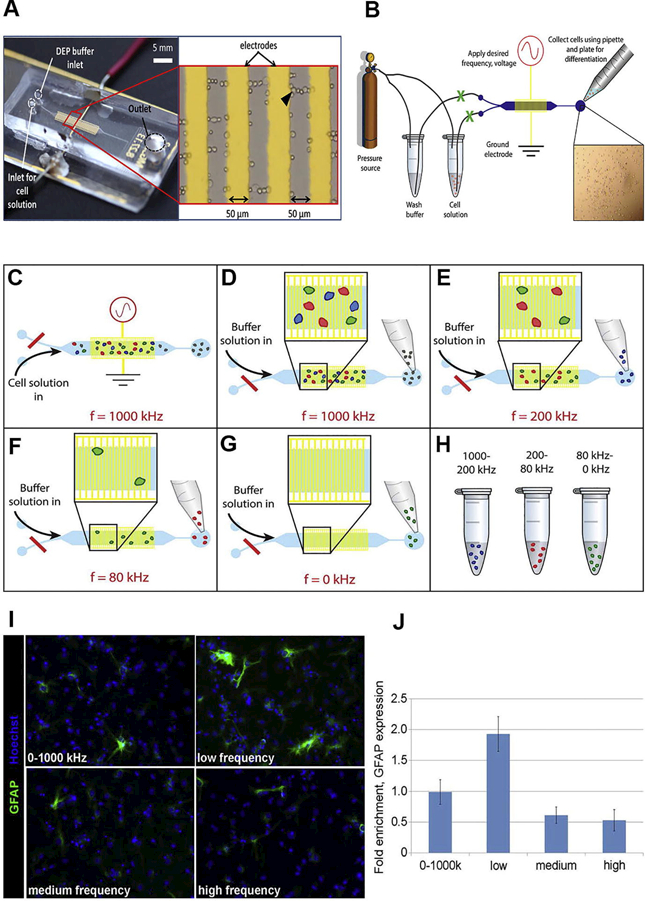
LCEA device, operation, and enrichment of APs from E12 NSPCs. (A) LCEA device consists of a DEP wash buffer inlet and cell solution inlet, electrode region (zoomed-in red box), and outlet channel for cell collection. Interdigitated electrodes are connected to a function generator with the red and black wires; 7 Vpp applied potential. Cells trap along electrode edges and sometimes create pearl chains between the electrodes. (B) LCEA fluid flow operation. A pressure source (compressed N2) was used to initiate fluid flow of the DEP buffer and cell solution. Tubing connects the pressure source to sample vials and the sample vials to the LCEA device. Valves (green X) between the sample vials and the device are opened/closed to enable or restrict fluid flow. With fluid flow enabled from the cell vial and the electric field on, cells move into the electrode region (gold) and untrapped cells are collected from the outlet using a pipette. For cell sorting, the cell solution inlet valve is closed and the DEP buffer inlet opened. The applied frequency is reduced in 100 kHz increments and as cells are released the fractions are collected from the outlet. (C-H) Cell sorting illustration: (C) cells loaded through cell solution inlet with DEP buffer inlet closed (red line). Viable cells are trapped at high frequency (1 MHz), and nonviable cells (gray) do not trap and flow to the outlet. (D) Untrapped cells are collected from the outlet with a pipette. (E) The applied frequency is reduced to 200 kHz, and a portion of the trapped cells are collected in the outlet (blue cells). (F) The applied frequency is changed to 80 kHz, and the released cells were collected (red cells). (G) The electric field is turned off and the remaining cells are released for collection (green cells). (H) Three different cell samples at specific frequencies are collected from steps (C-G). (I) Undifferentiated NSPCs were sorted into low, medium, and high frequency bins, differentiated and immunostained for GFAP to detect astrocytes formed from APs. The cell nuclei were stained with Hoechst (blue). (J) Stained cells were quantified and expressed as fold enrichment relative to the unsorted DEP buffer control. Reprinted with permission from [8].
The LCEA device sorting scheme is advantageous because a large number of cells can be sorted due to the size of the electrode array for trapping. Other advantages include the easy fabrication and use of the device. The fluid flow system driven by a pressure source is also simple to set up and operate. One disadvantage is that while the cell loading process was ideal to increase numbers of sorted cells, cell-cell interactions can alter their responses to the electric field, in part because cell clusters can alter the gradient of the electric field (i.e. cells that are grouped together may respond differently or release at different frequencies than individual cells). Another disadvantage is that although the fluid flow system is easy to set up and operate, it is difficult to maintain the low flow rates needed for DEP sorting. Designing a device that sorts a large number of cells to reduce or eliminate post-sorting cell expansion while limiting cell-cell interactions is a future goal of our group.
2.5 Comparison of sorting devices
Each device used to sort NSPCs has unique features, utilizes trap-and-release to sort the desired progenitor subpopulations, and touches on a few of our sorting wish-list items; Table 1 compares each device. The common advantages of these devices are that the electric field is non-harmful to the cells, AP and NP enrichment is achievable, and the devices are easily fabricated. In each device NSPCs come in contact with the electrodes during sorting. This contact does not reduce cell viability as the sorted cells are recovered and expanded in culture for post-sort differentiation. NSPCs viability remains above 80% post-sorting with these devices. NP enrichment of 1.7-fold was attained in the DACS device. AP enrichment was the highest in the LCEA device at 1.9-fold, and was slightly lower for the DEP microwell and DACS devices at 1.7-fold and 1.5-fold, respectively. Maximum enrichment for AP is 5-fold and maximum NP enrichment 3.3-fold. The devices described here show enrichment of APs and NPs on the basis of differences in membrane capacitance, and future device designs will focus on maximizing enrichment and cell throughput. The LCEA device is the most promising because of its enrichment potential and higher throughput. Each device is easily fabricated and assembled utilizing common thin film deposition and photolithography techniques. The evident disadvantage with each device is cell throughput, which is 100,000 cells/hr for the DEP microwell device, 6,000 cells/hr for the DACS device, and 150,000 cells/hr for the LCEA device; each falls well below the ideal sorting rate of 1.4×106 cells/hr for clinical translation. It takes several hours to collect cells, particularly for the DACS device, and very low numbers of cells are available for post-sorting analysis. To get around low cell throughput, NSPCs can be expanded post-sort and maintain enrichment, allowing for the generation of sufficient cell numbers for many applications, including cell transplantation [3]. However, DEP devices with higher throughput would enable minimal cell manipulation post-sorting and would reduce experimental times, which is ideal for transplantation therapy. Throughput can be increased by operating multiple devices in parallel or by creating a continuous sorter that does not rely on trap-and-release. The generation of DEP sorting devices with higher throughput and increased selectivity for enrichment will continue to accelerate the use of capacitance to sort NSPCs.
Table 1.
Comparison of DEP-based microfluidic devices used to sort Mouse NSPCs.
| DEP Microwell [8] | DACS [6,7] | LCEA [8] | |
|---|---|---|---|
|
Description (Sorting method) |
Cells are placed in a microwell and settle to electrode surface for 5 min. The electric field is applied with a frequency that traps 30% of cells by pDEP (frequency selected prior to sorting by rapidly building DEP spectra and converting to trapping curve). Trapped cells are washed with 40 µL of DEP buffer. Trapped cells are collected in sample tube. |
Cells entered the horizontal channel and were trapped on the electrode arrays by pDEP at f2. Non-trapped cells were removed with reversed fluid flow. The valves for the horizontal channel were closed and f1 was applied. Cells released at f1 were directed to the collection outlets. Cells collected were screened with additional electrodes to determine % cell trapping. Frequency bin collection method. |
The electrode array was saturated with cells trapped by pDEP at a high frequency. With DEP buffer flowing across the electrodes the applied frequency was reduced and released cells were collected in the outlet. This process was repeated until no cells were left. Cells were collected in frequency bins. |
| Simple fabrication | + | − | + |
|
Sample exposed to electric field |
+ | + | + |
| Programmable | − | + | + |
| Moving Parts | − | + | − |
| Low Cost | + | + | + |
| Applied Potential | 3 Vpp | 8 Vpp | 7 Vpp |
| DEP force area | 60 mm2 width = 2000 μm per well (15 wells total) length = 2000 μm per well (15 wells total) |
1.6 mm2 width = 500 μm length = 3150 μm |
9.8 mm2 width = 1500 μm length = 6540 μm |
|
Device Preparation Time (wash cycle) |
60 UV treatment 30 mins 70% ethanol 15 mins sterile H2O 15 mins DEP buffer 15 mins |
50 70% ethanol 15 mins sterile H2O 15 mins 5% BSA 5 mins DEP buffer 15 mins |
60 70% ethanol 15 mins sterile H2O 15 mins DEP buffer 15 mins 5% BSA 15 mins |
| Trapping time | ~5 mins | 60 sec (initial to remove dead cells) specific trapping time not given |
5 mins |
| Cell viable | NR | NR | ~86–91% |
| Enrichment | ~1.4-fold (APs) | 1.7-fold (300–400 kHz, NPs) [7] 1.5-fold (0–200 kHz, APs) [7] ~1.4-fold (neurons) [6] |
~1.9-fold (APs) |
| Cell throughput | No microfluidic channels; in terms of number of cells collected per hour ~100K cells/hr |
6000 cells/hr [8] |
150,000 cells/hr |
“+”, “−”, NR means yes, no, and not reported, respectively. Simple fabrication criteria based on whether device has moving parts; DACS has pneumatic valves. Viability is not reported for NSPCs sorted with the DEP microwell device; however, viability is reported in a separate survival study using the same device as ~90–100% (60 sec electric field exposure) and ~85–100% (5 min electric field exposure) [33]. Viability is also not reported for the DACS device, but viable cells were obtained post-sorting and differentiated to assess cell phenotype [7].
3. Hints for troubleshooting
Several issues can arise when running a DEP sorting experiment. The purpose of this section is to provide useful tips to work through issues that may be encountered. However, the best way to troubleshoot DEP experiments is practice. The more familiar someone is with their DEP system the easier it is to overcome problems.
3.1 Fabrication
-
○
It is important to deposit titanium and gold (for electrodes) at slow deposition rates (~0.7 /sec) to achieve uniformity. Depositing at faster rates will decrease the integrity of your titanium/gold thin films and the electrodes may have incomplete connections or the electrodes may lift off from the glass substrate after a few uses. Make sure glass slides are cleaned thoroughly with acetone, isopropanol, and methanol (soak for 10 mins in each and dry with air prior to entering clean room).
-
○
During electrode patterning with a UV lamp make sure the photomask is in tight contact with the photoresist to avoid patterning electrodes with rugged edges. Rough edge (nonideal) versus smooth edge (ideal) electrodes can change the shape of the electric field lines (selectivity is dependent on electric field lines/gradient). Sandwich the gold-plated glass slide coated with positive photoresist with the mask and place both between two pieces of glass. Clamp the edges of the glass pieces to reduce the contact distance between the mask and the positive photoresist. The orientation of the mask must be so that the ink is in direct contact with the photoresist (emulsion down).
-
○
If a good seal is not made between the PDMS and glass substrate your microfluidic device will leak. Leaking will affect the flow rate and pressure inside your device. To avoid this situation, make sure the side of the PDMS with channel features is completely clean. Tape can be used to remove small particles like lent. Both the channel side of the PDMS and the electrode side of the glass substrate should be cleaned in this manner. Secondly, it is important to monitor the pressure inside the plasma cleaner chamber once its closed. Targeting 300 mbar inside the chamber allows the glass and PDMS surfaces to be optimally treated with plasma, so that once they are pressed together a good seal is formed.
-
○
After the device is assembled check electrode connections. Add fluid (i.e. water, DEP buffer, etc.) and use a multimeter to check continuity, voltage, and resistance.
3.2 Device Operation Testing
-
○
When liquid initially enters a dry microdevice bubble(s) can be a problem. Bubbles can enter the device during the rinsing steps, where the device is washed with 70% ethanol, H2O, 5% BSA-PBS solution, and the DEP buffer solution (as described in section 2.4 “DEP devices for sorting NSPCs”). These solutions are supplied to the device via syringes and if air is present in the syringe upon liquid entry bubbles are created. To circumvent this, ethanol is rinsed through the device until bubble(s) are no longer present. Sometimes pressure is applied by pressing on the device to force bubble(s) toward the inlet or outlet for removal. This is not recommended as this manually applied pressure may damage the electrodes.
-
○
Before beginning a DEP, experiment calibration steps are recommended. The DEP force a cell experiences in a microdevice is dependent on several parameters like applied voltage, frequency, cell concentration, buffer conductivity, and flow rate; having good operating parameters figured out in advance will increase sorting success rate. For example, test the fluid flow characteristics of the microdevice using either water with colored dyes (for visualization) and/or polystyrene beads. Polystyrene beads dielectric properties differ from those of cells (beads display nDEP at low conductivities [28]) but they are a good first step for visualizing approximate cell trajectory. Once comfortable with operating parameters and device function, perform initial trials with non-precious cells before moving to cells of interests.
-
○
Clumped cells resulting from inadequate cell dissociation may clog the device. Clogging may also be caused by cell death, in which cellular contents leak and stick to microchannel surfaces and change fluid flow patterns. Once a device is clogged, it is rinsed repeatedly with DEP buffer, H2O and/or ethanol to remove the clog, after which the initial device washing must be repeated before reintroducing cells into the device for continued sorting. Rinsing the device again is critical because if cells come into contact with residual ethanol their viability may be altered.
3.3 Cell Preparation
-
○
Processing NSPCs for a DEP experiment is an important step. It is crucial to make sure the cells are completely dissociated; incomplete dissociation results in clumped cells, which decreases cell concentration for sorting or analysis (target concentration is 1–2×106 cells/mL) and clogs devices. Low cell concentration directly affects throughput as low concentrations extend experimental time.
-
○
It is critical to ensure that the population of cells to be sorted has high viability. If cells prepared for sorting are below 70% live cells, sorting does not work well.
-
○
Viability of the cells in the DEP buffer must be tested to ensure no reduction in viable cells during the sorting procedure. For long sorts, cells can be dissociated and transferred into DEP buffer for sorting at multiple points during the procedure to reduce the length of time in the DEP buffer. Mouse and human NSPCs remain viable in DEP buffer for at least 4 hours [3,5]. For a detailed analysis of cell viability after exposure to DEP electric fields, see [33].
3.4 Cell sorting
-
○
For our experiments the sorting frequency for astrocyte progenitor selection was set as the frequency at which 30% of the cells are trapped as determined from the trapping curve. The sorting frequency maybe higher or lower than the 30% point depending on the cell population to be sorted. If you notice over the course of an experiment that the appropriate percentage of the cells are no longer trapping at the sorting frequency, it could be due to the amount of time the cells have remained in the DEP buffer. Some cells may lyse and release ions after being in DEP buffer for extended periods (6 hours or more). The release of ions will increase the overall conductivity of the DEP buffer and change the cells’ responses to the sorting frequency. To prevent this, a small number of cells can be processed at a time for DEP experiments, with iterative cell processing steps to generate more cells as needed.
-
○
Electrode aging should be considered in DEP experiments. The amount of time one microfluidic device is in use should be tracked in order to determine when to switch to a new device. Electrode aging problems manifest as a change in the sorting frequency or visibly diminished electrode quality. If a decrease occurs in the sorting frequency experiment-to-experiment, it may be an indication of aged electrodes and the device should be replaced with a newly fabricated or unused device. We recommend retiring a device after 3–6 months of consistent use.
-
○
Tubing and syringes are used to supply cells and buffer solutions to microfluidic devices. During an experiment, if the number of cells flowing through the device is inconsistent, it may be attributed to cell settling in the tubing and syringe. To prevent this, add a small volume of cell suspension at a time to the syringe during sorting, we recommend ~30 μL. Also, cell samples should be mixed periodically to maintain cell suspension.
3.5 Cell analysis post-sorting
-
○
Low enrichment is possible when using the DEP microwell device (or a similarly designed device). This is due to the manual washing steps with DEP buffer to remove untrapped cells. To achieve the highest enrichment with a DEP microwell device, wash away untrapped cells very gently by adding DEP buffer in small increments (20 μL increments could even be reduced to 10 μL increments). Add the DEP buffer along the wall of the microwell to avoid disturbing the trapped cells.
-
○
Low enrichment may also be attributed to experimental parameters such as the applied voltage, sort frequency, and the DEP buffer conductivity. These parameters can be tuned to optimize the appropriate DEP force necessary to increase the sorting purity for the cells of interest.
-
○
If many dead cells are visible after sorting, the best method to preserve cell health is to collect sorted cells in a vial containing normal growth medium. Since the vial will now contain a mixture of DEP buffer and growth medium, gently centrifuge the sample and aspirate the supernatant to remove diluted DEP buffer and resuspend the cells in fresh medium prior to cell plating. This should reduce cell death. Cells can also be cultured after sorting in media with higher than normal growth factor concentrations (we use 2X) to expand cultures and maintain high cell viability.
4. Conclusions
NSPCs have therapeutic potential but it is important to effectively sort subpopulations to begin testing their potential to treat neurological diseases and injuries (AP-biased populations may have higher efficacy than NP-biased populations and vice versa). DEP is a promising label-free cell sorting technology that separates cells differing in whole cell membrane capacitance values. Here we describe the use of DEP to enrich subpopulations of cells from NSPCs, including sorting parameters and 3 different DEP-based sorting devices: the DEP microwell, DACS and LCEA. While these devices have been successfully utilized to enrich APs and NPs from NSPCs, additional improvements made to DEP sorting devices will further increase throughput and purity.
5. Acknowledgements:
The authors would like to thank Dr. Abraham P. Lee, who designed many of the DEP microfluidic devices described herein. This work was supported in part by the National Science Foundation Postdoctoral Research Fellowship in Biology DBI-1612261 (TNGA), National Science Foundation CAREER Award IOS-1254060 (LAF), the California Institute for Regenerative Medicine (CIRM) RT1–01074 and RB5–07254 (LAF), and the NIH National Center for Research Resources and the National Center for Advancing Translational Sciences through Grant UL1 TR000153 (Pilot Grant to LAF).
6. Appendix A
7. Appendix B
Trapping curves generated with the 3DEP Analyzer provide the DEP spectra of a cell population, which yields two useful pieces of information: (1) a normalized cell trapping curve that can be used to select sorting frequencies as described above, and (2) an idea of population heterogeneity determined by fitting a linear trend line to the transient response of the trapping curve (~10–100 kHz region). Trapping curves can reflect population heterogeneity since a homogeneous population of cells would experience pDEP at the same frequency while a heterogeneous population would have sets of cells experiencing pDEP at multiple frequencies [5]. For example, the trapping curves and slopes for primary astrocytes, mNSPCs and primary neurons may differ since astrocytes are a more homogeneous population than either of the other two cell types. Although there are subtypes of astrocytes, in general a population of astrocytes is fairly homogenous [39]. NSPC cultures contain progenitor cells, stem cells and a few differentiated cells [40], while primary neurons derived from E12 mouse cortex contain a variety of neuron subtypes [41]. The trapping curves of primary neurons and NSPCs have a more gradual slope as compared to that of primary astrocytes (Fig. B.1A). In Figure B.1B, the slopes were quantified as 0.81, 0.80, and 1.17 for primary neurons, NSPCs, and primary astrocytes, respectively. Although the astrocyte curve was steeper, there was no significant difference between the slopes of all 3 cell types. These data complement previous findings that the slope of DEP trapping curves reflect the heterogeneity of a cell population [5].
Footnotes
List of Abbreviations1
- (DEP)
- Dielectrophoresis
- (NSPCs)
- neural stem and progenitor cells
- (AP)
- astrocyte progenitor
- (NP)
- neuron progenitor
- (LCEA)
- large capacity electrode array
- (DACS)
- dielectrophoresis assisted continuous sorter
- (HSC)
- hematopoietic stem cell
- (MSC, ADSC)
- mesenchymal/adipose-derived stem cell
- (AC)
- alternating current
- (DC)
- direct current
- (Cmem)
- membrane capacitance
- (pDEP)
- positive dielectrophoresis
- (nDEP)
- negative dielectrophoresis
- (PDMS)
- polydimethylsiloxane
- (GFAP)
- glial fibrillary acidic protein
- (FACS)
- fluorescence activated cell sorting
- (BSA)
- bovine serum albumin
- (PBS)
- phosphate buffer solution
Selectivity can be manipulated by electrode configuration.
>Maximum enrichment may vary experimentally because the number of astrogenic and neurogenic progenitors in the starting unsorted population may fluctuate.
8. References
- 1.Lindvall O, Kokaia Z, Stem cells for the treatment of neurological disorders, Nature 441 (2006) 1094–1096. [DOI] [PubMed] [Google Scholar]
- 2.Gage FH, Mammalian neural stem cells, Science 287 (2000) 1433–1438. [DOI] [PubMed] [Google Scholar]
- 3.Labeed FH, Lu J, Mulhall HJ, Marchenko SA, Hoettges KF, Estrada LC, Lee AP, Hughes MP, Flanagan LA, Biophysical characteristics reveal neural stem cell differentiation potential, PLoS ONE 6 (2011) e25458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian XM, Shen Q, Goderie SK, He WL, Capela A, Davis AA, Temple S, Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells, Neuron 28 (2000) 69–80. [DOI] [PubMed] [Google Scholar]
- 5.Flanagan LA, Lu J, Wang L, Marchenko SA, Jeon NL, Lee AP, Monuki ES, Unique dielectric properties distinguished stem cells and their differentiated progeny, Stem Cells 26 (2008) 656–665. [DOI] [PubMed] [Google Scholar]
- 6.Prieto JL, Lu J, Nourse JL, Flanagan LA, Lee AP, Frequency discretization in dielectrophoretic assisted cell sorting arrays to isolate neural cells, Lab Chip 12 (2012) 2182–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nourse JL, Prieto JL, Dickson AR, Lu J, Pathak MM, Tombola F, Demetriou M, Lee AP, Flanagan LA, Membrane biophysics define neuron and astrocyte progenitors in the neural lineage, Stem Cells 32 (2014) 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon MG, Ling Y, Arulmoli J, McDonnell LP, Akil A, Nourse JL, Lee AP, Flanagan LA, Increasing label-free stem cell sorting capacity to reach transplantation-scale throughput, Biomicrofluidics 8 (2014) 064106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talary MS, Mills KI, Hoy T, Burnette AK, Pethig R, Dielectrophoretic separation and enrichment of CD34+ cell subpopulation from bone marrow and peripheral blood stem cells, Med Biol Eng Comput 33 (1995) 235–237. [DOI] [PubMed] [Google Scholar]
- 10.Stephens M, Talary MS, Pethig R, Burnett AK, Mills KI, The dielectrophoresis enrichment of CD34+ cells from peripheral blood stem cell harvests, Bone Marrow Transplant, 18 (1996) 777–782. [PubMed] [Google Scholar]
- 11.Vykoukal J, Vykoukal DM, Freyberg S, Alt EU, Gascoyne PR, Enrichment of putative stem cells from adipose tissue using dielectrophoretic field-flow fractionation, Lab Chip 8 (2008) 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagnaninchi PO, Drummond N, Real-time label-free monitoring of adipose-derived stem cell differentiation with electric cell-substrate impedance sensing, Proc Natl Acad Sci USA 108 (2011) 6462–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hildebrandt C, Buth H, Cho SB, Impidjati H Thielecke, Detection of the osteogenic differentiation of mesenchymal stem cells in 2D and 3D cultures by electrochemical impedance spectroscopy, J Biotech 148 (2010) 83–90. [DOI] [PubMed] [Google Scholar]
- 14.Song H, Rosano J, Wang Y, Garson C, Prabhakarpandian B, Pant K, Klarmann G, Perantoni A, Alvarez L, Lai E, Continuous-flow sorting of stem cells and differentiation products based on dielectrophoresis, Lab Chip 15 (2015) 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho S, Gorjup E, Thielecke H, Chip-based time-continuous monitoring of toxic effects on stem cell differentiation, Ann Anat 191 (2009) 145–152. [DOI] [PubMed] [Google Scholar]
- 16.Hirota Y, Hakoda M, Relationship between dielectric characteristic by DEP levitation and differentiation activity for stem cells, Key Eng Mater 459 (2011) 84–91. [Google Scholar]
- 17.Muratore M, Srsen V, Waterfall M, Downes A, Pethig R, Biomarker-free dielectrophoretic sorting of differentiation myoblast multipotent progenitor cells and their membrane analysis by raman spectroscopy, Biomicrofluidics 6 (2013) 034113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Velugotla S, Pells S, Mjoseng H, Duffy C, Smith S, De Sousa P, Pethig R, Dielectrophoresis based discrimination of human embryonic stem cells from differentiating derivatives, Biomicrofluidics 6 (2012) 044113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsikritsis D, Shi H, Wang Y, Velugotla S, Sren V, Elfick A, Downes A, Label-free biomarkers of human embryonic stem cell differentiation to hepatocytes, Cytom Part A 89A (2016) 575–584. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, Basu S, Laue E, Seshia AA, Single cell studies of mouse embryonic stem cell (mESC) differentiation by electrical impedance measurements in a microfluidic device, Biosens Bioelectron 81 (2016) 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyun KA, Jung HI, Microfluidic devices for the isolation of circulating rare cells: a focus on affinity-based, dielectrophoresis, and hydrophoresis, Electrophoresis 34 (2013) 1028–1041. [DOI] [PubMed] [Google Scholar]
- 22.Pethig R, Review article – dielectrophoresis: status of the theory, technology, and applications, Biomicrofluidics 4 (2010) 022811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velev OD, Gangwal S, Petsev DN, Particle-localized AC and DC manipulation and electrokinetics, Annu Rep Prog Chem Sect C 105 (2009) 213–246. [Google Scholar]
- 24.Minerick AR, DC dielectrophoresis in lab-on-a-chip devices, Encyclopedia of Microfluidics and Nanofluidics (2008) 331–334.
- 25.Mulhall HJ, Labeed FH, Kazmi B, Costea DE, Hughes MP, Lewis MP, Cancer, pre-cancer and normal oral cells distinguished by dielectrophoresis, Anal Bioanal Chem 401 (2011) 2455–2463. [DOI] [PubMed] [Google Scholar]
- 26.Pohl HA, Dielectrophoresis: the behavior of neutral matter in nonuniform electric fields, Cambridge University Press, New York, 1978. [Google Scholar]
- 27.Salmanzadeh A, Elvington ES, Roberts PC, Schmelz EM, Davalos RV, Sphingolipid metabolites modulate dielectric characteristics of cells in a mouse ovarian cancer progression model, Integr Biol 5 (2013) 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.G Adams TN, Leonard KM, Minerick AR, Frequency sweep rate dependence on the dielectrophoretic response of polystyrene beads and red blood cells, Biomicrofluidics 7 (2013) 064114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams TNG, Turner PA, Janorkar AV, Zhao F, Minerick AR, Characterizing the dielectric properties of human mesenchymal stem cells and the effects of charged elastin-like polypeptide copolymer treatment, Biomicrofluidics 8 (2014) 054109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinsen OG, Grimnes S, Schwan HP, Interface phenomena and dielectric properties of biological tissue, Encyclopedia Surf Colloid Sci 20 (2002) 2643 – 2652. [Google Scholar]
- 31.Chen CS, Pohl HA, Biological dielectrophoresis - the behavior of lone cells in a nonuniform electric field, Ann N Y Acad Sci 238 (1974) 176–185. [DOI] [PubMed] [Google Scholar]
- 32.Salamanzadeh A, Davalos RV, Chapter 3 Electrokinetics and rare-cell detection, in: Labeed FH, Fatoyinbo HO (Eds.), Microfluidics in Detection Science: Lab-on-a-chip Technologies, RSC Publishing, Cambridge, 2015, pp. 61–83. [Google Scholar]
- 33.Lu J, Barrios CA, Dickson AR, Nourse JL, Lee AP, Flanagan LA, Advancing practical usage of microtechnology: a study of the functional consequences of dielectrophoresis on neural stem cells, Integr Biol 4 (2012) 1223–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummings BJ, Uchida N, Tamaki SJ, Salazar DL, Hooshmand M, Summers R, Gage FH, Anderson AJ, Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice, Proc Natl Acad Sci U.S.A 102 (2005) 14069–14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O, Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury, J Neurosci 25 (2005) 4694–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cusulin C, Monni E, Ahlenius H, Wood J, Brune JC, Lindvall O, Kokaia Z, Embryonic stem cell-derived neural stem cells fuse with microglia and mature neurons, Stem Cells 30 (2012) 2657–2671. [DOI] [PubMed] [Google Scholar]
- 37.Glass JD, Boulis NM, Johe K, Rutkove SB, Federici T, Polak M, Kelly C, Feldman EL, Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients, Stem Cells 30 (2012) 1144–1151. [DOI] [PubMed] [Google Scholar]
- 38.Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, Caverzasi E, Gaetano L, Mandelli ML, Ryan T, Perry R, Farrell J, Jeremy RJ, Ulman M, Huhn SL, Barkovich AJ, Rowitch DH, Neural stem cell engraftment and myelination in the human brain, Sci Transl Med 4 (2012) 155ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Ambrosio R, Wenzel J, Schwartzkroin PA, McKhann GM, Janigro D, Functional specialization and topographic segregation of hippocampal astrocytes, J Neurosci 18 (1998) 4425–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campos LS, Leone DP, Relvas JB, Brakebusch C, Fassler R, Suter U, ffrench-Constant C, beta 1 integrins activate a MAPK signaling pathway in neural stem cells that contributes to their maintenance, Development 131 (2004) 3433–3444. [DOI] [PubMed] [Google Scholar]
- 41.Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, Temple S, The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells, Nature Neurosci 9 (2006) 743–751. [DOI] [PubMed] [Google Scholar]