

Abstract
Heme proteins utilize the heme cofactor, an iron porphyrin, to perform a diverse range of reactions including dioxygen binding and transport, electron transfer, and oxidation/oxygenations. These reactions share several, key metalloporphyrin intermediates, typically derived from dioxygen and its congeners such as hydrogen peroxide, and which are comprised of metal-dioxygen, metal-superoxo, metal-peroxo, and metal-oxo adducts. A wide variety of synthetic metalloporphyrinoid complexes have been synthesized to generate and stabilize these intermediates, and then employed to determine the spectroscopic features, structures, and reactivities of such species in controlled and well-defined environments. In this review, we summarize recent findings on the reactivity of these species with common porphyrinoid scaffolds employed for biomimetic studies. The proposed mechanisms of action are emphasized. The review is organized by structural type of metal-oxygen intermediate, and broken into subsections based on the metal (manganese, iron) and porphyrinoid ligand (porphyrin, corrole, corrolazine).
TABLE OF CONTENTS FIGURE
1.1. Introduction
Porphyrins and related macrocycles have been well studied because of their role as biological ligands. The heme cofactor, which consists of an iron-bound porphyrin, is at the active site of many metalloproteins and performs a diverse range of chemistry such as oxygen transport and storage, electron transfer, and oxidation/oxygenation reactions (using O2/H2O2 as the oxidant and in many cases the O-atom source).1-3
Examples of monooxygenation reactions catalyzed by heme enzymes include aromatic and alkane hydroxylations, olefin epoxidations, as well as N-, S- or O-dealkylations. One class of enzymes that performs many of these transformations, which use dioxygen (O2) as the oxidant and O-atom source, is a large class of thiolate-ligated heme enzymes called Cytochrome P450s (CYPs), which are found in most mammalian tissues and organs as well as other organisms such as plants, bacteria, and yeast. Heme-dependent dioxygenases, which incorporate both oxygen atoms into the substrate, are a smaller class of enzymes that oxidize tryptophan or other indole derivatives to give N-formylkynurenine products. While there is a lot of information regarding the mechanism of CYP, less is known about the mechanism of dioxygenases.1,4 Peroxidases, catalases, and choroperoxidases (CPOs) all utilize hydrogen peroxide as oxidant and substrate but differ in their functions and proximal ligand; peroxidase heme iron is bound by histidine, catalase heme iron is bound by tyrosinate, and CPO heme is bound by cysteinate. Peroxidases utilize hydrogen peroxide (H2O2) to oxidize such substrates as phenols and aromatic amines. Catalases perform the disproportionation of hydrogen peroxide to dioxygen and water. Heme chloroperoxidase is a halogenating enzyme that uses hydrogen peroxide to chlorinate aliphatic substrates; however, it is functionally tolerant of other substrates in that it is also capable of catalyzing peroxidase, catalase, and P450-type reactions. Finally, heme degradation is performed by heme oxygenase enzymes, in which the oxygenation of the porphyrin ligand itself occurs.
All of the enzymes discussed here utilize only a few common intermediates derived from dioxygen or hydrogen peroxide in their mechanistic cycles. These intermediates are metal-superoxo (O2•−), - peroxo (O22−), - hydroperoxo (OOH−), and - oxo (O2−) species (Scheme 1). Synthetic analogs target these intermediates and allow for the structural and spectroscopic features of these species to be analyzed. The development of synthetic analogs of these intermediates provides systems in which bond-making and bond-breaking events that occur at the metal can be studied in well-controlled environments. In this review, we focus on the biomimetic reactivity of synthetic oxygen-derived metalloporphyrinoid compounds.
Scheme 1.
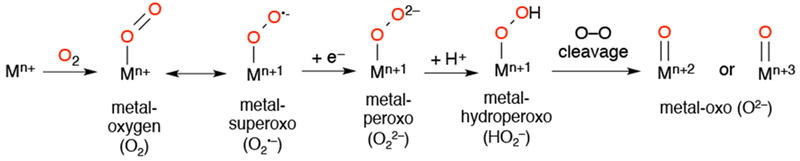
Common Metal-Oxygen Intermediates Utilized by Metalloproteins
The environment surrounding the heme in the active site varies depending on the system. The iron center is coordinated axially on the proximal side by an N, O, or S donor derived from the amino acids histidine, tryptophan, tyrosine, or cysteine. The identity of the amino acids surrounding the distal side of the metalloporphyrin also play important roles, such as providing hydrogen bond donors or acceptors that can interact with the reactive intermediates during the catalytic cycle. These structural features combine to provide selectivity and control reactivity of the metalloenzyme, allowing for a wide variety of reaction outcomes (Scheme 2). For example, within the broad class of heme proteins, myoglobin and CYP utilize heme iron at their active sites, but have entirely different functions and O2 reactivities due to differences in the chemical environment surrounding the heme sites. Myoglobin is a simple dioxygen- binding and transport protein. At the active site, the heme is ligated by an axial histidine nitrogen, and contains a nearby histidine on the distal side, which provides selectivity for O2 over other toxic small molecules such as carbon monoxide (CO). In CYP, a monooxygenase, the heme iron is ligated by a cysteinate on the proximal side, and several residues and a hydrogen-bonded water network on the distal side control O2 binding, cleavage, and subsequent substrate oxidation. Scheme 3 illustrates the “push/pull” effects of these interactions in CYP that influence this reactivity.1
Scheme 2.
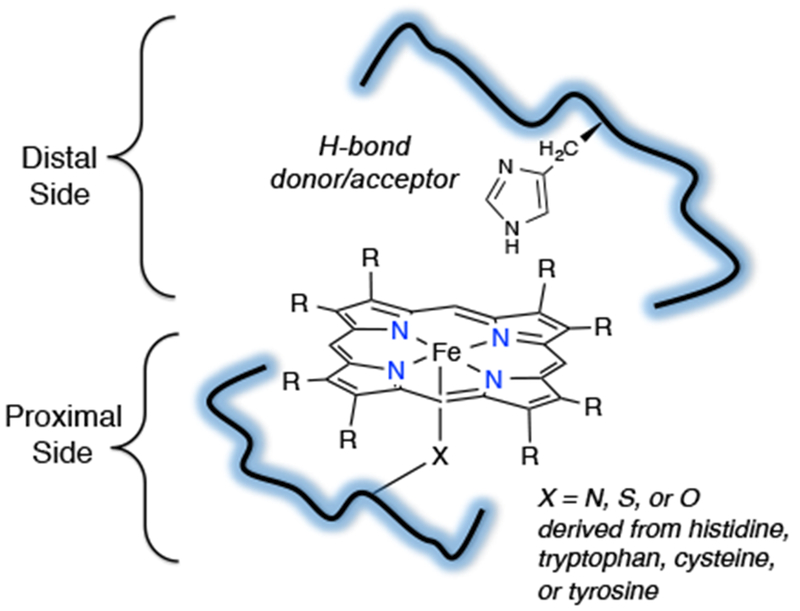
Structural Factors that Affect Reactivity in Heme Proteins
Scheme 3.

Push/Pull Effects of Axial Ligand and Distal Residues on O–O Bond Cleavage in Cytochrome P450
Because the heme cofactor utilizes the porphyrin framework, synthetic metalloporphyrin complexes have proved useful as model systems to study the factors that control the reactivity of heme proteins. Porphyrins are heterocyclic macrocycles composed of four pyrrole units joined by methine bridges. They are aromatic, 18-π electron systems, and this high degree of conjugation allows for intense absorption bands in the visible region. Physicochemical properties of the porphyrin ring, such as electronic effects, steric effects, and solubility, can be varied by substituting at the meso- positions of the macrocycle or the β- positions of the pyrrole unit. The porphyrinoid family encompasses the common structural types shown in Chart 1. This family includes ring-contracted analogs in which one meso position is missing from the framework (as in corroles and corrolazines). Ring contracted analogs such as subporphyrins, subphthalocyanines, and subporphyrazines, which are missing a pyrrole unit, are also members of the porphyrinoid family, but are only known as the boron-bound derivatives.5,6 Ring-expanded porphyrins are another large category within the porphyrinoid family, in which one or more meso- positions or pyrrole units are added to the framework. There are some excellent reviews on the synthesis of ring-expanded porphyrinoid compounds and their reactivities including articles by Dolphin,7 Sessler,8 and most recently Osuka.9 In this review, we focus on porphyrins and the contracted corrole and corrolazine systems, which have been used in a range of biomimetic studies over the past several years.
Chart 1.
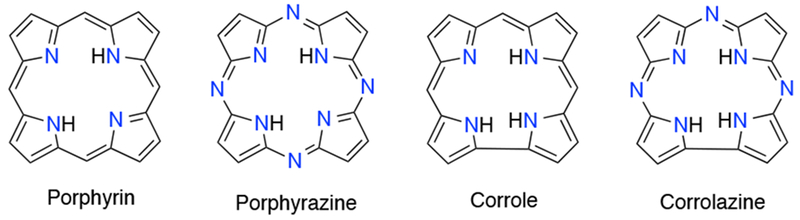
Core Structures of Common Members of the Porphyrinoid Family Used for Biomimetic Studies
In metalloporphyin models of heme proteins, the “active site” has been synthetically constructed in levels by building out from the porphyrin framework. The first level of synthetic design is to modify the porphyrin substituents on the periphery of the ring. The second level is to change the first coordination sphere of the central metal atom by adding axial ligands. The third level is to add steric encumbrance on functional groups orthogonal to the macrocycle plane, which may interact with oxygen-derived intermediates through non-covalent interactions such as hydrogen-bond donors or acceptors. An example of the latter comes from the synthesis of so-called “Pacman” porphyrins.10 The fourth level of synthetic modification is through the encapsulation of the metalloporphyrin in a larger scaffold such as MOFs, dendrimers, or short peptides.11,12 Finally, the ability to encapsulate the metalloporphyin in artificial or engineered proteins has seen a recent surge in effort.13-16
In many cases, the reactive intermediates of interest (e.g. metal-oxo, metal-O2) in metalloporphyrin systems are transient species at room temperature and must be generated or trapped at low temperatures for characterization and study. Ring-contracted porphyrinoid complexes such as corrole and corrolazine (Chart 1) provide enhanced thermal stabilization for some of these intermediates and allow for their direct characterization or even isolation in some cases. Corroles contain a direct pyrrole-pyrrole linkage and thus differ by the deletion of a meso-carbon atom relative to porphyrins. Fully deprotonated, they are trianionic (3−) ligands, and the higher charge and smaller cavity size provide a stabilizing environment for metal ions in higher oxidation states (≥ 3). Corrolazines are also ring-contracted like corroles, but contain meso-nitrogen atoms in place of meso-carbons. The biomimetic reactivity of these three ligand systems (porphyrins, corroles, and corrolazines) will be covered here. The synthesis of corroles, corrolazines, and their metalated forms has been recently reviewed in the Handbook of Porphyrin Science (2011).17 Herein recent developments on the biomimetic reactivity of such systems are reviewed. The review is organized by structural type of metalloporphyrinoid species. Complexes are restricted to those able to participate in one or more of the biomimetic reaction types defined in the following section.
1.2. Biomimetic reaction types
Shown in Chart 2 are several biomimetic reactions performed by metalloporphyrin complexes. Oxygen atom transfer (OAT) is a two-electron process which results in the net transfer of an oxygen atom to a substrate and can occur by at least two different mechanisms, with two of the most common for metal-oxo species shown in Scheme 4. The first of these is a concerted mechanism, in which the oxygen atom is transferred in one step. Another common mechanism is a stepwise mechanism, in which the substrate is oxidized by one electron either by an outer-sphere or inner-sphere oxidation, followed by fast oxygen transfer. Typically this reaction is facile when a substrate is nucleophilic and the oxidant is an electrophilic oxo complex. The substrate for OAT is often an alkene, sulfide, or phosphine, resulting in an epoxide, sulfoxide, or phosphine oxide, respectively.
Chart 2.
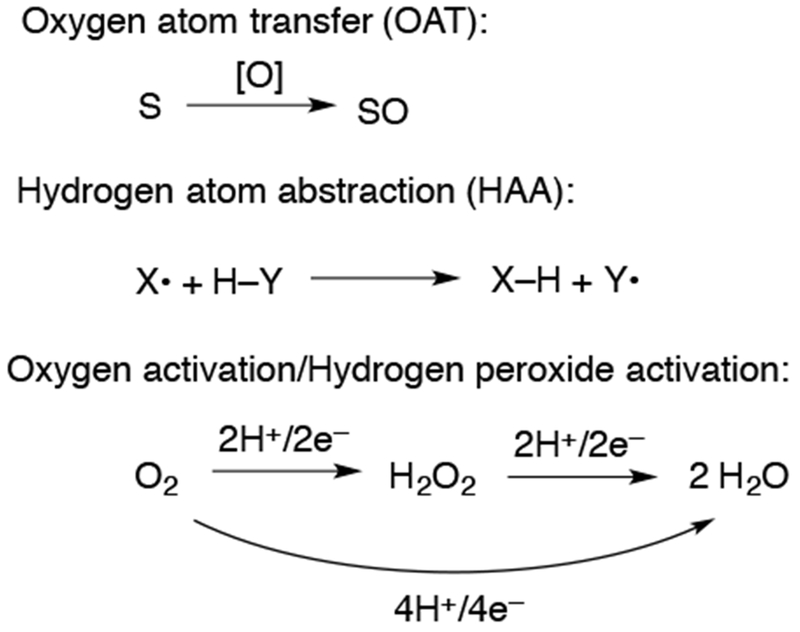
Biomimetic Reactions Performed by Metalloporphyrinoid Complexes
Scheme 4.
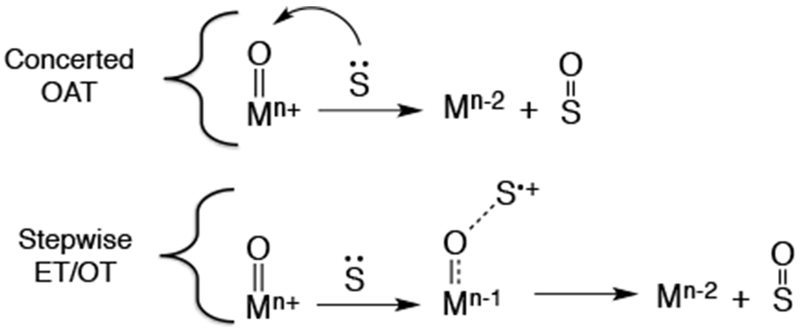
Possible Mechanisms for Oxygen Atom Transfer from a Metal-Oxo Complex
Hydrogen-atom abstraction is the net transfer of an H-atom, which can occur by several different mechanisms. H-atom abstraction is usually proposed as the first step in the oxidation of inert C–H bonds by high-valent metal-oxo species in biological systems, followed by rebound of the newly formed carbon radical with a metal-hydroxo intermediate. Mechanistically, understanding H-atom abstraction can be complex because H• transfer can occur in a step-wise fashion (proton transfer followed by electron transfer or vice versa) or in a concerted fashion. The kinetics and thermodynamics are intertwined, and the thermodynamic parameters (pKa, E°) associated with H-atom transfer are illustrated in the square scheme in Scheme 5. The square scheme is similar to a thermodynamic Hess cycle, in which the sum of the ΔG°s of the horizontal (from the E° of ET) and vertical (from the pKa of PT) steps adds up to give the ΔG° of the diagonal step.
Scheme 5.
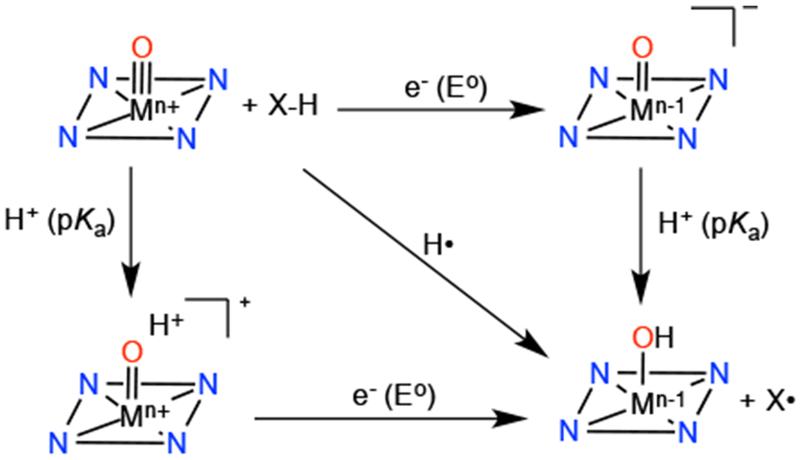
Square Scheme Depicting H-atom Transfer to a Metal-Oxo Complex
There are many examples of hydroxylation reactions by high-valent metal-oxo porphyrin species. These reactions typically are initiated by H-atom abstraction of a substrate by the metal-oxo species to give a high-valent metal hydroxide and a substrate radical, followed by oxygen rebound in which the substrate radical “combines” with the hydroxyl radical from the metal-hydroxide. Thus far, there are no examples in the biomimetic literature, in which this second step, involving oxygen rebound, has been examined directly via reaction of a high-valent metal-hydroxide complex with an organic radical to give an ROH product.
Oxygen (O2) activation is another common biomimetic reaction. This umbrella term encompasses O2 binding/transport (reversible O2 activation), the two-electron/two-proton reduction of O2 to hydrogen peroxide (H2O2), and the four-electron/four-proton reduction of O2 to water. The binding and activation of H2O2 is related to this process, and shares common metal-oxygen intermediates. The extent of O2 activation by a metal porphyrinoid complex is controlled by many different factors, including the metal identity, oxidation state, and ligand electronic properties, as well as any exogenous or ligand-appended hydrogen-bond donor groups in the secondary coordination sphere.
2. Metal-Oxo Porphyrinoid Complexes as Models for Biological Oxidations
2.1. Iron-Oxo Complexes
2.1.1. Iron-Oxo Porphyrins
Reactions carried out by iron-oxo porphyrins have received considerable attention because of their synthetic utility in the oxidation of organic substrates and their relevance to enzymatic heme-catalyzed processes such as those found in cytochrome P450, peroxidases, and catalases. The key reactive intermediates are postulated as high-valent iron-oxo porphyrin species defined in the biological context as Cpd I (FeIV(O)(porphyrin•+)), formally a FeV(O) complex, and Cpd II ((FeIV(O)(porphyrin)). The terms “compound I” and “compound II” were historically applied to the reactive ferryl species found in horseradish peroxidases (HRP), but are now used to describe isoelectronic iron-oxo porphyrin species in other heme enzymes.18 Studies of synthetic high-valent iron-oxo porphyrin complexes have provided valuable insights into the reactivity of these species because of the relative ease of control of the ligand steric and electronic properties, and reaction environment, as compared to heme enzymes. In 1979, Groves et al. demonstrated alkene epoxidation and alkane hydroxylation using iodosylbenzene (PhIO) with an FeIII(Cl)(TPP) (TPP = tetraphenylporphyrin) catalyst at room temperature.19 One of the proposed intermediates for this catalytic oxidation was an iron-oxo complex. An early analog of the proposed catalytic intermediate came from the oxidation of FeIII(Cl)(TMP) (TMP = tetramesitylporphyrin) with 1.5 equiv of mCPBA at −78 °C to form FeIV(O)(TMP•+), a porphyrin π-radical cation complex (Scheme 6).20 The large downfield shifts in the 1H NMR peaks of the mesityl groups and the UV-visible spectrum suggested a porphyrin π-radical cation. The small Mossbauer isomer shift of 0.05 mm/s (ΔEQ = 1.49 mm/s) was in the range expected for an iron(IV) species. This complex is also reactive with olefins to form epoxides even at low temperatures. When the oxidation reaction was performed in the presence of H218O, almost complete (99%) 18O incorporation in the epoxide was observed.
Scheme 6.
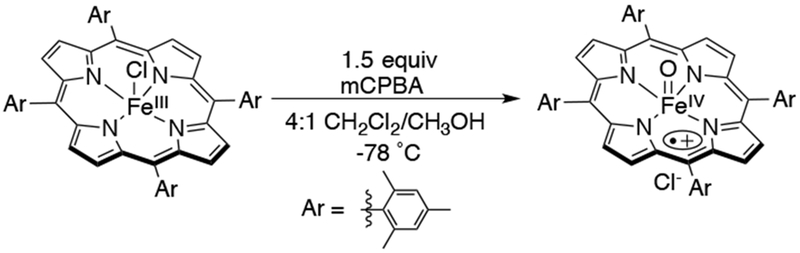
Synthesis of FeIV(O)(TMP•+), an Early Compound I Analog
After these initial reports, a number of studies on iron-oxo porphyrins were made in efforts to define possible mechanisms of substrate oxidation by heme enzymes. Excellent reviews on the reactivity of iron-oxo porphyrin complexes can be found in the literature, including those from Fujii (2000,21 200222, 201623) Meunier et al. (2004),24 Groves (2006),25 Newcomb (2006),26 Nam (2007),27 Costas (2011),28 Ray and Meyer (2012),29 and Rutkowska-Zbik (2014)30. Dedicated chapters in The Porphyrin Handbook (2000),18,31-34 Handbook of Porphyrin Science,35 and other compilations36-38 can also be found. This section will focus on recently published works in this area.
Although iron complexes with the tetrakis(meso-phenyl) porphyrin general structure led to some metastable iron-oxo complexes that mimic cytochrome P450 compound I reactivity, their catalytic activities were plagued with porphyrin decomposition and μ-oxo dimer formation. These problems were overcome by introducing alkyl, halogen, or other substituents to either the meso-aryl or the β-pyrrole positions of the porphyrin ring. The result was increased catalytic activity and stability of the porphyrin ligand towards decomposition.38,39 For example, Groves has reported high rates for C-H hydroxylation in aqueous solution for FeIV(O)(TMPyP•+) (TMPyP = tetrakis(N-methyl-4-pyridinium)porphyrin) (Scheme 7).40 Reaction of FeIV(O)(TMPyP•+) with xanthene gave the hydroxylation product, 9-xanthydrol (90%), and performing the reaction in the presence of 47.5 % H218O led to 21% 18O incorporation. This incorporation is consistent with an oxygen-rebound scenario, where a high-valent FeIV(O)(porphyrin•+) species abstracts an H-atom from xanthene to give a carbon radical, and an FeIV(OH) rebound intermediate. This species “rebounds” the •OH group to the carbon radical in the solvent cage to form the alcohol product and the resting ferric state. The rate constant for this reaction was found to be 3.6 × 106 M−1 s−1, with a modest kinetic isotope effect of 2.1. A variety of C-H hydroxylation substrates (fluorene-4-carboxylic acid, 4-isopropyl and 4-ethylbenzoic acid) were tested as well, and the hydroxylation rates correlate well with the substrate BDEs (C-H). The BDE(O-H) for FeIV(O-H)(TMPyP) was estimated from a Brønsted–Evans–Polanyi relationship to be ~100 kcal/mol, much larger than the values estimated for FeIV(O-H)(TMP) (92 kcal/mol),41 and FeIII(O-H)(TPFPP) (~86 kcal/mol)42. The high kinetic reactivity of this complex can be traced to the redox and basicity tuning of the iron-oxo porphyrin, expected from a porphyrin ligand with electronegative and positively charged substituents. It was further suggested that the increase in reactivity could be due to a spin-state crossing phenomena occurring in the course of the reaction.
Scheme 7.
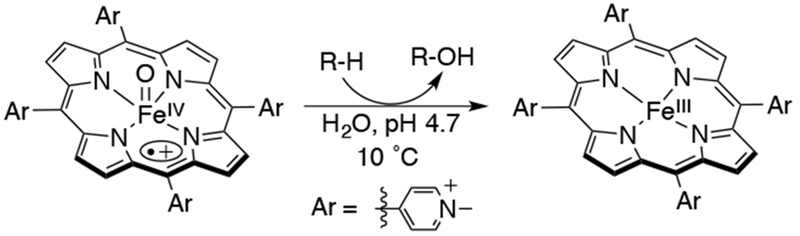
Reactivity of FeIV(O)(TMPyP•+), a Highly Reactive Compound I Model Complex
An alternative method of producing high valent iron-oxo species was reported by the groups of Zhang, Fung, and Newcomb.41 In this method, FeIII(ClO4)(TMP) was oxidized to the metastable FeIV(ClO4)2(TMP) which, when irradiated with 355 nm laser light, gave a new transient species which could be characterized only by UV-visible spectroscopy (Scheme 8). Preparative reaction of this transient iron-oxo species with cyclooctene gave cyclooctene epoxide in 28% yield. This species also reacts with alkanes and benzylic C-H substrates with rates up to 5 orders of magnitude larger than the cpd I analog FeIV(O)(TMP•+). For this reason, this species was speculated to be an FeV(O)(TMP) complex on the basis of its reactivity. Although this speculation is reasonable from kinetic data, this oxidation state assignment should remain tentative until appropriate spectroscopic evidence such as EPR, Mössbauer, or X-ray absorption spectroscopy is presented. These laser flash photolysis (FLP) studies were further extended to other porphyrin analogs, TPP (tetraphenylporphyrin) and OEP (octaethylporphyrin), and the order of reactivity was found to be OEP>TPP>TMP (Scheme 8). These putative transient iron-oxo species, characterized by their unique UV-visible spectra, exhibited similar rate constants to the similarly generated MnV(O) porphyrins. Rate constants for benzylic C-H bond cleavage correlated with the C-H BDEs, and Hammett correlations with para-substituted styrenes gave slopes between −0.5 and −0.7, reflecting the electrophilic character of these reactions.43
Scheme 8.
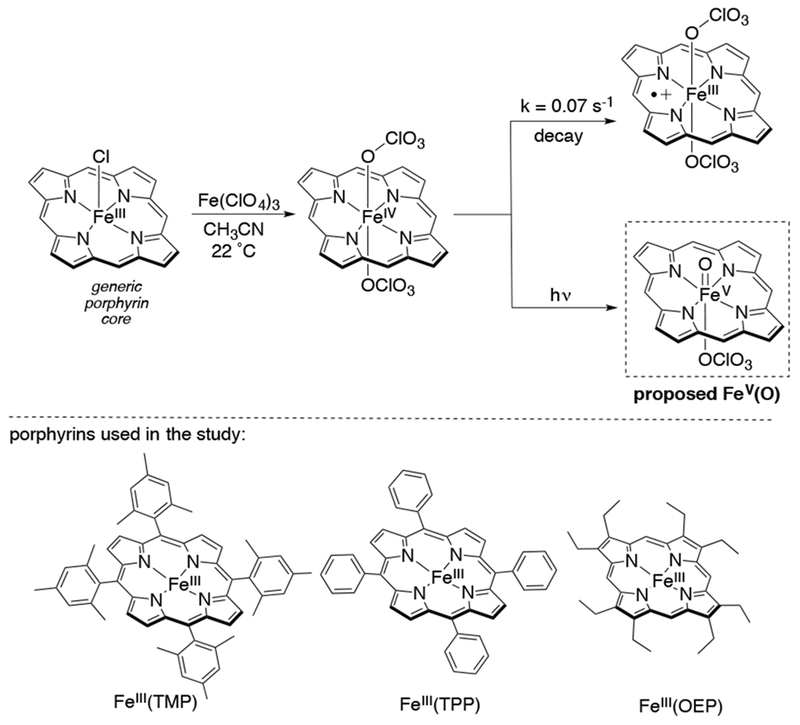
Synthesis of a Proposed FeV(O) Intermediate by Laser-Flash Photolysis, and Different Porphyrin Ligands Used in the Study
Zhang and coworkers recently reported a method of generating FeIV(O)(porphyrin•+) or FeIV(O)(porphyrin) (porphyrin = TMP, TPP, TPFPP) complexes by simple visible light irradiation of FeIII(OBrO2)(porphyrin) complex.44 The photolysis reaction is proposed to go through the heterolytic cleavage of the O-Br bond, forming a putative FeV(O) complex which then relaxes to an FeIV(O)(porphyrin•+). This was observed in electron-rich porphyrins such as TMP and TPP. With FeIII(TPFPP) (TPFPP = tetrakis(pentafluorophenyl)porphyrin), however, a final FeIV(O)(TPFPP) product was observed, which is proposed to result from the FeV(O)(TPFPP) comproportionating with residual FeIII(TPFPP). The formation of cpd I and cpd II analogs was further confirmed by comparable rate constants obtained in the HAT and OAT reactions of iron-oxo porphyrins generated using visible light generation vs addition of mCPBA. DFT studies show that an FeV(O) ground state is favored in vacuo. However, the FeIV(O)(porphyrin•+) configuration is favored in the solvent or protein environment.45
Newcomb and coworkers have shown that single-turnover hydroxylation reactions of ethyl benzene and benzyl alcohol with FeIV(O)(TMP•+), generated from the addition of m-CPBA to FeIII(TMP) in CH3CN, give large temperature-dependent KIEs, which are ascribed to H-atom tunneling.46 The substrates were oxidized in the benzylic C-H position, to give 1-phenylethanol and benzaldehyde respectively, as determined by gas chromatography (GC).
It has been shown that axial ligands of heme enzymes also play an important role in their reactivity. In work from Fujii and coworkers, the factors that affect H-tunneling processes were explored by studying the C-H hydroxylations of xanthene and tetralin with FeIV(O)(TMP•+)(L) with different axial ligands (L = NO3−, Cl−, imidazole) (Scheme 9).47 The FeIV(O)(TMP•+)(L) complex was generated with excess O3 (ozone) in CH2Cl2 at low temperatures (−20 to −95 °C). Non-linear Arrhenius plots were observed, providing strong evidence for the participation of H-tunneling in the rate-limiting H-transfer process. In addition, the authors showed that H-tunneling is controlled by temperature, the BDE of the C-H substrate, and the reactivity and Fe-O bond strength of the FeIV(O)(TMP•+)(L) complex.
Scheme 9.
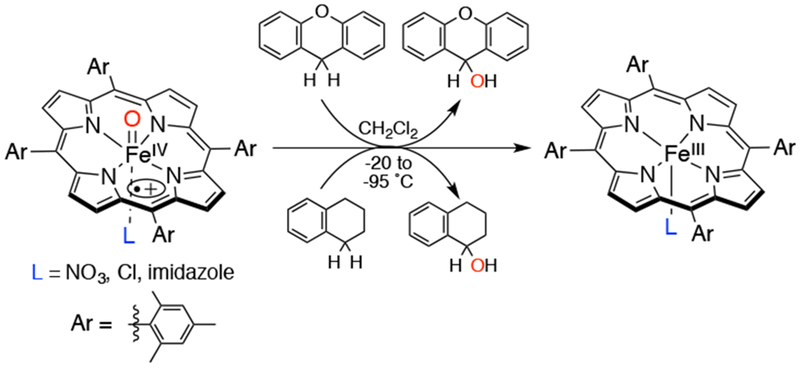
Reactivity of a Series of FeIV(O)(TMP•+)(L) Complexes with Benzylic C-H Substrates Xanthene and Tetralin
The strong electron-donation from the cysteinyl thiolate ligand in cytochrome P450s is proposed to influence the driving force for C-H bond cleavage.48 Efforts have been made to synthesize thiolate-ligated heme models with the goal of studying the role of thiolate ligation on reactivity, and these are summarized in a review.49 The facile oxidation of thiolate ligands to disulfide remains an outstanding difficulty in the synthesis of thiolate-ligated heme models, and this is usually prevented by using a large excess of the thiolate ligand or by covalently attaching the thiolate group to the porphyrin scaffold.38,49-51 A stable thiolate-ligated complex (called SR complex, Swan Resting) was prepared by Higuchi and co-workers by covalently attaching an alkane-thiolate group to the porphyrin, and bulky pivaloyl groups were added to protect the sulfur atom from oxidation, and amide NH groups for hydrogen-bonding with the sulfur atom (Scheme 10).52 However, although the SR complex is known to react with oxygen atom donors and peroxyacids, it does not form a stable dioxygen adduct due to the absence of a binding pocket. In addition to the instability of thiolate-ligated heme-dioxygen adducts, the increasingly complicated synthesis of these model complexes has limited the number of examples of these systems in the literature.
Scheme 10.
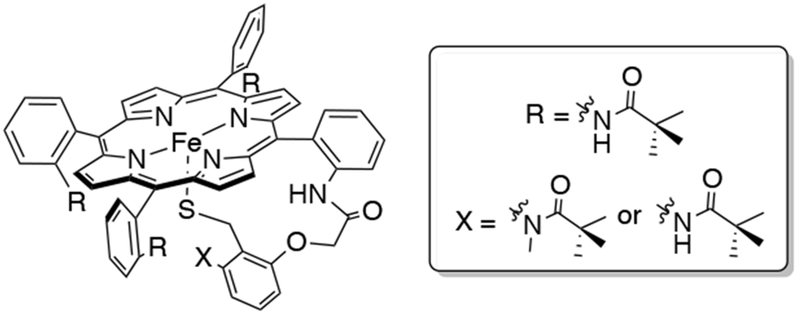
Swan Resting (SR) Complex, an Example of a Thiolate-Ligated Heme Model Complex, and its Derivatives
Substitution of a thiolate ligand (RS−) with a sulfonate (RSO3−) ligand reduces the charge density at the metal and tunes the FeIII/II redox couple to a more positive potential close to that found in P450cam.53 Woggon and van Eldik showed that the cpd I analog FeIV(O)(porphyrin•+)(RSO3−) can be generated with m-CPBA under saturation kinetics, which suggests the formation of an acyl-peroxo adduct intermediate, eventually converting to the cpd I analog (Scheme 11).54 There has been speculation that an FeIII(OOH)(porphyrin) complex (compound 0), the precursor to compound I, can act as a second oxidant for certain substrates in cytochrome P450. Thus, the generation of an FeIII-acylperoxo complex with m-CPBA can be utilized as a model to study the possible involvement of an FeIII-OOH species in heme enzyme oxidations.
Scheme 11.
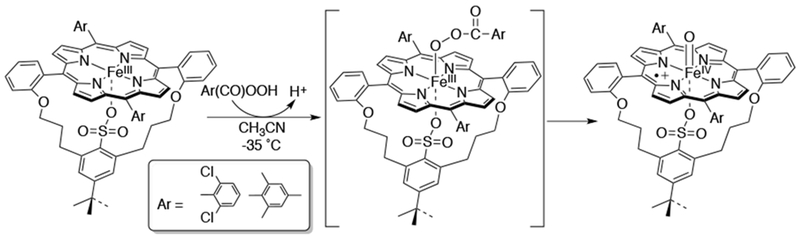
Conversion of an RSO3-ligated FeIII(acylperoxo)(porphyrin) Intermediate into a Cpd I Analog FeIV(O)(RSO3)(porpyrin•+)
Nam and coworkers have shown that FeIV(O)(TMP•+) is the active oxidant in olefin epoxidation with cyclohexene, aromatic ring oxidation with anthracene, C–H bond activation with dihydroanthracene, and alcohol oxidation with benzyl alcohol in CH3CN/CH2Cl2 (1:1 v/v) at −40 °C. These results contrast with the poor oxidizing ability of the FeIII(m-CPBA) adduct of this complex.55 Similarly, van Eldik and coworkers reported kinetic evidence showing that oxygen atom transfer for FeIII(m-CPBA)(TMP) was orders of magnitude slower than the FeIV(O)(TMP•+) complex in epoxidation and sulfoxidation reactions.56
Heme synthetic models with axial ligands covalently tethered to the porphyrin ligand can provide insights into effects of axial ligation on reactivity. However, the synthesis of these tethered systems are usually difficult and low-yielding. An alternative approach is by adding exogenous ligands to the cpd I and cpd II analogs. Large increases in cyclooctene epoxidation rates were observed upon addition of axial ligands to the FeIV(O)(porph•+) (porph = TMP, TMTMP) (TMTMP = 2,7,12,17-tetramethyl-3,8,13,18-tetramesitylporphyrin) complex, and the epoxidation rates increase in the order of L = NO3− < Cl− < 3-fluoro-4-nitrophenolate<imidazole.57 Interestingly, the measured redox potentials for FeIV(O)(porph)(L)/FeIV(O)(porph•+)(L) follow the opposite trend, i.e., a positive shift was observed with anionic axial ligands (more oxidizing) and a negative shift was observed with neutral ligands (less oxidizing).58 The reactivity of FeIV(O)(L)(porphyrin•+) correlates instead with the FeIII/FeII redox potential of the reaction product, FeIII(L)(porphyrin), suggesting that axial ligands affect the reactivity of FeIV(O)(porphyrin•+) complexes by stabilizing the reaction product.59 In a separate study, the reactivity of FeIV(O)(TMP•+) in olefin epoxidation, aromatic hydroxylation, alcohol oxidation and alkane hydroxylation was measured in the presence of para-substituted pyridine N-oxide derivatives (para-Y-C5H5NO, Y = -OCH3, -CH3 -H, -Cl) as axial donors. It was found that the binding of these axial ligands lead to enhanced reactivities in all of the oxidation reactions.60 The most electron-donating pyridine N-oxide (p-OCH3-C5H5NO) gives the most reactive FeIV(O)(TMP•+)(L) complex in both O-atom transfer and H-atom transfer reactions. This increased reactivity was attributed to a stronger Fe(O-H) bond formed in HAT reactions, and a weaker reactant Fe=O bond broken in OAT reactions.
DFT calculations on the effects of various anionic axial ligands on aromatic hydroxylation with FeIV(O)(porphyrin•+) have shown that stronger axial ligands give rise to higher pKas of the corresponding FeIV(OH)(porphyrin) complex, while electronic effects are less affected. Moreover, the barrier for aromatic hydroxylation appear to be proportional to the strength of the O-H bond formed in the FeIV(OH) complex.61 A similar trend was observed in alkane hydroxylation,62 with the strength of the C-H bond of the substrate also correlating with the reaction barrier. For sulfoxidation63 and alkene epoxidation64,65 reactions however, the barrier height correlates with the ionization potential of the substrate, supporting an ET rate-determining step in the oxidation process. Calculations were also performed to determine the source of regioselectivity in the oxidation of ethylbenzene by FeIV(O)(porphyrin•+) to form 1-phenylethanol (alkane hydroxylation product) and p-ethylphenol (aromatic hydroxylation product). The calculations indicate that an axial acetonitrile ligand favors aromatic hydroxylation, whereas a Cl− axial ligand favors alkane hydroxylation.66 The anionic ligand, such as Cl−, makes the oxidant less electrophilic due to orbital mixing which disfavors the aromatic hydroxylation mechanism and favors the alternative alkane hydroxylation mechanism.
A high-valent terminal iron-oxo species generated from a nitrido-bridged-diiron porphyrin complex was trapped and characterized, and was found to oxidize C-H bonds as strong as that of methane (BDE = 104 kcal/mol).67 Addition of m-CPBA to [(TPP)FeIII(N)FeIV(TPP)] in CH2Cl2 at −80 °C results in formation of a peroxo complex which undergoes heterolytic cleavage to form the diiron, terminal oxo species [(TPP)(m-CBA)FeIV(N)FeIV(O)(TPP•+)]−, characterized by UV-vis, ESI-MS, EPR and Mössbauer spectroscopies (Scheme 12 and Figure 1). This species was found to have superior oxidizing properties towards alkanes when compared with the model complex FeIV(O)(TPP•+), and KIEs of about 3 support a rate-limiting C-H cleavage. To avoid uncertainties as a result of the possible oxidation of organic solvent by strongly oxidizing species, heterogeneous oxidation of CH4 was performed starting with the [(TPP)FeIII(N)FeIV(TPP)] catalyst combined with silica and dried at 50 °C. The supported catalyst, m-CPBA and methane gas were added in an autoclave reactor heated to 60 °C. Methane was oxidized to HCOOH (formic acid) from GC and LC analyses. This report showed the enhancement of C-H bond oxidations induced by an N-bridging ligand on an iron-oxo complex.
Scheme 12.
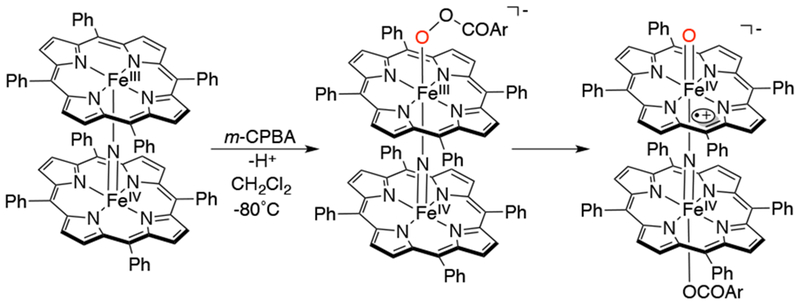
Proposed Mechanism for the Formation of [(TPP)(m-CBA)FeIV(N)FeIV(O)(TPP•+)]−
Figure 1.
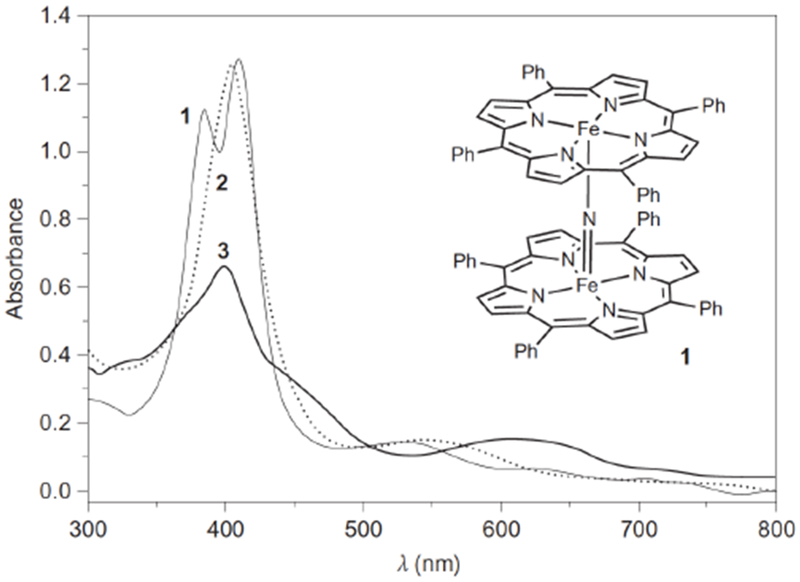
UV-vis spectral changes from the starting [(TPP)FeIII(N)FeIV(TPP)] (1), formation of the mcpba adduct (2), and [(TPP)(m-CBA)FeIV(N)FeIV(O)(TPP•+)]− (3) in CH2Cl2 at −80 °C. Reprinted by permission from Macmillan Publishers Ltd: Nature Chemistry, Nature Chemistry 4, 1024–1029 (2012), copyright 2012.
Chloroperoxidase (CPO) and myeloperoxidase (MPO) are heme enzymes that catalyze the oxidation of Cl− with H2O2, and are responsible for the biosynthesis of chlorinated compounds in microorganisms.68,69 Studies have suggested that a cpd I intermediate oxidizes Cl− to form a transient FeIII(OCl) species. Studies from the Fujii group have shown different ways of accessing FeIII(OCl) species from an FeIII(TPFP) complex (Scheme 13). Addition of Cl− ions to a solution of FeIV(O)(tpfp•+) at −90 °C leads to reduction of the Fe complex, giving FeIV(O)(tpfp). The Cl− ion is proposed to be oxidized to chlorine radical (Cl•).70 A study from Fujii58 showed that the redox couple for FeIV(O)(tpfp)(ClO4−)+/0 is 1.39 V vs SCE in dichloromethane at −60 °C. This finding indicates that the iron-oxo porphyrin complex should be capable of oxidizing chloride ion (1.11 V vs SCE in H2O).71 Single turn-over chlorination reactions with organic substrates led to modest yields of chlorinated products. In an alternative route, protonation of FeIV(O)(TPFP•+) with TFA results in the tautomerization of this complex to FeIII(L’)(TPFP++) (L’ = anion other than Cl−), and further addition of Cl− ions lead to conversion to FeIII(L’)(Cl)(TPFP-Cl), a meso-chloro-isoporphyrin.72 This complex can chlorinate electron-rich benzene and alkene derivatives catalytically through an electrophilic aromatic substitution (for benzene) or electrophilic addition mechanism (for alkenes). An FeIII(OCl) species was independently formed by addition of 4 equiv of OCl− to FeIII(OH)(TPFP) in 1:1 CH2Cl2-CH3CN at −60°C.73 Based on spectroscopic data, the species was characterized as FeIII(OCl)2(TPFP)−, which eventually decomposed to FeIV(O)(TPFP) (Scheme 13). Imidazole derivatives of this complex were also synthesized. The O-atom transfer and chlorination reactivity of the FeIII(OCl) complexes were tested with thioanisole, cyclohexene and 1,3,5-trimethoxybenzene substrates. The imidazole derivatives were found to have attenuated reactivity compared to the bis-hypochlorite analog. These biomimetic studies find their importance in comparison with CPO/MPO heme enzymes, where FeIII(OCl) species are indicated, but spectroscopic evidence for such species is scarce.
Scheme 13.
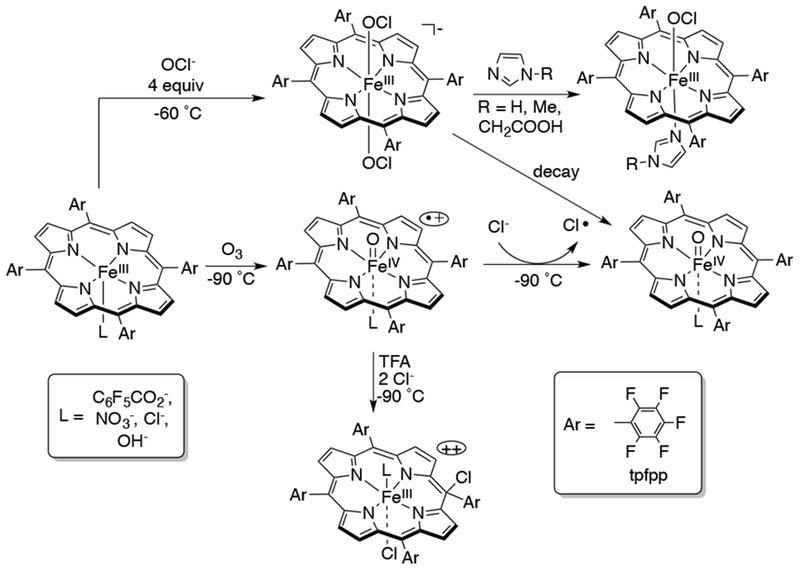
Generation of FeIII(OCl)2(porph)−, FeIV(O)(porph•+), and FeIV(O)(porph) Species and Their Interconversions
The last step in perchlorate (ClO4−) degradation by microbes is the decomposition of chlorite (ClO2−) to dioxygen (O2) and chloride (Cl−) mediated by the heme enzyme chlorite dismutase. From stopped-flow and EPR experiments, the mechanism was proposed to go through a cpd I intermediate.74,75 This process was mimicked outside of a protein with water-soluble porphyrins, where chlorite is decomposed to chloride and chlorate in a 1:2 stoichiometry (Scheme 14).76,77 Based on experimental observations and kinetic studies, two competing cycles were proposed. The first FeIV(O)P•+/FeIII cycle produces Cl− and O2, as in the heme enzyme. However, the FeIV(O)P•+ can also comproportionate with FeIII to form FeIV(O)P. The second competing cycle involves an O-atom transfer from FeIV(O)P to ClO2− to form FeII (P) and ClO3−.
Scheme 14.
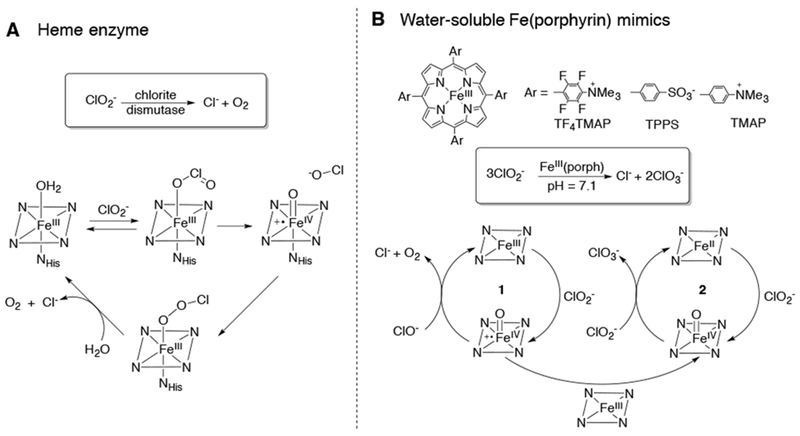
Proposed Mechanisms for Chlorite Dismutase and Water-Soluble Porphyrins
Aromatic hydroxylation is another reaction studied extensively with iron porphyrin models. Its mechanism differs from alkane hydroxylation in that small KIEs and 1,2-hydride shifts (NIH shift) are observed, precluding the usual oxygen rebound mechanism proposed for alkane hydroxylations. Mechanistic information on aromatic hydroxylation of a series of para-substituted benzene derivatives with FeIV(O)(TPFPP•+) was obtained by the groups of Jang and Nam.78 A large negative Hammett slope (ρ) of −8.0 and a KIE value of ~0.8 were observed, consistent with the formation of a cationic σ adduct upon addition of an electrophilic iron-oxo species. Fujii and coworkers studied the ability of a series of FeIV(O)(porphyrin•+) with electron-withdrawing porphyrins to hydroxylate aromatic substrates (benzene, anisole, naphthalene), and were able to show the involvement of an electron transfer process in these reactions.79 Plots of (RT/F) ln(k2) vs ΔE1/2 (where k2 is the second-order rate constant of aromatic hydroxylation and E1/2 is the redox potential for the cpd I/II couple) were constructed, and revealed slopes that are in between 0.5 and 1.0, indicating that the electron transfer process is coupled to the subsequent O–C bond formation (instead of proton transfer, which is not found to be in the rate-limiting step). Direct electron transfer between the substrate and FeIV(O)(porphyrin•+) can be ruled out because the reaction is highly endergonic (ΔGET >> 0). However, this uphill electron transfer step is followed by a downhill O-C bond formation, making the over-all process thermodynamically favorable. The proposed mechanism (Scheme 15) brings together and reasonably explains the previously observed results.
Scheme 15.
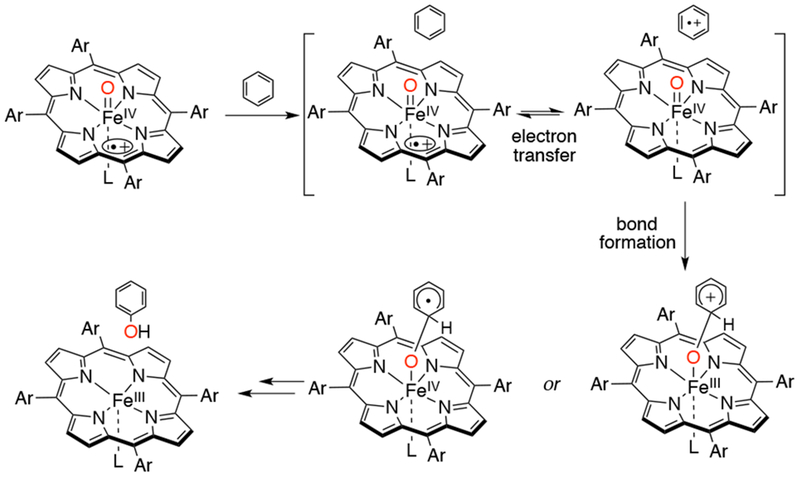
Proposed Mechanism of Aromatic Hydroxylation with FeIV(O)(porphyrin•+) Complexes Showing an Electron Transfer Process Coupled to the Subsequent Bond Formation Step
N-oxygenation and N-dealkylation reactions are some of the many oxidations performed by compound I. The mechanism for N-dealkylation is proposed to go through either an H-atom transfer mechanism or a rate-limiting electron transfer step coupled with a proton transfer step (Scheme 16). DFT calculations showed that both N-oxygenation and N-dealkylation proceed from the low spin state of compound I, and the computed KIEs for the rate-determining HAT step fits well with experimental data.80
Scheme 16.
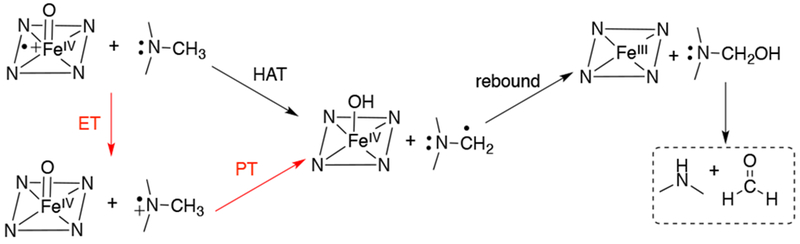
Proposed Mechanisms for Oxidative N-demethylation by Compound I, FeIV(O)(porphyrin•+)
Aside from exogenous substrates, the high-valent iron porphyrin complex is also capable of performing heme degradation reactions as observed in the enzyme heme oxygenase. One of the proposed reaction pathways for the first step of this reaction is the formation of a transient cpd-I (FeIV(O)(porphyrin•+)), through the heterolytic cleavage of an FeIII(OOH)(porphyrin) species. Karlin and co-workers have described a heme oxygenase model system in which an iron(III) tetraarylporphyrin complex is selectively oxidized to the final verdoheme-like product using a sacrificial oxidant, cerium(IV) ammonium nitrate (CAN), in the presence of water (Scheme 17).81 A combination of UV-vis, EPR, and ESI-MS observations provided evidence for the formation of 1) an isoporphyrin complex resulting from the attack of water/hydroxide on a cpd-I analog, 2) a porphyrin degradation product resembling benzoyl biliverdin, and 3) a verdoheme-like product as well as a benzoyl fragment. From 18O labeling experiments, water was shown to be the O-atom source in the final oxidized products. In addition, the intermediacy of a cpd-II-like complex (FeIV(O)(porphyrin)) was ruled out, as no further oxidation with this complex was detected, while direct observation of the conversion of cpd-I (generated at −80 °C) to the isoporphyrin complex was observed.
Scheme 17.
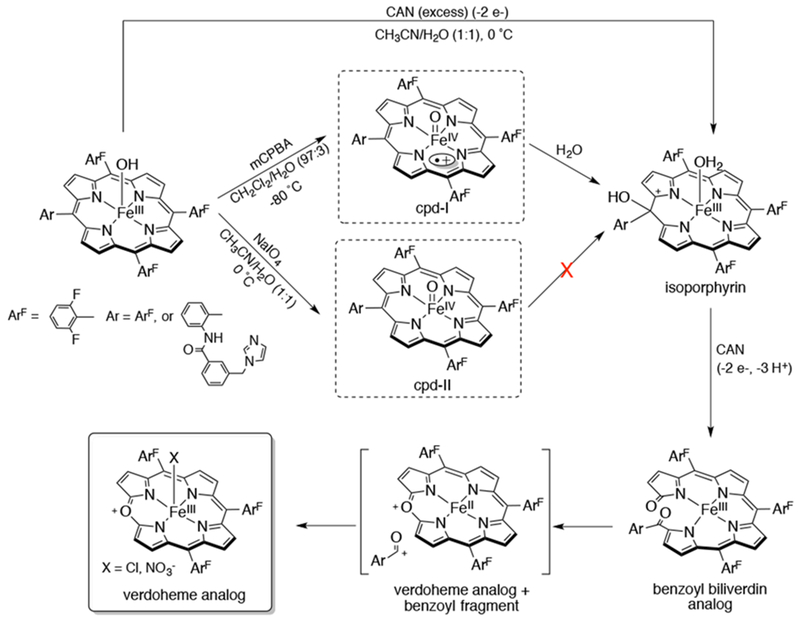
Proposed Mechanism for the Selective Oxidation of FeIII(OH)(porphyrin) with CAN to a Verdoheme-like Complex via Formation of Intermediates Resembling Cpd-I, Isoporphyrin, and Benzoyl Biliverdin
Iron(IV)-oxo porphyrins, one-electron reduced analogs of iron(IV)-oxo porphyrin π-radical cation complexes, were first synthesized in 1980 by Balch and La Mar by the addition of nitrogen bases (N-methylimidazole, pyridine, piperidine) to FeIII peroxo-bridged porphyrins at −80 °C (Scheme 18).82,83 The FeIV(O) complex is EPR silent and shows paramagnetic proton resonances, while its magnetic susceptibility and UV-visible absorption spectrum resembled that of compound II horseradish peroxidase.
Scheme 18.
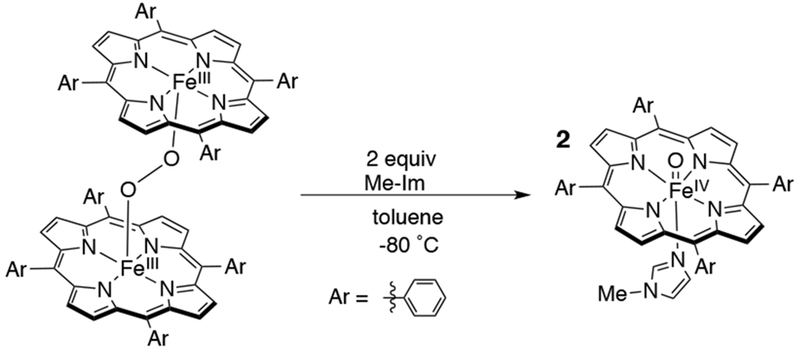
Synthesis of FeIV(O)(TPP), an Early Compound II Analog
Other than homolytic cleavage of the O-O bond of FeIII peroxo-bridged porphyrins, iron(IV)-oxo porphyrins can be prepared from the starting FeIII porphyrin by a) chemical oxidation with mCPBA or PhIO under certain conditions, b) electrochemical oxidation and c) addition of hydroperoxides (see ref. 38 and related references therein). Iron(IV)-oxo porphyrins were initially considered as poor oxidants.83 However, it has been shown that iron(IV)-oxo porphyrins react with substituted styrenes to give epoxide, although with poor selectivity towards cis vs trans epoxide formation.84 Nam and coworkers reported the reactivity of FeIV(O)(porphyrin) complexes with benzylic C-H substrates and NADH analogues to give the corresponding oxidized products (Scheme 19).42 A linear correlation is observed between the rate constant log k2’ and the C-H substrate BDEs, in addition to large KIEs of up to 21 for DHA and xanthene, providing evidence for an H-atom abstraction mechanism. Among the three porphyrins tested, there is a modest increase in HAT and hydride transfer reaction rates with the more electron-deficient porphyrins.
Scheme 19.
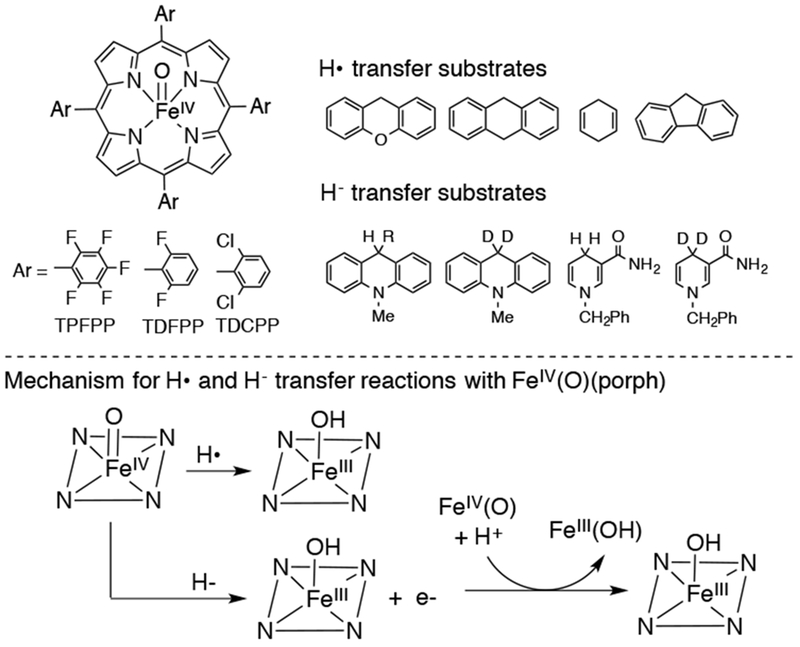
Reactivity of a Series of Iron(IV)-Oxo Porphyrins with H-atom and Hydride Donors
A DFT study on the comparative reactivity of FeIV(O)(porphyrin•+) and FeIV(O)(porphyrin) with 10-methyl-9,10-dihydroacridine show that FeIV(O)(porphyrin•+) reacts with a much lower barrier.85 The main difference, however, is that FeIV(O)(porphyrin) reacts via hydrogen atom transfer, while FeIV(O)(porphyrin•+) reacts via hydride transfer. This difference in mechanism was rationalized to be a result of differences in electron abstraction abilities of these two oxidants.
The ability to selectively produce cpd 0, I, and II with FeIII(TMP) under different conditions was exploited by van Eldik and coworkers to compare directly their reactivity towards O-atom transfer, H-atom transfer, and hydride transfer reactions at low temperatures.86 As expected, the FeIV(O)(TMP•+) (cpd I) complex turned out to be the most reactive species. Interestingly, for hydride transfer reactions, the reactivity order was found to be cpd II > cpd 0 > cpd I. In the initial electron-transfer step, Cpd II is reduced to an iron (III)–oxo species, which would be the most basic intermediate and would therefore promote the subsequent proton abstraction step. A similar study from the same group on the comparison of reactivity of FeIV(O)(TMP•+)(2-MeIm) and FeIV(O)(TMP)(2-MeIm) also confirmed the high reactivity of the cpd I analog towards epoxidation, H-atom abstraction, and heteroatom oxidation.87 Compound I and II mimics were also stabilized in aqueous solution using the water soluble porphyrin FeIII(TMPS), and their formation was found to be pH-dependent.88 Combined temperature and pressure measurements have shown that, for selected substrates, oxidation with the Cpd II mimic is an enthalpy-controlled process whereas it is entropy-controlled for Cpd I.
Iron(IV)-oxo porphyrins were also shown to carry out oxidative N-dealkylation of N,N-dialkylanilines via a rate-limiting electron transfer coupled with proton transfer (ET-PT.89 The large negative slopes obtained from the plots of log kobs vs Hammett parameter σ (ρ = −3.3) and E°ox of para-substituted N,N-dimethylanilines (slope = −5.0) suggest a rate-limiting electron transfer. In addition, the use of several mechanistic probes and a modest inter- and intramolecular KIE of 2.8 and 4.2 all pointed to an ET-PT mechanism. An FeIV(O) porphyrin complexed with a per-o-methylated ß-cyclodextrin dimer was found to oxidize ROCH3 to ROCH2OH, which further decomposes to ROH and HCHO, and the corresponding FeII(porphyrin) complex (Scheme 20).90 An initial H-atom abstraction followed by an oxygen rebound mechanism similar to compound I has been proposed for this system. Work from the same group showed that an FeIV(O) porphyrin complexed to a per-o-methylated ß-cyclodextrin dimer was capable of a direct oxygen transfer reaction to the sulfide bond found intramolecularly on the linker.91
Scheme 20.
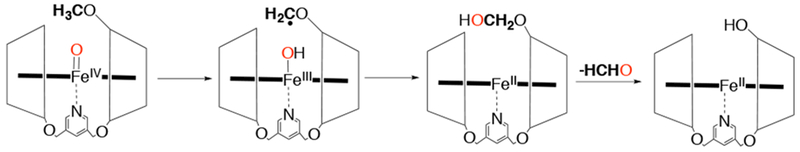
Proposed Mechanism for Intramolecular O-demethylation Mediated by an FeIV(O) Complex Encapsulated in a Per-O-methylated Cyclodextrin Dimer
As shown previously, there have been several instances of iron(IV)-oxo porphyrin complexes acting as competent oxidants for various substrates. A DFT electronic structure analysis of alkane hydroxylation with FeIV(O)(porph) and its hydrosulfide-ligated analog was performed, with methane as a model substrate for alkanes. DFT calculations on the ground triplet and excited quintet spin state surfaces revealed that H-atom abstraction is the rate-determining step, and a two-state reactivity (TSR) mechanism is plausible for C-H activation with FeIV(O)(porph), but not for the [FeIV(O)(porph)(SH)]− analog, where C-H activation can only occur on the triplet surface.92
It was shown by pH-jump stopped-flow UV-vis spectroscopy that iron(IV)-oxo porphyrins can be protonated twice to form the iron(III)-aquo porphyrin•+ valence tautomer (Scheme 21).93 The two-proton pKa values of sulfonated, water-soluble FeIV(O) porphyrins were identified and it was found that the most basic FeIV(O) complex had the largest C-H bond cleavage rates. For FeIV(O)(TMPS•+), a KIE of 1.80 was observed with xanthene-d2, and a solvent KIE of 1.65 was seen with D2O, giving a combined substrate-solvent isotope effect of 2.2. A mechanism which involves both the substrate proton and the solvent proton was proposed which accounts for all of the observed data (Scheme 21).
Scheme 21.

A) Proposed Mechanism for Solvent-Proton Assisted C-H Bond Cleavage by a Sulfonated, Water-soluble Iron(IV)-Oxo Porphyrin π-Radical Cation Complex in the Presence of an Exogenous Proton Source B) Valence Tautomer Equilibrium Observed upon Protonation of Iron(IV)-Oxo Porphyrins C) Water-soluble Porphyrins Used in the Study
2.1.2. Iron-Oxo Corroles and Corrolazines
The ring contracted porphyrinoid compounds corroles and corrolazines are designed to stabilize high-valent transition metals. Although a number of high-valent complexes have been prepared with these porphyrinoid ligands, the iron-oxo corroles and corrolazines are rare and less studied than the iron-oxo porphyrins.94 The synthesis of well-characterized, high-valent, mononuclear iron-oxo corroles is challenging in part because of the propensity of iron corroles to form μ-oxo dimers upon oxidation of Fe(III) corrole.95-97 In addition, the inherent non-innocence in iron(IV) corroles obscures their electronic structure assignment, with both FeIV(corrole) (S = 1) and FeIII(corrole•+) (S = 1) states as limiting possibilities.98,99
The use of laser flash photolysis (LFP) – induced ligand cleavage of MnIV(OX) (X = ClO2, NO2) corroles to generate MnV(O) corroles was extended by Newcomb and Zhang to the analogous FeIV(OX) (X = ClO2, NO2 or FeIV) corroles100-102 to generate a proposed FeV(O) corrole (Scheme 22). This species was formulated as FeV(O), as opposed to FeIV(O)(corrole•+), because of its increased reactivity compared to the analogous FeIV(O)(porphyrin•+). However, the inability to characterize this highly reactive complex by methods other than UV-vis spectroscopy has precluded definitive oxidation state assignment. The proposed species shows an intense Soret band that decays after 20 ms at 22 °C, which is similarly observed from the reaction of FeIII corrole with excess mCPBA, and displays high oxidative reactivity towards alkenes (cyclohexene, cyclooctene). A KIE of 3.6 for the oxidation reaction of ethylbenzene was also observed, suggesting that C-H bond cleavage is the rate-determining step.
Scheme 22.
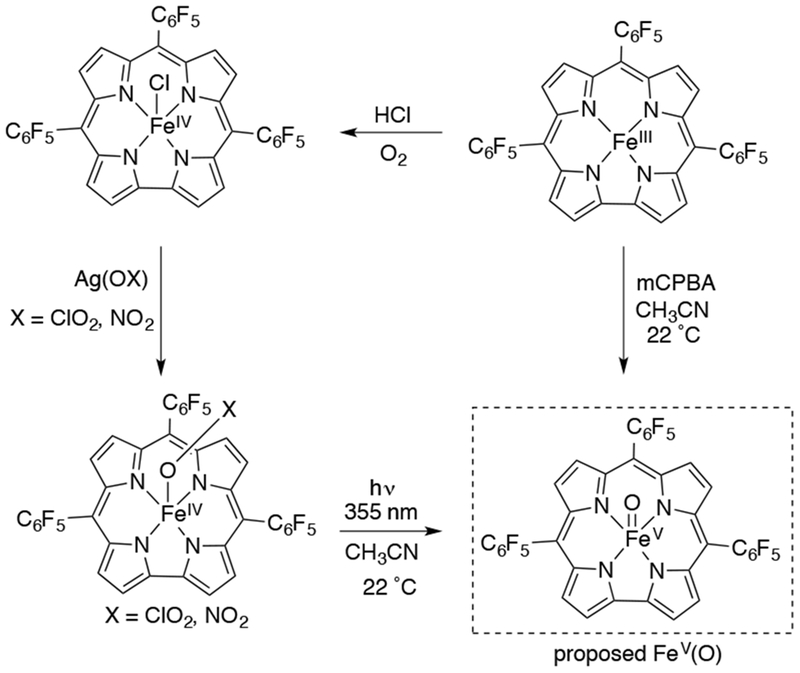
Formation of a Proposed FeV(O) Corrole
The isoelectronic FeIV(O)(corrole•+) has been proposed to be energetically accessible based on calculations,103 which also found the FeV(O)(corrole) complex to be equienergetic. Neither of these species have been experimentally observed. However, more recent DFT calculations from Valentine, Nam, and de Visser104 find that the FeIV(O)(corrole•+) valence tautomer is lower in energy than the FeV(O)(corrole). The H-atom abstraction reactivity of this complex towards a broad range of C-H bond substrates was explored by DFT calculations, and it was shown to react via a single, dominant low spin surface. These results are in stark contrast with the FeIV(O)(porphyrin•+) analog, which is computationally predicted to react via a two-state reactivity surface.
Iron(IV)-oxo corroles, which are compound II analogs, are proposed as an intermediate in the catalytic decomposition of peroxynitrite (ONOO−) with water-soluble sulfonated FeIII corroles.105 An FeIII(corrole)-mediated O-O bond cleavage of peroxynitrite forms nitrogen dioxide (N2O) and a hydroxo or oxo-iron(IV) corrole, which was found to decay via multiple pathways of similar rates of about 106 M−1 s−1 (Scheme 23). However, this highly reactive species has only been observed with stopped-flow UV-vis measurements. FeIII corroles are also shown to catalyze the oxidation of hydrocarbons with t-butylhydroperoxide,106 and xanthene-modified and hangman FeIV(Cl) corroles are shown to catalyze the disproportionation of hydrogen peroxide via the catalase reaction.107,108 In both cases, the formation of an FeIV(O) corrole intermediate is invoked, but no direct evidence was provided.
Scheme 23.
Proposed Mechanism for the Catalytic Decomposition of Peroxynitrite with a Water-soluble FeIII Corrole, Showing an [FeIV(O)(corrole)]− Intermediate
In 2009, Goldberg and coworkers reported the synthesis and the first direct spectroscopic observation of a compound I analog in a corrole-like scaffold, an FeIV(O)(TBP8Cz•+) (TBP8Cz = octakis(tert-butylphenyl)corrolazine) complex.109 The FeIII precursor complex was synthesized by reaction of metal-free corrolazine with Fe(acac)3 in refluxing pyridine,110,111 to give FeIII(TBP8Cz). Addition of an O-atom transfer reagent (mCPBA, PFIB, or PhIO) to the FeIII precursor complex in 1:1 CH2Cl2/CH3OH at −78 °C resulted in the formation of FeIV (O)(TBP8Cz•+) (Scheme 24). The FeIV(O)(TBP8Cz•+) complex (Figure 2) has been invoked as the key oxidizing species in the catalytic oxidation of olefinic and benzylic C-H substrates with PFIB as oxidant and FeIII(TBP8Cz) as catalyst.109
Scheme 24.

Synthesis of FeIII(TBP8Cz) and FeIV(O)(TBP8Cz•+)
Figure 2.
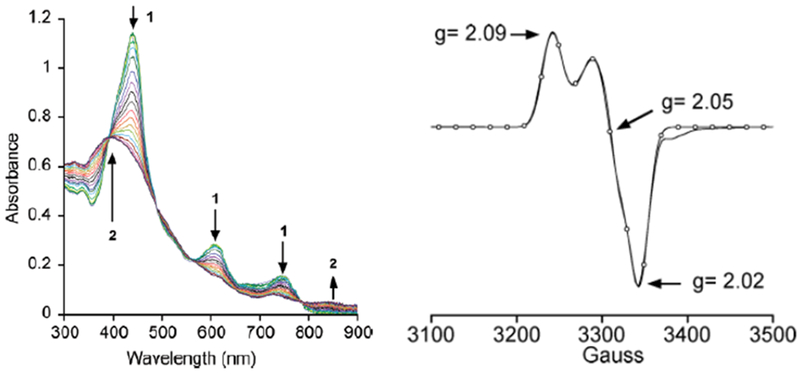
Left: Time-resolved UV-vis spectrum upon addition of PFIB to FeIII(TBP8Cz) in CH2Cl2/CH3OH at −78 °C. Right: X-band EPR spectrum of FeIV(O)(TBP8Cz•+) at 15 K. Reprinted with permission from J. Am. Chem. Soc., 2009, 131 (23), pp 8040–8048. Copyright 2009 American Chemical Society.
EPR110 and X-ray absorption spectroscopy (XAS) measurements112 of this iron-oxo corrolazine led to an assignment of an S = 1 FeIV metal center antiferromagnetically coupled to an S = ½ corrolazine π-radical cation, giving a total spin ground state of S = 1/2. Best fits to the EXAFS data showed a short Fe–O bond distance of 1.64 Å, in excellent agreement with the FeIV(O) assignment. The FeIV(O)(TBP8Cz•+) complex is stable at −78 °C for hours, but reverts to FeIII(TBP8Cz) at room temperature. This iron-oxo corrolazine complex is capable of O-atom transfer, H-atom abstraction, and electron transfer (Scheme 1).
Addition of PPh3 to this complex at −78 °C converts the species back to the starting FeIII complex. The ability of FeIV(O)(TBP8Cz•+) to cleave activated C-H bonds was studied.112 A KIE of 5.7 was obtained for xanthene, and a linear correlation between the rate constants (log k) and BDE(C-H) of the C-H substrates show that these reactions occur via a rate-determining H-atom transfer. However, the putative FeIV(OH) intermediate was not observed. A temperature dependence on the product distribution was observed for the reaction between FeIV(O)(TBP8Cz•+) and xanthene, where the oxygen-rebound product (xanthydrol) is favored at lower temperatures (−25 °C) over the radical dimerization (9,9’bixanthene) product. Thus, lowering the reaction temperature increases the efficiency of oxygen rebound.
2.2. Manganese-Oxo Complexes
2.2.1. Manganese-Oxo Porphyrins
Iron-oxo intermediates are found in the catalytic cycles of heme enzymes, and Mn has been used as an analog of Fe in models of these systems in part due to the enhanced stability of Mn-oxo species over the corresponding Fe-oxo species. While Mn(porphyrins) have yet to be identified as natural cofactors in metalloenzymes, synthetic manganese porphyrins have been used as catalysts in the biomimetic oxidation of organic substrates. These reactions usually proceed through a high-valent metal-oxo intermediate in mechanisms similar to those proposed for heme enzymes. Manganese-oxo porphyrins have been identified in the MnIV-oxo and MnV-oxo states. An early example of a well-characterized manganese(IV)-oxo porphyrin complex was reported in 1995, in which the MnIV(O) complex was synthesized from oxidation of the Mn(III)porphyrin with potassium peroxycarbonate, and structurally characterized by XAS.113 Typically, MnIV(O) porphyrin species are less reactive than their MnV(O) counterparts.31 Spectroscopic characterization of a MnV(O) porphyrin species remained elusive until relatively recently.114,115 The complex (MnV(O)(TMPyP) (TMPyP = meso-tetrakis(4-methylpyridinium)porphyrinato2−)) was generated in aqueous solution at 25 °C and characterized by UV-vis and 1H NMR spectroscopy. High-valent Mn-oxo porphyrins, including the previous example, have been reviewed in The Porphyrin Handbook (2000-2003)18,31 and Chemical Reviews116,117 and won’t be reiterated here.
In 2002, Nam and coworkers presented UV-vis and EPR evidence for the formation of a MnV(O) porphyrin complex from the reaction of MnIII(por) and H2O2 in aqueous solution (Scheme 26).118 The reaction between MnIII(TF4TMAP) (TF4TMAP = meso-tetrakis(2,3,5,6-tetrafluoro-N,N,N-trimethyl-4-aniliniumyl)porphyrinato2−) and H2O2 was highly pH dependent, with O–O bond homolysis proposed to occur at low pH to generate an MnIV(O)(TF4TMAP) and O–O bond heterolysis proposed at high pH to generate an MnV(O)(TF4TMAP). These two high-valent species could be differentiated by their UV-vis and NMR spectra. When alkyl or acyl hydroperoxide was used as oxidant, the electron-donating or -withdrawing properties of the oxidant controlled the mechanism of O–O cleavage, with the former cleaving homolytically to generate the MnIV(O) species and the latter cleaving heterolytically to give the MnV(O) species. The high-valent Mn-oxo species also had differing reactivity. The MnV(O)(TF4TMAP) was a very efficient epoxidizing agent, whereas the lower-valent MnIV(O)(TF4TMAP) was unable to oxidize alkenes. This result was consistent with earlier findings that a MnIV(O)(TM-2-PyP) (TM-2-PyP = meso-tetrakis(N-methyl-2-pyridyl)porphyrinato2−) complex was unable to oxidize olefins.115 Electrochemical evidence for MnV(O)(TF4TMAP) generated from H2O2 and MnIII has been obtained.119
Scheme 26.
Synthesis of High-Valent Manganese-Oxo Species Depends on the Identity of the Oxidant
A dimanganese complex of a dimeric tetraaryl porphyrin was reported by Naruta and coworkers to generate a dinuclear MnV(O) complex (Scheme 27).120 When two equivalents of mCPBA were added to the MnIII2 porphyrin dimer [Mn2(DTMP)(OH)] (TMP = meso-(tetramesityl) porphyrinato2−) in CH2Cl2/CH3CN (1:1 v/v), a new species assigned as an (MnV(O))2 species was generated. This new species was stable in the presence of excess tetrabutylammonium hydroxide; however, when excess triflic acid was added to the complex in the presence of 10% water, fast reduction to a dinuclear manganese(III) species occurred with simultaneous evolution of O2. A 92% yield of O2 relative to the starting dimeric complex was obtained by mass spectrometry and when 18-O labeled water/hydroxide was used, 18O2 was observed. Based on the available data, the authors proposed that the (MnV(O))2 species underwent O–O bond formation to evolve dioxygen, either by nucleophilic attack of solvent water on an MnV(O)(H2O) species or by coupling the two MnV(O) units.
Scheme 27.
Oxygen Evolution from a Dimanganese-oxo Porphyrin Complex
Evidence for the formation of a MnV(O) porphyrin species has come from laser flash photolysis (LFP) studies.121 It is important to note that in the following photochemical studies, the high-valent Mn-oxo species were characterized mainly by transient UV-vis spectroscopy, and further characterization (e.g. Raman, XAS) is needed to confirm their identity. Irradiation of a MnIII(TPFPP) (TPFPP = meso-tetrakis(pentafluorophenyl)porphyrinato2−) in acetonitrile in the presence of the perchlorate anion resulted in the formation of a transient UV-vis spectrum for a new species assigned as MnV(O)(TPFPP) based on its reactivity. In the presence of alkene substrates, this species decayed back to MnIII(TPFPP) at a rate faster than the self-decay process. The new species was able to oxidize stilbene to stilbene oxide with high stereoselectivity in a >95:5, cis:trans ratio. Furthermore, when ethylbenzene and ethylbenzene-d10 were used as substrates, a kinetic isotope effect of 2.3 was obtained, implicating C–H cleavage in the rate determining step. The LFP generation method was subsequently applied to a wider series of manganese porphyrin complexes (MnIII(TPFPP), MnIII(TPP), and MnIII(TMPyP)) in which the electron-donating properties were varied (Scheme 28).122 When LFP was initiated in the presence of NO3− or ClO3−, a species assigned as an MnIV(O)(por) complex was obtained, which results from the homolytic cleavage of the N–O or Cl–O bonds. LFP in the presence of ClO4− resulted in the formation of a species assigned as an MnV(O)(por) complex, which results from heterolytic Cl–O cleavage. The presence of water further stabilized the higher-valent species. The reactivity of the postulated MnV(O)(por) species were studied with various substrates. Smooth two-electron chemistry was observed, with no intermediacy of an MnIV(O)(por) species. The reaction rates with a given substrate depended on the electronics of the porphyrin ligand. The most electron-withdrawing porphyrin was TPFPP, and the MnV(O)(TPFPP) species was the most reactive, followed by the MnV(O)(TMPyP) and MnV(O)(TPP) species. The MnIV(O)(por) species were found to be less reactive than the MnV(O)(por) species. Furthermore, the effect of the electronics of the ligand in reactions with substrates was found to be reversed relative to the MnV(O)(por). Thus, the rates of reaction were found to be as follows: MnIV(O)(TPP) > MnIV(O)(TMPyP) > MnIV(O)(TPFPP). This surprising trend was rationalized by the rapid disproportionation equilibrium of MnIV(O)(por) to form MnV(O)(por) and MnIII(por)(X), with the MnV(O)(por) performing the actual oxidation of the substrate. The disproportionation equilibrium controls the amount of MnV(O)(por) in solution and therefore the “apparent” rate of the reaction. A related photochemical method of generating high-valent Mn-oxo porphyrins was reported for a dinuclear MnIII complex. Upon visible light irradiation, bis-porphyrin dimanganese(III)-μ-oxo complexes could be photo-disproportionated to MnIV(O) porphyrin and MnII porphyrin.123
Scheme 28.
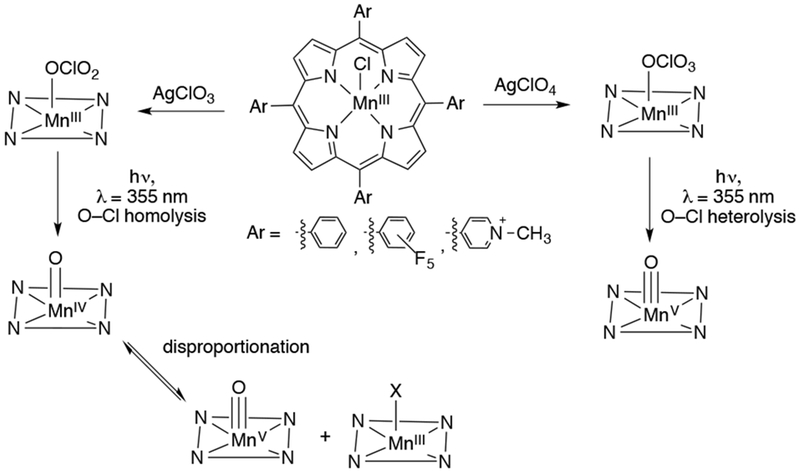
Dependence of O–Cl Bond Cleavage on Starting Manganese Porphyrin Axial Ligand
The effects of oxo-hydroxo tautomerism (Scheme 29) on the reactivity of MnV(O) porphyrins were studied in two isomeric porphyrin complexes.124 It was found that MnV(O)(X)(4-TMPyP) was more reactive with Br− ions to form hypobromite (BrO−) than the less electron-rich MnV(O)(X)(2-TMPyP). This result is due to the higher basicity of the MnV(O)(4-TMPyP) complex, resulting in the presence of more MnV(O)(H2O) (which is the active oxidizing species) rather than MnV(O)(OH) in this complex. This finding is consistent with the idea that an anionic axial donor would make the MnV(O) less electrophilic. Density functional theory (DFT) calculations of the possible spin states (singlet, triplet, and quintet) of the three possible MnV-oxo isomers (dioxo, oxo-hydroxo, and oxo-aquo complexes) reveal a singlet ground state for all three complexes. However, for the oxo-hydroxo and oxo-aquo complexes, the triplet and quintet states are relatively close in energy to the singlet ground state. This small energy difference could allow for a possible spin-crossing event during oxygen transfer, leading to enhanced rates of reaction with substrates.
Scheme 29.
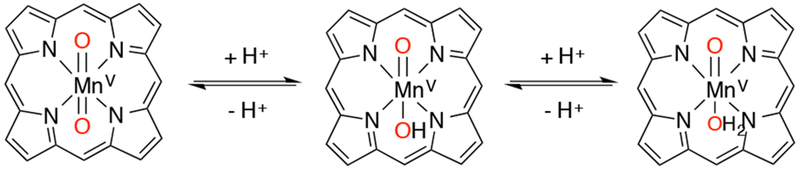
pH-Dependent Oxo-Hydroxo Tautomerism in High-Valent Manganese-Oxo Porphyrins
The importance of spin states on Mn-oxo reactivity has been studied extensively by DFT. A DFT study on C–H hydroxylation by MnV(O)(X)(TPP) (X = O2−, OH−, and H2O) suggested that a rebound mechanism is possible in the triplet and quintet states, but not along the singlet pathway.125 The higher selectivity for the rebound mechanism in the triplet and quintet states was attributed to an increased oxyl radical character found in these higher spin-states, which caused the reaction to occur in two distinct one-electron steps, with a spin cross-over from singlet to triplet in the initial step.126 The extent of oxyl radical character influenced the bond order of the Mn–O interaction. The bond order was found to increase with protonation of the axial ligand X (X = O2−, OH−, and H2O) according to the following series MnV(O)2<MnV(O)(OH)<MnV(O)(H2O).127 However, the multiplicity of the ground states and the extent of oxyl character were highly dependent on the functional used.126 Furthermore, the effect of the axial ligand in the calculations was found to decrease the singlet-triplet and singlet-quintet gaps along this series, and this gap is the dominant factor in controlling the calculated reactivity, with the MnV(O)(H2O) species as the most reactive, as found experimentally.124,128 The protonated bis-hydroxy complex MnV(OH)(OH)(por) was also found to be a viable, potent C–H hydroxylation oxidant by DFT.129,130 Recently, an ab initio benchmarking study on the spin-state energetics and oxyl character of Mn-oxo porphyrins was reported, in which the authors suggest that the best electronic description of these species can be achieved with pure functionals.131
N-donor axial ligands are common additives to C–H bond activation reactions catalyzed by Mn-porphyrins, for which MnV(O)-porphyrin intermediates are often proposed. Addition of axial N-donors is in part motivated by the prominent role played by the axial ligands (e.g. histidine, tyrosine, cysteine) in heme enzymes. A DFT study on the effect of Mn–O bond length versus axial N–donor strength found that the Mn–O bond lengthens with decreasing Mn–Naxial bond distance, indicating that the stronger the axial ligand, the weaker the Mn–O bond, which in turn leads to predicted enhanced reactivity.132 Furthermore, the ability of the axial N-donor to interact with the meso-phenyl rings of an Mn(O)(por) through non-covalent interactions, such as hydrogen-bonding, was also an indicator of reactivity.133 DFT calculations showed that the ortho-C–F bonds in the phenyl rings of MnV(O)(TPFPP) were able to hydrogen-bond with the C–H groups of axially ligated pyridine derivatives, providing a stronger Mn–Naxial interaction, and a lower barrier for C–H activation.
An unusual substrate for MnV(O) porphyrin is the Br− anion. It was shown that MnV(O)(TDMImP) (TDMImP = meso-tetrakis(dimethylimidazolium)porphyrinato2−) was capable of oxidizing the bromide ion to form hypobromite (OBr−), and this reaction was reversible.134 The equilibrium for the reaction MnV(O)(TDMImP) + Br− ⇌ MnIII(TDMImP) + OBr− could be controlled by changing the pH. The formation of the MnV(O)(TDMImP) and Br− was favored under basic conditions, whereas the formation of MnIII(TDMImP) and the O–Br bond was favored under acidic conditions.
Nam, Solomon, and coworkers synthesized a high-valent Mn porphyrin complex by reaction of Mn(III) porphyrins with one of various oxidants including mCPBA, iodosylarenes, and hydrogen peroxide. This product was formulated as an MnV(O) complex based on UV-vis, EPR, 1H and 19F NMR, resonance Raman, and XAS.135 The XAS data led to identification of an Mn–O distance of 1.68 Å for the proposed MnV(O)(TDCPP) (TDCPP = meso-tetrakis(2,6-dichlorophenyl)porphyrinato2−) and indicated that the central Mn was likely 6-coordinate with an axial −OH ligand. However, the Mn–O distance of 1.68 Å was significantly longer than other MnV–O distances, including that reported for a related MnV(O) corrolazine.136 Reactions of the high-valent Mn complex with various substrates were analyzed kinetically. A linear Hammett plot was obtained for reactions with para-substituted thioanisoles of varying electron-donating or electron-withdrawing substituents. The negative slope for this plot indicated that the Mn complex was electrophilic in nature. When the reaction with cyclooctene and cyclooctane was examined, no oxidized organic products were observed. The authors postulated that, in light of the EXAFS results showing that an axial ligand was present, an anionic −OH ligand could decrease the electrophilicity of the proposed MnV(O)(TDCPP) complex and stabilize it. However, the structure of this complex was not assigned correctly and it was subsequently reformulated as a trans-dioxo MnV porphyrin complex, based on reinterpretation of the spectroscopic data and independent generation of another example of a trans-dioxo MnV porphyrin complex.137
The formal bonding picture of MnV(O) porphyrins, known since the 1960s, predicted that mono-oxo complexes should be stable species, but trans-dioxo MnV complexes would be inaccessible. However, an example of a trans-dioxo MnV was reported by Groves and Spiro in 2007. Trans-dioxo MnV-oxo porphyrins were generated by exploiting pH-dependent oxo-hydroxo tautomerism.138 Under highly basic conditions, a series of trans-dioxo MnV(O) porphyrin complexes were generated in organic solvent. These species were half- or fully-labelled isotopically by using 18O-sources, allowing for the key identification of the symmetric and asymmetric Mn–O stretches by resonance Raman and IR spectroscopy. A correlation could be obtained between the force constant for Mn–O stretches and Mn–O bond lengths for a series of comparable manganese-oxo complexes. This correlation, known as Badger’s rule, allowed for a bond length prediction of ~1.7 Å for the Mn=O double bonds.
The isolated trans-dioxo complexes were unreactive towards olefins, but addition of one equivalent of trifluoroacetic acid caused instantaneous reaction with cyclooctene at −70 °C. The authors suggested that the acid protonated one of the oxo ligands and lowered the net negative charge on the [MnV(O)2]− complex, thereby enhancing its electrophilicity. While unreactive with olefins, trans-dioxo MnV-oxo porphyrins were found to perform hydride abstraction from dihydronicotinamide adenine dinucleotide (NADH) analogues, such as 10-methyl-9,10-dihydroacridine (AcrH2).139 The hydride abstraction rates were sensitive to the electron-donating properties of the AcrH2 derivative, as well as the electronics of the porphyrin ligand, with the most electron-deficient porphyrin having the fastest hydride transfer rates. Large deuterium kinetic isotope effects were also observed for the AcrH2 substrates. The kinetic data indicated that the hydride transfer reaction was occurring through a mechanism involving initial rate-determining proton-coupled electron transfer followed by fast electron transfer.
Manganese-porphyrins were found to photocatalytically oxygenate AcrH2, exploiting the facile reaction between MnV(O)(por) and this hydride donor.140 A mechanism was invoked in which the resting state of the catalyst, the MnIII(por), is irradiated and generates a photoexcited [MnIII]* which reacts with O2 to generate a MnIV-superoxo species (Scheme 30). The superoxo species cleaves the C–H bond of the AcrH2 to generate a MnIV-hydroperoxo species and the acridinyl radical AcrH•. The O–O bond of the MnIV-OOH species goes through oxygen rebound homolysis to generate the MnV(O) species and a hydroxylated AcrH–OH molecule. The MnIII(por) catalyst is regenerated by the further oxidation of the AcrH–OH molecule to the ketone, acridone, or by the oxidation of another AcrH2 molecule.
Scheme 30.
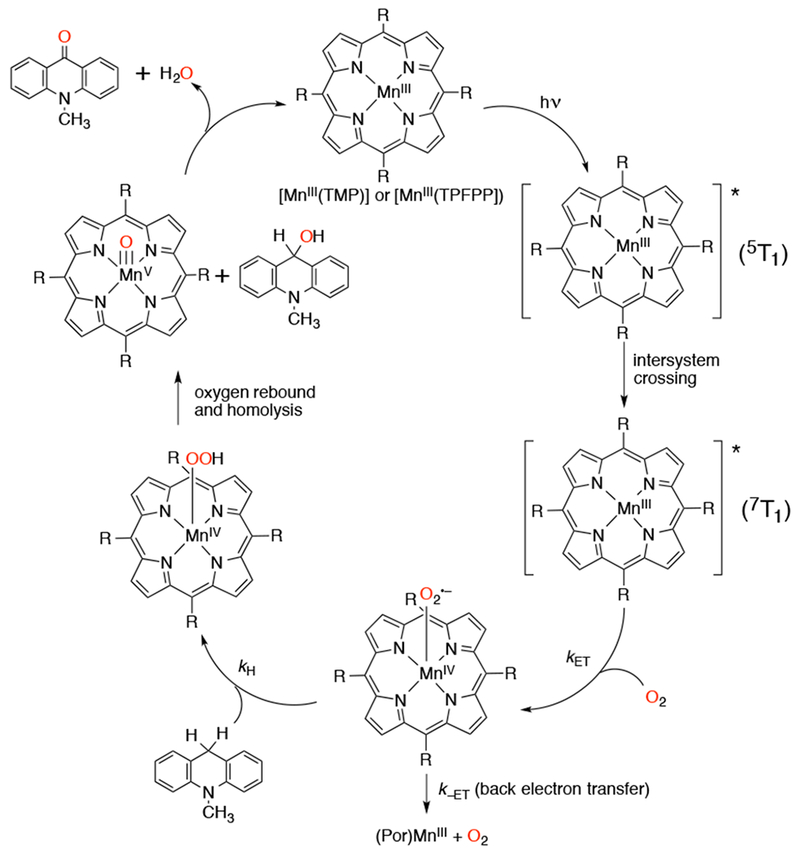
Proposed Mechanism of Photocatalytic Oxygenation of AcrH2 by Manganese Porphyrins
Early work on high-valent Mn–oxo porphyrins showed that MnV(O) and MnIV(O) porphyrins had distinct reactivity, with the lower valent MnIV(O) complexes being less reactive in oxidations such as epoxidation of alkenes.31 A comparison of the differing reactivity in OAT and HAT reactions for trans-dioxo MnV and MnIV(O) generated in aqueous solutions was described by Fukuzumi and Nam. The trans-dioxo complex MnV(O)2(tf4tmap) (tf4map = meso-tetrakis(2,3,5,6-tetrafluoro-N,N,N-trimethyl-4-aniliniumyl)porphyrinato2−), and the oxo-hydroxo complex, MnIV(O)(OH)(tf4tmap), were generated in H2O by oxidation with H2O2 and tert-butyl hydroperoxide, respectively, and characterized by UV-vis and EPR spectroscopies. The two high-valent Mn(O) complexes were reacted with various C–H substrates and O-atom acceptor reagents and the kinetics of these reactions were analyzed (Scheme 31).141 It was found that only the MnV(O)2 complex was capable of OAT to thioanisole substrates, and this reaction gave a MnIII product and the corresponding methyl phenyl sulfoxide. A linear Hammett plot for para-substituted thioanisole derivatives showed a negative slope which indicated a mechanism where the nucleophilic sulfide substrate attacks the electrophilic MnV(O)2. The MnIV(O)(OH) species was able to perform OAT to triarylphosphines, a highly nucleophilic substrate, to give a MnII product and triphenylphosphine oxide. When C–H substrates were added to MnV(O)2(tf4map), the product of the H-atom abstraction reaction was MnIV(O)(OH)(tf4tmap). When C–H substrates were added to the independently generated MnIV(O)(OH)(tf4tmap), slow formation of MnIII(tf4tmap) was observed. No substrate hydroxylation was observed for either Mn-oxo species. A mechanism was suggested from DFT calculations by showing that the energy barrier for the oxygen rebound step is higher than that for the escape of the carbon radical from the cage. These DFT calculations along the reaction coordinate showed that oxygen rebound was disfavored, especially for a dioxo-manganese complex MnV(O)2 relative to the oxo-hydroxo complex MnV(O)(OH), which suggests that the strongly bound oxo (O2−) ligand may play a role in controlling the reaction barrier associated with oxygen rebound.142 The H-atom abstraction rate constants for both MnV(O)2 and MnIV(O)(OH) complexes were found to be linearly dependent on the strength of the C–H bond, with the stronger C–H bonds giving slower reaction rates, and large kinetic isotope effects (kH/kD = 8-24) for the deuterated substrates were also observed. These data support C–H cleavage as the rate-determining step and implicate a hydrogen atom transfer mechanism for both high-valent Mn-oxo species. The authors conclude that the reaction between H-atom donor and MnIV(O)(OH)(tf4tmap) was direct, and, under these conditions, does not proceed via a disproportionation pathway as proposed earlier by Newcomb.122 In contrast, when the hydride donor AcrH2 was added to a different MnIV(O) complex, MnIV(O)(TMP), a disproportionation mechanism was invoked in which the MnIV(O) is in equilibrium with MnIII and MnV(O) (Scheme 32). The MnV(O) species was proposed to be the active oxidant in the reaction.143 The reaction of MnIV(O)(TMP) with ferrocene derivatives, which are one-electron reductants, was proposed to go through a direct electron-transfer mechanism, and did not involve disproportionation. DFT calculations provided some insight into the different reaction pathways observed for MnV(O) vs. MnIV(O) with AcrH2.144 In the case of MnIV(O), an H-atom abstraction is more thermodynamically favored than overall hydride transfer, whereas with MnV(O), hydride abstraction is more favored. The MnV(O) was described as the more potent oxidant with that substrate. The different reaction pathways of these MnIV(O) porphyrins were highly controlled by the nature of the substrate.
Scheme 31.
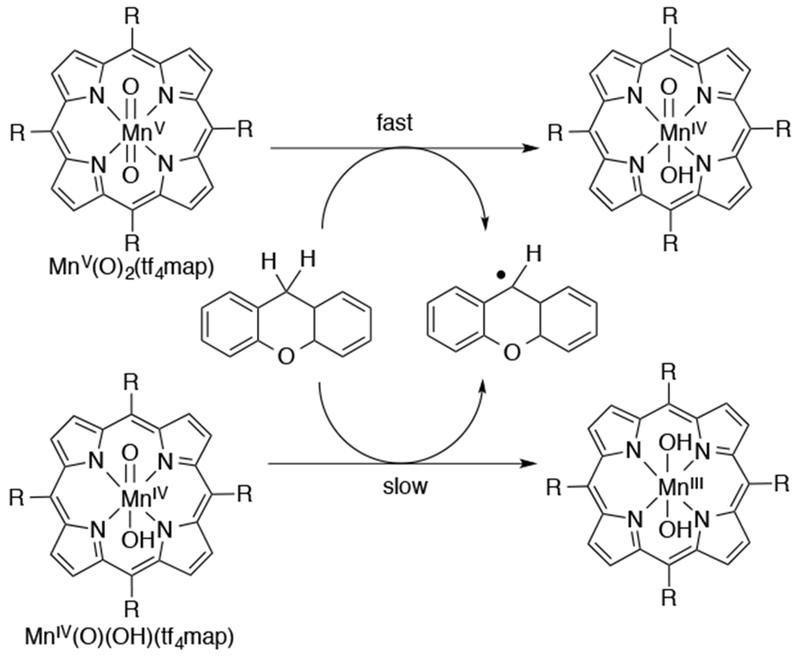
Reactivity Comparison of MnV(O) and MnIV(O) Porphyrins
Scheme 32.
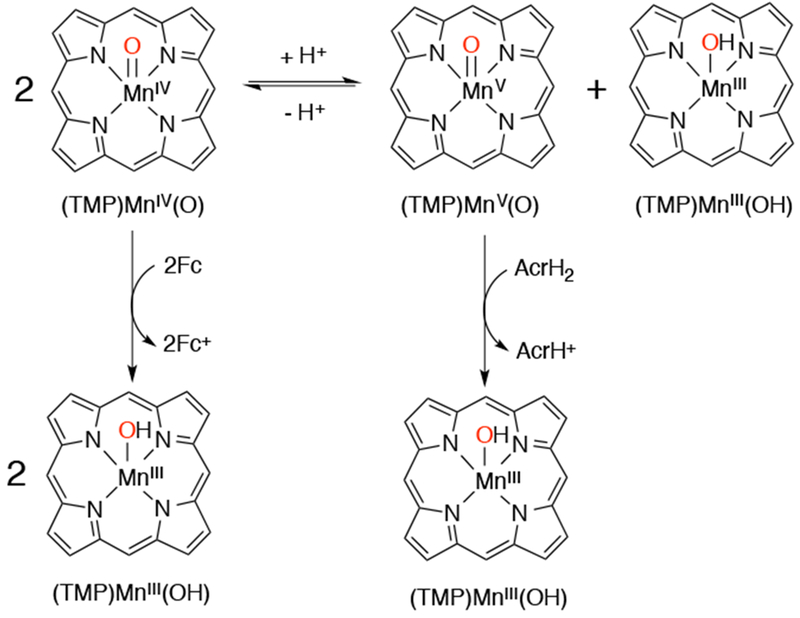
Substrate Dependence on Reactivity of a MnIV(O) Porphyrin
The reactivity of MnV(O) porphyrins with substrates is complicated by the possible presence of axial ligands (H2O, OH−, or O2−) that are present in buffered aqueous solution. A “naked” 5-coordinate MnV(O)(TPFPP) complex was generated from MnIII(TPFPP) and iodosylbenzene in the gas phase in methanol by electrospray ionization and the reactivity was monitored by Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometry.145,146 The MnV(O)(TPFPP) was capable of OAT to olefins to give the reduced MnIII(TPFPP) complex. The rate of OAT depended on the ionization energy of the olefin, with the fastest reactions occurring for olefins with lower ionization energies. Furthermore, selectivity of the gaseous MnV(O)(TPFPP) complex for Z isomers over E isomers of the alkene was also observed. In negative ion mode, the gas-phase trans-dioxo MnV(O)2(TPFPP) complex could be generated, and was found to be completely unreactive towards olefins and sulfides, as found previously in aqueous solution.138 The MnV(O)(TPFPP) complex was also found to oxidize p-substituted thioanisoles and perform N-dealkylation of N,N-dialkylamines.147 The sulfoxidation reaction rates were found to depend on the electronics of the substrate. A negative linear correlation between the Hammett σ-constant of the p-substituted thioanisole and the log of the rate constants could be obtained. The reaction rates between MnV(O)(TPFPP) and p-substituted dimethylanilines were also linearly correlated with the Hammett σ+-parameters of the substrates, and the slope of the line had a negative value. In the case of this substrate, an initial electron-transfer mechanism from the dimethylaniline substrate to the MnV(O) species was proposed. The reaction rates slowed with increasing ionization energy of the dimethylaniline, providing further support for this mechanism.
High-valent Mn-oxo porphyrins are typically generated via O-atom transfer from an appropriate activated O-atom transfer reagent (mCPBA, PhIO) to the MnIII(por). However, Fukuzumi and coworkers showed that water can be used as the O-atom source in the presence of a one-electron oxidant, [Ru(by)3]3+.148 Oxidation of a series of MnIII-porphyrins in acetonitrile with added water generated either MnIV(O) or MnV(O) porphyrins, depending on the axial ligands present in the starting MnIII(por). If Cl− was present, the MnIV(O) species was formed, whereas if H2O was the axial ligand, the MnV(O) species was formed. The MnIII-porphyrin/oxidant/water system could also serve as a catalyst for epoxidation and alkane hydroxylation reactions, in which the MnV(O)(por) species is implicated as the active oxidant (Scheme 33).
Scheme 33.
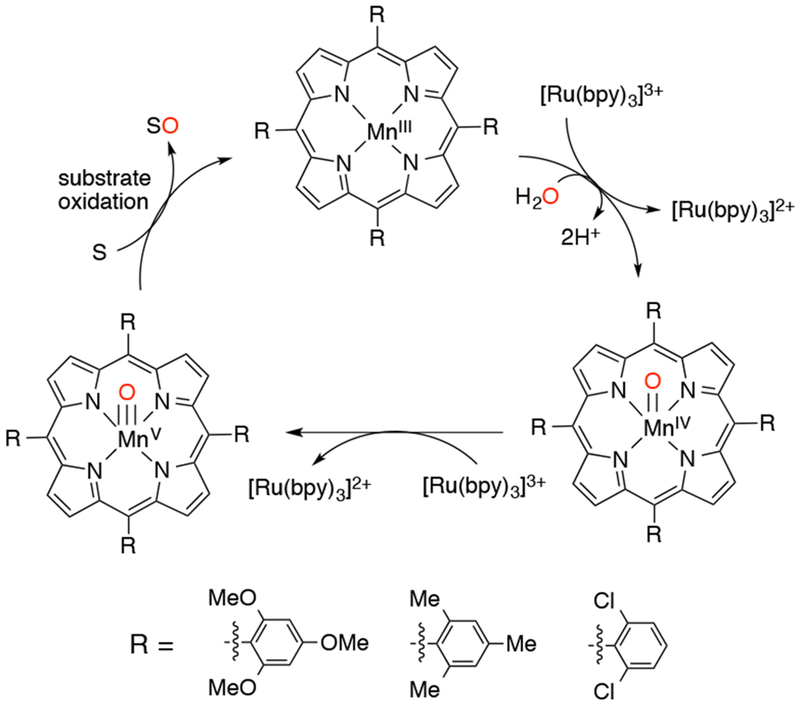
Water as the O-atom Source in Manganese Porphyrin-Catalyzed Oxidations
Another unique set of oxidants, the mild, single-oxygen atom donors tetra-n-butylammonium periodate (n-Bu4NIO4) and tetra-n-butylammonium hydrogen monosulfate (n-Bu4NHSO5), were found to generate high-valent Mn-oxo porphyrins.149,150 The stability and formation of the high-valent Mn(O) species in CH2Cl2 were found to be highly dependent on the sterics and electronics of an added alcohol co-solvent (Scheme 34). It was found that the rate of formation of MnV(O) species was faster for small linear alcohols than for the larger ones. This observation indicated that the hydrogen bonding between the alcohols and the periodate ion coordinated to Mn facilitated the heterolytic cleavage of the I–O bond. Furthermore, the authors found that the stronger the acidity of the alcohol, the better the hydrogen-bonding interaction. The stability of the MnV(O) species was enhanced generally by the presence of alcohols, but especially in the presence of bulkier alcohols. These results pointed to hydrogen bonding between the Mn=O unit and the H–O of the alcohols.
Scheme 34.
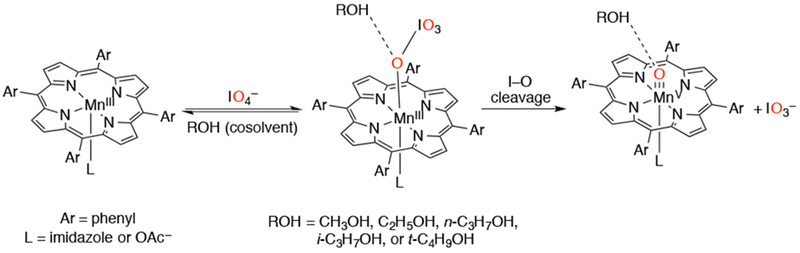
Synthesis of an MnV(O) Porphyrin Complex with Periodate as Oxygen Atom Donor in the Presence of Alcohol Cosolvents
Manganese(V)-oxo porphyrins have been found to oxygenate chloride ions, leading to a variety of halogenation chemistry with these molecules. A water soluble manganese porphyrin MnIII(TDMImP) was found to catalytically generate chlorine dioxide (ClO2) from chlorite ion (ClO2−).151 The proposed mechanism (Scheme 35) involved oxygen transfer from ClO− or ClO2− to MnIII(TDMImP) to give MnV(O)2(TDMImP), which can oxidize a molecule of ClO2− to give MnIV(O)(TDMImP) and one molecule of ClO2. The MnIV(O)(TDMImP) intermediate then reacts with ClO2− to give another ClO2 molecule. To provide support for this mechanism, chlorite ion added to authentic MnIV(O)(TDMImP) underwent facile reaction under the catalytic conditions to produce ClO2 and the MnIII(TDMImP) catalyst. The rate-determining step was determined to be the initial oxygen atom transfer to MnIII(TDMImP) from the chlorite ion to give the high-valent MnV(O)2 species.152 Halogenation of unactivated C–H bonds was observed with a Mn-porphyrin catalyst, and the presence of high-valent MnV(O) species has been observed in the UV-vis spectra during the reactions, although the direct C–H cleavage step has not been directly observed.153
Scheme 35.
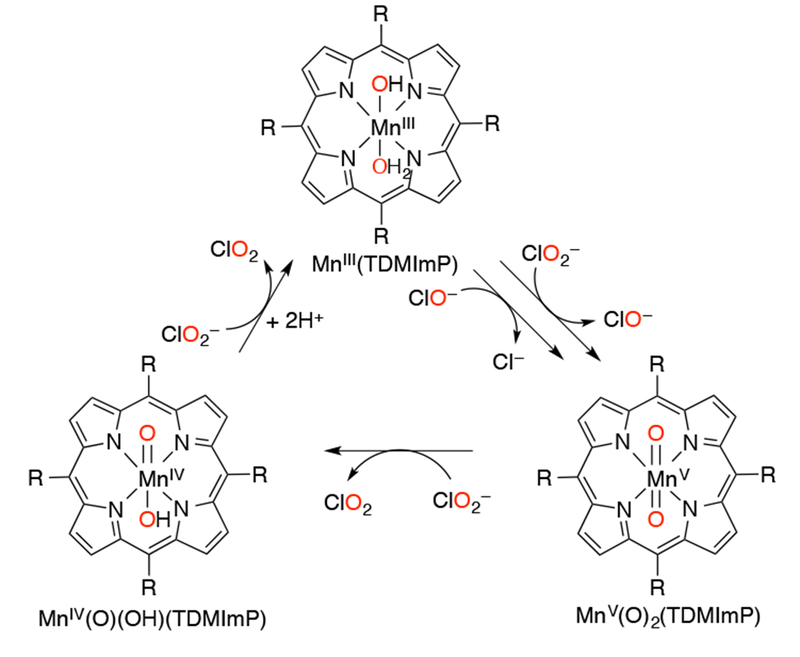
Catalytic Chlorite Oxidation by a Mn(III) Porphyrin
2.2.2. Manganese-Oxo Corroles and Corrolazines
Manganese-oxo corroles have been studied as meta-stable analogues of their reactive porphyrin counterparts. Relatively stable MnV(O) corroles can be synthesized from the MnIII precursor and an appropriate O-atom donor. These species are low-spin diamagnetic S = 0 complexes and the lifetime of the MnV(O) depends on the corrole ligand and solvent. Manganese-oxo corroles have been found to participate in the catalytic oxidation of substrates, and the reaction pathways depend highly on the electronic nature of the ligand. Some recent reviews have covered the reactivity of these complexes, including a chapter in the Handbook of Porphyrin Science in 2011,17 which covers high-valent transition metal corroles and corrolazines, and a Coordination Chemistry Review154 in 2013, which exclusively covers recent advances in manganese corrole chemistry. Since those reviews were published, several reports on the reactivity of Mn-oxo corroles have further advanced the field.
A survey of the reactivity of a series of MnV(O) corroles with a series of alkene substrates revealed a strong dependence of the rate constant as well as the mechanism of OAT on the solvent.155,156 The product of decomposition, which typically changes with ligand identity, was also found to be solvent dependent. The more electron-rich MnV(O)-corroles decomposed to MnIV(X) species in toluene and dichloromethane, but in dimethylformamide and dimethylacetamide, the product of decomposition was an MnIII corrole. In toluene and dichloromethane, the rate of decomposition was faster for the electron-deficient corroles, but in dimethylformamide and dimethylacetamide the trend was reversed. Addition of alkene to the self-decay reaction of MnV(O) was found to greatly accelerate them, with the most electron-rich and least-substituted alkenes giving the fastest rates. The OAT reaction rates between alkene and MnV(O)corroles in either CH2Cl2 or C6H5CH3 depended on the electron-donating properties of the corrole, with the most electron-deficient corroles giving the fastest rates. This trend in OAT rates was reversed in dimethylformamide and dimethylacetamide. The authors attributed this reversed trend to a change in mechanism from direct OAT in toluene and dichloromethane to a disproportionation mechanism in dimethylformamide and dimethylacetamide, in which the MnV(O) disproportionates to an MnIV(X) and an MnVI(O) species, which could be the actual oxidizing agent.
The effect of peripheral substituents on the corrole ring on the reactivity of manganese-oxo corroles has been studied. The effect of β-bromination on the OAT reactivity of a series of A3 and A2B - meso-phenyl substituted MnV(O) corroles was determined by Chang, Liu, and coworkers.157 It was found that the brominated MnV(O) corroles performed OAT at rates much higher than their non-brominated analogues. When the meso positions contained ortho-ortho′-dibromophenyl groups, the steric protection of these substituents provided enhanced stability for the MnV(O) species. DFT calculations provided insight into the mechanism of various β-substituted (with Br, H, or CH3) high-valent Mn-oxo corrole complexes reacting with dimethylsulfide, an O-atom acceptor. Liu and coworkers studied different oxidation levels of the corroles by DFT including [MnIV(O)(tpfc)]− (tpfc = 5,10,15-tris(pentafluorophenyl)corrolato3−), MnV(O)(tpfc), and the one electron-oxidized [MnV(O)(tpfc•+)]+ complexes.158 The calculated barriers suggested that the OAT reactivity followed the order: [MnV(O)(tpfc•+)]+ > MnV(O)(tpfc) > [MnIV(O)(tpfc)]−, indicating that the overall charge on the complex plays an important role in the mechanism. Furthermore, the more electron-deficient corrole ligand gave a more reactive complex, consistent with earlier DFT work159 as well as experimental studies. Spin states were also important for determining the energies of the barriers for the reaction between MnV(O)(tpfc) and DMS. The barrier for the triplet state, best described as high-spin MnV(O), was found to be lower in energy than the singlet state, implicating the possibility of a spin crossover event along the reaction pathway. This result is consistent with earlier calculations from Cao and coworkers that found that while the ground state of the starting MnV(O) corrole is the singlet, the OAT triplet pathway has a lower barrier and therefore might play a significant role in the reaction.160
High-valent manganese-oxo and -imido corroles were shown to participate in the two-electron reduction of O2 to H2O2, serving as precursors to the catalytically active manganese(III) corrole (Scheme 36).161 The addition of trifluoroacetic acid to a high-valent MnV-imido complex, MnV(NAr)(tpfc) (NAr = 2,6,-dichlorophenylimido2−), resulted in hydrolysis by residual water and production of a complex assigned as a protonated Mn-oxo corrole, [MnV(OH)(tpfc)]+. However, direct evidence for the structure of this complex was not obtained. This complex can be reduced rapidly to MnIII(tpfc) by the reductant octamethylferrocene. The reduced MnIII(tpfc) reacts with O2 to give a putative MnIV-superoxo species, which can obtain a proton and electron from TFA and octamethylferrocene, respectively, giving an MnIV-hydroperoxo species. Rather than undergoing O–O bond cleavage, it was suggested that the MnIV-hydroperoxo species is protonated and hydrogen peroxide is released. Reduction of the resulting MnIV(tpfc) allows for the catalytic cycle to begin again. Others have observed the formation of MnV(O) corroles from MnIII(corroles) during the metallation of free-base corroles with MnII sources in the presence of O2, although the mechanism was not elucidated.162
Scheme 36.
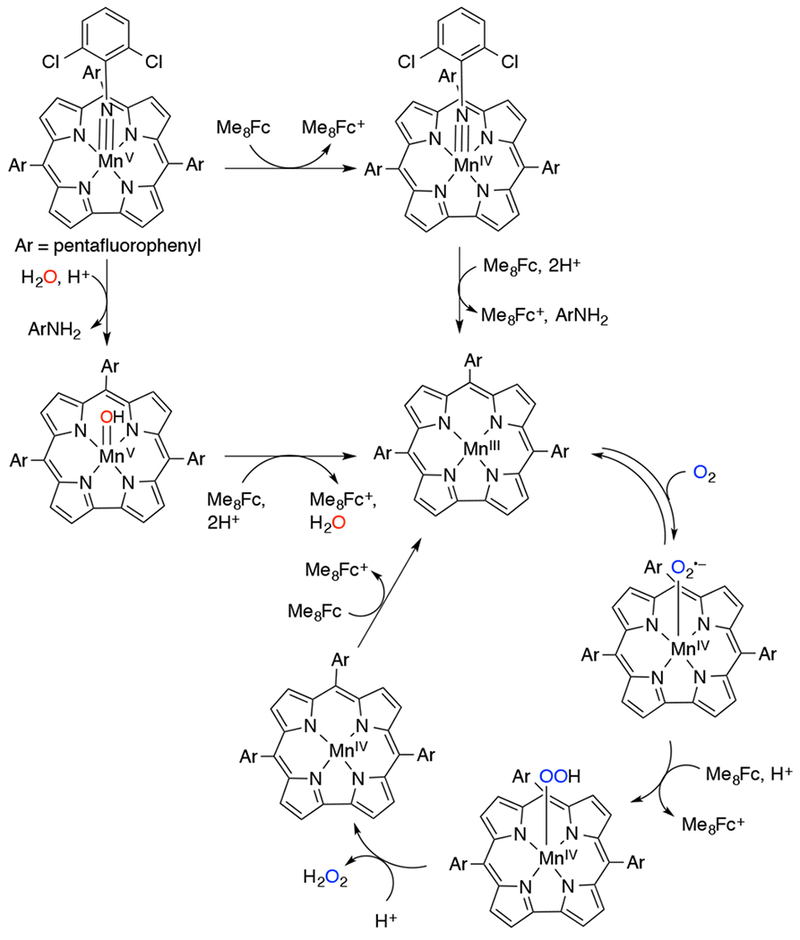
Proton-Coupled Reduction of MnV-imido Corrole to Give a MnIII Corrole
The addition of trifluoroacetic acid to MnV(O)(tpfc) by Fukuzumi, Abu-Omar, and coworkers generated an electronic isomer of this complex, characterized as an MnIV(OH)(tpfc•+) species, in which the oxo ligand was protonated.163 The comparative reactivity of these two valence tautomers was studied (Scheme 37). There was little or no difference for the two complexes in H-atom transfer rates with a substituted phenol as H-atom donor. However, OAT from MnV(O)(tpfc) to thioanisole was extremely facile, whereas the MnIV(OH)(tpfc•+) species was unreactive with thioanisole in the same time frame. In contrast, the electron-transfer reactivity exhibited the opposite trend, with the MnIV(OH)(tpfc•+) complex having the faster rates with the reductant ferrocene.
Scheme 37.
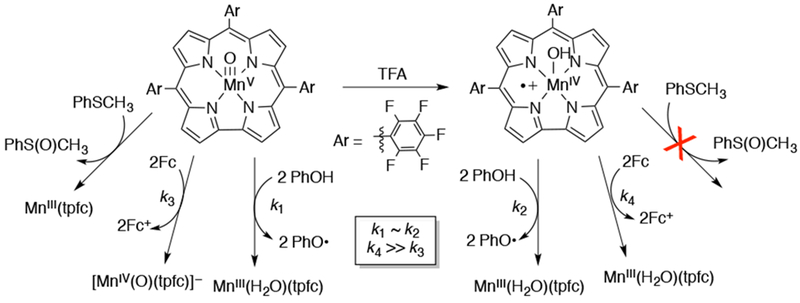
Comparison of Reactivity for Two High-Valent Manganese-Oxo Corrole Valence Tautomers
Another method of entry into high-valent Mn-oxo corroles was found by Zhang and coworkers, in which visible light irradiation of MnIV(BrO3−)(tpc) (tpc = triphenylcorrolato3−) and MnIV(BrO3−)(tpfc) complexes resulted in Br–O bond homolysis to give the MnV(O)-corroles (Scheme 38).164 This kind of light-induced O–X bond homolysis was used previously by Newcomb and coworkers.121,122 The reactivity of the photo-generated species was then studied with various organic substrates. The products of the reaction of photo-generated MnV(O)(tpfc) with organic substrates were highly solvent-dependent, with MnIII(tpfc) as the product in CH3CN and MnIV (tpfc) as the product in CH2Cl2. In contrast, the reaction of the more electron-rich MnV(O)(tpc) with organic substrates gave MnIV(tpc) as the only product regardless of solvent. Despite the complexity of products, the kinetics of the reactions of MnV(O) species with organic substrates all followed a simple second-order rate law. The reaction of MnV(O)(tpfc) with para-substituted thioanisoles resulted in a linear Hammett plot with a negative slope, which indicated a mechanism in which nucleophilic thioanisole reacted with an electrophilic Mn-oxo complex. When the rates of MnV(O)(tpfc) and MnV(O)(tpc) are compared, the MnV(O)(tpc) was generally the faster oxidant. Because the tpc ligand is more electron-rich than tpfc, and the reactivity of electrophilic metal-oxo species is generally enhanced with a more electron-poor ligand, this result was unexpected. The reactivity was therefore rationalized by the disproportionation of MnV(O) to MnIV(X) and MnVI(O), with MnVI(O) proposed as the actual oxidant (Scheme 39). This idea also explains the ability of MnV(O) to promote the facile 2-electron oxidations of alkene and sulfide substrates to give the MnIV(X) species, which is only one oxidation state below MnV(O). However, direct experimental evidence of the putative MnVI(O) oxidant remains elusive.
Scheme 38.
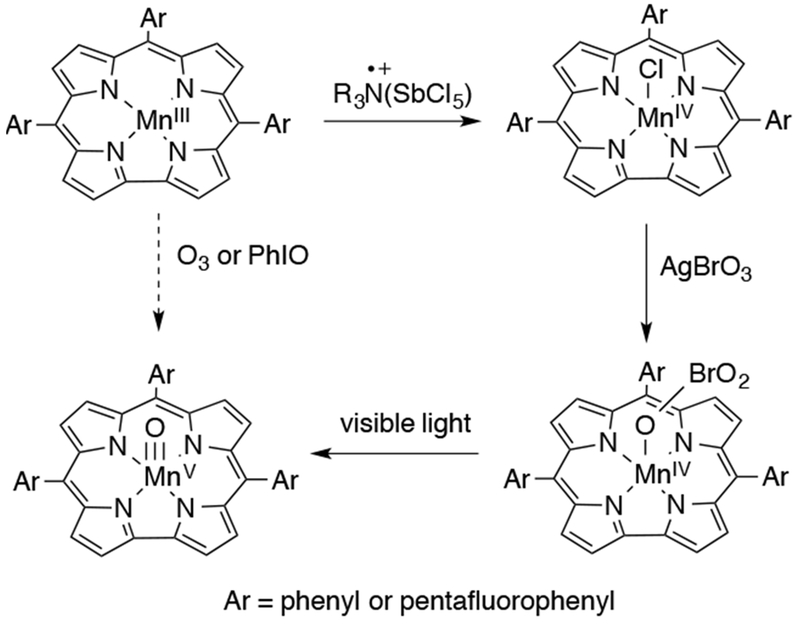
Generation of MnV(O) Corroles by Br–O Bond Homolysis of Mn-Bromate Complexes
Scheme 39.
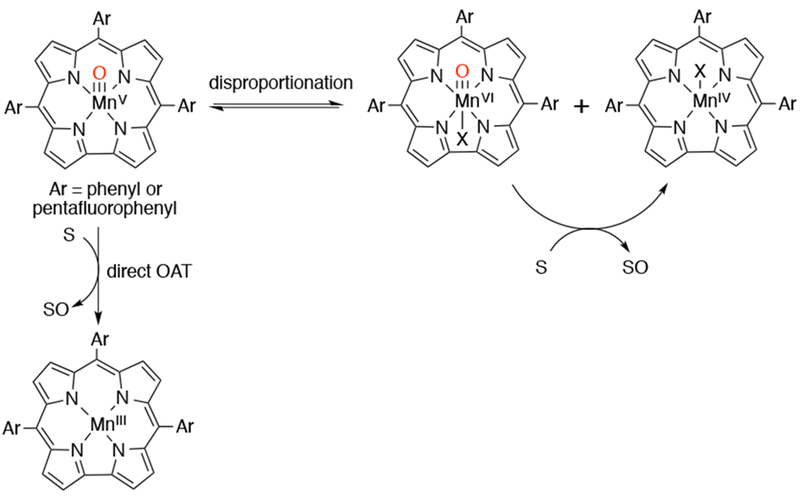
Disproportionation of MnV(O) Corrole to Give MnIV(X) and MnVI(O) Corrole Complexes
The effects of axial ligation on high-valent manganese-oxo corroles have been studied by ligands with tethered nitrogenous-bases.165 The reactivity between the 6-coordinate acetamido-, pyridyl-, and imidazolyl- appended MnV(O) corroles and alkenes was found to be enhanced relative to the 5-coordinate MnV(O)corrole analogue. The reaction rates increased with increasing axial donor strength in the order: acetamido < pyridyl < imidazolyl. It was postulated that the coordination of the axial ligand destabilizes the MnV(O) species, which then increases the rate of OAT. These appended corroles were also capable of catalytic epoxidation, and the turnover frequency (TOF) also increased with the strength of the axial donor, following the same trend as the stoichiometric reactions.
The chemistry of high-valent manganese-oxo corrolazines was reviewed in 2011 in the Handbook of Porphyrin Science17 and a survey of recent work from Goldberg and coworkers on these complexes appeared in 2015 in an Accounts of Chemical Research article.166 A summary of work since these previous reviews is given here.
Although a number of spectroscopic methods had been used to characterize a high-valent manganese-oxo corrolazine complex MnV(O)(TBP8Cz) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3−) since the initial report of this complex in 2001, it was not until 2015 that the structure was determined by single crystal x-ray diffraction (XRD).136 The structure revealed a short Mn–O bond distance of 1.55 Å and the Mn ion was displaced 0.55 Å relative to the plane of the four pyrrole nitrogens. Importantly, the Mn ion was five-coordinate, in contrast to Mn(O) porphyrin complexes, which typically have a sixth ligand.
The effect of modifying the peripheral aryl substituents at the β-position of the corrolazine ring had subtle but measurable effects on the reactivity of the MnV(O) complexes.167 The one-electron reduction potential of MnV(O)(MeOP8Cz) (MeOP8Cz = octakis(p-methoxyphenyl)corrolazinato3−) (E1/2 = −0.57 V vs Fc+/Fc) was shifted negative compared to MnV(O)(TBP8Cz) (E1/2 = −0.53 V vs Fc+/Fc), indicating a slightly more electron rich metal center for the methoxy derivative. A Hammett study of the OAT reaction of MnV(O)(MeOP8Cz) with various p-substituted triarylphosphines to give triarylphosphine oxides revealed a more negative slope than with the MnV(O)(TBP8Cz) derivative, consistent with MnV(O)(MeOP8Cz) as a less electrophilic oxidant. In H-atom abstraction, the MnV(O)(MeOP8Cz) gave slightly faster rates than the MnV(O)(TBP8Cz) complex, and this result was rationalized by an enhanced basicity for the putative [MnIV(O)(MeOP8Cz)]− species which compensates for the more negative redox potential of the MnV(O) complex.
The corrolazine scaffold provides additional opportunities for modification through attachment of electrophiles to the meso-nitrogen atoms. An MnIII(TBP8Cz) complex could be singly- or doubly- protonated on the meso-nitrogens using triflic acid (HOTf).168 The mono- protonated [MnIII(TBP8CzH)]+ complex was the resting state of a catalyst capable of oxidizing the benzyl C–H bonds of toluene derivatives. One proposed intermediate in the catalytic cycle was a protonated high-valent MnV(OH)(TBP8Cz) complex. Addition of HOTf to independently synthesized MnV(O)(TBP8Cz) was found to generate the valence tautomer MnIV(OH)(TBP8Cz•+). This species oxidized substrates very slowly; however, when excess HOTf was added, it was capable of rapid C–H oxidation by a PCET mechanism consistent with this species being competent during catalysis. From these results, the proposed catalytic cycle is shown in Scheme 40, whereby the photoexcited protonated manganese corrolazine complex [MnIII(OTf)(TBP8CzH)]+ reacts with dioxygen to generate a putative MnIV-superoxo species, which abstracts an H• from a toluene derivative (hexamethylbenzene) to generate a putative MnIV-hydroperoxo species and the substrate radical. O–O bond cleavage of the MnIV(OOH) species occurs in concert with oxygen rebound to the substrate radical to give the high-valent manganese-oxo species, MnIV(OH)(TBP8Cz•+), and hydroxylated organic product. The MnIV(OH)(TBP8Cz•+), in the presence of a second H+ equivalent, reacts with another equivalent of hexamethylbenzene to give another equivalent of hydroxylated product along with regeneration of the resting state of the catalyst.
Scheme 40.
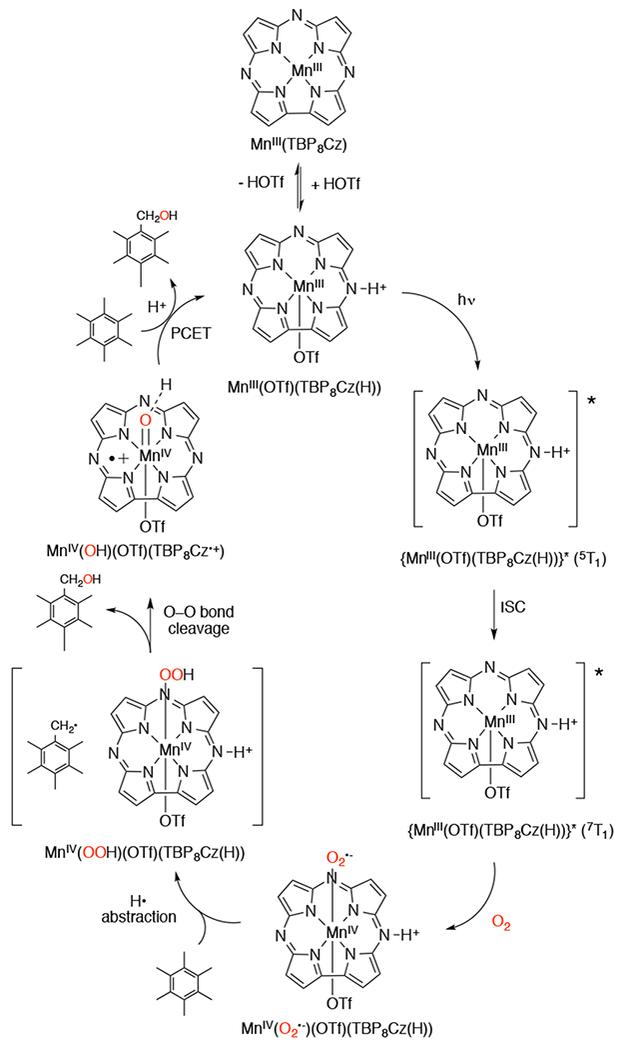
Mechanism for Photocatalytic Oxygenation of the Toluene Derivative Hexamethylbenzene by a Protonated Manganese Corrolazine Complex with Dioxygen
Theoretical calculations have been used as insightful tools into the nature of high-valent metal-oxo species. DFT calculations are popular for studying these types of complexes, but pitfalls in these methods remain, especially with respect to describing spin state energetics. Ghosh and coworkers used a higher level of theory to examine the low-lying excited states of MnV(O) corroles and corrolazines using ab initio methods. The calculations uniformly indicated that a low-spin (S = 0) d2 ground state was favored for these compounds, and confirmed that the corrole and corrolazine macrocycles are unambiguously innocent in the ground states.169 The next low-lying excited states exhibited a high-spin MnV configuration, with the MnIV(O) macrocycle-π-cation-radical states much higher in energy. Furthermore, the authors proposed that the meso-nitrogens of the corrolazine do not impart special stability to the ligand, but rather the steric bulk provided by the eight tert-butylphenyl β-substituents allow for the enhanced stability of the MnV(O) species relative to the corrole.
DFT calculations have provided some insight into the reactivity of high-valent Mn-oxo corrolazines. The oxidation of C–H bonds by the low-spin MnV(O)(TBP8Cz) showed dramatic rate accelerations in the presence of anionic axial donors.170 In a separate computational study, Shaik and coworkers proposed that the origin of the rate enhancement was due to a low-energy triplet transition state, as the most likely C–H bond cleavage pathway.171 Higher-level NEVPT2:CAS calculations have shown that the energetically favored ground state is the singlet state for the same six-coordinate complexes, and showed that the singlet-triplet gap is much larger than predicted by the former DFT calculations. The six-coordinate complexes [MnV(O)(X)(TBP8Cz)]− (X = CN−, F−) also exhibited dramatically enhanced OAT rates relative to the non-axially ligated MnV(O)(TBP8Cz) in reactions with sulfide substrates, including para-substituted thioanisole derivatives.172,173 Calculations on this OAT reaction predicted that the six-coordinate complex [MnV(O)(X)(TBP8Cz)]− reacted through the low-spin singlet pathway, avoiding spin-crossover during the reaction mechanism.174 Experimental data showed a rare V-shaped Hammett plot for OAT, and this result could only be reproduced by calculations on the singlet pathway (Figure 3). When the Hammett plot was calculated for the reaction along the triplet pathway, a linear Hammett plot was obtained. This result provided an experimental test for distinguishing spin state pathways.
Figure 3.
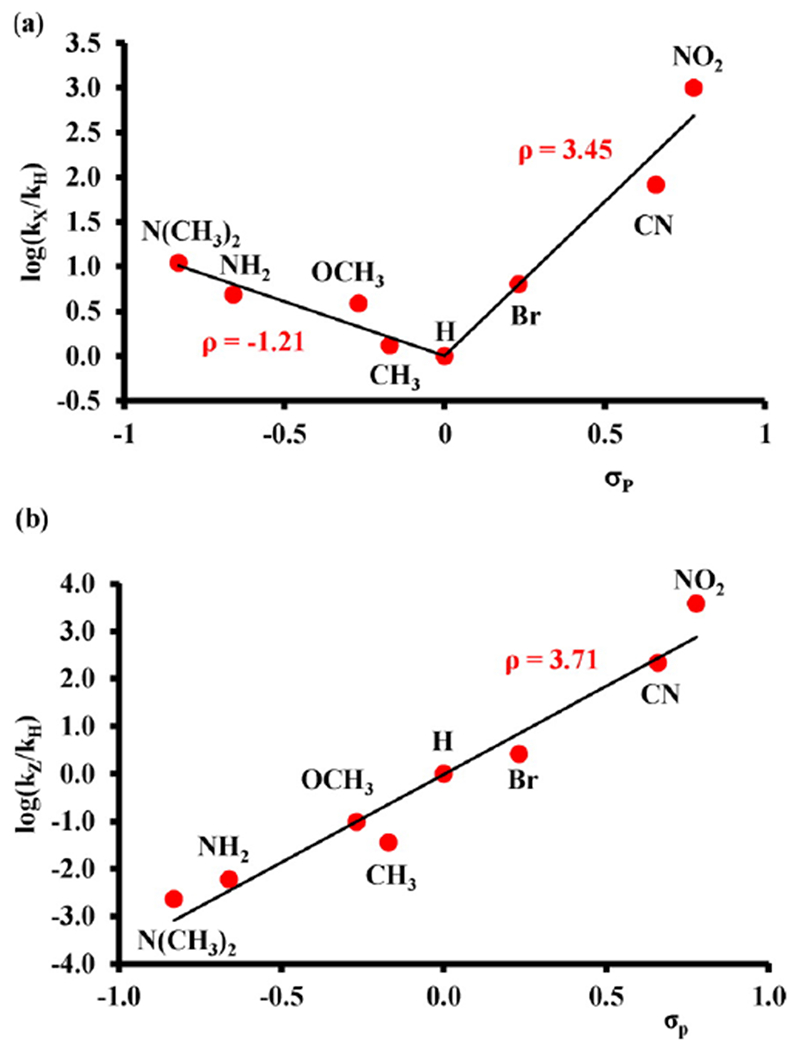
Computational Hammett plot for the reaction of singlet and triplet [Mn(O)(H8Cz)(CN)]− with para-Z-substituted thioanisole derivatives. Data calculated at RIJCOSX-TPSSh-D3/def2-QZVPP/ZORA//RIJCOSX-B3LYP-D3/SDD/BS2 and includes zero-point, thermal, and solvent corrections. (a) Correlation for singlet spin barriers. (b) Correlation for triplet spin barriers. Reproduced with permission from reference 174. Copyright 2016 American Chemical Society.
3. High-Valent Metal-Hydroxo Complexes
For discrete, isolated synthetic complexes of Mn or Fe, we are not aware of any high-valent (oxidation state = +4 or +5) metal-hydroxo porphyrinoid species. The analogous porphyrin and corrole metal(IV)-halogen (chloride, bromide, iodide, fluoride) complexes have been isolated, but have not exhibited biomimetic reactivity.154,175,176
4. Metal-Oxygen Complexes
4.1. Iron-Oxygen Complexes
4.1.1. Iron-Oxygen Porphyrins
Many studies of heme-dioxygen chemistry have focused on functional mimics for myoglobin and hemoglobin. Several reviews exist that cover dioxygen binding and activation by synthetic iron porphyrin complexes, including spectroscopic and structural features of the dioxygen adducts as well as their reactivity with proton/electron sources. A comprehensive review was published in Chemical Reviews in 1994 by Momenteau and Reed, covering synthesis and characterization of heme dioxygen adducts and some reactivity.177 Extensive auto-oxidation reactivity of these species was summarized in a Coordination Chemistry Reviews article by Shikama in 1988.178 In The Porphyrin Handbook (2000), Watanabe described the reactivity of ferric porphyrin peroxo complexes, and Collman et al. summarized the literature of functional synthetic analogues of terminal oxidases.33,179 In 2004, a Chemical Review article by Collman180 summarized the literature on reversibly-formed synthetic heme-dioxygen adducts as functional analogs of the heme enzymes Cytochrome c oxidase, myoglobin, and hemoglobin. The same year, a Chemical Review article by Karlin181 summarized the literature on synthetic heme-copper dioxygen chemistry as models for the active site of Cytochrome c oxidase. It is important to note that this review contained information on well-defined mononuclear heme-dioxygen adducts as a starting point for heme-copper dioxygen chemistry. In 2013, a Coordination Chemistry Reviews article by Ohta and Naruta described their recent progress in understanding the vibrational properties of mononuclear heme-peroxo complexes with regards to their influence on the reactivity of these species.182 In this section, we focus on recent progress in the reactivity of well-defined dioxygen adducts of mononuclear iron porphyrins since the latest review (2012-present).
A report by Ivanović-Burmazović and coworkers showed that the formation of iron-O2 porphyrin adducts in DMSO is reversible (Scheme 41).183 The addition of excess potassium superoxide to FeII(tBuTPP) (tBuTPP = meso-tetrakis(p-tert-butylphenyl)porphyrinato2−) generated the peroxo species [FeIII(O22−)(tBuTPP)]−. This process could be reversed by addition of a proton source, triflic acid (HOTf). The HOTf attacked excess free superoxide in the solution, which shifted the equilibrium back towards the FeII(tBuTPP) complex, according to Scheme 41. This result lies in contrast to other reports in which the presence of acid has been found to induce O–O cleavage to give the high-valent Fe-oxo species rather than disproportionation. By calculation, DMSO was found to have a strong influence on the O2 binding mode and therefore the electronic structure of the Fe-O2 adduct. However, potassium ion, present in solution from the superoxide source, was found to exert very little influence on these factors.
Scheme 41.
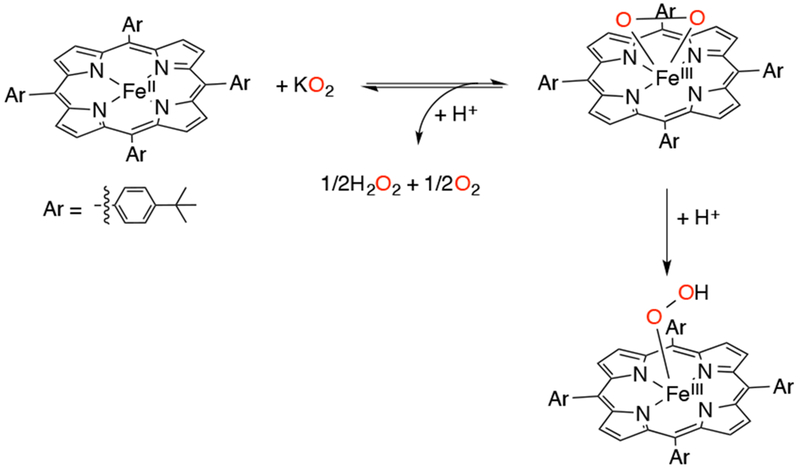
Effect of Acid Addition on a Peroxo Adduct of an Iron-Porphyrin
A study by van Eldik examined the influence of axial ligand and spin state on the O–O bond cleavage and reactivity of Fe-hydroperoxo porphyrin complexes.184 The proposed five-coordinate, high-spin [FeIII(TPFPP)(OOH)] was observed to give FeIV(O)(TPFPP), a Cpd-II-like species, suggesting homolytic cleavage of the O–O bond, while the proposed six-coordinate, low-spin [FeIII(TPFPP)(OH)(OOH)]− was observed to give an FeIV(O)(TPFPP•+) species, suggesting heterolytic cleavage of the O–O bond. The authors attributed this difference in reactivity to the spin-states of the reactant and products for each reaction. The reactivity of the [FeIII(TPFPP)(OH)(OOH)]− complex with various substrates was tested. No reaction was observed upon addition of olefins and sulfides to this complex, and addition of triphenylphosphine resulted only in displacement of the hydroxide ligand rather than oxidation (Scheme 42).
Scheme 42.
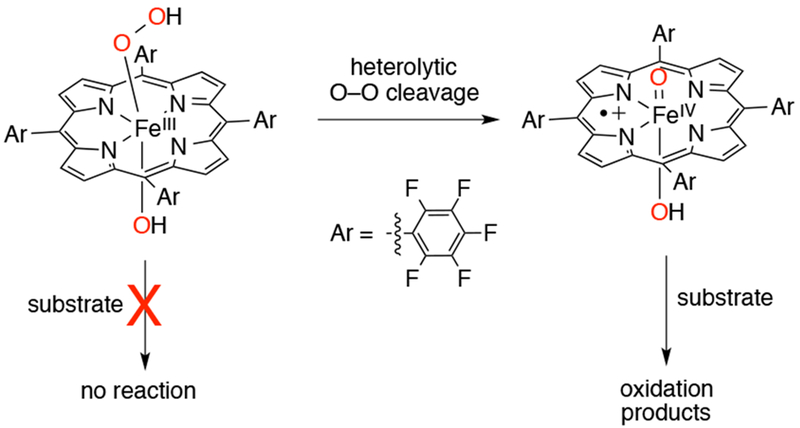
Comparative Reactivity of a Ferric-Hydroperoxide Porphyrin and the O–O Bond Cleavage Product, a Cpd-I-like Species
DFT calculations suggest that a doubly-protonated FeIII-(H2O2) porphyrin complex could be a competent oxygen-atom donor for sulfide substrates.185 The reaction was found to proceed via proton-coupled heterolytic cleavage of the O–O bond in the FeIII(H2O2)(Por) complex. The mechanism was initiated by nucleophilic attack of the distal oxygen by the sulfur lone pair of dimethyl sulfide, an electron-rich sulfur substrate. In tandem with the heterolytic cleavage, the FeIII(OH)(Por) abstracts the proton from the protonated sulfoxide product to give dimethylsulfoxide and FeIII(OH2)(Por). An alternative mechanism was ruled out, in which the O–O bond of FeIII(H2O2)(Por) is cleaved homolytically to generate an [FeIII + •OH] and a hydroxyl radical, •OH. The •OH abstracts an H-atom from [FeIII + •OH] to give FeIV(O)(porphyrin π-cation-radical) (Cpd-I), which can then perform sulfoxidation. However, the homolytic cleavage step was found to have a much higher activation barrier than the heterolytic/sulfur attack mechanism, indicating that the heterolytic mechanism will undergo sulfoxidation much faster than Cpd-I can be generated (Scheme 43). While an FeIII(H2O2) species has not been observed in heme enzyme catalysis, the peroxide shunt is often invoked in mechanistic studies where hydrogen peroxide is used to generate the high-valent iron-oxo species.
Scheme 43.
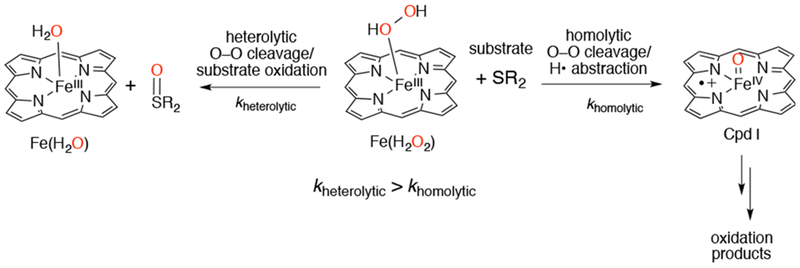
DFT Calculations on the Heterolytic vs Homolytic Cleavage Mechanisms in Oxidation of Sulfide Substrates by an Fe(H2O2)-Porphyrin Complex
Spectroscopic detection of a ferrous-superoxo porphyrin could be achieved by cryo-generation (Scheme 44).186 Irradiation of a ferric-superoxo complex with a γ-ray source at 77 K resulted in one electron reduction to the ferrous superoxo species, which was characterized by EPR and resonance Raman spectroscopies. DFT calculations also confirmed that the reduction of the FeIII(O2•−) species occurred at the iron center rather than the superoxo-ligand. When the ferrous-superoxo species was warmed to 193 K, the ferric-hydroperoxide species was observed, as characterized previously by Naruta and coworkers,182 indicating that the FeII(O2•−) species was basic enough to scavenge a proton even in the absence of an exogenous acid source. This work gave insight into the thermodynamics of the transformation between ferric-superoxo and ferric-hydroperoxo, which differ by a hydrogen atom, and both of which are implicated in the catalytic cycles of heme monooxygenase enzymes.
Scheme 44.
Cryo-Reduction of a Ferric-Superoxo to the Ferrous-Superoxo Porphyrin Complex
The influence of the secondary coordination sphere on the reactivity of a ferric-superoxo porphyrin was investigated (Scheme 45).187 An imidazole as an axial ligand and a carboxylic acid group as a proton donor were tethered to the porphyrin ligand. As a control, the secondary coordination sphere effects of a similar ligand containing an ethoxy carbonyl group instead of the carboxylic acid were compared. When the iron-superoxo species was generated by addition of O2 to the ferrous iron in the ligand without the proton donor, addition of exogenous reductant and acid resulted in controlled formation of the ferric hydroperoxo species. However, when O2 was added to the ferrous iron porphyrin containing the pendant proton donor, some ferric-hydroperoxide was observed, in addition to the ferric-superoxo species. The presence of ferric-hydroperoxide from the oxygenation reaction, in the absence of added proton and electron donors, implied that the reduced ferrous porphyrin itself could act as an H+/e− donor, but only if the carboxylic acid group was present.
Scheme 45.
Comparison of the Reactivity of a Ferric Superoxo Porphyrin in the Presence and Absence of a Ligand-tethered Proton Donor
4.1.2. Iron-Oxygen Corroles and Corrolazines
To our knowledge, there are no examples of well defined iron-(hydro)peroxo or iron–superoxo corrole or corrolazine complexes.
4.2. Manganese-Oxygen Complexes
4.2.1. Manganese-Oxygen Porphyrins
In heme enzymes, the binding and activation of dioxygen or hydrogen peroxide at the metal center are typically the initial steps in the catalytic cycles. Cleavage of the O–O bond leads to the generation of high-valent metal-oxo species. There are very few examples of well-defined manganese-superoxo or –(hydro)peroxo porphyrins.188 This may be due to the relative stability of the O–O bond-cleaved product, a high-valent manganese-oxo species, in these systems. However, those rare Mn–superoxo or – (hydro)peroxo species that have been observed can be generated by either O2, H2O2, or ROOH binding and activation. Some reviews on Mn-superoxo/peroxo porphyrin species include a section of a chapter by Koga and Tabushi in Biomimetic Chemistry from the Advances in Chemistry (1980) series37 and a section of a chapter by Groves and coworkers in the book Biomimetic Oxidations Catalyzed by Transition Metal Complexes (2000).189 In this section, we focus on the reactivity of the Mn–O–O adducts, which is intimately tied to the extent of O–O bond activation.
The first example of a Mn(porphyrin)(O2) adduct was reported in 1975. Addition of O2 at −79 °C to MnII(TPP) resulted in the reversible formation of a five-coordinate dioxygen-bound Mn(O2)(TPP) species.190 Debate about the electronic structure and mode of dioxygen binding followed, with some suggesting an MnIV(O22−)(Por)(O22− = peroxide2−) with the peroxide ligand binding to the Mn in a side-on fashion191-194 and others an MnIII(O2•−)(Por) (O2−• = superoxide1−) with the superoxide ligand binding to Mn in an end-on fashion.195,196 The one-electron reduced [Mn(O2)(Por)]− adduct generated from addition of potassium superoxide to MnII (generated from outer sphere reduction of MnIII(Por) by another equivalent of potassium superoxide) was subjected to a similar debate regarding geometry and electronic structure.197,198 Another way to generate the [Mn(O2)(Por)]− species was found when O2 was added to MnIII(por) in the presence of hydroxide.199 Ultimately, a crystal structure of an [Mn(O2)(TPP)]− adduct showed that a peroxide ligand was bound to MnIII(TPP) in the side-on mode, resolving the debate. This binding mode for the reduced complex also served as precedent for the oxidation state assignment of the neutral MnIV(O22−)(TPP) complex.200
The equilibrium constant for the binding of O2 to Mn(II) porphyrins was found to depend on the electron-donating or electron-withdrawing nature of the porphyrin ligand.201 The effect of neutral axial ligands (L) bound to MnII(por) complexes trans to the O2 binding site was studied and a correlation between the proton affinities of the series of ligands and the equilibrium constants for O2 binding was found.202
The acylation of an oxygenated manganese-porphyrin complex could be achieved at low temperature by addition of acylchloride to Mn(II) or Mn(III) porphyrins in the presence of O2 or superoxide, respectively.203 The Mn-acyl-peroxo species was capable of the oxidation of cyclooctene to cyclooctene oxide. Addition of hydroxide ion to the acyl-peroxo species and warming of the reaction solution induced O–O bond cleavage to give the high-valent Mn-oxo species, which was also able to undergo epoxidation of cyclooctene.
Changing the electronics of the peroxyacid allowed for tuning of the O–O bond cleavage reactivity of an acylperoxo-Mn(TMP) adduct, but the relative rates of O–O cleavage to form the high valent Mn-oxo species were strongly dependent on the presence or absence of hydroxide ion.204 In the presence of hydroxide, the more electron-rich peroxyacids were more reactive than the electron-poor peroxyacids. In the absence of hydroxide ion, the opposite trend was observed. These differences in reactivity were attributed to a change in mechanism from homolytic to heterolytic O–O cleavage in the presence and absence of hydroxide, respectively.
A manganese(III)peroxycarbonate complex could be formed from the reaction of [Mn(O2)(Por)]− adducts with carbon dioxide.205 Homolytic or heterolytic O–O bond cleavage of the peroxycarbonate adducts was dependent on the solvent, temperature, and porphyrin ligand. Formation of [Mn(O2CO2)(Por)]− adducts was achieved by addition of potassium superoxide to either MnII(TPP), MnII(TPFPP), or MnII(TPivP) (TPivP = meso-tetrakis(α-pivalamidophenyl)porphyrinato2−), followed by addition of carbon dioxide at − 70 °C. Depending on the solvent, O–O cleavage occurred to give one of two high-valent Mn(O) species. In THF, a ligating solvent, homolytic O–O cleavage to give the MnIV(O) was favored, whereas in toluene heterolysis to give the MnV(O) was preferred.
The reactivity of peroxomanganese(III) porphyrins with organic substrates was studied and compared with other metal-peroxo complexes.206 It was found that [MnIII(O2)(Por)]− acted as a nucleophile of moderate strength. The peroxo complex [MnIII(O2)(Por)]− was able to oxidize an extremely electron-deficient alkene, tetracyanoethylene, in addition to acyl halides and CO2. However, [MnIII(O2)(Por)]− was unable to oxidize other olefins, including some electron-deficient olefins and all electron-rich olefins tested, as well as triphenylphosphine and butyllithium. It was suggested that the relative stability of this complex was due to strong orbital overlap between the metal and O2 ligand.
The mechanism of heterolytic O–O bond cleavage of a MnIII(OOH)(TDMImP) was studied.207 The O–O bond cleavage step was found to be rate determining and followed a concerted “push-pull” mechanism, in which an axial −OH was partially deprotonated and provided the “push”, while the terminal oxygen in the −OOH ligand was partially protonated and acted as a “pull” to release an H2O molecule into the solvent medium. This “push-pull” model has also been invoked to explain the heterolytic O–O bond cleavage to generate Cpd-I during the catalytic cycle of Cytochrome P450s and other heme enzymes.
4.2.2. Manganese-Oxygen Corroles
Well-defined manganese-peroxo corrole complexes are rare, while manganese-superoxo corrole complexes are unknown. The following examples constitute the only evidence for these species to date, and their reactivity is still not well understood.
In 2009, Åkermark, Sun, and coworkers showed evidence for the formation of O2 by addition of hydroxide to an MnV(O) corrole complex (Scheme 46).208 Nucleophilic attack of hydroxide on MnV(O)(tnpc) (tnpc = 5,10,15-tris(4-nitrophenyl)corrolato3−) initially generated a putative MnIII hydroperoxy complex, which was not observed. Formal H• abstraction by an unidentified oxidant from this complex gave an MnIV species, which was assigned as an MnIV-peroxo complex based on UV-vis and mass spectrometry. Further oxidation through an unknown mechanism results in the MnIII corrole and release of dioxygen. Formation of O2 was quantitated by online gas analysis, and addition of H218O to the hydroxide reaction resulted in mixed-labeled Mn-peroxo complex as well as mixed-label dioxygen (16,18O2), confirming that O–O bond formation occurs in this system.
Scheme 46.
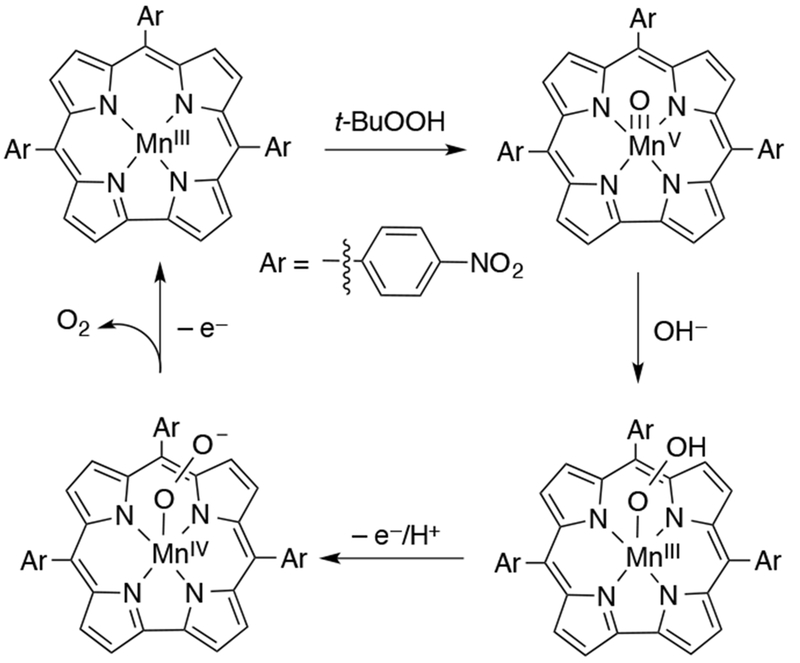
O-O bond Formation from Nucleophilic Attack of Hydroxide on a High-Valent Metal-Oxo Corrole
Following this work, Nam and coworkers provided evidence for the reversible interconversion of an MnV(O) corrole and an Mn-peroxo corrole complex (Scheme 47).209 The addition of H2O2 to MnIII (tfmpc) (tfmpc = 5,10,15-tris(trifluoromethylphenyl)corrolato3−) in the presence of hydroxide resulted in an intermediate assigned as an MnIV(O2)− species based on UV-vis, EPR, and mixed labeling mass spectrometry studies using 18O-labeled reagents. The addition of hydroxide to MnV(O)(tfmpc) also resulted in the formation of this new species, and could be reversed back to the starting MnV(O) complex with the addition of perchloric acid. The mechanism of interconversion is not understood with regard to electron-count, since MnV(O) and hydroxide should give an MnIII(peroxo) complex and H2O2 addition to MnIII (tfmpc) should give the same MnIII species. The overall pathway is shown in Scheme 47.
Scheme 47.
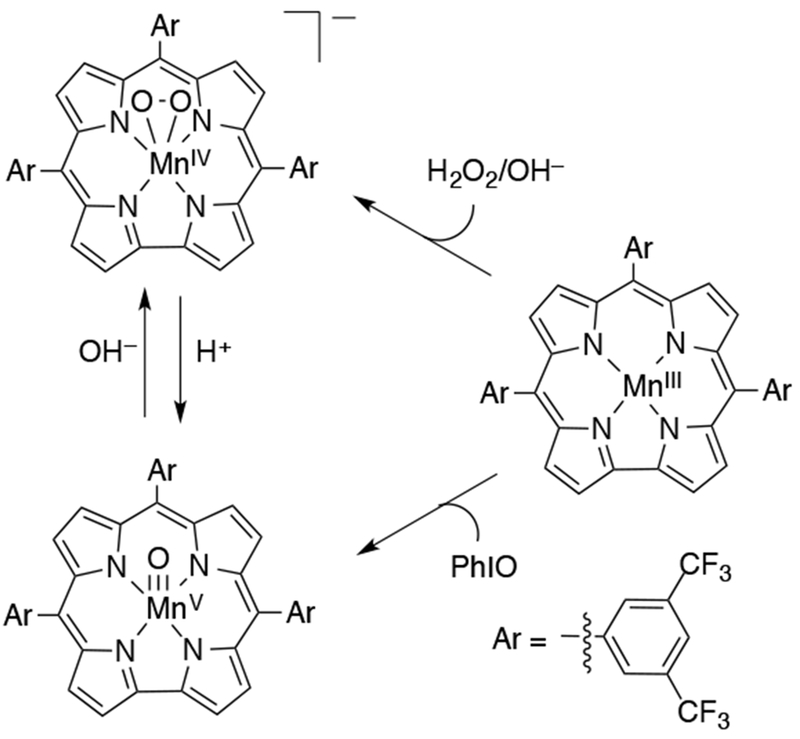
Reversible Interconversion of a Manganese-oxo and Manganese-peroxo Corrole Complex in the Presence of Acid or Base
5. Conclusions and Final Remarks
In this review, we discussed the biomimetic reactivity of iron and manganese -oxo and -dioxygen intermediates in porphyrinoid systems. The reactivity of these species depends heavily on the identity of the porphyrinoid ligand (either porphyrin, corrole, or corrolazine), the metal identity and oxidation state, as well as the type of substrate. These results are analogous to natural systems that depend on tuning environment to control reactivity. Experimental and theoretical studies both gave crucial insights into the fundamental reaction mechanisms.
While biomimetic reactivity of metal-oxo and metal-dioxygen intermediates has been extensively studied, we still foresee interesting challenges ahead. For example, the direct observation of the combination of an organic radical and a high-valent metal-hydroxide compound has not yet been achieved. Understanding the factors that select for rebound chemistry over other reactions (such as dehydrogenation) could allow for insight into the design of metalloporphyrinoid catalysts that undergo selective hydroxylations with substrates that contain many possible sites of oxidation. The design principles from these studies could also give insight into C–H halogenation performed by heme chloroperoxidase. High-valent iron-oxo corrole complexes and their reactivity are notably absent from the literature. Only one corresponding high-valent iron-oxo corrolazine has been characterized. The field is open for ligand development to access high-valent mononuclear Fe(O) corroles and corrolazines. There is still much to learn about metal-oxygen species and what controls the extent of O2 activation, the O–O bond formation/cleavage steps, and the mechanism of O–O bond formation/cleavage. Manganese or iron corroles could allow for the study of these species, but there are few examples in the literature showing that they can form stable metal-oxygen complexes. The syntheses of these species would allow for unknown reactivity to be revealed.
Achieving answers to these questions and opening up the field to new reactivity could give valuable insight into the natural systems and provide a foundational basis for the development of new metalloporphyrinoid-mediated reactions.
Scheme 25.
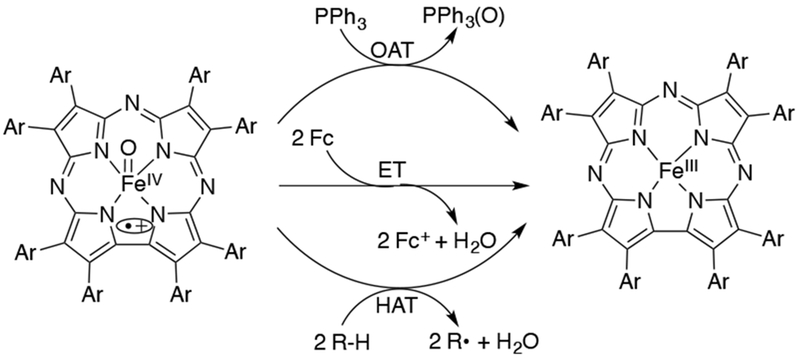
O-atom Transfer, H-atom Abstraction, and Electron Transfer Reactions of FeIV(O)(corrolazine•+)
Acknowledgements
We thank the NIH for financial support (GM101153 to D.P.G.). R.A.B. is grateful for the Harry and Cleio Greer Fellowship. J.P.T.Z. is grateful for The Glen E. Meyer ‘39 Fellowship.
Author Biographies
Regina A. Baglia received her B.S. degree in biochemistry in 2011 from Temple University, in Philadelphia, PA, where she performed undergraduate research as a Diamond Research Scholar in the Department of Chemistry with Prof. Michael J. Zdilla. She received her PhD in chemistry at The Johns Hopkins University in 2016, where she worked with Prof. David P. Goldberg. While at Hopkins, she was awarded the George E. Owens Graduate Fellowship, an E2SHI fellowship, and the Harry and Cleio Greer fellowship.
Jan Paulo T. Zaragoza was born in Manila, Philippines in 1990. He received his B.S. in Biochemistry from the University of the Philippines—Manila in 2010. He is currently pursuing his doctoral studies under the supervision of Prof. David P. Goldberg at the Johns Hopkins University. His research involves the study of high-valent metal-oxo corroles and corrolazines as synthetic models for heme enzyme active sites.
David P. Goldberg received his B.A. degree from Williams College in 1989, and his Ph.D. degree in Inorganic Chemistry in the research group of Prof. Stephen J. Lippard. Following his Ph.D. in 1995, he was an NIH Postdoctoral Fellow in the group of Prof. Brian Hoffman at Northwestern University. He began his independent career at Johns Hopkins University in the department of Chemistry in 1998, where he is currently a Professor of Chemistry. His research interests include synthetic inorganic chemistry, models of heme and nonheme metalloenzymes, dioxygen and nitric oxide activation, and other fundamental transformations important to synthetic and biological oxidation/reduction catalysis.
Footnotes
The authors declare no competing financial interest.
References
- (1).Sono M; Roach MP; Coulter ED; Dawson JH Heme-Containing Oxygenases Chem. Rev 1996, 96, 2841–2888. [DOI] [PubMed] [Google Scholar]
- (2).Poulos TL Heme Enzyme Structure and Function Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mansuy D; Battioni P In The Porphyrin Handbook; >Kadish KM, >Smith JM, >Guilard R, Eds.; Academic Press: San Diego, CA, USA, 2000; Vol. 4, p 1–15. [Google Scholar]
- (4).Efimov I; Basran J; Thackray SJ; Handa S; Mowat CG; Raven EL Structure and Reaction Mechanism in the Heme Dioxygenases Biochemistry 2011, 50, 2717–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Claessens CG; González-Rodríguez D; Rodríguez-Morgade MS; Medina A; Torres T Subphthalocyanines, Subporphyrazines, and Subporphyrins: Singular Nonplanar Aromatic Systems Chem. Rev 2014, 114, 2192–2277. [DOI] [PubMed] [Google Scholar]
- (6).Shimizu S Recent Advances in Subporphyrins and Triphyrin Analogues: Contracted Porphyrins Comprising Three Pyrrole Rings Chem. Rev 2017, 117, 2730–2784. [DOI] [PubMed] [Google Scholar]
- (7).Jasat A; Dolphin D Expanded Porphyrins and Their Heterologs Chem. Rev 1997, 97, 2267–2340. [DOI] [PubMed] [Google Scholar]
- (8).Sessler JL; Seidel D Synthetic Expanded Porphyrin Chemistry Angew. Chem., Int. Ed 2003, 42, 5134–5175. [DOI] [PubMed] [Google Scholar]
- (9).Tanaka T; Osuka A Chemistry of meso-Aryl-Substituted Expanded Porphyrins: Aromaticity and Molecular Twist Chem. Rev 2017, 117, 2584–2640. [DOI] [PubMed] [Google Scholar]
- (10).Rosenthal J; Nocera DG In Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: 2008, p 483–544. [Google Scholar]
- (11).Anderson JS; Gallagher AT; Mason JA; Harris TD A Five-Coordinate Heme Dioxygen Adduct Isolated within a Metal–Organic Framework J. Am. Chem. Soc 2014, 136, 16489–16492. [DOI] [PubMed] [Google Scholar]
- (12).Li W-S; Aida T Dendrimer Porphyrins and Phthalocyanines Chem. Rev 2009, 109, 6047–6076. [DOI] [PubMed] [Google Scholar]
- (13).Lu Y; Berry SM; Pfister TD Engineering Novel Metalloproteins: Design of Metal-Binding Sites into Native Protein Scaffolds Chem. Rev 2001, 101, 3047–3080. [DOI] [PubMed] [Google Scholar]
- (14).Prier CK; Arnold FH Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes To Create New Catalysts J. Am. Chem. Soc 2015, 137, 13992–14006. [DOI] [PubMed] [Google Scholar]
- (15).Dydio P; Key HM; Nazarenko A; Rha JY-E; Seyedkazemi V; Clark DS; Hartwig JF An artificial metalloenzyme with the kinetics of native enzymes Science 2016, 354, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bhagi-Damodaran A; Michael MA; Zhu Q; Reed J; Sandoval BA; Mirts EN; Chakraborty S; Moënne-Loccoz P; Zhang Y; Lu Y Why copper is preferred over iron for oxygen activation and reduction in haem-copper oxidases Nat Chem 2017, 9, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McGown AJ; Badiei YM; Leeladee P; Prokop KA; DeBeer S; Goldberg DP In Handbook of Porphyrin Science; >Kadish KM, >Smith JM, >Guilard R, Eds.; World Scientific Press: Singapore, 2011; Vol. 14, p 525–599. [Google Scholar]
- (18).Weiss R, Gold A, Trautwein AX, and Terner J In The Porphyrin Handbook; >Kadish KM, >Smith KM and >Guilard R, Ed.; Academic Press: San Diego, CA, USA, 2000; Vol. 4, p 65–96. [Google Scholar]
- (19).Groves JT; Nemo TE; Myers RS Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene J. Am. Chem. Soc 1979, 101, 1032–1033. [Google Scholar]
- (20).Groves JT; Haushalter RC; Nakamura M; Nemo TE; Evans BJ High-valent iron-porphyrin complexes related to peroxidase and cytochrome P-450 J. Am. Chem. Soc 1981, 103, 2884–2886. [Google Scholar]
- (21).Watanabe Y; Fujii H In Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations; >Meunier B, Ed.; Springer: Berlin; New York, 2000, p 62. [Google Scholar]
- (22).Fujii H Electronic structure and reactivity of high-valent oxo iron porphyrins Coord. Chem. Rev 2002, 226, 51–60. [Google Scholar]
- (23).Fujii H In Heme Peroxidases; >Raven E, >Dunford B, Eds.; The Royal Society of Chemistry: 2016, p 181–217. [Google Scholar]
- (24).Meunier B; de Visser SP; Shaik S Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (25).Groves JT High-valent iron in chemical and biological oxidations J. Inorg. Biochem 2006, 100, 434–447. [DOI] [PubMed] [Google Scholar]
- (26).Pan Z; Zhang R; Newcomb M Kinetic studies of reactions of iron(IV)-oxo porphyrin radical cations with organic reductants J. Inorg. Biochem 2006, 100, 524–532. [DOI] [PubMed] [Google Scholar]
- (27).Nam W High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions Acc. Chem. Res 2007, 40, 522–531. [DOI] [PubMed] [Google Scholar]
- (28).Costas M Selective C–H oxidation catalyzed by metalloporphyrins Coord. Chem. Rev 2011, 255, 2912–2932. [Google Scholar]
- (29).Hohenberger J; Ray K; Meyer K The biology and chemistry of high-valent iron-oxo and iron-nitrido complexes Nat Commun 2012, 3, 720. [DOI] [PubMed] [Google Scholar]
- (30).Rutkowska-Zbik D; Drzewiecka-Matuszek A; Witko M The Influence of Structural Parameters on the Reactivity of Model Complexes for Compound II: A Mini Review Topics in Catalysis 2014, 57, 946–952. [Google Scholar]
- (31).Groves JT; Shalyaev K; Lee J In The Porphyrin Handbook; >Kadish KM, >Smith KM, >Guilard R, Eds.; Academic Press: San Diego, CA, USA, 2000; Vol. 4, p 17–40. [Google Scholar]
- (32).Suslick KS In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: San Diego, 2000; Vol. 4, p 41–64. [Google Scholar]
- (33).Watanabe Y In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: San Diego, CA, USA, 2000; Vol. 4, p 97–118. [Google Scholar]
- (34).Meunier B; Robert A; Pratviel G; Bernadou J In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: San Diego, CA, USA, 2000; Vol. 4, p 119–188. [Google Scholar]
- (35).de Visser SP; Nam W High-valent iron-oxo porphyrins in oxygenation reactions Handbook of Porphyrin Science; Kadish KM, Smith KM, Guilard R, Eds 2010, 85–140.
- (36).McLain JL; Lee J; Groves JT In Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Meunier B, Ed.; Imperial College Press: London, 2000, p 91–169. [Google Scholar]
- (37).Tabushi I; Koga N In Biomimetic Chemistry; American Chemical Society: 1980; Vol. 191, p 291–306. [Google Scholar]
- (38).Nam W In Comprehensive Coordination Chemistry II; Meyer TJ, Ed.; Pergamon: Oxford, 2003, p 281–307. [Google Scholar]
- (39).Dolphin D; Traylor TG; Xie LY Polyhaloporphyrins: Unusual Ligands for Metals and Metal-Catalyzed Oxidations Acc. Chem. Res 1997, 30, 251–259. [Google Scholar]
- (40).Bell SR; Groves JT A Highly Reactive P450 Model Compound I J. Am. Chem. Soc 2009, 131, 9640–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Pan Z; Zhang R; Fung LWM; Newcomb M Photochemical Production of a Highly Reactive Porphyrin–Iron–Oxo Species Inorg. Chem 2007, 46, 1517–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jeong YJ; Kang Y; Han A-R; Lee Y-M; Kotani H; Fukuzumi S; Nam W Hydrogen Atom Abstraction and Hydride Transfer Reactions by Iron(IV)–Oxo Porphyrins Angew. Chem., Int. Ed 2008, 47, 7321–7324. [DOI] [PubMed] [Google Scholar]
- (43).Pan Z; Wang Q; Sheng X; Horner JH; Newcomb M Highly Reactive Porphyrin–Iron–Oxo Derivatives Produced by Photolyses of Metastable Porphyrin–Iron(IV) Diperchlorates J. Am. Chem. Soc 2009, 131, 2621–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chen T-H; Asiri N; Kwong KW; Malone J; Zhang R Ligand control in the photochemical generation of high-valent porphyrin-iron-oxo derivatives Chem. Commun 2015, 51, 9949–9952. [DOI] [PubMed] [Google Scholar]
- (45).Radoń M; Broclawik E; Pierloot K DFT and Ab Initio Study of Iron-Oxo Porphyrins: May They Have a Low-Lying Iron(V)-Oxo Electromer? J. Chem. Theory Comput 2011, 7, 898–908. [DOI] [PubMed] [Google Scholar]
- (46).Pan Z; Horner JH; Newcomb M Tunneling in C–H Oxidation Reactions by an Oxoiron(IV) Porphyrin Radical Cation: Direct Measurements of Very Large H/D Kinetic Isotope Effects J. Am. Chem. Soc 2008, 130, 7776–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cong Z; Kinemuchi H; Kurahashi T; Fujii H Factors Affecting Hydrogen-Tunneling Contribution in Hydroxylation Reactions Promoted by Oxoiron(IV) Porphyrin π-Cation Radical Complexes Inorg. Chem 2014, 53, 10632–10641. [DOI] [PubMed] [Google Scholar]
- (48).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450 Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tani F; Matsu-ura M; Nakayama S; Naruta Y Synthetic models for the active site of cytochrome P450 Coord. Chem. Rev 2002, 226, 219–226. [Google Scholar]
- (50).Hessenauer-Ilicheva N; Franke A; Wolak M; Higuchi T; van Eldik R Spectroscopic and Mechanistic Studies on Oxidation Reactions Catalyzed by the Functional Model SR Complex for Cytochrome–P450: Influence of Oxidant, Substrate, and Solvent Chem. - Eur. J 2009, 15, 12447–12459. [DOI] [PubMed] [Google Scholar]
- (51).Hessenauer-Ilicheva N; Franke A; Meyer D; Woggon W-D; van Eldik R Mechanistic Insight into Formation of Oxo-Iron(IV) Porphyrin π-Cation Radicals from Enzyme Mimics of Cytochrome P450 in Organic Solvents Chem. - Eur. J 2009, 15, 2941–2959. [DOI] [PubMed] [Google Scholar]
- (52).Suzuki N; Higuchi T; Urano Y; Kikuchi K; Uekusa H; Ohashi Y; Uchida T; Kitagawa T; Nagano T Novel Iron Porphyrin–Alkanethiolate Complex with Intramolecular NH¬∑¬∑¬∑S Hydrogen Bond: Synthesis, Spectroscopy, and Reactivity J. Am. Chem. Soc 1999, 121, 11571–11572. [Google Scholar]
- (53).Meyer DN; Woggon W-D Synthesis and Characterization of a New Family of Iron Porphyrins CHIMIA International Journal for Chemistry 2005, 59, 85–87. [Google Scholar]
- (54).Hessenauer-Ilicheva N; Franke A; Meyer D; Woggon W-D; van Eldik R Low-Temperature Rapid-Scan Detection of Reactive Intermediates in Epoxidation Reactions Catalyzed by a New Enzyme Mimic of Cytochrome P450 J. Am. Chem. Soc 2007, 129, 12473–12479. [DOI] [PubMed] [Google Scholar]
- (55).Han A-R; Jin Jeong Y; Kang Y; Yoon Lee J; Sook Seo M; Nam W Direct evidence for an iron(iv)-oxo porphyrin π-cation radical as an active oxidant in catalytic oxygenation reactions Chem. Commun 2008. [DOI] [PubMed] [Google Scholar]
- (56).Franke A; Fertinger C; van Eldik R Which Oxidant Is Really Responsible for P450 Model Oxygenation Reactions? A Kinetic Approach Angew. Chem., Int. Ed 2008, 47, 5238–5242. [DOI] [PubMed] [Google Scholar]
- (57).Takahashi A; Kurahashi T; Fujii H Effect of Imidazole and Phenolate Axial Ligands on the Electronic Structure and Reactivity of Oxoiron(IV) Porphyrin π-Cation Radical Complexes: Drastic Increase in Oxo-Transfer and Hydrogen Abstraction Reactivities Inorg. Chem 2009, 48, 2614–2625. [DOI] [PubMed] [Google Scholar]
- (58).Takahashi A; Kurahashi T; Fujii H Redox Potentials of Oxoiron(IV) Porphyrin π-Cation Radical Complexes: Participation of Electron Transfer Process in Oxygenation Reactions Inorg. Chem 2011, 50, 6922–6928. [DOI] [PubMed] [Google Scholar]
- (59).Takahashi A; Yamaki D; Ikemura K; Kurahashi T; Ogura T; Hada M; Fujii H Effect of the Axial Ligand on the Reactivity of the Oxoiron(IV) Porphyrin π-Cation Radical Complex: Higher Stabilization of the Product State Relative to the Reactant State Inorg. Chem 2012, 51, 7296–7305. [DOI] [PubMed] [Google Scholar]
- (60).Kang Y; Chen H; Jeong YJ; Lai W; Bae EH; Shaik S; Nam W Enhanced Reactivities of Iron(IV)-Oxo Porphyrin π-Cation Radicals in Oxygenation Reactions by Electron-Donating Axial Ligands Chemistry - A European Journal 2009, 15, 10039–10046. [DOI] [PubMed] [Google Scholar]
- (61).Kumar D; Sastry GN; de Visser SP Axial Ligand Effect On The Rate Constant of Aromatic Hydroxylation By Iron(IV)–Oxo Complexes Mimicking Cytochrome P450 Enzymes J. Phys. Chem. B 2012, 116, 718–730. [DOI] [PubMed] [Google Scholar]
- (62).de Visser SP Trends in Substrate Hydroxylation Reactions by Heme and Nonheme Iron(IV)-Oxo Oxidants Give Correlations between Intrinsic Properties of the Oxidant with Barrier Height J. Am. Chem. Soc 2010, 132, 1087–1097. [DOI] [PubMed] [Google Scholar]
- (63).Kumar D; Sastry GN; de Visser SP Effect of the Axial Ligand on Substrate Sulfoxidation Mediated by Iron(IV)–Oxo Porphyrin Cation Radical Oxidants Chem. - Eur. J 2011, 17, 6196–6205. [DOI] [PubMed] [Google Scholar]
- (64).Kumar D; Latifi R; Kumar S; Rybak-Akimova EV; Sainna MA; de Visser SP Rationalization of the Barrier Height for p-Z-styrene Epoxidation by Iron(IV)-Oxo Porphyrin Cation Radicals with Variable Axial Ligands Inorg. Chem 2013, 52, 7968–7979. [DOI] [PubMed] [Google Scholar]
- (65).Kumar D; Karamzadeh B; Sastry GN; de Visser SP What Factors Influence the Rate Constant of Substrate Epoxidation by Compound I of Cytochrome P450 and Analogous Iron(IV)-Oxo Oxidants? J. Am. Chem. Soc 2010, 132, 7656–7667. [DOI] [PubMed] [Google Scholar]
- (66).de Visser SP; Tahsini L; Nam W How Does the Axial Ligand of Cytochrome P450 Biomimetics Influence the Regioselectivity of Aliphatic versus Aromatic Hydroxylation? Chem. - Eur. J 2009, 15, 5577–5587. [DOI] [PubMed] [Google Scholar]
- (67).Kudrik EV; Afanasiev P; Alvarez LX; Dubourdeaux P; Clémancey M; Latour J-M; Blondin G; Bouchu D; Albrieux F; Nefedov SE; Sorokin AB An N-bridged high-valent diiron–oxo species on a porphyrin platform that can oxidize methane Nat Chem 2012, 4, 1024–1029. [DOI] [PubMed] [Google Scholar]
- (68).Dunford HB; Lambeir A-M; Kashem MA; Pickard M On the mechanism of chlorination by chloroperoxidase Arch. Biochem. Biophys 1987, 252, 292–302. [DOI] [PubMed] [Google Scholar]
- (69).Harrison JE; Schultz J Studies on the chlorinating activity of myeloperoxidase J. Biol. Chem 1976, 251, 1371–1374. [PubMed] [Google Scholar]
- (70).Cong Z; Kurahashi T; Fujii H Oxidation of Chloride and Subsequent Chlorination of Organic Compounds by Oxoiron(IV) Porphyrin π-Cation Radicals Angew. Chem., Int. Ed 2011, 50, 9935–9939. [DOI] [PubMed] [Google Scholar]
- (71).Du J; Chen Z; Chen C; Meyer TJ A Half-Reaction Alternative to Water Oxidation: Chloride Oxidation to Chlorine Catalyzed by Silver Ion J. Am. Chem. Soc 2015, 137, 3193–3196. [DOI] [PubMed] [Google Scholar]
- (72).Cong Z; Kurahashi T; Fujii H Formation of Iron(III) meso-Chloro-isoporphyrin as a Reactive Chlorinating Agent from Oxoiron(IV) Porphyrin π-Cation Radical J. Am. Chem. Soc 2012, 134, 4469–4472. [DOI] [PubMed] [Google Scholar]
- (73).Cong Z; Yanagisawa S; Kurahashi T; Ogura T; Nakashima S; Fujii H Synthesis, Characterization, and Reactivity of Hypochloritoiron(III) Porphyrin Complexes J. Am. Chem. Soc 2012, 134, 20617–20620. [DOI] [PubMed] [Google Scholar]
- (74).Abu-Omar MM High-valent iron and manganese complexes of corrole and porphyrin in atom transfer and dioxygen evolving catalysis Dalton Trans 2011, 40, 3435–3444. [DOI] [PubMed] [Google Scholar]
- (75).Lee AQ; Streit BR; Zdilla MJ; Abu-Omar MM; DuBois JL Mechanism of and exquisite selectivity for O–O bond formation by the heme-dependent chlorite dismutase Proc. Natl. Acad. Sci 2008, 105, 15654–15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zdilla MJ; Lee AQ; Abu-Omar MM Concerted Dismutation of Chlorite Ion: Water-Soluble Iron-Porphyrins As First Generation Model Complexes for Chlorite Dismutase Inorg. Chem 2009, 48, 2260–2268. [DOI] [PubMed] [Google Scholar]
- (77).Zdilla MJ; Lee AQ; Abu-Omar MM Bioinspired Dismutation of Chlorite to Dioxygen and Chloride Catalyzed by a Water-Soluble Iron Porphyrin Angew. Chem., Int. Ed 2008, 47, 7697–7700. [DOI] [PubMed] [Google Scholar]
- (78).Kang M-J; Song WJ; Han A-R; Choi YS; Jang HG; Nam W Mechanistic Insight into the Aromatic Hydroxylation by High-Valent Iron(IV)-oxo Porphyrin π-Cation Radical Complexes J. Org. Chem 2007, 72, 6301–6304. [DOI] [PubMed] [Google Scholar]
- (79).Asaka M; Fujii H Participation of Electron Transfer Process in Rate-Limiting Step of Aromatic Hydroxylation Reactions by Compound I Models of Heme Enzymes J. Am. Chem. Soc 2016, 138, 8048–8051. [DOI] [PubMed] [Google Scholar]
- (80).Li C; Wu W; Cho K-B; Shaik S Oxidation of Tertiary Amines by Cytochrome P450—Kinetic Isotope Effect as a Spin-State Reactivity Probe Chem. - Eur. J 2009, 15, 8492–8503. [DOI] [PubMed] [Google Scholar]
- (81).Garcia-Bosch I; Sharma SK; Karlin KD A Selective Stepwise Heme Oxygenase Model System: An Iron(IV)-Oxo Porphyrin π-Cation Radical Leads to a Verdoheme-Type Compound via an Isoporphyrin Intermediate J. Am. Chem. Soc 2013, 135, 16248–16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Chin D-H; Balch AL; La Mar GN Formation of porphyrin ferryl (FeO2+) complexes through the addition of nitrogen bases to peroxo-bridged iron(III) porphyrins J. Am. Chem. Soc 1980, 102, 1446–1448. [Google Scholar]
- (83).Chin D-H; La Mar GN; Balch AL Role of ferryl (FeO2+) complexes in oxygen atom transfer reactions. Mechanism of iron(II) porphyrin catalyzed oxygenation of triphenylphosphine J. Am. Chem. Soc 1980, 102, 5945–5947. [Google Scholar]
- (84).Groves JT; Gross Z; Stern MK Preparation and Reactivity of Oxoiron(IV) Porphyrins Inorg. Chem 1994, 33, 5065–5072. [Google Scholar]
- (85).Tahsini L; Bagherzadeh M; Nam W; de Visser SP Fundamental Differences of Substrate Hydroxylation by High-Valent Iron(IV)-Oxo Models of Cytochrome P450 Inorg. Chem 2009, 48, 6661–6669. [DOI] [PubMed] [Google Scholar]
- (86).Fertinger C; Hessenauer-Ilicheva N; Franke A; van Eldik R Direct Comparison of the Reactivity of Model Complexes for Compounds 0, I, and II in Oxygenation, Hydrogen-Abstraction, and Hydride-Transfer Processes Chem. - Eur. J 2009, 15, 13435–13440. [DOI] [PubMed] [Google Scholar]
- (87).Ji L; Franke A; Brindell M; Oszajca M; Zahl A; van Eldik R Combined Experimental and Theoretical Study on the Reactivity of Compounds I and II in Horseradish Peroxidase Biomimetics Chem. - Eur. J 2014, 20, 14437–14450. [DOI] [PubMed] [Google Scholar]
- (88).Oszajca M; Franke A; Drzewiecka-Matuszek A; Brindell M; Stochel G; van Eldik R Temperature and Pressure Effects on C–H Abstraction Reactions Involving Compound I and II Mimics in Aqueous Solution Inorg. Chem 2014, 53, 2848–2857. [DOI] [PubMed] [Google Scholar]
- (89).Nehru K; Seo MS; Kim J; Nam W Oxidative N-Dealkylation Reactions by Oxoiron(IV) Complexes of Nonheme and Heme Ligands Inorg. Chem 2007, 46, 293–298. [DOI] [PubMed] [Google Scholar]
- (90).Kitagishi H; Kurosawa S; Kano K Intramolecular Oxidative O-Demethylation of an Oxoferryl Porphyrin Complexed with a Per-O-methylated β-Cyclodextrin Dimer Chem. Asian J 2016, 11, 3213–3219. [DOI] [PubMed] [Google Scholar]
- (91).Ueda T; Kitagishi H; Kano K Intramolecular Direct Oxygen Transfer from Oxoferryl Porphyrin to a Sulfide Bond Inorg. Chem 2014, 53, 543–551. [DOI] [PubMed] [Google Scholar]
- (92).Rosa A; Ricciardi G Reactivity of Compound II: Electronic Structure Analysis of Methane Hydroxylation by Oxoiron(IV) Porphyrin Complexes Inorg. Chem 2012, 51, 9833–9845. [DOI] [PubMed] [Google Scholar]
- (93).Boaz NC; Bell SR; Groves JT Ferryl Protonation in Oxoiron(IV) Porphyrins and Its Role in Oxygen Transfer J. Am. Chem. Soc 2015, 137, 2875–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).McGown AJ; Badiei YM; Leeladee P; Prokop KA; DeBeer S; Goldberg DP In The Handbook of Porphyrin Science; Kadish KM, Smith KM, Guilard R, Eds.; World Scientific: New Jersey, 2011; Vol. 14, p 525–600. [Google Scholar]
- (95).Vogel E; Will S; Tilling AS; Neumann L; Lex J; Bill E; Trautwein AX; Wieghardt K Metallocorroles with Formally Tetravalent Iron Angew. Chem., Int. Ed. Engl 1994, 33, 731–735. [Google Scholar]
- (96).Gross Z High-valent corrole metal complexes J. Biol. Inorg. Chem 2001, 6, 733–738. [DOI] [PubMed] [Google Scholar]
- (97).Simkhovich L; Mahammed A; Goldberg I; Gross Z Synthesis and Characterization of Germanium, Tin, Phosphorus, Iron, and Rhodium Complexes of Tris(pentafluorophenyl)corrole, and the Utilization of the Iron and Rhodium Corroles as Cyclopropanation Catalysts Chem. - Eur. J 2001, 7, 1041–1055. [DOI] [PubMed] [Google Scholar]
- (98).Ye S; Tuttle T; Bill E; Simkhovich L; Gross Z; Thiel W; Neese F The Electronic Structure of Iron Corroles: A Combined Experimental and Quantum Chemical Study Chem. - Eur. J 2008, 14, 10839–10851. [DOI] [PubMed] [Google Scholar]
- (99).Walker FA; Licoccia S; Paolesse R Iron corrolates: Unambiguous chloroiron(III) (corrolate)2–• π-cation radicals J. Inorg. Biochem 2006, 100, 810–837. [DOI] [PubMed] [Google Scholar]
- (100).Harischandra DN; Zhang R; Newcomb M Photochemical Generation of a Highly Reactive Iron–Oxo Intermediate. A True Iron(V)–Oxo Species? J. Am. Chem. Soc 2005, 127, 13776–13777. [DOI] [PubMed] [Google Scholar]
- (101).Harischandra DN; Lowery G; Zhang R; Newcomb M Production of a Putative Iron(V)–Oxocorrole Species by Photo-Disproportionation of a Bis-Corrole–Diiron(IV)–μ-Oxo Dimer: Implication for a Green Oxidation Catalyst Organic Letters 2009, 11, 2089–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Zhang R; Vanover E; Chen T-H; Thompson H Visible light-driven aerobic oxidation catalyzed by a diiron(IV) μ-oxo biscorrole complex Applied Catalysis A: General 2013, 464–465, 95–100. [Google Scholar]
- (103).Wasbotten I; Ghosh A Theoretical Evidence Favoring True Iron(V)-Oxo Corrole and Corrolazine Intermediates Inorg. Chem 2006, 45, 4910–4913. [DOI] [PubMed] [Google Scholar]
- (104).Latifi R; Valentine JS; Nam W; de Visser SP Predictive studies of H-atom abstraction reactions by an iron(iv)–oxo corrole cation radical oxidant Chem. Commun 2012, 48, 3491–3493. [DOI] [PubMed] [Google Scholar]
- (105).Avidan-Shlomovich S; Gross Z Reaction mechanism for the highly efficient catalytic decomposition of peroxynitrite by the amphipolar iron(iii) corrole 1-Fe Dalton Trans 2015, 44, 12234–12243. [DOI] [PubMed] [Google Scholar]
- (106).Biswas AN; Pariyar A; Bose S; Das P; Bandyopadhyay P Mild oxidation of hydrocarbons catalyzed by iron corrole with tert-butylhydroperoxide Catal. Commun 2010, 11, 1008–1011. [Google Scholar]
- (107).Schwalbe M; Dogutan DK; Stoian SA; Teets TS; Nocera DG Xanthene-Modified and Hangman Iron Corroles Inorg. Chem 2011, 50, 1368–1377. [DOI] [PubMed] [Google Scholar]
- (108).Zyska B; Schwalbe M Synthesis of sterically hindered xanthene-modified iron corroles with catalase-like activity Chem. Commun 2013, 49, 3799–3801. [DOI] [PubMed] [Google Scholar]
- (109).McGown AJ; Kerber WD; Fujii H; Goldberg DP Catalytic Reactivity of a Meso-N-Substituted Corrole and Evidence for a High-Valent Iron–Oxo Species J. Am. Chem. Soc 2009, 131, 8040–8048. [DOI] [PubMed] [Google Scholar]
- (110).Kerber WD; Ramdhanie B; Goldberg DP H2O2 Oxidations Catalyzed by an Iron(III) Corrolazine: Avoiding High-Valent Iron–Oxido Species? Angew. Chem., Int. Ed 2007, 46, 3718–3721. [DOI] [PubMed] [Google Scholar]
- (111).Kurahashi S; Ikeue T; Sugimori T; Takahashi M; Mikuriya M; Handa M; Ikezaki A; Nakamura M Formation and characterization of five- and six-coordinate iron(III) corrolazine complexes J. Porphyrins Phthalocyanines 2012, 16, 518–529. [Google Scholar]
- (112).Cho K; Leeladee P; McGown AJ; DeBeer S; Goldberg DP A High-Valent Iron–Oxo Corrolazine Activates C–H Bonds via Hydrogen-Atom Transfer J. Am. Chem. Soc 2012, 134, 7392–7399. [DOI] [PubMed] [Google Scholar]
- (113).Charnock JM; Garner CD; Trautwein AX; Bill E; Winkler H; Ayougou K; Mandon D; Weiss R Characterization of an Oxo(porphyrinato)manganese(IV) Complex by X-ray Absorption Spectroscopy Angew. Chem., Int. Ed. Engl 1995, 34, 343–346. [Google Scholar]
- (114).Groves JT; Lee J; Marla SS Detection and Characterization of an Oxomanganese(V) Porphyrin Complex by Rapid-Mixing Stopped-Flow Spectrophotometry J. Am. Chem. Soc 1997, 119, 6269–6273. [Google Scholar]
- (115).Jin N; Groves JT Unusual Kinetic Stability of a Ground-State Singlet Oxomanganese(V) Porphyrin. Evidence for a Spin State Crossing Effect J. Am. Chem. Soc 1999, 121, 2923–2924. [Google Scholar]
- (116).Meunier B; de Visser S. l. P.; Shaik S Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- (117).Meunier B Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage Chem. Rev 1992, 92, 1411–1456. [Google Scholar]
- (118).Nam W; Kim I; Lim MH; Choi HJ; Lee JS; Jang HG Isolation of an Oxomanganese(V) Porphyrin Intermediate in the Reaction of a Manganese(III) Porphyrin Complex and H2O2 in Aqueous Solution Chem. - Eur. J 2002, 8, 2067–2071. [DOI] [PubMed] [Google Scholar]
- (119).Albin V; Bedioui F First electrochemical evidence of existence of an oxomanganese(V) porphyrin intermediate in the reaction of manganese(III) porphyrin and hydrogen peroxide as a model of enzyme mimetics Electrochem. Commun 2003, 5, 129–132. [Google Scholar]
- (120).Shimazaki Y; Nagano T; Takesue H; Ye B-H; Tani F; Naruta Y Characterization of a Dinuclear MnV=O Complex and Its Efficient Evolution of O2 in the Presence of Water Angew. Chem., Int. Ed 2004, 43, 98–100. [DOI] [PubMed] [Google Scholar]
- (121).Zhang R; Newcomb M Laser Flash Photolysis Formation and Direct Kinetic Studies of Manganese(V)-Oxo Porphyrin Intermediates J. Am. Chem. Soc 2003, 125, 12418–12419. [DOI] [PubMed] [Google Scholar]
- (122).Zhang R; Horner JH; Newcomb M Laser Flash Photolysis Generation and Kinetic Studies of Porphyrin–Manganese–Oxo Intermediates. Rate Constants for Oxidations Effected by Porphyrin–MnV–Oxo Species and Apparent Disproportionation Equilibrium Constants for Porphyrin–MnIV–Oxo Species J. Am. Chem. Soc 2005, 127, 6573–6582. [DOI] [PubMed] [Google Scholar]
- (123).Kwong KW; Winchester CM; Zhang R Photochemical generation of manganese(IV)-oxo porphyrins by visible light photolysis of dimanganese(III) μ-oxo bis-porphyrins Inorg. Chim. Acta 2016, 451, 202–206. [Google Scholar]
- (124).De Angelis F; Jin N; Car R; Groves JT Electronic Structure and Reactivity of Isomeric Oxo-Mn(V) Porphyrins: Effects of Spin-State Crossing and pKa Modulation Inorg. Chem 2006, 45, 4268–4276. [DOI] [PubMed] [Google Scholar]
- (125).Balcells D; Raynaud C; Crabtree RH; Eisenstein O The rebound mechanism in catalytic C-H oxidation by MnO(tpp)Cl from DFT studies: electronic nature of the active species Chem. Commun 2008, 744–746. [DOI] [PubMed] [Google Scholar]
- (126).Sameera WMC; McGrady JE The role of substrate in unmasking oxyl character in oxomanganese complexes: the key to selectivity? Dalton Trans. 2008, 6141–6149. [DOI] [PubMed] [Google Scholar]
- (127).Li H-B; Tian SX; Yang J Theoretical Study of the Stepwise Protonation of the Dioxo Manganese(V) Porphyrin J. Phys. Chem. B 2008, 112, 15807–15812. [DOI] [PubMed] [Google Scholar]
- (128).Balcells D; Raynaud C; Crabtree RH; Eisenstein O A Rational Basis for the Axial Ligand Effect in C–H Oxidation by [MnO(porphyrin)(X)]+ (X = H2O, OH−, O2−) from a DFT Study Inorg. Chem 2008, 47, 10090–10099. [DOI] [PubMed] [Google Scholar]
- (129).Balcells D; Raynaud C; Crabtree RH; Eisenstein O C-H oxidation by hydroxo manganese(v) porphyrins: a DFT study Chem. Commun 2009, 1772–1774. [DOI] [PubMed] [Google Scholar]
- (130).Silaghi-Dumitrescu R High-valent metalloporphyrins in hydrocarbon activation: metal(v)-oxo or metal(v)-hydroxo? New J. Chem 2010, 34, 1830–1833. [Google Scholar]
- (131).Venturinelli Jannuzzi SA; Phung QM; Domingo A; Formiga ALB; Pierloot K Spin State Energetics and Oxyl Character of Mn-Oxo Porphyrins by Multiconfigurational ab Initio Calculations: Implications on Reactivity Inorg. Chem 2016, 55, 5168–5179. [DOI] [PubMed] [Google Scholar]
- (132).Kavousi H; Rezaeifard A; Raissi H; Jafarpour M A DFT investigation of axial N-donor ligands effects on the high valent manganese-oxo meso-tetraphenyl porphyrin J. Porphyrins Phthalocyanines 2015, 19, 651–662. [Google Scholar]
- (133).Rezaeifard A; Kavousi H; Raissi H; Jafarpour M Significant hydrogen-bonding effect on the reactivity of high-valent manganese(V)–oxo porphyrins in C–H bond activation: A DFT study J. Porphyrins Phthalocyanines 2015, 19, 1197–1203. [Google Scholar]
- (134).Lahaye D; Groves JT Modeling the haloperoxidases: Reversible oxygen atom transfer between bromide ion and an oxo-Mn(V) porphyrin J. Inorg. Biochem 2007, 101, 1786–1797. [DOI] [PubMed] [Google Scholar]
- (135).Song WJ; Seo MS; DeBeer George S; Ohta T; Song R; Kang M-J; Tosha T; Kitagawa T; Solomon EI; Nam W Synthesis, Characterization, and Reactivities of Manganese(V)–Oxo Porphyrin Complexes J. Am. Chem. Soc 2007, 129, 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Baglia RA; Prokop-Prigge KA; Neu HM; Siegler MA; Goldberg DP Mn(V)(O) versus Cr(V)(O) Porphyrinoid Complexes: Structural Characterization and Implications for Basicity Controlling H-Atom Abstraction J. Am. Chem. Soc 2015, 137, 10874–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Gross Z The Groves–Spiro Dioxomanganese(V) Story Angew. Chem., Int. Ed 2008, 47, 2737–2739. [DOI] [PubMed] [Google Scholar]
- (138).Jin N; Ibrahim M; Spiro TG; Groves JT Trans-dioxo Manganese(V) Porphyrins J. Am. Chem. Soc 2007, 129, 12416–12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Lee JY; Lee Y-M; Kotani H; Nam W; Fukuzumi S High-valent manganese(v)-oxo porphyrin complexes in hydride transfer reactions Chem. Commun 2009, 704–706. [DOI] [PubMed] [Google Scholar]
- (140).Jung J; Ohkubo K; Goldberg DP; Fukuzumi S Photocatalytic Oxygenation of 10-Methyl-9,10-dihydroacridine by O2 with Manganese Porphyrins J. Phys. Chem. A 2014, 118, 6223–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Arunkumar C; Lee Y-M; Lee JY; Fukuzumi S; Nam W Hydrogen-Atom Abstraction Reactions by Manganese(V)–and Manganese(IV)–Oxo Porphyrin Complexes in Aqueous Solution Chem. - Eur. J 2009, 15, 11482–11489. [DOI] [PubMed] [Google Scholar]
- (142).Cho K-B; Nam W A theoretical study into a trans-dioxo MnV porphyrin complex that does not follow the oxygen rebound mechanism in C-H bond activation reactions Chem. Commun 2016, 52, 904–907. [DOI] [PubMed] [Google Scholar]
- (143).Fukuzumi S; Fujioka N; Kotani H; Ohkubo K; Lee Y-M; Nam W Mechanistic Insights into Hydride-Transfer and Electron-Transfer Reactions by a Manganese(IV)–Oxo Porphyrin Complex J. Am. Chem. Soc 2009, 131, 17127–17134. [DOI] [PubMed] [Google Scholar]
- (144).Latifi R; Tahsini L; Karamzadeh B; Safari N; Nam W; de Visser SP Manganese substituted Compound I of cytochrome P450 biomimetics: A comparative reactivity study of MnV-oxo versus MnIV-oxo species Arch. Biochem. Biophys 2011, 507, 4–13. [DOI] [PubMed] [Google Scholar]
- (145).Crestoni ME; Fornarini S; Lanucara F Oxygen-Atom Transfer by a Naked Manganese(V)–Oxo–Porphyrin Complex Reveals Axial Ligand Effect Chem. - Eur. J 2009, 15, 7863–7866. [DOI] [PubMed] [Google Scholar]
- (146).Chiavarino B; Crestoni ME; Fornarini S; Lanucara F Probing bare high-valent transition oxo-metal complexes: an electrospray ionization Fourier transform ion cyclotron resonance study of reactive intermediates Eur J Mass Spectrom (Chichester, Eng) 2010, 16, 407–414. [DOI] [PubMed] [Google Scholar]
- (147).Lanucara F; Crestoni ME Biomimetic Oxidation Reactions of a Naked Manganese(V)–Oxo Porphyrin Complex Chem. - Eur. J 2011, 17, 12092–12100. [DOI] [PubMed] [Google Scholar]
- (148).Fukuzumi S; Mizuno T; Ojiri T Catalytic Electron-Transfer Oxygenation of Substrates with Water as an Oxygen Source Using Manganese Porphyrins Chem. - Eur. J 2012, 18, 15794–15804. [DOI] [PubMed] [Google Scholar]
- (149).Mohajer D; Abbasi M Solvent Effects on Catalytic Epoxidation of Alkenes by Tetra-n-butylammonium Periodate and (Tetraarylporphyrinato)manganese(III) Eur. J. Inorg. Chem 2008, 2008, 3218–3224. [Google Scholar]
- (150).Mohajer D; Jahanbani M A UV–vis study of the effects of alcohols on formation and stability of Mn(por)(O)(OAc) complexes Spectrochim. Acta, Part A 2012, 91, 360–364. [DOI] [PubMed] [Google Scholar]
- (151).Umile TP; Groves JT Catalytic Generation of Chlorine Dioxide from Chlorite Using a Water-Soluble Manganese Porphyrin Angew. Chem., Int. Ed 2011, 50, 695–698. [DOI] [PubMed] [Google Scholar]
- (152).Umile TP; Wang D; Groves JT Dissection of the Mechanism of Manganese Porphyrin-Catalyzed Chlorine Dioxide Generation Inorg. Chem 2011, 50, 10353–10362. [DOI] [PubMed] [Google Scholar]
- (153).Liu W; Groves JT Manganese Catalyzed C–H Halogenation Acc. Chem. Res 2015, 48, 1727–1735. [DOI] [PubMed] [Google Scholar]
- (154).Liu H-Y; Mahmood MHR; Qiu S-X; Chang CK Recent developments in manganese corrole chemistry Coord. Chem. Rev 2013, 257, 1306–1333. [Google Scholar]
- (155).Yu L; Wang Q; Dai L; Li W-Y; Chen R; Mahmood MHR; Liu H-Y; Chang C-K Solvent effects on oxygen atom transfer reaction between manganese(V)-oxo corrole and alkene Chin. Chem. Lett 2013, 24, 447–449. [Google Scholar]
- (156).Wang Q; Zhang Y; Yu L; Yang H; Mahmood MH; Liu H-Y Solvent effects on the catalytic activity of manganese(III) corroles J. Porphyrins Phthalocyanines 2014, 18, 316–325. [Google Scholar]
- (157).Mahmood MH; Wang H-H; Liu H-Y; Chang C-K Oxygen atom transfer reactions from sterically encumbered brominated (oxo)manganese(V) corroles to styrene J. Porphyrins Phthalocyanines 2015, 19, 1238–1250. [Google Scholar]
- (158).He J; Xu Z-G; Xu X; Gong L-Z; Mahmood MH; Liu H-Y Reactivity of (oxo)manganese(V) corroles in one-electron redox state: insights from conceptual DFT and transition state calculations J. Porphyrins Phthalocyanines 2013, 17, 1196–1203. [Google Scholar]
- (159).He Jing, X. Z-G, Zeng Yun-Xiu, Xu Xuan, Yu Lan, Wang Qi, Liu Hai-Yang Effect of Substituents on Mn–O Bond in Oxo-Manganese(V) Corrole Complexes Acta Phys. -Chim. Sin 2012, 28, 1658–1664. [Google Scholar]
- (160).Zhu C; Liang J; Wang B; Zhu J; Cao Z Significant effect of spin flip on the oxygen atom transfer reaction from (oxo)manganese(v) corroles to thioanisole: insights from density functional calculations Phys. Chem. Chem. Phys 2012, 14, 12800–12806. [DOI] [PubMed] [Google Scholar]
- (161).Jung J; Liu S; Ohkubo K; Abu-Omar MM; Fukuzumi S Catalytic Two-Electron Reduction of Dioxygen by Ferrocene Derivatives with Manganese(V) Corroles Inorg. Chem 2015, 54, 4285–4291. [DOI] [PubMed] [Google Scholar]
- (162).Guntner A; Faschinger F; Aichhorn S; Müllegger S; Schöfberger W Observation of High-Valent Manganese(V)–Corrole Complexes During Metalation of meso-Functionalized A3-Corroles under Aerobic Conditions Synlett 2015, 26, 2180–2184. [Google Scholar]
- (163).Bougher CJ; Liu S; Hicks SD; Abu-Omar MM Valence Tautomerization of High-Valent Manganese(V)-Oxo Corrole Induced by Protonation of the Oxo Ligand J. Am. Chem. Soc 2015, 137, 14481–14487. [DOI] [PubMed] [Google Scholar]
- (164).Ka WK; Ngo FL; Ranburger D; Malone J; Zhang R Visible light-induced formation of corrole-manganese(V)-oxo complexes: Observation of multiple oxidation pathways J. Inorg. Biochem 2016, 163, 39–44. [DOI] [PubMed] [Google Scholar]
- (165).Ng N-C; Mahmood MHR; Liu H-Y; Yam F; Yeung L-L; Chang C-K Appended corrole manganese complexes: Catalysis and axial-ligand effect Chin. Chem. Lett 2014, 25, 571–574. [Google Scholar]
- (166).Neu HM; Baglia RA; Goldberg DP A Balancing Act: Stability versus Reactivity of Mn(O) Complexes Acc. Chem. Res 2015, 48, 2754–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Joslin EE; Zaragoza JPT; Baglia RA; Siegler MA; Goldberg DP The Influence of Peripheral Substituent Modification on PV, MnIII, and MnV(O) Corrolazines: X-ray Crystallography, Electrochemical and Spectroscopic Properties, and HAT and OAT Reactivities Inorg. Chem 2016, Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Jung J; Neu HM; Leeladee P; Siegler MA; Ohkubo K; Goldberg DP; Fukuzumi S Photocatalytic Oxygenation of Substrates by Dioxygen with Protonated Manganese(III) Corrolazine Inorg. Chem 2016, 55, 3218–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Zhao H; Pierloot K; Langner EHG; Swarts JC; Conradie J; Ghosh A Low-Energy States of Manganese–Oxo Corrole and Corrolazine: Multiconfiguration Reference ab Initio Calculations Inorg. Chem 2012, 51, 4002–4006. [DOI] [PubMed] [Google Scholar]
- (170).Prokop KA; de Visser SP; Goldberg DP Unprecedented Rate Enhancements of Hydrogen-Atom Transfer to a Manganese(V)–Oxo Corrolazine Complex Angew. Chem., Int. Ed 2010, 49, 5091–5095. [DOI] [PubMed] [Google Scholar]
- (171).Janardanan D; Usharani D; Shaik S The Origins of Dramatic Axial Ligand Effects: Closed-Shell MnVO Complexes Use Exchange-Enhanced Open-Shell States to Mediate Efficient H Abstraction Reactions Angew. Chem., Int. Ed 2012, 51, 4421–4425. [DOI] [PubMed] [Google Scholar]
- (172).Neu HM; Yang T; Baglia RA; Yosca TH; Green MT; Quesne MG; de Visser SP; Goldberg DP Oxygen-Atom Transfer Reactivity of Axially Ligated Mn(V)–Oxo Complexes: Evidence for Enhanced Electrophilic and Nucleophilic Pathways J. Am. Chem. Soc 2014, 136, 13845–13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Neu HM; Quesne MG; Yang T; Prokop-Prigge KA; Lancaster KM; Donohoe J; DeBeer S; de Visser SP; Goldberg DP Dramatic Influence of an Anionic Donor on the Oxygen-Atom Transfer Reactivity of a MnV–Oxo Complex Chem. - Eur. J 2014, 20, 14584–14588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Yang T; Quesne MG; Neu HM; Cantú Reinhard FG; Goldberg DP; de Visser SP Singlet versus Triplet Reactivity in an Mn(V)–Oxo Species: Testing Theoretical Predictions Against Experimental Evidence J. Am. Chem. Soc 2016, 138, 12375–12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Liu W; Huang X; Cheng M-J; Nielsen RJ; Goddard WA; Groves JT Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin Science 2012, 337, 1322–1325. [DOI] [PubMed] [Google Scholar]
- (176).Simkhovich L; Gross Z Halogeno-Coordinated Iron Corroles Inorg. Chem 2004, 43, 6136–6138. [DOI] [PubMed] [Google Scholar]
- (177).Momenteau M; Reed CA Synthetic Heme-Dioxygen Complexes Chem. Rev 1994, 94, 659–698. [Google Scholar]
- (178).Shikama K Stability properties of dioxygen-iron(II) porphyrins: an overview from simple complexes to myoglobin Coord. Chem. Rev 1988, 83, 73–91. [Google Scholar]
- (179).Collman JP; Boulatov R; Sunderland CJ In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: San Diego, CA, USA, 2003; Vol. 11, p 1–49. [Google Scholar]
- (180).Collman JP; Boulatov R; Sunderland CJ; Fu L Functional Analogues of Cytochrome c Oxidase, Myoglobin, and Hemoglobin Chem. Rev 2004, 104, 561–588. [DOI] [PubMed] [Google Scholar]
- (181).Kim E; Chufán EE; Kamaraj K; Karlin KD Synthetic Models for Heme– Copper Oxidases Chem. Rev 2004, 104, 1077–1134. [DOI] [PubMed] [Google Scholar]
- (182).Ohta T; Liu J-G; Naruta Y Resonance Raman characterization of mononuclear heme-peroxo intermediate models Coord. Chem. Rev 2013, 257, 407–413. [Google Scholar]
- (183).Duerr K; Troeppner O; Olah J; Li J; Zahl A; Drewello T; Jux N; Harvey JN; Ivanovic-Burmazovic I Solution behavior of iron(iii) and iron(ii) porphyrins in DMSO and reaction with superoxide. Effect of neighboring positive charge on thermodynamics, kinetics and nature of iron-(su)peroxo product Dalton Trans. 2012, 41, 546–557. [DOI] [PubMed] [Google Scholar]
- (184).Franke A; Fertinger C; van Eldik R Axial Ligand and Spin-State Influence on the Formation and Reactivity of Hydroperoxo–Iron(III) Porphyrin Complexes Chem. - Eur. J 2012, 18, 6935–6949. [DOI] [PubMed] [Google Scholar]
- (185).Wang B; Li C; Cho K-B; Nam W; Shaik S The FeIII(H2O2) Complex as a Highly Efficient Oxidant in Sulfoxidation Reactions: Revival of an Underrated Oxidant in Cytochrome P450 J. Chem. Theory Comput 2013, 9, 2519–2525. [DOI] [PubMed] [Google Scholar]
- (186).Ohta T; Liu J-G; Nagaraju P; Ogura T; Naruta Y A cryo-generated ferrous-superoxo porphyrin: EPR, resonance Raman and DFT studies Chem. Commun 2015, 51, 12407–12410. [DOI] [PubMed] [Google Scholar]
- (187).Nagaraju P; Ohta T; Liu J-G; Ogura T; Naruta Y The secondary coordination sphere controlled reactivity of a ferric-superoxo heme: unexpected conversion to a ferric hydroperoxo intermediate by reaction with a high-spin ferrous heme Chem. Commun 2016, 52, 7213–7216. [DOI] [PubMed] [Google Scholar]
- (188).Sahu S; Goldberg DP Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective J. Am. Chem. Soc 2016, 138, 11410–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).McLain JL; Lee J; Groves JT In Biomimetic Oxidations Catalyzed by Transition Metal Complexes; Published by Imperial College Press and distributed by World Scientific Publishing Co.: 2011, p 91–169. [Google Scholar]
- (190).Weschler CJ; Hoffman BM; Basolo F Synthetic oxygen carrier. Dioxygen adduct of a manganese porphyrin J. Am. Chem. Soc 1975, 97, 5278–5280. [DOI] [PubMed] [Google Scholar]
- (191).Hoffman BM; Weschler CJ; Basolo F The dioxygen adduct of meso-tetraphenylporphyrinmanganese(II), a synthetic oxygen carrier J. Am. Chem. Soc 1976, 98, 5473–5482. [DOI] [PubMed] [Google Scholar]
- (192).Hoffman BM; Szymanski T; Brown TG; Basolo F The dioxygen adducts of several manganese(II) porphyrins. Electron paramagnetic resonance studies J. Am. Chem. Soc 1978, 100, 7253–7259. [Google Scholar]
- (193).Hanson LK Axial ligand effects on iron and manganese porphyrins: Extended hückel calculations of cyt p450 analogs and of O2 binding to iron and manganese Int. J. Quantum Chem 1979, 16, 73–87. [Google Scholar]
- (194).Urban MW; Nakamoto K; Basolo F Infrared spectra of molecular oxygen adducts of (tetraphenylporphyrinato)manganese(II) in argon matrixes Inorg. Chem 1982, 21, 3406–3408. [Google Scholar]
- (195).Dedieu A; Rohmer MM Oxygen binding to manganese porphyrin. An ab initio calculation J. Am. Chem. Soc 1977, 99, 8050–8051. [DOI] [PubMed] [Google Scholar]
- (196).Newton JE; Hall MB Generalized molecular orbital calculations on transition-metal dioxygen complexes: model for manganese porphyrin Inorg. Chem 1985, 24, 2573–2577. [Google Scholar]
- (197).Valentine JS; Quinn AE Reaction of superoxide with the manganese(III) tetraphenylporphine cation Inorg. Chem 1976, 15, 1997–1999. [Google Scholar]
- (198).Shirazi A; Goff HM Characterization of superoxide-metalloporphyrin reaction products: effective use of deuterium NMR spectroscopy J. Am. Chem. Soc 1982, 104, 6318–6322. [Google Scholar]
- (199).Nakagaki PC; Calderwood TS; Bruice TC Relevance of the reaction of a manganese(III) chelate with hydroxide ion to photosynthesis: Reaction of hydroxide ion with 5,10,15,20-tetrakis(2,4,6-trimethylphenyl)porphinatomanganese(III) in ligating and nonligating solvents Proc. Natl. Acad. Sci 1988, 85, 5424–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).VanAtta RB; Strouse CE; Hanson LK; Valentine JS Peroxo(tetraphenylporphinato)manganese(III) and chloro(tetraphenylporphinato)manganese(II) anions. Synthesis, crystal structures, and electronic structures J. Am. Chem. Soc 1987, 109, 1425–1434. [Google Scholar]
- (201).Jones RD; Summerville DA; Basolo F Manganese(II) porphyrin oxygen carriers. Equilibrium constants for the reaction of dioxygen with para-substituted meso-tetraphenylporphinatomanganese(II) complexes J. Am. Chem. Soc 1978, 100, 4416–4424. [Google Scholar]
- (202).Basolo F; Jones RD; Summerville DA Equilibrium Constants for the Axial Coordination of meso-Tetraphenylporphinatomanganese(II) and -chromium(III) Complexes. Acta Chemica Scandinavica 1978, 32A, 771–780. [Google Scholar]
- (203).Groves JT; Watanabe Y; McMurry TJ Oxygen activation by metalloporphyrins. Formation and decomposition of an acylperoxymanganese(III) complex J. Am. Chem. Soc 1983, 105, 4489–4490. [Google Scholar]
- (204).Groves JT; Watanabe Y Heterolytic and homolytic oxygen-oxygen bond cleavage reactions of acylperoxomanganese(III) porphyrins Inorg. Chem 1986, 25, 4808–4810. [Google Scholar]
- (205).Schappacher M; Weiss R Formation of manganese(IV)-oxo-porphyrin derivatives by decomposition of peroxycarbonate complexes Inorg. Chem 1987, 26, 1189–1190. [Google Scholar]
- (206).Sisemore MF; Selke M; Burstyn JN; Valentine JS Metalloporphyrin Peroxo Complexes of Iron(III), Manganese(III), and Titanium(IV). Comparative Studies Demonstrating That the Iron(III) Complex Is Extremely Nucleophilic Inorg. Chem 1997, 36, 979–984. [DOI] [PubMed] [Google Scholar]
- (207).Jin N; Lahaye DE; Groves JT A “Push–Pull” Mechanism for Heterolytic O–O Bond Cleavage in Hydroperoxo Manganese Porphyrins Inorg. Chem 2010, 49, 11516–11524. [DOI] [PubMed] [Google Scholar]
- (208).Gao Y; Åkermark T; Liu J; Sun L; Åkermark B Nucleophilic Attack of Hydroxide on a MnV Oxo Complex: A Model of the O–O Bond Formation in the Oxygen Evolving Complex of Photosystem II J. Am. Chem. Soc 2009, 131, 8726–8727. [DOI] [PubMed] [Google Scholar]
- (209).Kim SH; Park H; Seo MS; Kubo M; Ogura T; Klajn J; Gryko DT; Valentine JS; Nam W Reversible O–O Bond Cleavage and Formation between Mn(IV)-Peroxo and Mn(V)-Oxo Corroles J. Am. Chem. Soc 2010, 132, 14030–14032. [DOI] [PubMed] [Google Scholar]