

Visual Abstract

Key Words: atrial endocardium, endothelium, mechanical stretch, transient receptor potential channels
Abbreviations and Acronyms: Ab, antibody; AE, atrial endocardium; AF, atrial fibrillation; APB, aminoethoxydiphenyl borate; Ca2+, calcium; [Ca2+]i, intracellular global Ca2+; CM, cardiomyocyte; Dil-Ac-LDL, dil acetylated−low-density lipoprotein; ET, endothelin; HUVEC, human umbilical vein endothelial cell; iPS, induced pluripotent stem; OAG, 1-oleoyl-2-acetyl-sn-glycerol; TAC, thoracic aortic constriction; Tet, tetanus toxin; TRPC, transient receptor potential channel
Highlights
-
•
TRPC-6 is present in atrial endocardial endothelium in humans, pigs, and mice.
-
•
Endocardial TRPC-6 channels act as atrial mechanosensors, showing changes in localization, expression levels, and activity in response to applied mechanical stretch in a time-dependent manner.
-
•
The atrial endocardial endothelium exerts TRPC-6-dependent paracrine effects that increase the amplitude of myocardial Ca2+ transients under normal physiological conditions. These positive inotropic effects are lost in settings of acute stretch.
-
•
Endocardial TRPC-6 participates in a regulatory feedback loop that acutely protects against stretch-induced myocardial Ca2+ overload. However, with persistent stretch, reduced expression of endocardial TRPC-6 and irregular Ca2+ transient periodicity may contribute to an increased arrhythmia propensity.
Summary
Mechanoelectrical feedback may increase arrhythmia susceptibility, but the molecular mechanisms are incompletely understood. This study showed that mechanical stretch altered the localization, protein levels, and function of the cation-selective transient receptor potential channel (TRPC)-6 in atrial endocardial cells in humans, pigs, and mice. In endocardial/myocardial cross-talk studies, addition of media from porcine atrial endocardium (AE) cells altered the calcium (Ca2+) transient characteristics of human-induced pluripotent stem cell-derived cardiomyocytes. These changes did not occur with media from stretched AE cells. Our data suggested that endocardial TRPC-6-dependent paracrine signaling may modulate myocardial Ca2+ homeostasis under basal conditions and protect against stretch-induced atrial arrhythmias.
Transient receptor potential channels (TRPCs) are non-voltage-gated cation channels that contribute to mechanosensory transduction in diverse tissue types. The channels have 4 subunits that consist of 6 transmembrane domains, a pore domain, and cytoplasmic N- and C-terminal domains that form homo- or heterotetramers. There are 6 subfamilies, including the canonical TRPC subfamily that has 7 members: TRPC-1, TRPC-2, TRPC-4/5, and TRPC-1/3/6 (1).
TRPC-6 is highly selective for calcium (Ca2+) over other cations and has been implicated in Ca2+-dependent processes in the peripheral vasculature and the heart. TRPC-6 channels are activated by ligand binding to Gαq/11-protein coupled receptors or receptor tyrosine kinases (“receptor-operated Ca2+ entry”), and mechanical stimuli such as stretch, flow and osmotic pressure 2, 3, 4, 5. Whether TRPC-6 channels are directly or indirectly gated by mechanical factors is controversial 3, 6, 7, although a recent patch clamp study that examined TRPC-6 activity of liposome-reconstituted channels suggested that these channels are inherently mechanosensitive (8). Channel activity is also regulated by post-translational modification at several key glycosylation and phosphorylation sites 9, 10. TRPC-6 is highly expressed in vascular smooth muscle cells, where it mediates vasoconstrictor and proliferative responses to increased intra-arterial pressure 4, 11, 12. In ventricular cardiomyocytes, TRPC-6 is believed to contribute to excitation-contraction coupling, stretch-induced membrane depolarization, and pathological myocardial hypertrophy (13). Transgenic mice with TRPC-6 overexpression show exaggerated ventricular hypertrophy when subjected to transverse aortic constriction (TAC) (14). In contrast, mice that express a dominant negative TRPC-6 are relatively protected from TAC-induced hypertrophy (15). TRPC-6 is also present in sinus node cells, cardiac myofibroblasts, intracardiac ganglia, and in coronary artery smooth muscle cells, pointing to pleiotropic roles extrinsic to cardiomyocytes (CMs) 16, 17, 18, 19.
Ca2+ influx through TRPC-6 channels has been implicated in endothelial permeability and initiation of inflammatory signaling pathways in the peripheral vasculature (20). However, little is known about TRPC-6 in intracardiac endothelium. The endocardial endothelium is a thin monolayer in continuity with vascular endothelium that lines the interior surface of the cardiac chambers, providing an interface between circulating blood and the myocardium. Vascular endothelial cells are highly sensitive to stretch, with sustained stretch causing endothelial dysfunction as well as having adverse effects on underlying smooth muscle cells (21). Similarly, the ventricular endocardium is stretch-responsive, with local and paracrine effects that modify myocardial contractility and rhythmicity (22). Mechanical stretch is known to be a determinant of atrial size and function, and mechanoelectrical feedback has been linked to arrhythmia susceptibility 23, 24. However, whether the atrial endocardium (AE) might have a role in this process has not been established.
In this study, we investigated the hypothesis that endocardial TRPC-6 is required for mechanical stretch responses in the atrium. The aims of our study were: 1) to characterize TRPC-6 localization, protein levels, and activity in the AE at baseline and under conditions of mechanical stretch; and 2) to determine the consequences of stretch-induced endocardial TRPC-6 activation on myocardial function. Our data provided new insight into the roles of the AE and TRPC-6 channels in cardiac mechanotransduction and defined a novel cross-talk mechanism between AE and contractile CMs aside the Frank-Starling mechanism.
Methods
An expanded Methods section is provided in the Supplemental Appendix.
Isolation and culture of primary atrial endocardial endothelial cells
Atrial tissue was obtained from healthy adult Landrace pigs, and AE cells were isolated by exposure to 0.1% collagenase (BD Biosciences, Bedford, Massachusetts) in E199 medium (Sigma Aldrich, St Louis, Missouri) for 45 min at 37°C. Isolated cells were pelleted, placed on gelatin- and/or fibronectin-coated, 6-well plates in E199 medium supplemented with 20% fetal bovine serum, 1% endothelial cell growth factor (Sigma Aldrich), and incubated at 37°C for a another 6 h. Protocols were approved by the Garvan Institute St. Vincent’s Hospital Animal Ethics Committee.
Immunofluorescence analysis
Immunofluorescence labeling was performed in AE and human umbilical vein endothelial cells (HUVECs) (Clonetics, Lonzer Walkersville Inc., Walkersville, Maryland) plated on silicone stretch chambers, and in frozen tissue sections obtained from the hearts of anesthetized 12-week-old male wild-type mice (C57BL/6L) 14 days after TAC. Right atrial appendage tissue samples were also collected from 8 male Caucasian patients with (n = 4) and without (n = 4) a history of atrial fibrillation (AF) who were undergoing cardiothoracic surgical procedures. Protocols were approved by the Garvan Institute St. Vincent’s Hospital Animal Ethics Committee and the St. Vincent’s Hospital Human Research Ethics Committee. Cells and tissue sections were fixed then immunolabeled with primary antibody (Ab) (see the Supplemental Appendix for Ab information), washed in 1 × phosphate-buffered saline and incubated with secondary AlexaFluor-488/568/594 Ab (1:200 dilution) (Thermo Fisher Scientific, Waltham, Massachusetts). Samples were mounted on glass coverslips and analyzed under a confocal microscope (Zeiss LSM DUO 7, Carl Zeiss, Oberkochen, Germany) at magnification ×20.
Quantitative real-time polymerase chain reaction analysis
Total RNA was isolated from primary AE or control HUVEC cells using an RNeasy kit (Qiagen, Venlo, the Netherlands). One microgram of total RNA from each sample was reverse transcribed with the Super Script III First Strand Synthesis Super Mix Kit (Invitrogen, Carlsbad, California). The relative abundance of selected cDNA fragments was determined using SYBR-Green Master Mix (Roche, Basel, Switzerland), to which forward and reverse primers were added. Accumulation of polymerase chain reaction products and the threshold cycle were determined using the Light-Cycler 480 analyzer and analysis software (Applied Biosystems, Foster City, California).
Protein analysis
Total protein extracts and nuclear and cytosolic protein fractions were prepared from primary AE cells using the NE-PER kit (Pierce Biotechnology Inc., Rockford, Illinois) as per manufacturer’s instructions. Proteins were separated by sodium dodecyl sulphate−polyacrylamide gel electrophoresis using 10% gels and transferred to a polyvinylidene difluoride membrane (GE Healthcare, Little Chalfont, United Kingdom) overnight before hybridization with primary protein-specific Ab and horseradish peroxidase-conjugated goat antirabbit or antimouse secondary Ab (1:2,000; Amersham Biosciences Corp., Piscataway, New Jersey). Western blots were normalized to β-tubulin and quantified with ImageJ software.
Mechanical stretch protocol
AE and HUVEC cells were grown to confluence on gelatin and/or fibronectin pre-coated silicone chambers, placed on a stretch-base apparatus, and subjected to cyclic unidirectional stretch with a frequency of 1 Hz. A confocal microscope-mounted single-chamber stretch unit was used for 1- to 15-min stretch periods, and a stretch chamber apparatus (STREX ST-040, B-Bridge International, Mountain View, California) was used for stretch periods of 1 to 24 h. Acute stretch was defined as ≤4 h, and chronic stretch as 4 to 24 h.
Intracellular global Ca2+ measurement
Cell membrane-permeant, Ca2+ labeling dye fluo-4/acetoxymethyl ester (Fluo-4/AM, 5 μM; Molecular Probes) was added to the live cells followed by incubation for 20 min, at room temperature in the dark. Excess dye was washed off, and the cells were allowed additional incubation at 37°C for 10 to 15 min in complete E199 media. Before Ca2+ measurements, the growth media was replaced with physiological salt solution devoid of Ca2+: 0 Ca2+PSS (140 mM sodium chloride, 5 mM potassium chloride, 1 mM magnesium chloride, 10 mM glucose, 2 mM ethyleneglycotetraacetic acid, 5 mM hydroxyethyl piperazine ethanesulfonic acid, pH 7.4). Cells were then exposed to cyclic stretch, and Ca2+ influx in the cells was initiated by addition of 1 mM Ca2+ chloride to the cell bathing media. Fluorescence intensity within the cell was recorded with the Zeiss LSM 7 confocal laser scanning system. In channel-inhibition studies, cells were pre-treated with channel inhibitors: GsMTx-4 (5 μM, for 1 h), nifedipine (10 μM, for 10 min), 2-aminoethoxydiphenyl borate (2-AP) (50 μM, for 5 min) and a custom-made pore-blocking TRPC-6 Ab (1:100, for 1 h). The specificity and dose-dependent inhibitory action of the Ab on TRPC-6 channels was previously demonstrated (25).
Preconditioning studies
AE cells were bathed in hydroxyethyl piperazine ethanesulfonic acid–buffered Tyrode’s solution and exposed to cyclic stretch for 0, 1, 4 and 24 h. Pre-conditioned media was collected and added to human-induced pluripotent stem (iPS) cell-derived CMs (Cor.4u, Axiogenesis AG, Cologne, Germany) that were preloaded with Ca2+ labeling dye Fluo-4/AM. Ca2+ transients were video recorded using the Zeiss LSM 7 confocal laser scanning system at an average speed of 30 ms/frame, and were analyzed using Clampfit version 10.0 software (Molecular Devices, Sunnyvale, California).
Statistical analysis
Differences in continuous variables across multiple groups were assessed using 1-way analysis of variance, with the Student t-test used to compare subsets of 2 groups. Data were expressed as mean ± SEM (unless indicated otherwise). A p value <0.05 was considered statistically significant.
Results
TRPC-6 localization in normal and dilated atria
Patterns of TRPC-6 localization were determined by immunostaining in mice with and without TAC-induced ventricular hypertrophy (Supplemental Figures 1A to 1D). In sham-operated mice, TRPC-6 was present in the CM sarcolemma, T-tubules, and intercalated disks in both the atria (Supplemental Figure 1E) and ventricles (Supplemental Figure 1F). In mice 14 days post-TAC, the overall distribution of myocardial TRPC-6 was similar to sham-operated mice (Supplemental Figures 1E and 1F). As previously reported (14), TRPC-6 transcript levels were increased in the ventricular myocardium post-TAC (data not shown). In sham-operated animals, TRPC-6 was seen not only in the atrial myocardium, but also along the AE membrane where it co-localized with the endothelial cell-surface marker PECAM-1 (Figure 1A, top panel and inset). In contrast, in the dilated atria of TAC mice, endocardial TRPC-6 was primarily present in and around the nucleus (Figure 1A, bottom panel and inset). These TAC-induced changes were not observed for the related mechanosensitive TRPC family member, TRPC-1 (Supplemental Figure 2). We also evaluated TRPC-6 localization in human atrial tissue samples (Supplemental Table 1). TRPC-6 was located along the AE cell surface in patients with normal-sized atria and no AF (Figure 1B, top panel and inset). In patients with atrial dilatation, all of whom also had AF, endocardial TRPC-6 was mostly accumulated intracellularly (Figure 1B, bottom panel and inset).
Figure 1.

Distribution of TRPC-6 in Dilated Atria
(A) Immunofluorescence evaluation of transient receptor potential channel (TRPC-6) (green) in atrial tissue from mice subjected to thoracic aortic constriction (TAC) (bottom row) and in sham-operated animals (top row). In sham-operated mice, endocardial TRPC-6 is present as a thin green line, co-localizing with the endothelial-specific cell-membrane marker PECAM-1 (red). Fourteen days post-TAC, endocardial TRPC-6 was mostly located around and within the nuclei. Similar re-distribution of endocardial TRPC-6 was seen in human right atrial tissue samples (B) from patients with (bottom row) and without (top row) a history of atrial fibrillation (AF). Cell nuclei are outlined by 4′,6-diamidino-2-phenylindole (blue). Scale bar = 10 μm.
A novel porcine primary AE cell culture model
To further evaluate the effects of atrial stretch, we established a novel primary AE cell culture model using tissue obtained from normal porcine atria. We verified the general endothelial cell phenotype of these AE cells by comparison with the well-characterized HUVECs using standard criteria 26, 27. Detailed description of AE cell isolation and characterization is included in Supplemental Figures 3 to 5.
Stretch alters AE cell morphology
Vascular endothelial cells showed adaptive responses within minutes after applied mechanical stretch, with subsequent changes dependent on the duration of the stretch stimulus (21). We subjected AE cells to unidirectional cyclic stretch (10% elongation, 1-Hz frequency [0.5-s stretch – 0.5-s relaxation per cycle]), ranging from 1 min to 24 h. Within 1 h of stretch, cells became elongated and aligned perpendicular to the direction of stretch (Supplemental Figure 6). Internal stress fibers also aligned in a similar orientation. These changes were progressive and became uniform after 4 h of stretch. By 24 h, 90% of the cells also exhibited marked hypertrophy.
Effects of stretch on AE TRPC-6 expression
With immunofluorescence labeling, TRPC-6 was demonstrated at the AE cell membrane, with some staining also present around the nucleus in the vicinity of the endoplasmic reticulum under baseline conditions (Figure 2A, left column). There were no effects on TRPC-6 localization with acute (15 min) stretch (Figure 2A, middle column). However, application of chronic (24 h) stretch was associated with redistribution of TRPC-6 from the cell periphery into the cytosol, where there was strong perinuclear and nuclear staining (Figure 2A, right column). Western blotting showed that protein levels of TRPC-6 in AE cells increased within the first hour of stretch, after which they started to decline, with significant differences seen at 24 h compared with baseline levels (Figure 2B). Immunoblotting of nuclear and cytoplasmic extracts from control and 24-h stretched AE cells confirmed the nuclear translocation of TRPC-6 (Supplemental Figure 7A). Stretch-induced changes in cellular localization were not seen for TRPC-1 (Supplemental Figure 8A), TRPC-3 (data not shown), L-type Ca2+ channel, Cav1.2 (Supplemental Figure 8B), ryanodine receptor (Supplemental Figure 9A), or SERCA2a (Supplemental Figure 9B).
Figure 2.

Effects of Stretch on TRPC-6 Expression and Localization in AE Cells
(A) Fluorescence immunostaining of TRPC-6 (green), F-actin (red), and nuclear 4′,6-diamidino-2-phenylindole (DAPI) stain (blue) in control (nonstretched) atrial endocardium (AE) cells (left column), and in cells stretched for 15 min (middle column) and 24 h (right column). In the control cells and cells subjected to 15-min stretch, TRPC-6 is localized at the cell membrane and other intracellular locations. In cells stretched for 24 h, TRPC-6 accumulates in the cell interior, mostly in and around the nucleus. Scale bar = 20 μm. (B) Western blot evaluation of TRPC-6 protein levels in AE cells stretched for 0, 1, 4, and 24 h. Representative blots (left panel) and quantification from 3 different TRPC-6 immunoblots (right panel) are shown. β-tubulin immunolabeling was used as loading control. See Supplemental Figure 7B for full Western blots. All values are mean ± SEM. *p < 0.001 versus control cells. Abbreviation as in Figure 1.
TRPC-6 activity in AE cells is determined by the duration of stretch
We next sought to determine the effects of stretch on TRPC-6 activity in AE cells. The Ca2+ conducting activity of TRPC-6 was indirectly evaluated by intracellular global [Ca2+]i Fluo-4 fluorescence studies, with Ca2+ influx into cells induced by addition of 1 mM Ca2+ to the Ca2+-free external media. There were no significant changes in Ca2+ uptake when cells were stretched acutely (5 min) at 5% elongation and then kept statically; however [Ca2+]i levels increased markedly at 10% elongation (Figure 3A and 3B, Supplemental Video 1). Addition of the L-type Ca2+ channel inhibitor, nifedipine, had a modest effect on this stretch response, but greater reductions in [Ca2+]i were seen with GsMTx-4 and 2-APB (Figure 3C). Both GsMTx-4 and 2-APB inhibit TRPC-6 (1), but can also act on other mechanosensitive channels (GsMTx-4) or store-operated channels (2-APB). To evaluate the specific effects of TRPC-6 inhibition, we used a custom made and previously validated pore-blocking TRPC-6 Ab (25), and we found that Ab administration substantially reduced the stretch-induced increase in [Ca2+]i levels (Figure 3D).
Figure 3.
Ca2+ Measurements in AE Cells Exposed to Acute Unidirectional Cyclic Stretch
(A) Increases in intracellular calcium levels ([Ca2+]i) due to external Ca2+ entry were demonstrated by augmentation of Fluo-4 signals in stretched AE cells (right panels) but not in control cells (left panels). Fluorescence recordings were initiated after acute cyclic stretch holding at maximum distension while confocal imaging. (B) Quantitative time curves of Ca2+ entry into AE cells in control cells (n = 37), and in cells stretched at 5% (n = 58) and 10% (n = 72) intensity. Bar graph shows mean increase in [Ca2+]i at different stretch intensities. (C) Representative time curves of Ca2+ entry after 10% stretch in control (n = 45) and stretched AE cells pre-treated with inhibitors of mechanosensitive (GsMTx-4; n = 55), store-operated (2-aminoethoxydiphenyl borate [2-APB]; n = 60) and voltage-gated (nifedipine; n = 55) Ca2+ channels. Bar graphs for mean data are shown. (D) Addition of TRPC-6-specific pore-blocking (TRPC-6 Ab) attenuated Ca2+ influx in 10% stretched cells (control: n = 40; 10% stretch: n = 36; 10% stretch + TRPC-6 Ab: n = 38). For (B to D), internal stores were pre-emptied with thapsigargin to rule out contributions from internal endoplasmic reticulum Ca2+ release to F/F0. Data are shown as mean ± SEM. *p < 0.001 versus control cells; ❖p < 0.001 versus 10% stretch. See Supplemental Video 1. Abbreviations as in Figures 1 and 2.

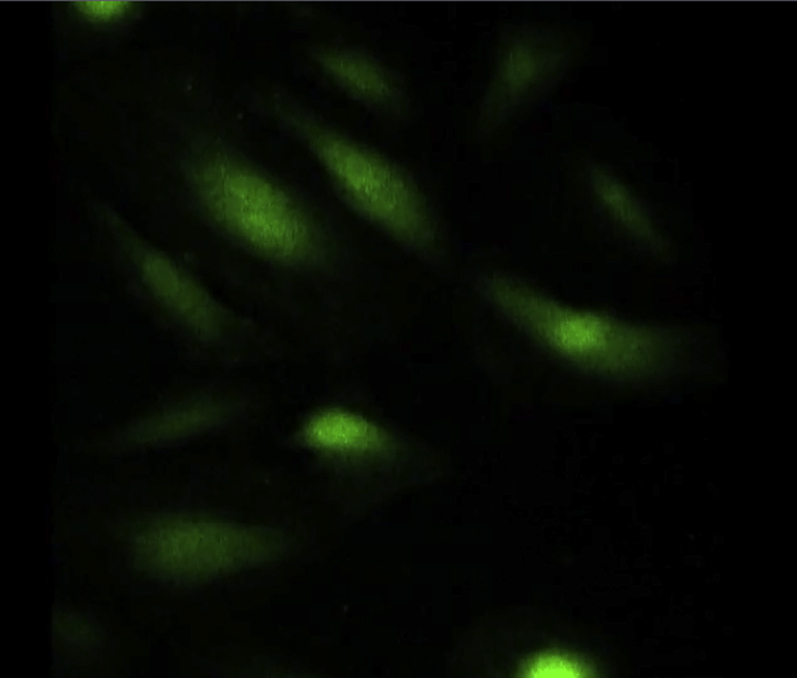
Ca2+ influx in Fluo-4 labeled AE cells, stretched for 5 min at 10% elongation.
In contrast to acute stretch, Ca2+ uptake was almost completely abolished in cells subjected to chronic (24 h) stretch (Figure 4B). This could be the result of several factors, including: 1) direct inactivation of TRPC-6 due to stretch-induced conformational changes of the channel and/or disrupted interactions with proteins that control its mechanosensitivity; and/or 2) downregulation of TRPC-6 expression in the AE cell membrane. The first possibility was ruled out by application of the TRPC-6-specific activator, hyperforin (10 μM), which was able to activate channels and cause Ca2+ influx in both nonstretched and 24-h stretched cells (Figure 4B). Hyperforin treatment also allowed visualization of the TRPC-6 Ca2+ conducting effect and Ca2+ influx dynamics at the AE cell membrane (Figure 4A). In nonstretched cells, Ca2+ uptake via TRPC-6 was initially seen as a continuous, intense green line along the cell surface, followed by cytosolic redistribution of the Ca2+ signal (Figure 4A top panel, Supplemental Video 2). After chronic (24 h) stretch, Ca2+ influx through TRPC-6 in elongated cells was restricted to small, discrete areas that formed an interspersed punctate pattern along the cell membrane (Figure 4A, bottom panel, Supplemental Video 3). These changes were not seen when hyperforin was co-administered with TRPC-6 pore-blocking Ab (Supplemental Video 4). These appearances could be indicative of depleted levels of TRPC-6 at the cell membrane and/or an increased distance between the individual channels or channel aggregates caused by the increased membrane perimeter. Addition of the membrane-permeable di-acyl glycerol-analogue, 1-oleoyl-2-acetyl-sn-glycerol (OAG; 100 μM), also increased Ca2+ uptake in both nonstretched and stretched cells (Figure 4B). This provided further evidence for functional stretch-sensitive channels, including TRPC-6, because OAG activates the channels by changing the bilayer curvature (7). OAG is a potent TRPC-6 activator, but is also known to activate TRPC-3 and TRPC-7, which may contribute in part to the more pronounced Ca2+ uptake under these experimental conditions (12).
Figure 4.
Effects of Chronic (24 h) Cyclic Stretch on [Ca2+]i in AE Cells
(A) Control (top panel) and stretched (bottom panel) AE cells were stained with Fluo-4 following administration of hyperforin to activate TRPC-6. In control cells, strong and continuous Ca2+ influx through the evenly distributed TRPC-6 channels at the cell surface is indicated (white arrows, top panel inset). In chronically stretched cells, Ca2+ influx at the cell membrane is restricted to small, localized areas that form a green punctate pattern (white arrows, bottom panel inset). (B) Bar graph showing the effects of chronic stretch, hyperforin, and 1-oleoyl-2-acetyl-sn-glycerol (OAG), a mechanosensitive channel activator, on the [Ca2+]i (respective cell numbers = 26, 30, 32, 25, 33, 40, in the order shown in graph). Ca2+ uptake by TRPC-6 and mechanosensitive Ca2+ channels, in general, is lower in the 24-h stretched AE cells compared with the non-stretched (control) cells. (C) Representative TRPC-6 immunoblot of biotin-labeled/streptavidin-immunoprecipitated cell membrane fraction (M) and total cell lysate (T) from 0-, 1-, and 24-h stretched AE cells, showing depleted levels of TRPC-6 in the cell membrane fraction of the 24-h stretched cells. Bar graph shows quantification of TRPC-6 levels from 3 separate experiments. β-tubulin immunolabeling was used as loading control. See Supplemental Figure 7C for full Western blots. All values are mean ± SEM. *p < 0.001 versus control cells. See Supplemental Videos 2, 3, and 4. Abbreviations as in Figures 1, 2, and 3.

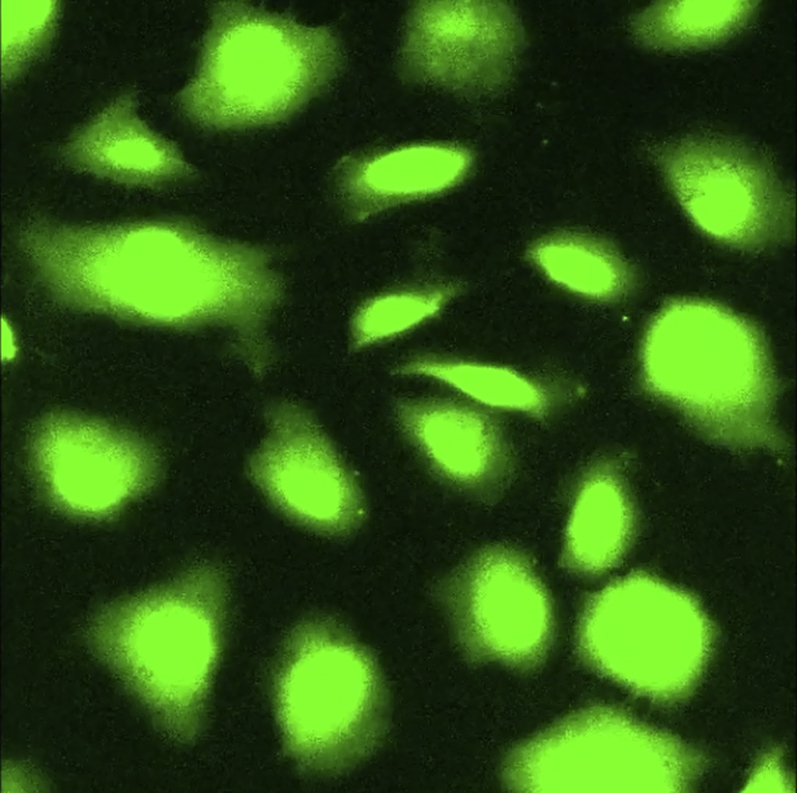
Ca2+ uptake in Fluo-4 labeled and hyperforin treated non-stretched AE cells.
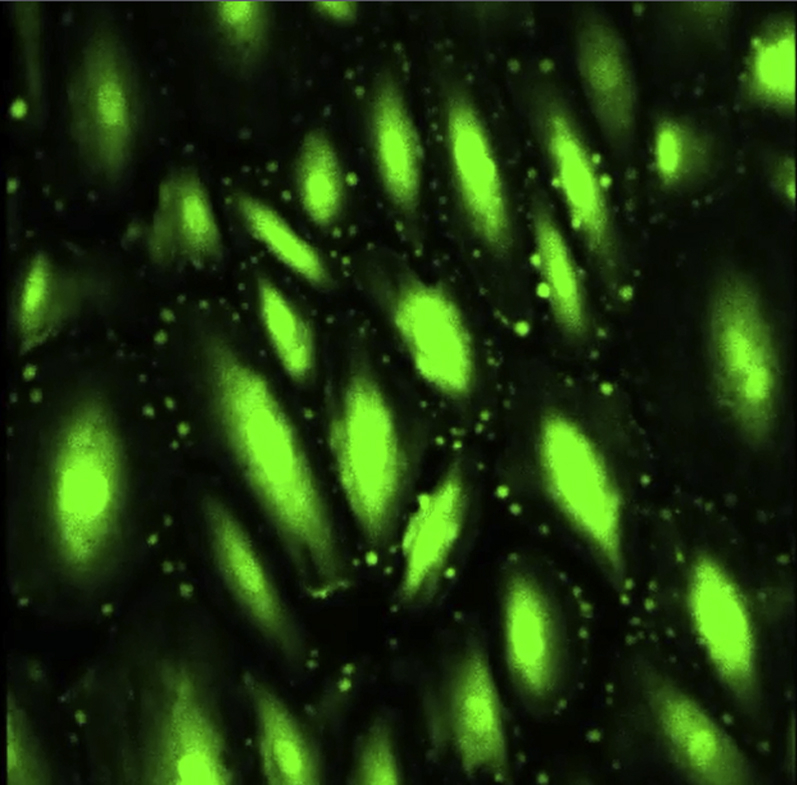
Ca2+ uptake in Fluo-4 labeled and hyperforin treated 24 h stretched AE cells.
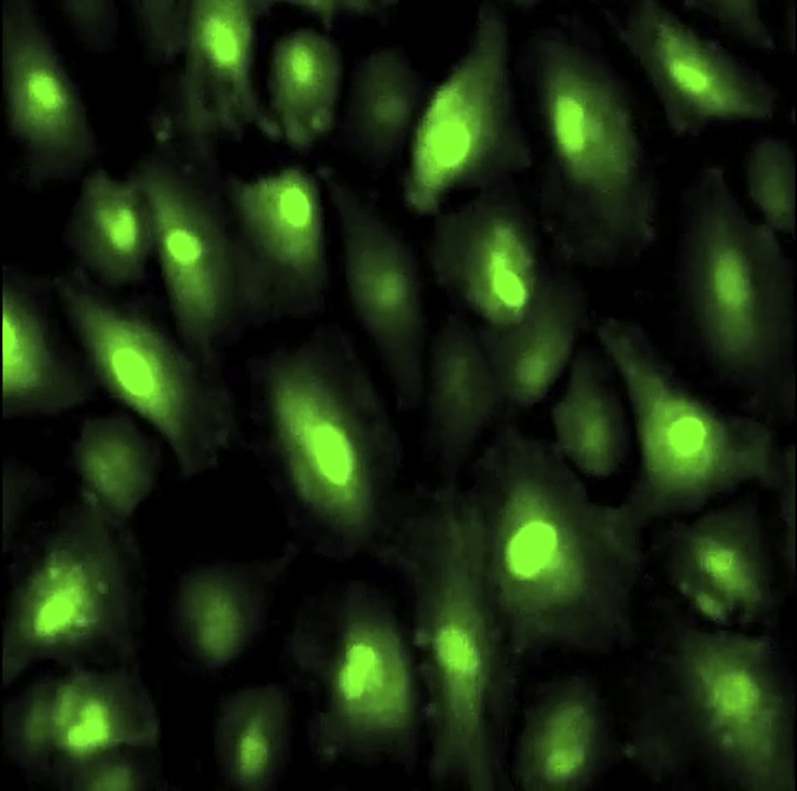
Ca2+ uptake in Fluo-4 labeled and hyperforin treated 24 h stretched AE cells in which TRPC6 activity was inhibited by pore-blocking anti-TRPC6 antibody.
Because the hyperforin and OAG experiments indicated that functional TRPC-6 channels were present, we next sought to determine whether the reduced Ca2+ influx in chronically stretched cells was due to reduced TRPC-6 cell membrane expression. A biotinylation assay and Western blotting demonstrated that there was significant depletion of TRPC-6 in the cell membrane of AE cells after chronic (24 h) stretching (Figure 4C).
TRPC-6 trafficking defects in AE cells subjected to chronic stretch
The reduced abundance of TRPC-6 at the cell surface in AE cells after 24-h stretch could be caused by impaired exocytosis and/or increased endocytosis. TRPC-6 exocytosis is mediated by the growth factor−activated phosphoinositide-3-kinase pathway that promotes channel packing in transport vesicles that contain vesicle-associated membrane protein 2 (28). Exocytosis can be blocked by tetanus toxin (Tet) treatment that cleaves vesicle-associated membrane protein 2 (Supplemental Figure 10B). In nonstretched cells, Tet resulted in TRPC-6 accumulation in the cellular interior that was similar to the pattern observed after 24-h stretch (Supplemental Figure 10A). Subsequent treatment of these cells with hyperforin failed to induce TRPC-6 activity as seen in the control nonstretched cells, which confirmed the lack of functional TRPC-6 at the cell surface (Supplemental Figure 10C).
To evaluate endocytosis, we looked at the cellular uptake of dil acetylated-low density lipoprotein (Dil-Ac-LDL). After 24-h stretch, there was increased intracellular accumulation of Dil-Ac-LDL compared with non-stretched AE cells. There were no differences in Dil-Ac-LDL staining between stretched and nonstretched cells when endocytosis was blocked using dynasore, a dynamin inhibitor (Supplemental Figures 11A and 11B). Dynasore treatment also partially rescued TRPC-6 mislocalization in the stretched cells; channel internalization from the cell membrane was prevented, but translocation into the nucleus was not (Supplemental Figure 11C, arrows). Collectively, these data show that chronic stretch is associated with reduced exocytosis and increased endocytosis in AE cells, indicating that stretch-induced reductions in TRPC-6 expression at the cell surface are most likely due to defective trafficking.
Effects of TRPC-6 and stretch on paracrine functions of the AE
To determine whether AE cells might have paracrine effects that could affect myocardial function, we performed AE myocardial cross-talk studies in which Ca2+ transient dynamics were evaluated in cultured human iPS cell−derived CMs before and after the addition of media collected from nonstretched and stretched AE cells.
When media from nonstretched AE cells was added to spontaneously beating CMs, Ca2+ transient amplitude increased by a mean 37% compared with control CMs (Figures 5A and 5B, Supplemental Table 2, Supplemental Video 5). There was also an increase in the time to peak [Ca2+]I and decay time, and a reduction in transient frequency. When media from nonstretched cells incubated with pore-blocking TRPC-6 Ab was added to CMs, there was a further reduction in Ca2+ transient frequency, but no significant differences in amplitude or time to peak compared with untreated nonstretched cells. In a separate control experiment, we confirmed that the latter was not due to a direct nonparacrine action of any residual TRPC-6 Ab in the pre-conditioning media (Figure 5B, Supplemental Table 2). These findings suggested that the AE modulates CM Ca2+ handling under basal conditions and that TRPC-6 might contribute to these paracrine effects.
Figure 5.
Ca2+ Transient Measurements in Spontaneously Beating iPS Cell-Derived CMs Preconditioned With Media Collected From Nonstretched and Stretched AE Cells
(A) Representative Ca2+ transient traces from cardiomyocytes (CMs) bathed in Tyrode’s solution (control cells) or pre-conditioned with media collected from AE cells with or without stretch and addition of TRPC-6 pore-blocking Ab. (B) Bar graphs showing Ca2+ transient amplitude and frequency in CMs pre-conditioned with media from AE cells with or without stretch and addition of TRPC-6 pore-blocking Ab. All values are mean ± SEM. *p < 0.025 versus control cells; ❖p < 0.001 versus nonstretched cells (without Ab). See Supplemental Videos 5, 6, and 7. iPS = induced pluripotent stem; other abbreviations as in Figures 1 and 2.

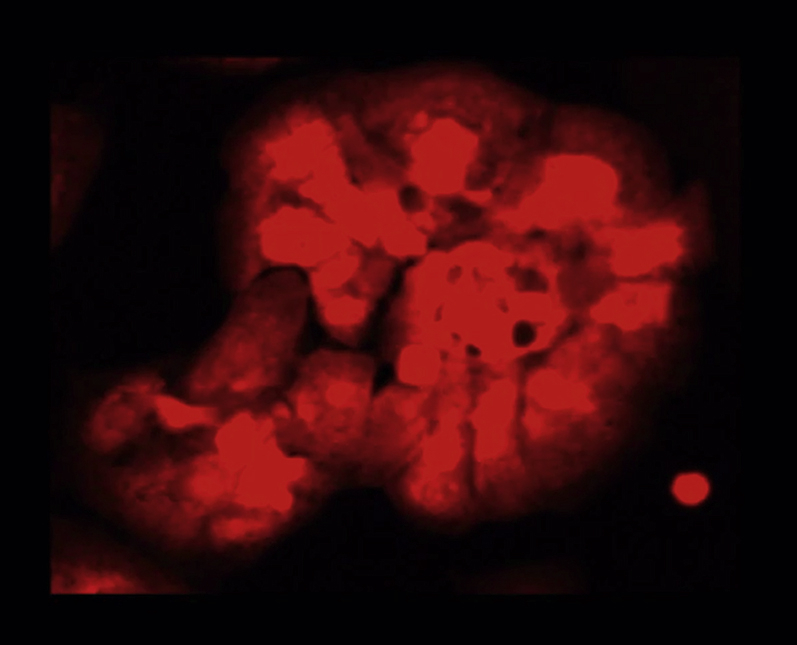
Ca2+ transients in Fluo-4 labeled cardiomyocytes preconditioned with media from non-stretched AE cells.
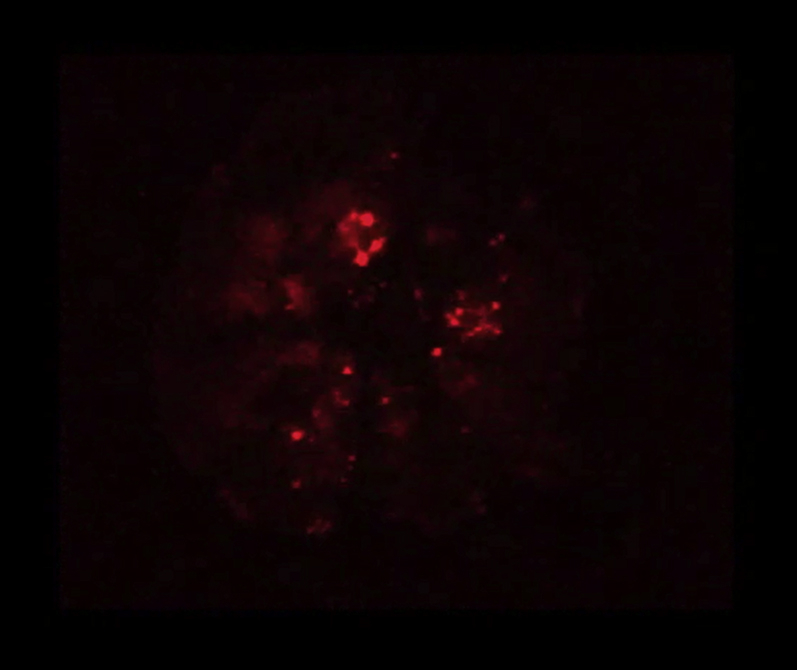
Ca2+ transients in Fluo-4 labeled cardiomyocytes preconditioned with media from 4 h stretched AE cells.
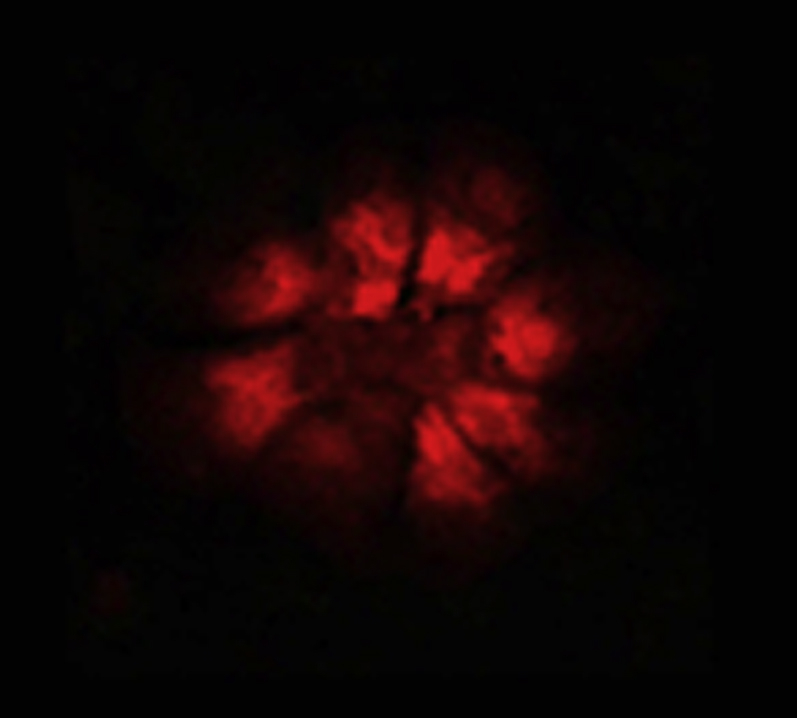
Ca2+ transients in Fluo-4 labeled cardiomyocytes preconditioned with media from 24 h stretched AE cells.
Unexpectedly, addition of media from AE cells that had been stretched for 4 h resulted in Ca2+ transient characteristics that were similar to control cells (Figures 5A and 5B, Supplemental Table 2, Supplemental Video 6). Pre-treating AE cells with TRPC-6 pore-blocking Ab almost completely reversed these stretch effects, resulting in Ca2+ transients that resembled those with media from Ab-treated nonstretched cells. These findings demonstrated that the normal paracrine function of the AE was lost during stretch and suggested that TRPC-6 has an important role in mediating these stretch effects.
To start to investigate what these AE-released paracrine factors might be, we studied the expression and functional effects of endothelin (ET)-1, a 21 amino acid peptide, which under normal conditions is synthesized exclusively by endothelial cells, but acts on the ETA/ETB receptors most densely expressed on the CMs and atrioventricular conducting system (29). ET-1 has been shown to have potent inotropic properties (29). Levels of ET-1 mRNA in AE cells were evaluated by quantitative real-time polymerase chain reaction analysis. Compared with non-stretched AE cells, modest reductions in ET-1 expression were seen after 1-h (15% decrease) and 4-h (20% decrease) stretch (Figure 6A). Under the same stretch conditions, TRPC-6 protein levels were significantly elevated (Figure 6B). However, after 24-h stretch, ET-1 expression was increased (35% vs. nonstretched cells) in parallel with down-regulation of TRPC-6 (Figure 6B). These results pointed to a potential negative feedback loop linking ET-1 and TRPC-6 expression. Addition of the TRPC-6 pore-blocking Ab markedly reduced ET-l levels in both nonstretched and stretched AE cells (70% decrease vs. nonstretched cells) (Figure 6A), providing evidence that TRPC-6 was required for ET-1 production under baseline conditions and in stretch responses.
Figure 6.

Paracrine Effect of AE-Secreted ET-1 on CM Ca2+ Transients Under Basal and Stretched Conditions
Bar graphs showing (A) endothelin (ET)-1 mRNA levels in stretched AE cells (n = 3), and (B) mean TRPC-6 expression in the cell membrane of control and stretched AE cells (n = 3). (C) Ca2+ transient traces in CMs under basal conditions (control cells) and in cells pre-conditioned with media from nonstretched and stretched AE cells with or without addition of 1 nM of ET-1 peptide. (D) Mean Ca2+ transient amplitude and frequency in CM bathed in Tyrode’s solution (control cells) or pre-conditioned with media from nonstretched and stretched AE cells, with or without addition of 1 nM of ET-1 peptide. (E) Ca2+ transient traces in CMs pre-treated with BQ-123 and pre-conditioned with media from non-stretched and 24-h stretched AE cells. All values are mean ± SEM. *p < 0.05 versus control cells; ❖p < 0.025 versus nonstretched; §p < 0.001 versus 4-h stretch. BQ-123, ET-1 receptor (ETA) inhibitor; other abbreviations in Figures 1, 2, and 5.
To determine whether ET-1 might have a role in AE stretch-induced paracrine effects, we first showed that addition of 1-nM synthetic ET-1 to cultured CMs in basal Tyrode’s solution increased the amplitude and reduced the frequency of Ca2+ transients, replicating the effects of media from nonstretched AE cells (Figures 6C and 6D, Supplemental Table 3). We speculated that the reduced ET-1 expression after 4-h stretch might have been contributing to the observed lack of effect on Ca2+ transients. When we added ET-1 (1 nM) to media from 4-h stretched AE cells, Ca2+ transient amplitude and frequency were similar to those seen with nonstretched cell media. In contrast to 4-h stretch, media from 24-h stretched AE cells increased the amplitude of Ca2+ transients, as seen for control cells with added ET-1 and for nonstretched cells. However, it also caused marked irregularity in the periodicity of Ca2+ transients (Figures 5A and 5B, Supplemental Video 7). Complete inhibition of ET-1 by addition of the ETA receptor inhibitor, BQ-123, to media from control, nonstretched, and 24-h stretched AE cells resulted in decreased Ca2+ transient amplitude as seen with 4-h stretch, but also arrhythmic transient periodicity (Figures 6D and 6E, Supplemental Table 3). The reduced Ca2+ transient frequency seen in BQ-123−treated control cells suggested that this inhibitor might act on ETA-related pathways in addition to those activated by ET-1.
Discussion
Data from our own group and others have shown that TRPC-6 is diffusely present in ventricular and atrial CMs and is involved in development of pathological myocardial hypertrophy 14, 15, 30, 31. We focused on an unexplored role of TRPC-6 in AE function. We found that TRPC-6 is present in AE cells and that its cellular localization, levels, and activity are altered by mechanical stretch in a time-dependent manner. In endocardial/myocardial cross-talk studies, media from nonstretched AE cells increased the amplitude and reduced the frequency of Ca2+ transients in iPS cell−derived CMs, which suggested that under basal conditions, AE cells have paracrine effects that modulate CM function. When AE cells were subjected to mechanical stretch (4 h), these paracrine effects were lost. Collectively, our data provided new insights into the roles of AE and TRPC-6 in normal CM biology and in stretch responses.
Our current understanding of cardiovascular endothelial function is largely based on studies of the peripheral vasculature, and to a lesser extent, the cardiac ventricles. The endothelium has an important barrier function, separating the circulating blood from smooth muscle and myocardial cells. Ca2+ signaling is a critical determinant of endothelial integrity, and is required for release of factors that prevent intraluminal thrombosis, as well as autocrine and paracrine factors that regulate vasomotor tone and myocardial contractility (22). In the peripheral vasculature, Ca2+ influx through TRP channels, including TRPC-6, has been shown to regulate endothelial permeability in response to inflammatory and ischemic stimuli, as well as contributing to flow-mediated and agonist-induced vasodilation (32). Stretch-activated cation channels have been identified previously in endocardial cells of porcine right atria, but their precise identity has not been determined (33). We show that TRPC-6 is present in AE cells of humans, pigs, and mice.
Using a novel porcine primary AE cell line, we found that AE cells were extremely sensitive to cyclic stretch, showing changes in cell morphology and function within minutes of stimulation. Protein levels of TRPC-6 in stretched AE cells initially increased (within the first hour), then progressively decreased in parallel with channel internalization. The relative depletion of TRPC-6 at the cell membrane with chronic stretch was associated with defective trafficking, with evidence for decreased exocytosis and increased endocytosis. Similar patterns of AE TRPC-6 internalization were also observed in the dilated atria of mice subjected to ventricular pressure overload and in humans with AF. Previous studies demonstrated the importance of TRPC-6 localization to its function. In vascular endothelial cells, the arachidonic acid metabolites, epoxyeicosatrienoic acids, facilitated TRPC-6 translocation to the caveolin-1−rich cell membrane areas that led to enhanced Ca2+ influx (34). Alpha-1A−adrenergic receptors that overexpressed CMs also showed increased translocation of TRPC-6 from the cytosol to the plasma membrane and [Ca2+]i overload (25). The nuclear localization of TRPC-6 staining was particularly notable. Ca2+ and potassium channels were demonstrated previously in nuclear membrane invaginations in endocardial cells and believed to contribute to nuclear transport (35). Whether TRPC-6 contributes to this and/or other Ca2+-dependent nuclear processes, including transcriptional regulation, remains to be determined. Any potential roles of TRPC-6 in nuclear function might be independent of its activity at the cell surface because blocking endocytosis in chronically stretched cells did not prevent its nuclear translocation.
To exhibit direct mechanosensitivity, ion channel activity needs to be altered by a membrane parameter (i.e., bilayer thickness, curvature, and/or a lateral pressure profile) that changes with mechanical deformation (36). The attenuation of stretch-induced [Ca2+]i by GsMTx-4 suggested that TRPC-6 might be a direct sensor of mechanical force, because GsMTx-4 acts by disrupting the channel-membrane bilayer interaction 7, 8. In our study, TRPC-6 activity in AE cells was determined by the duration of the stretch stimulus, with acute stretch associated with increases in TRPC-6 expression and concomitant global [Ca2+]i levels, which was consistent with channel activation. In contrast, chronic stretch resulted in TRPC-6 silencing and decreased local [Ca2+]i at the cell membrane that was indirectly caused by a channel trafficking defect.
Stretch-induced changes in global [Ca2+]i would be expected to have intrinsic effects on AE function, as well as potentially affecting myocardial function. For decades, it has been recognized that endocardial cells in the heart can regulate myocardial development, growth, contractile performance, and rhythmicity (22). These effects are believed to be mediated in part by factors secreted from the endothelium, including ET-1, nitric oxide, prostacyclin, and angiotensin II. We found that addition of media from nonstretched and stretched AE cells to cultured CMs altered the Ca2+ transient characteristics in different ways, which pointed to the presence of TRPC-6-dependent release of paracrine factors. Pre-conditioning CMs with media from nonstretched AE cells increased the Ca2+ transient amplitude, whereas media from 4-h stretched cells decreased Ca2+ transients in a manner previously reported in muscle strips or isolated whole hearts with damaged or complete endocardial denudation 37, 38. Our findings suggested that TRPC-6 might be involved in an AE-mediated feedback loop that sustains myocardial Ca2+ levels and contractile function under physiological conditions. In accordance with the Frank-Starling law, the heart responds to acute myocardial volume load by increasing myocardial [Ca2+]I and contractile force. Our data suggested that, in settings of stretch, the AE might initially have a counter-regulatory role that mitigates against myocardial [Ca2+]I overload. With persistent stretch, electrical and molecular remodeling of AE might further modify its paracrine effects on myocardial [Ca2+]I and result in increased arrhythmia susceptibility.
ET-1 is at least one of a number of possible factors that might be involved in this endocardial/myocardial regulatory mechanism. We showed that AE cells express ET-1 under basal conditions and that addition of ET-1 within physiological range (29) could stabilize Ca2+ transients in cultured CMs. When AE cells were stretched, ET-1 levels initially decreased (1- to 4-h stretch) then increased (24-h stretch). A reciprocal relationship between TRPC-6 activity and ET-1 secretion was evident, which was suggested in a proposed model of endothelium and/or myocardium cross-talk (Figure 7). Whether AE has similar TRPC-6-dependent effects on myocardial function in the intact atrium remains to be determined.
Figure 7.

Proposed Model of TRPC-6-Dependent Endocardial/Myocardial Feedback Loop
Paracrine effects of the AE on myocardial Ca2+ transient characteristics differ under baseline conditions and with varying duration of stretch, and are dependent on reciprocal changes in endocardial TRPC-6 activity and endothelin-1 secretion. Abbreviations as in Figures 1, 2, 3 and 6.
The atrial walls are subjected to cyclical variation in intracavity pressure and volume under normal physiological conditions. Acute atrial stretch can be induced by factors such as cardiac arrhythmias or heart failure, whereas chronic stretch occurs in pathological conditions that result in persistent elevation of atrial pressure and/or volume, such as left ventricular hypertrophy or dilatation and aortic or mitral valve disease. Mechanoelectrical feedback refers to the electrophysiological changes that occur in response to mechanical stress in the heart. In the atria, ventricles, and pulmonary veins, acute stretch has been shown to shorten the action potential duration and effective refractory period, as well as increasing premature depolarizations and arrhythmia vulnerability 23, 39. With chronic stretch, there is electrical and structural remodeling of the myocardium that can further alter impulse propagation and arrhythmia susceptibility (23). Stretch-activated channels have been proposed to contribute to mechanoelectrical feedback in atrial and ventricular CMs 40, 41. TRPC-6 has been identified as a candidate stretch-activated channel that mediates these cardiomyocyte effects (42). Our data raised the intriguing possibility that endocardial TRPC-6 might have a role in initial stages of the mechanotransduction cascade by acting as a stretch sensor and by modulating Ca2+ transient properties in CMs.
Our data challenged the prevailing view that the myocardium is the primary mediator of mechanical stretch responses in the heart. Elucidation of the intricately connected and inseparable functions of the endocardium and myocardium will be vital for a better understanding of the pathophysiology of stretch-induced atrial arrhythmias and the development of biologically-targeted therapies. TRPC-6-blocking drugs were proposed for treatment of cardiac hypertrophy and its arrhythmic complications 10, 13, 31, 43, but the rationale for these drugs was primarily based on studies of TRPC-6 function in the myocardium, with little account of the role of this channel in other cardiac compartments. As for many other ion channels, there might be a narrow therapeutic window, and extremes of TRPC-6 inhibition or activation might have deleterious effects.
Conclusions
Our data highlight the key role of AE TRPC6 channels in atrial responses to mechanical stretch. The localization, expression levels, and activity of AE TRCP6 channels and paracrine effects on myocardial function are determined by the duration of the stretch stimulus.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: These data provided proof of concept and opened a new avenue of investigation into the role of endocardial endothelium in atrial mechanoelectrical feedback. A better understanding of the mechanisms in which mechanical stretch stimuli acutely alter atrial function, and induce electrical and structural atrial remodeling, might provide new opportunities for prevention and treatment of stretch-induced arrhythmias. Increased endocardial TRPC-6 activity in settings of acute stretch might contribute to a regulatory feedback loop that protects against myocardial Ca2+ overload. If stretch persists, reductions of endocardial TRPC-6 might have pro-arrhythmogenic effects.
TRANSLATIONAL OUTLOOK: Further clinical studies are required to determine the extent to which endocardial TRPC-6 regulates myocardial Ca2+ homeostasis in the normal intact atrium, how AE function is altered in disease states, and whether TRPC-6-targeted therapies are effective.
Acknowledgments
The authors thank P. MacDonald for porcine atrial tissue, R. Graham and M. Mohl for the TRPC-6 Ab, S. Mann for assistance with setting up the iPS cell culture, and K. Dhital, K. Tan, and K. Ramesh for human myocardial tissue collection.
Footnotes
Dr. Fatkin was supported by a National Health and Medical Research Council (NHMRC) Program Grant (APP1074386), NHMRC Senior Research Fellowship (APP1025008), St. Vincent’s Clinic Foundation Grant, and the Estate of the Late RT Hall. Drs. Martinac, Friedrich, Feneley, and Nikolova-Krstevski were supported by a NHMRC Project Grant (APP1079398). Dr. Nikolova-Krstevski was supported by a St. Vincent’s Clinic Foundation Grant. Dr. Friedrich was supported by the German Academic Exchange Service (DAAD-Go8 57214153, 57058305) and Deutsche Forschungsgemeinschaft grant FR2993/23-1. Dr. Martinac was supported by the Group-of-Eight.
All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Boris Martinac and Diane Fatkin contributed equally to this work and are joint senior authors.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Contributor Information
Boris Martinac, Email: b.martinac@victorchang.edu.au.
Diane Fatkin, Email: d.fatkin@victorchang.edu.au.
Appendix
References
- 1.Hamill O.P., Maroto R. TRPCs as MS channels. Curr Top Membr. 2007;59:191–231. doi: 10.1016/S1063-5823(06)59009-X. [DOI] [PubMed] [Google Scholar]
- 2.Singh I., Knezevic N., Ahmmed G.U., Kini V., Malik A.B., Mehta D. Galphaq-TRPC-6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282:7833–7843. doi: 10.1074/jbc.M608288200. [DOI] [PubMed] [Google Scholar]
- 3.Inoue R., Jensen L.J., Jian Z. Synergistic activation of vascular TRPC-6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res. 2009;104:1399–1409. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 4.Zhang D.X., Gutterman D.D. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. 2011;57:133–139. doi: 10.1097/FJC.0b013e3181fd35d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson C., Dryer S.E. A mutation in TRPC-6 channels abolishes their activation by hypoosmotic stretch but does not affect activation by diacylglycerol or G protein signaling cascades. Am J Physiol Renal. 2014;306:F1018–F1025. doi: 10.1152/ajprenal.00662.2013. [DOI] [PubMed] [Google Scholar]
- 6.Gottlieb P., Folgering J., Maroto R. Revisiting TRPC1 and TRPC-6 mechanosensitivity. Pflugers Arch. 2008;455:1097–1103. doi: 10.1007/s00424-007-0359-3. [DOI] [PubMed] [Google Scholar]
- 7.Spassova M.A., Hewavitharana T., Xu W., Soboloff J., Gill D.L. A common mechanism underlies stretch activation and receptor activation of TRPC-6 channels. Proc Natl Acad Sci USA. 2006;103:16586–16591. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikolaev Y.A., Rohde P.R., Laver D.R., Martinac B. Mechanosensitivity of TRPC-6 ion channels reconstituted into liposomes. J Biophysics. 2016;110(Suppl):610a–611a. [Google Scholar]
- 9.Kinoshita H., Kuwahara K., Nishida M. Inhibition of TRPC-6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res. 2010;106 doi: 10.1161/CIRCRESAHA.109.208314. 1849–U153. [DOI] [PubMed] [Google Scholar]
- 10.Koitabashi N., Aiba T., Hesketh G.G. Cyclic GMP/PKG-dependent inhibition of TRPC-6 channel activity and expression negatively regulates cardiomyocyte NFAT activation novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beech D.J., Muraki K., Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol. 2004;559:685–706. doi: 10.1113/jphysiol.2004.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soboloff J., Spassova M., Xu W., He L.P., Cuesta N., Gill D.L. Role of endogenous TRPC-6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J Biol Chem. 2005;280:39786–39794. doi: 10.1074/jbc.M506064200. [DOI] [PubMed] [Google Scholar]
- 13.Eder P., Molkentin J.D. TRPC channels as effectors of cardiac hypertrophy. Circ Res. 2011;108:265–272. doi: 10.1161/CIRCRESAHA.110.225888. [DOI] [PubMed] [Google Scholar]
- 14.Kuwahara K., Wang Y.G., McAnally J. TRPC-6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X., Eder P., Chang B.J., Molkentin J.D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci USA. 2010;107:7000–7005. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju Y.K., Chu Y., Chaulet H. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ Res. 2007;100:1605–1614. doi: 10.1161/CIRCRESAHA.107.152181. [DOI] [PubMed] [Google Scholar]
- 17.Davis J., Burr A.R., Davis G.F., Birnbaumer L., Molkentin J.D. A TRPC-6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–715. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calupca M.A., Locknar S.A., Parsons R.L. TRPC-6 immunoreactivity is colocalized with neuronal nitric oxide synthase in extrinsic fibers innervating guinea pig intrinsic cardiac ganglia. J Comp Neurol. 2002;450:283–291. doi: 10.1002/cne.10322. [DOI] [PubMed] [Google Scholar]
- 19.Jung S., Strotmann R., Schultz N., Plant T.D. TRPC-6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- 20.Ahmmed G.U., Malik A.B. Functional role of TRPC channels in the regulation of endothelial permeability. Pflugers Arch. 2005;451:131–142. doi: 10.1007/s00424-005-1461-z. [DOI] [PubMed] [Google Scholar]
- 21.Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209–H1224. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 22.Brutsaert D.L. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 23.Ravens U. Mechano-electric feedback and arrhythmias. Prog Biophys Mol Biol. 2003;82:255–266. doi: 10.1016/s0079-6107(03)00026-9. [DOI] [PubMed] [Google Scholar]
- 24.Kohl P., Sachs F., Franz M.R. 2 ed. Oxford University Press; New York: 2011. Cardiac mechano-electric coupling and arrhythmias. [Google Scholar]
- 25.Mohl M.C., Iismaa S.E., Xiao X.H. Regulation of murine cardiac contractility by activation of alpha(1A)-adrenergic receptor-operated Ca(2+) entry. Cardiovasc Res. 2011;91:310–319. doi: 10.1093/cvr/cvr081. [DOI] [PubMed] [Google Scholar]
- 26.Chang M.W.F., Grillari J., Mayrhofer C. Comparison of early passage, senescent and hTERT immortalized endothelial cells. Exp Cell Res. 2005;309:121–136. doi: 10.1016/j.yexcr.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Kuruvilla L., T R.S., Kartha C.C. Immortalization and characterization of porcine ventricular endocardial endothelial cells. Endothelium. 2007;14:35–43. doi: 10.1080/10623320601177353. [DOI] [PubMed] [Google Scholar]
- 28.Xie J., Cha S.K., An S.W., Kuro O.M., Birnbaumer L., Huang C.L. Cardioprotection by Klotho through downregulation of TRPC-6 channels in the mouse heart. Nat Commun. 2012;3:1238. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mebazaa A., Mayoux E., Maeda K. Paracrine effects of endocardial endothelial cells on myocyte contraction mediated via endothelin. Am J Physiol. 1993;265:H1841–H1846. doi: 10.1152/ajpheart.1993.265.5.H1841. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Y., Huang H.X., Liu P. Expression and localization of TRPC proteins in rat ventricular myocytes at various developmental stages. Cell Tissue Res. 2014;355:201–212. doi: 10.1007/s00441-013-1733-4. [DOI] [PubMed] [Google Scholar]
- 31.Seo K., Rainer P.P., Hahn V.S. Combined TRPC3 and TRPC-6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci USA. 2014;111:1551–1556. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao X.Q., Garland C.J. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853–863. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 33.Hoyer J., Distler A., Haase W., Gogelein H. Ca2+ influx through stretch-activated cation channels activates maxi K+ channels in porcine endocardial endothelium. Proc Natl Acad Sci USA. 1994;91:2367–2371. doi: 10.1073/pnas.91.6.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fleming I., Rueben A., Popp R. Epoxyeicosatrienoic acids regulate trp channel-dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscl Throm Vasc Biol. 2007;27:2612–2618. doi: 10.1161/ATVBAHA.107.152074. [DOI] [PubMed] [Google Scholar]
- 35.Bkaily G., Avedanian L., Jacques D. Nuclear membrane receptors and channels as targets for drug development in cardiovascular diseases. Can J Physiol Pharmacol. 2009;87:108–119. doi: 10.1139/Y08-115. [DOI] [PubMed] [Google Scholar]
- 36.Perozo E., Kloda A., Cortes D.M., Martinac B. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat Struct Biol. 2002;9:696–703. doi: 10.1038/nsb827. [DOI] [PubMed] [Google Scholar]
- 37.Smith J.A., Shah A.M., Lewis M.J. Factors released from endocardium of the ferret and pig modulate myocardial-contraction. J Physiol. 1991;439:1–14. doi: 10.1113/jphysiol.1991.sp018653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brutsaert D.L., Meulemans A.L., Sipido K.R., Sys S.U. Effects of damaging the endocardial surface on the mechanical performance of isolated cardiac muscle. Circ Res. 1988;62:358–366. doi: 10.1161/01.res.62.2.358. [DOI] [PubMed] [Google Scholar]
- 39.Ravelli F. Mechano-electric feedback and atrial fibrillation. Prog Biophys Mol Biol. 2003;82:137–149. doi: 10.1016/s0079-6107(03)00011-7. [DOI] [PubMed] [Google Scholar]
- 40.Bode F., Sachs F., Franz M.R. Tarantula peptide inhibits atrial fibrillation. Nature. 2001;409:35–36. doi: 10.1038/35051165. [DOI] [PubMed] [Google Scholar]
- 41.Friedrich O., Wagner S., Battle A.R., Schurmann S., Martinac B. Mechano-regulation of the beating heart at the cellular level–mechanosensitive channels in normal and diseased heart. Prog Biophys Mol Biol. 2012;110:226–238. doi: 10.1016/j.pbiomolbio.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 42.Inoue R., Jian Z., Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Therapeut. 2009;123:371–385. doi: 10.1016/j.pharmthera.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Rowell J., Koitabashi N., Kass D.A. TRP-ing up heart and vessels: canonical transient receptor potential channels and cardiovascular disease. J Cardiovasc Transl. 2010;3:516–524. doi: 10.1007/s12265-010-9208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.