

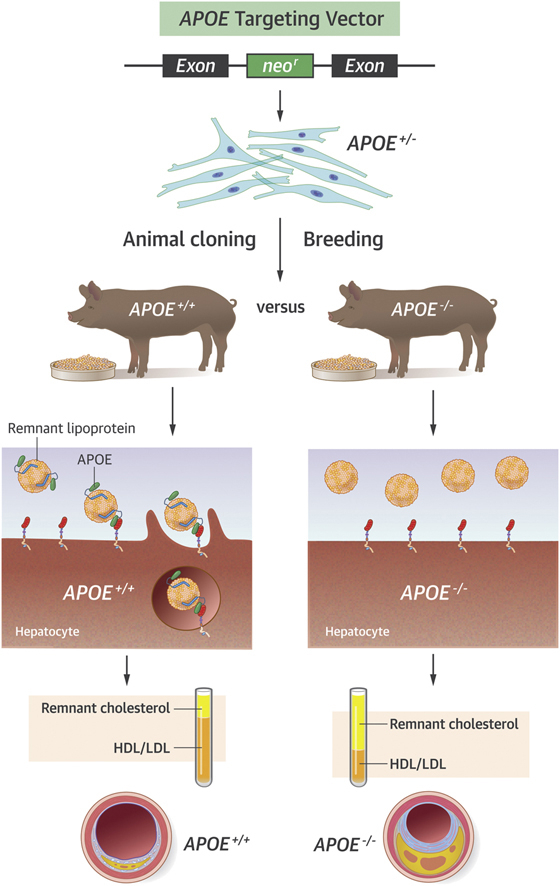
Visual Abstract
Key Words: apolipoprotein E, atherosclerosis, pig, remnant cholesterol dysbetalipoproteinemia
Abbreviations and Acronyms: APOB, apolipoprotein B; APOE, apolipoprotein E; cDNA, complementary DNA; HFHC, high-fat high-cholesterol; IDL, intermediate-density lipoprotein; LAD, left anterior descending (coronary artery); LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; LF, low-fat; Neo, neomycin; rAAV, recombinant adeno-associated virus; SMC, smooth muscle cell; VLDL, very-low-density lipoprotein
Highlights
-
•
APOE-deficient Yucatan minipigs were created by recombinant adeno-associated virus mediated gene targeting in porcine fibroblasts followed by somatic cell nuclear transfer.
-
•
APOE−/− minipigs displayed increased plasma cholesterol and accumulation of APOB48-containing chylomicron remnants on low fat-diet, which was significantly accentuated upon feeding a high-fat, high-cholesterol diet.
-
•
APOE−/− minipigs showed accelerated progressive atherosclerosis but not xanthoma formation indicating that remnant lipoproteinemia does not induce early lesions but is atherogenic in pre-existing atherosclerosis.
Summary
Deficiency of apolipoprotein E (APOE) causes familial dysbetalipoproteinemia in humans resulting in a higher risk of atherosclerotic disease. In mice, APOE deficiency results in a severe atherosclerosis phenotype, but it is unknown to what extent this is unique to mice. In this study, APOE was targeted in Yucatan minipigs. APOE−/− minipigs displayed increased plasma cholesterol and accumulation of apolipoprotein B-48–containing chylomicron remnants on low-fat diet, which was significantly accentuated upon feeding a high-fat, high-cholesterol diet. APOE−/− minipigs displayed accelerated progressive atherosclerosis but not xanthoma formation. This indicates that remnant lipoproteinemia does not induce early lesions but is atherogenic in pre-existing atherosclerosis.
The most efficient animal model of experimental atherosclerosis is the apolipoprotein E (APOE)–deficient mouse (1) which develops severe hypercholesterolemia and rapid, fibroatheromatous atherosclerosis on normal chow diet. APOE is synthesized in hepatocytes and several other cell types, and serves as a ligand for low-density receptor-related protein 1–mediated clearance of apolipoprotein B-48 (APOB-48)–containing chylomicron remnants and low-density lipoprotein receptor (LDLR)–mediated clearance of very-low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) (2). Due to hepatic editing of Apob mRNAs, mice secrete APOB-48–containing VLDLs, making them particularly dependent on APOE-mediated clearance mechanisms (3). However, loss of several other APOE-mediated functions may contribute to the severe atherosclerosis phenotype of the Apoe−/− mice. APOE has been shown to dampen inflammation, inhibit smooth muscle cell (SMC) proliferation and migration, and to be involved in reverse cholesterol transport (4). Lipoproteins from Apoe−/− mice also more readily produce foam cells in vitro for reasons that are not well understood (5). In humans, genetic variants that increase remnant lipoproteins similar to those seen with APOE-deficiency not only increase the risk of ischemic heart disease but also the plasma level of C-reactive protein (CRP). This is not seen with gene variants that increase low-density lipoprotein (LDL); possibly reflecting a pro-inflammatory effect of remnant lipoproteins in the arterial wall (6).
Several lines of hypercholesterolemic minipigs exist with spontaneous or genetically engineered defects in LDLR-mediated hepatic LDL uptake (7). These include familial hypercholesterolemia-Bretoncelles-Meishan pigs with a spontaneous LDLR mutation (8), human D374Y gain-of-function proprotein convertase subtilisin/kexin type 9 (PCSK9) transgenic Yucatan minipigs (9), and most recently, LDLR knockout Yucatan minipigs (10). When fed a high-fat high-cholesterol diet, these lines accumulate LDL-sized and larger APOB-100–containing lipoproteins and they develop fibroatheromatous atherosclerotic plaques in major arteries. These lines are useful for many purposes, but rates of plaque progression are modest, especially in the coronary arteries, and the disease does not reach a symptomatic stage with plaque rupture or thrombotic events within time frames observed so far.
In the present study, we created Yucatan minipigs with targeted disruption of the APOE gene and assessed the impact of accumulating remnant lipoproteins on the development of atherosclerosis.
Methods
Detailed methods are available in the Supplemental Appendix.
Animals
The Danish Animal Experiments Inspectorate approved all procedures involving animals. Yucatan minipigs were originally purchased from Moran Farm Enterprises (Ballina, Ireland) and a breeding colony was established and maintained at Aarhus University. All animals were housed in a specific pathogen-free stable facility under standard conditions. The minipigs were fed a low-fat (LF) standard pig diet until 8 weeks of age and subsequently a high-fat high-cholesterol (HFHC) diet consisting of standard pig feed comprising 20% (w/w) of lard and 2% cholesterol (Sigma-Aldrich, St. Louis, Missouri) until euthanization at the age of 16 weeks (APOE−/+) or 52 weeks (APOE+/+ and APOE−/−).
Construction of recombinant adeno-associated virus/APOE knockout targeting vector
As human APOE, the porcine APOE gene consists of 4 exons and the porcine protein has an amino acid similarity of 70.3% compared to human APOE 11, 12. The recombinant adeno-associated virus (rAAV)/APOE knockout (KO) vector was constructed as previously described 13, 14. Two homology arms (∼1 kb each), flanking exon 3 of the porcine APOE gene, were amplified by polymerase chain reaction (PCR) using Yucatan minipig genomic DNA as template. These 2 homology arms were subsequently linked to a neomycin (neor)/zeomycin (zeor) resistance gene cassette by a 3-way fusion PCR. The neor/zeor cassette, flanked by loxP sites, was comprised in a 4-kb PvuI fragment isolated from a plasmid called pNeDaKO-Neo (pronounced ‘p-Need a Knockout-Neo’); a generous gift from Bert Vogelstein and Kenneth W. Kinzler, The Johns Hopkins University Medical Institutions, Baltimore, Maryland. The 3-way fusion PCR was performed as described by Kohli et al. (13) using a Platinum Pfx polymerase (Thermo Fisher Scientific, Waltham, Massachusetts) and the following PCR protocol: 1 cycle of 94°C for 1 min; 25 cycles of 94°C for 30 s, 59°C for 30 s, and 68°C for 4 min; 1 cycle of 68°C for 7 min. Fusion products were digested with NotI and ligated to a NotI-cleaved pAAV-MCS plasmid backbone (Stratagene, San Diego, California, cat. no. 240071-5) containing an ampicillin (amp) resistance gene and the viral inverted terminal repeat sequences. The final rAAV/APOE KO plasmid construct was verified by NotI digestions and sequencing. Successful gene targeting using the rAAV/APOE KO vector results in deletion of 295 bp in the APOE gene including the entire exon 3. PCR primers for generating the rAAV/APOE KO construct are listed in Supplemental Table S1.
Cloning and embryo transfer
A detailed description of the methods used to clone piglets from genetically modified fibroblasts has been published previously (15). Briefly, after partial digestion of zona pellucida with 3.3 mg/ml pronase, oriented bisection of matured oocytes was performed manually under stereomicroscope with a microblade (AB Technology, Pullman, Western Australia, Australia). Each ooplast was attached with an APOE KO genetically modified fibroblast and fused in fusion medium (0.3 mol mannitol, 0.1 mmol MgSO4 and 0.01% [w/v] polyvinyl alcohol) in a fusion chamber (BTX microslide 0.5-mm fusion chamber, model 450; BTX Instrument Division, Harvard Apparatus, Holliston, Massachusetts) with a single direct current pulse of 2.0 kV/cm for 9 μs. One hour later, each ooplast-APOE KO genetically modified cell pair was fused with another ooplast in activation medium (fusion medium supplemented with 0.1 mmol CaCl2) by a single direct current pulse of 0.86 kV/cm for 80 μs. After incubation in porcine zygote medium 3 supplemented with 5 mg/ml cytochalasin B and 10 mg/ml cycloheximide for 4 h, the reconstructed embryos were cultured individually in well-of-the-wells made in 4-well dishes filled with porcine zygote medium 3 medium. Morulae and blastocysts were collected on days 5 and 6 and surgically transferred to Danish Landrace sows on day 4 or 5 after heat, which were observed 5 days after weaning. Five embryo transfers were performed with an average of 96 reconstructed embryos transferred per recipient sow (16). Successful pregnancies were diagnosed in 4 of the 5 recipient sows by ultrasonography on day 28. A total of 34 live-born piglets were delivered by caesarian section or vaginal birth 24 hours after induction with prostaglandin. Three of the cloned founders survived the neonatal period and were used for further breeding.
Pathology
Animals were intubated following sedation with 20 mg etomidate (Janssen-Cilag A/S, Birkerød, Denmark) intravenously, anesthetized using propofol (infusion rate 150 mg/h, B. Braun Melsungen AG, Melsungen, Germany) and fentanyl (infusion rate 1 mg/h, Hameln Pharmaceuticals Ltd., Gloucester, United Kingdom), and mechanically ventilated. To prevent clot formation, the animals were anticoagulated with 10,000 IU heparin injection intravenously before being euthanized by exsanguination. Hearts, aortas and right iliofemoral arteries were excised and immersed in 4% phosphate-buffered formaldehyde for 24 h followed by immersion in cold phosphate-buffered saline.
Aortas were cut open longitudinally and divided into thoracic aorta and abdominal aorta at the site between the 6th and 7th intercostal arteries. Aortas and the right iliofemoral arteries were stained with Sudan IV (Sigma-Aldrich; 5 g/l in 96% ethanol for 5 min followed by 90 s washout in 96% ethanol) and en face images of the vessels were obtained using a digital scanner (Epson Perfection V600 Photo, Seiko Epson Corporation, Suwa, Nagano, Japan).
The percentage of intimal surface covered with lesions was determined by automated computer-assisted measurement of Sudan-IV stained (sudanophilic) intima using ImageJ 1.48v (National Institutes of Health, Bethesda, Maryland). Lesions in the abdominal aorta were traced manually using ImageJ since most lesions were less sudanophilic.
Left anterior descending (LAD) coronary arteries were excised from the myocardium and the proximal 3 cm were divided into five 5-mm segments. The transversal segment, containing the most raised lesion (by macroscopic inspection) in the region proximal to the aortic trifurcation, was also obtained from each pig. Segments were paraffin-embedded and sectioned for histological analysis. LAD sections were stained with hematoxylin and eosin, and the lesions in the abdominal aorta sections were stained with elastin-trichrome.
Lesion sizes in the LAD were measured by computer-assisted planimetry. For the aortic plaques, maximum plaque-thickness was measured from the internal elastic lamina to the lumen.
We performed immunohistochemical staining for smooth muscle α-actin (SMαA) to detect SMCs in abdominal aorta lesions. Endogenous peroxidase activity was blocked by incubating the sections for 10 min with Hydrogen Peroxidase Block (Thermo Fisher Scientific) followed by blocking for nonspecific protein binding using Protein Block for 10 min. Sections were incubated with monoclonal mouse anti-human smooth muscle alpha actin antibody (Dako Denmark A/S, Glostrup, Denmark, cat. no. M0851, clone 1A4 [71 mg/l]) diluted 1:200 for 1 h at room temperature and visualized using horseradish peroxidase-conjugated secondary antibodies (Abcam, Cambridge, United Kingdom, EXPOSE detection IHC kit, cat. no. ab80436), 3,3′-diaminobenzidine for 7 min, and hematoxylin counterstain following the manufacturer's recommendations.
All microscopic analyses were performed blinded. Lesions were categorized according to the classification introduced by Virmani et al. (17) as normal, xanthoma, or progressive atherosclerotic lesions (pathological intimal thickening or fibroatheroma).
Statistics
All analyses were performed using Prism 6 (GraphPad Software Inc.). To compare 2 groups, we used the Student t test when data were normally distributed and Mann-Whitney U test when data were not normally distributed. Area under the curve was calculated by the trapezoidal method and compared to test for differences in time serial measurements. Plaque types were categorized as either progressive or nonprogressive types, and the frequency of progressive plaques was compared between groups using Fisher's exact test. p <0.05 was considered significant. In all figures, an * indicates p < 0.05 and a ** indicates p < 0.01. Exact p values are given in figure legends.
Results
Targeted KO of the porcine APOE gene
APOE comprises 2 structural domains separated by a hinge region. The N-terminal domain (amino acids 1–191) contains the receptor binding region and the C-terminal domain (amino acids 225-299) harbors the lipid binding region (18). We used rAAV-mediated gene targeting 14, 19 to create Yucatan minipigs that lack APOE due to deletion of the entire exon 3 and disruption of the receptor binding region. An rAAV-APOE KO viral vector (Figure 1A) was generated to produce rAAV particles used for gene targeting in primary porcine fibroblasts isolated from newborn male Yucatan minipigs. The APOE+/− gene targeted cell clones were validated by Southern blotting using an APOE-specific probe located upstream of the targeted region confirming deletion of exon 3, and a neor-specific probe detecting the neor region of the targeting vector to validate the absence of unwanted random integrations of the vector in the gene targeted cell clones.
Figure 1.

APOE Gene Targeting
(A) Schematic representation of the endogenous APOE locus, the gene targeting vector and the targeted APOE locus. The exons of the endogenous porcine APOE gene are shown as black boxes. Expression of the neor cDNA cassette is driven by a PGK promoter, whereas expression of the zeor cDNA cassette used for bacterial selection is driven by an EM7 promoter. A region of 295 bp in APOE, comprising the entire exon 3 and its flanking regions, is upon successful gene targeting replaced by the PGK-neor-EM7-zeor cassette resulting in a targeted DNA-fragment of 5,530 bp when XmnI-digested genomic DNA is hybridized with the APOE- or neor-specific probe. Positions of the PCR screening primer pairs F1/R1 and F2/R2 are indicated by small horizontal arrows. XmnI restriction sites are indicated by vertical arrows and Southern blot probes (APOEprobe and neorprobe) are illustrated as black horizontal bars. (B) Representative Southern blot of genomic DNA isolated from cloned APOE+/− Yucatan minipigs. XmnI-digested genomic DNA was electrophoresed, blotted on to a nitrocellulose membrane, and hybridized with the APOE probe resulting in a 5.5 kb and a 11.9 kb band representing the targeted and the wild-type allele, respectively (upper panel). The genomic DNA was also hybridized with the neor probe, detecting the neor cassette, yielding only the expected targeted 5.5 kb band (lower panel). Lanes 1-6: Genomic DNA from representative cloned APOE+/− Yucatan minipigs. Lane 7: Yucatan APOE+/+ control minipig. (C) Reverse transcriptase PCR. Total RNA was isolated from liver tissue and used for first strand cDNA synthesis. RT-PCR was performed using primers specific for the APOE exon 3 region or the porcine β-actin gene ACTB. PGK = phosphoglycerate kinase; PCR = polymerase chain reaction; RT-PCR = reverse transcriptase polymerase chain reaction.
Cloning of APOE KO Yucatan minipigs
One of the APOE+/− gene targeted cell clones was used as nuclear donor cells for somatic cell nuclear transfer by handmade cloning. All piglets were genotypically validated as APOE+/− by Southern blotting (Figure 1B). Three APOE+/− founders of the 34 cloned live-born male piglets survived the neonatal period. All 3 founders were healthy, and cytogenetical analysis performed on 1 of them revealed a seemingly normal karyotype (Supplemental Figure 1). The 3 APOE+/− Yucatan boars were mated with wild-type Yucatan sows to obtain naturally bred F1 (APOE+/−) individuals. The F1 minipigs were interbred to obtain F2 APOE+/+, APOE+/−, and APOE−/− minipigs including 6 male APOE+/+, 6 male APOE−/−, 7 male APOE+/−, 7 female APOE+/+, 7 female APOE+/−, and 5 female APOE−/− minipigs which were included in the study. The expected level of APOE expression in the 3 different genotypes was validated by reverse transcriptase PCR performed on liver tissue isolated from the minipigs at euthanization (Figure 1C, Supplemental Figure 2).
Accumulation of chylomicron remnants
At 8 weeks of age, when the animals were fed a LF standard diet, APOE−/− minipigs exhibited significantly higher total cholesterol (4.28 ± 0.31 mmol/l) compared to both APOE+/− (1.60 ± 0.08 mmol/l) and APOE+/+(1.81 ± 0.08 mmol/l) (p <0.001). Size-exclusion chromatography (Figure 2A, left column) and ultracentrifugation (Figure 2B, left column) showed that this was explained by accumulation of lipoproteins of IDL/VLDL size and density. The APOE−/− phenotype was intensified after 8 weeks on HFHC diet (Figure 2A, right column and Figure 2B, right column). Western blot of APOB showed accumulation of APOB-48 in APOE−/− pigs on LF diet and accentuated APOB-48 accumulation after HFHC diet feeding (Figure 2C, Supplemental Figure 3). This indicates that the accumulating lipoproteins were chylomicron remnants because pigs, like humans, only produce APOB-48–containing lipoproteins in the small intestine (20).
Figure 2.

Characterization of Cholesterol Profiles and Inflammatory Markers
(A) Size-exclusion chromatography on plasma pools of cholesterol in APOE+/+, APOE+/−, and APOE−/− Yucatan male and female minipigs on low-fat (LF) diet (left column) and after 8 weeks on high-fat high-cholesterol (HFHC) diet (right column). (B) Ultracentrifugation characterization of cholesterol on plasma pools from APOE+/+ and APOE−/− Yucatan minipigs on LF diet (left) and after 8 weeks on HFHC diet (right). (C) Western blot of APOB in plasma from APOE+/+, APOE+/−, and APOE−/− Yucatan minipigs on LF diet (upper) and on HFHC diet (lower). (D) Enzymatic measurement of total cholesterol at 8 (on LF diet), 16, 26, 36, and 52 (all on HFHC diet) weeks of age. **p = 0.0022 for APOE+/+ male versus APOE−/− male minipigs and p = 0.0051 for APOE+/+ female versus APOE−/− female minipigs. (E) Enzymatic measurement of triglycerides at 8 (on LF diet), 16, 26, 36, and 52 (all on HFHC diet) weeks of age. **p = 0.0087 for APOE+/+ male versus APOE−/− male minipigs. (F) Ultracentrifugation characterization of cholesterol in plasma from APOE+/+, APOE+/−, and APOE−/− Yucatan minipigs on HFHC diet at 52 weeks of age. **p = 0.0022 for IDL/VLDL between APOE+/+ male and APOE−/− male minipigs and p = 0.0025 for IDL/VLDL between APOE+/+ female and APOE−/− female minipigs. p = 0.0303 for HDL and p = 0.0025 for LDL between APOE+/+ female and APOE−/− female minipigs. (G) C-reactive protein (CRP) (left) and haptoglobin (right) levels in APOE+/+ and APOE−/− Yucatan minipigs at 8, 16, and 52 weeks of age. *p = 0.0411 for APOE+/+ male versus APOE−/− male minipigs. (D to G)APOE+/+ males n = 6, APOE+/+ females n = 7, and APOE−/− males n = 6, and APOE−/− females n = 5. Error bars indicate SEM. HDL = high-density lipoprotein; IDL = intermediate-density lipoprotein; VLDL = very-low-density lipoprotein.
Persistent increase in remnant cholesterol
To induce atherosclerosis, APOE+/+ and APOE−/− minipigs were fed an HFHC diet until euthanization at the age of 52 weeks. At this age, mean weights were 55.2 ± 3.5 kg for male APOE+/+, 48.7 ± 2.9 kg for male APOE−/− (p = 0.331), 59.1 ± 3.1 kg for female APOE+/+, and 58.8 ± 4.04 kg for female APOE−/− minipigs (p = 0.899). Total cholesterol rose rapidly in APOE−/− minipigs after 8 weeks on HFHC diet and remained higher throughout the whole period at a level ≈18 mmol/l compared with APOE+/+ minipigs that reached a level ≈10 mmol/l (Figure 2D). The differences were statistically highly significant when comparing areas under the curve. Triglycerides were also numerically higher in APOE−/− minipigs, although only statistically significant in male minipigs (Figure 2E). Lipoprotein ultracentrifugation performed on plasma obtained at 52 weeks of age showed significantly increased IDL/VLDL sub-fractions of cholesterol in APOE−/− compared with APOE+/+ minipigs (Figure 2F). Female APOE−/− minipigs had significantly lower LDL and high-density lipoprotein (HDL) cholesterol compared with APOE+/+ female minipigs.
Remnant hyperlipoproteinemia not associated with increased inflammation
Recent genetic population studies in humans showed a link between gene variants causing higher levels of remnant lipoproteins and plasma levels of CRP in humans (6), but neither CRP nor haptoglobin, another acute phase reactant, was significantly different between APOE−/− and APOE+/+ minipigs (Figure 2G). Although levels increased with age in female minipigs, values were lower than those seen in pigs with infectious disease (21).
Accelerated atherosclerosis
Atherosclerosis was analyzed in several arterial beds in the HFHC-fed APOE−/− and APOE+/+ minipigs. Lesions in the LAD coronary artery were generally small and the mean intimal area was unaffected by the lack of APOE and the resulting remnant lipoproteinemia (Figures 3A to 3C). Thoracic aortas were largely covered by non-raised sudanophilic fatty streaks (Supplemental Figures 4A and 4B). The extent, judged by en face measurement (Figures 3D to 3F), did not differ between APOE−/− and APOE+/+ minipigs. In contrast, in both the abdominal aortas and iliofemoral arteries where wild-type minipigs develop more advanced atherosclerosis, development of lesions were significantly accelerated in APOE−/− minipigs showing 2.7-fold (Figure 3G) and 2.8-fold (Figure 3H, Supplemental Figures 4C and 4D) increases in median surface coverage in the 2 arterial beds, respectively.
Figure 3.

Lesion Quantification
(A) Average cross-sectional intima area in left anterior descending (LAD) coronary artery. (B, C) Representative examples (green dots in A) of lesions in the LAD of an APOE+/+(B) and an APOE−/−(C) Yucatan minipig. Arrows indicate examples of foam cells. Scale bar = 100 μm. Both are examples of xanthomas with intima containing foam cells. (D, E) Representative examples (red dots in F and G) of en face stained aortic lesions from an APOE+/+(D) and an APOE−/−(E) Yucatan minipig. In both, abdominal aorta is depicted left and thoracic aorta depicted right. Red areas (stained with Sudan IV) represent lipid-rich areas of the aortic intima. (F) Surface covered with red-stained area in the thoracic aorta (mainly non-raised lesions). (G) Surface covered with red-stained area in the abdominal aorta (mainly raised lesions). **p = 0.0020. (H) Surface covered with red-stained area in the iliofemoral arteries. **p = 0.0059. In A, F to H: APOE+/+ n = 13 and APOE−/− n = 11. Bars indicate median.
We further characterized lesion types in histological sections from the LAD and abdominal aortas (Figure 4A). In serial sections of LADs, the most advanced lesion identified in each minipig was a xanthoma in all APOE+/+ minipigs except 1 female that had a fibroatheroma and was an outlier in all atherosclerosis measurements. In APOE−/− minipigs, the most advanced lesion types were xanthomas (73%) and pathological intimal thickening (27%). The frequency of progressive lesions (pathological intimal thickening and fibroatheroma) was not significantly different between APOE+/+ and APOE−/− minipigs.
Figure 4.

Lesion Characterization
(A) Plaque-classification of the most advanced lesion in the LAD coronary artery of each animal. (B) Plaque classification of the most raised lesion in close proximity to the aortic trifurcation. **p = 0.0059. (C) Thickness of the most raised lesion in close proximity to the aortic trifurcation. Bars indicate median. **p = 0.012. (D) Area stained positive for SMαA in the most raised lesion in close proximity to the aortic trifurcation. Bars indicate mean. (E, F) Representative examples (red dots in C and D) of elastin trichrome stained lesions in the most raised lesion in close proximity to the aortic trifurcation of an APOE+/+(E) and an APOE−/−(F) Yucatan minipig. Scale bar = 500 μm. (G, H) Same lesions as in E and F, respectively, immunohistochemically stained for SMαA. (A to D)APOE+/+ n = 13 and APOE−/− n = 11. SMαA = smooth muscle α-actin; other abbreviation as in Figure 3.
Histology of the most raised lesion identified in the distal part of the abdominal aorta (Figures 4B, 4E, and 4F) showed normal or adaptive intimal thickening (23%), xanthomas (31%), pathological intimal thickenings (38%), and fibroatheromas (8%, n = 1) in APOE+/+ minipigs. In APOE−/− minipigs, this was significantly skewed towards more advanced lesion stages with all showing progressive atherosclerotic lesions, 73% of which were pathological intimal thickenings and 27% of which were fibroatheromas. Lesion thickness of the abdominal aorta plaque was also significantly increased in APOE−/− compared with APOE+/+ minipigs (Figure 4C). All fibroatheromas were of the thick fibrous cap type and no thrombotic complications were observed.
Finally, as APOE has been shown to regulate SMC proliferation in mice, we addressed the possibility that exaggerated SMC accumulation could explain the acceleration of advanced atherosclerosis. Sections of lesions in the abdominal aortas were stained for smooth muscle α-actin, but the percentage of SMαA–positive area did not differ between APOE−/− and APOE+/+ minipigs (Figures 4D, 4G, and 4H)— neither did it differ when correlated to lesion size (data not shown).
Discussion
Defects or deficiency of APOE cause familial dysbetalipoproteinemia in humans, characterized by accumulations of VLDL, IDL, and chylomicron remnants and a higher risk of premature atherosclerotic disease (22). Whereas the human disease rarely becomes symptomatic before adulthood, the same genetic defect in mice is aggressive with rapid development of severe atherosclerosis. This is partly explained by the higher reliance on APOE for lipoprotein clearance in mice compared to humans (3), and consequently the much higher plasma cholesterol concentration resulting from its loss. However, it is also possible that a particular atherogenicity of the accumulating lipoproteins or loss of intrinsic anti-atherosclerotic effects of APOE identified in the mouse may contribute (4). The Apoe−/− mouse model is widely applied for experimental atherosclerosis because of its rapid lesion development, but it has an unusual etiology of atherosclerosis and its translational value is controversial (23).
In the present study, we created a model of dysbetalipoproteinemia in minipigs, where the distribution of lipoprotein classes and the lack of hepatic APOB editing are closer to human physiology, and examined the impact of remnant lipoproteinemia caused by APOE KO on the development of atherosclerosis. We found that, as in APOE-deficient humans, heterozygotes generally had normal lipid levels, but homozygotes showed large accumulations of APOB-48 remnant-type lipoproteins. Surprisingly, despite the severe remnant lipoproteinemia, which caused an approximate doubling of plasma total cholesterol, the effect on atherosclerosis development was modest and only present in the most atherosclerosis-susceptible arterial beds. APOE deficiency accelerated atherosclerosis in the abdominal aorta and iliofemoral arteries, which are predilection sites for progressive atherosclerosis in pigs and humans, but no effect was detected in thoracic aortas and the LAD where lesions were restricted to mainly xanthomas. In this way, the phenotype of APOE−/− minipigs was clearly different from that of D374Y-PCSK9 transgenic minipigs, which we have previously described using a similar diet composition, genetic background and follow-up time as in the present study. D374Y-PCSK9 minipigs exhibited similar increases in plasma cholesterol, but cholesterol was mainly carried by APOB-100–containing LDLs and larger lipoproteins, and atherosclerosis was significantly accelerated in all arterial beds examined, including the thoracic aorta and the LAD (9). The coronary atherosclerosis phenotype of the APOE−/− minipig model was also much milder than reported for several other porcine models, in which atherosclerosis is driven by increases in APOB100-containing lipoproteins, such as the diabetic Yorkshire model and pigs with spontaneous or induced defects in LDL-clearance 7, 24. Part of the difference to those models may also lie in the induction of diabetes, differences in diet composition, including the use of cholic acid to accentuate atherosclerosis, and potential differences in atherosclerosis susceptibility of the background strain.
Our findings indicate that the remnant lipoproteinemia of APOE−/− minipigs is not efficient in initiating atherosclerosis. However, it may contribute to further progression of pre-existing lesions, either alone or together with the loss of direct anti-atherogenic effects of APOE. One explanation for the lack of effect in vascular beds with early atherosclerosis could be abolishment of APOE-mediated binding of lipoproteins to intimal proteoglycans. APOE, similar to APOB, contains motifs that facilitate lipoprotein retention in the vascular wall (25). Loss of this binding type may diminish the atherogenicity of APOE-deficient lipoproteins during initiation of atherosclerosis, but the effect is likely lost with further plaque development as has previously been shown for APOB (26).
Study limitations
Further studies are needed to understand by which mechanisms APOE deficiency accelerates progressive atherosclerotic lesions. We addressed the possibilities that remnant lipoproteins could exert a pro-inflammatory effect in the body, as suggested by human genetic studies (6), or that loss of APOE would lead to increased SMC proliferation, but found no evidence that this was the underlying explanation.
Conclusions
We showed that targeted gene KO of APOE in minipigs causes severe remnant lipoproteinemia similar to human familial dysbetalipoproteinemia. We found that this increased progressive atherosclerotic lesions but not xanthoma formation. Further research in this model may pave the way for better understanding of the pathophysiological effects of remnant lipoproteinemia in atherosclerosis.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Remnant cholesterol increases atherosclerosis and risk of ischemic heart disease. Existing large-animal models of dyslipidemia and atherosclerosis, both diet-induced as well as genetically modified animal models, display cholesterol profiles resembling familial hypercholesterolemia. We here report the first large-animal model of APOE-deficiency and remnant lipoproteinemia modelling the dyslipidemia seen in patients with familial dysbetalipoproteinemia.
TRANSLATIONAL OUTLOOK: The human-like features of this animal model, in terms of size, lipoprotein metabolism, and histopathological features of atherosclerosis, may pave the way for better understanding of dysbetalipoproteinemia and the mechanisms involved in atherogenecity of remnant lipoproteins observed in humans.
Acknowledgments
The authors would like to thank Bert Vogelstein and Kenneth W. Kinzler, Johns Hopkins University, Baltimore, Maryland, for kindly providing the pNeDaKO plasmid vector and Dr. R. Jude Samulski and the UNC Vector Core Facility, Chapel Hill, North Carolina, for the rAAV packaging. They would also like to thank Peter M. Kragh and Lisbeth D. Schrøder, Trine S. Petersen, Christian Knudsen, Lisa Maria Røge, Dorte Qualmann, Zahra P. Nasr, Anette M. Pedersen, Janne Adamsen, Klaus Villemoes, and Ruth Kristensen for skilled technical assistance.
Footnotes
Supported by grants from The Danish Heart Association, Copenhagen, Denmark, The Novo Nordisk Foundation, Hellerup, Denmark, The Lundbeck Foundation, Copenhagen, Denmark, The Danish Council for Independent Research—Technology and Production Sciences, Copenhagen, Denmark, and Graduate School of Health, Aarhus University, Aarhus, Denmark. The CNIC is supported by the Spanish Ministry of Economy, Industry and Competitiveness (MINECO) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (MINECO award SEV-2015-0505). Aarhus University owns a patent on porcine APOE targeting with Drs. Bentzon, Sørensen, and Bolund listed as inventors (EP 2134847). Drs. Sørensen, Callesen, and Bolund are members of the EU COST Action BM1308 “Sharing advances on large animal models—SALAAM.” Dr. Bolund is the Chief Scientific Officer of PixieGene. Drs. Bentzon and Sørensen contributed equally to this work.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Contributor Information
Jacob F. Bentzon, Email: jacobfog.bentzon@cnic.es.
Charlotte B. Sørensen, Email: cbs@clin.au.dk.
Appendix
References
- 1.Plump A.S., Smith J.D., Hayek T. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 2.Véniant M.M., Zlot C.H., Walzem R.L. Lipoprotein clearance mechanisms in LDL receptor-deficient “apo-B48- only” and “apo-B100-only” mice. J Clin Invest. 1998;102:1559–1568. doi: 10.1172/JCI4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim E., Cham C.M., Véniant M.M., Ambroziak P., Young S.G. Dual mechanisms for the low plasma levels of truncated apolipoprotein B proteins in familial hypobetalipoproteinemia. Analysis of a new mouse model with a nonsense mutation in the Apob gene. J Clin Invest. 1998;101:1468–1477. [PMC free article] [PubMed] [Google Scholar]
- 4.Getz G.S., Reardon C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50:156–161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu D., Sharan C., Yang H. Apolipoprotein E-deficient lipoproteins induce foam cell formation by downregulation of lysosomal hydrolases in macrophages. J Lipid Res. 2007;48:2571–2578. doi: 10.1194/jlr.M700217-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Varbo A., Benn M., Tybjaerg-Hansen A., Nordestgaard B.G. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128:1298–1309. doi: 10.1161/CIRCULATIONAHA.113.003008. [DOI] [PubMed] [Google Scholar]
- 7.Shim J., Al-Mashhadi R.H., Sørensen C.B., Bentzon J.F. Large animal models of atherosclerosis — new tools for persistent problems in cardiovascular medicine. J Pathol. 2016;238:257–266. doi: 10.1002/path.4646. [DOI] [PubMed] [Google Scholar]
- 8.Thim T., Hagensen M.K., Drouet L. Familial hypercholesterolaemic downsized pig with human-like coronary atherosclerosis: a model for preclinical studies. EuroIntervention. 2010;6:261–268. doi: 10.4244/EIJV6I2A42. [DOI] [PubMed] [Google Scholar]
- 9.Al-Mashhadi R.H., Sorensen C.B., Kragh P.M. Familial hypercholesterolemia and atherosclerosis in cloned minipigs created by DNA transposition of a human PCSK9 gain-of-function mutant. Sci Transl Med. 2013;5:166ra1. doi: 10.1126/scitranslmed.3004853. [DOI] [PubMed] [Google Scholar]
- 10.Davis B.T., Wang X.-J., Rohret J.A. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan Miniature Pigs. PLoS ONE. 2014;9:e93457. doi: 10.1371/journal.pone.0093457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anna Brzozowska, Unni Grimholt, Mari Ann Kulseth, Inger Wold, Sissel Rogne The sequence of porcine apolipoprotein E (APOE) cDNA. DNA Seq Mapp. 1993;4:207–210. doi: 10.3109/10425179309015633. [DOI] [PubMed] [Google Scholar]
- 12.Ramsoondar J.J., Rucker E.B., Vasquez J.C. Isolation and genetic characterization of the porcine apolipoprotein E gene. Anim Genet. 1998;29:43–47. doi: 10.1046/j.1365-2052.1998.00273.x. [DOI] [PubMed] [Google Scholar]
- 13.Kohli M., Rago C., Lengauer C., Kinzler K.W., Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers C.S., Hao Y., Rokhlina T. Production of CFTR -null and CFTR- Δ F508 heterozygous pigs by adeno-associated virus–mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118:1571–1577. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y., Ostrup O., Li J. Increased blastocyst formation of cloned porcine embryos produced with donor cells pre-treated with Xenopus egg extract and/or digitonin. Zygote. 2012;20:61–66. doi: 10.1017/S096719941000064X. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt M., Kragh P.M., Li J. Pregnancies and piglets from large white sow recipients after two transfer methods of cloned and transgenic embryos of different pig breeds. Theriogenology. 2010;74:1233–1240. doi: 10.1016/j.theriogenology.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 17.Virmani R., Kolodgie F.D., Burke A.P., Farb A., Schwartz S.M. Lessons from sudden coronary death. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 18.Mahley R.W., Weisgraber K.H., Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimerʼs disease to AIDS. J Lipid Res. 2009:183–188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Y., Li J., Liu Y., Lin L., Du Y. High efficiency of BRCA1 knockout using rAAV-mediated gene targeting: developing a pig model for breast cancer. Transgenic Res. 2011;1:975–988. doi: 10.1007/s11248-010-9472-8. [DOI] [PubMed] [Google Scholar]
- 20.Black D.D., Davidsont N. Intestinal apolipoprotein synthesis and secretion in the suckling pig. J Lipid Res. 1989;30:207–218. [PubMed] [Google Scholar]
- 21.Heegaard P.M., Klausen J., Nielsen J.P. The porcine acute phase response to infection with Actinobacillus pleuropneumoniae. Haptoglobin, C-reactive protein, major acute phase protein and serum amyloid A protein are sensitive indicators of infection. Comp Biochem Physiol B Biochem Mol Biol. 1998;119:365–373. doi: 10.1016/s0305-0491(97)00362-3. [DOI] [PubMed] [Google Scholar]
- 22.Mahley R.W., Huang Y., Rall S.C., Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. 1999;40:1933–1949. [PubMed] [Google Scholar]
- 23.Pasterkamp G., Van Der Laan S.W., Haitjema S. Human validation of genes associated with a murine atherosclerotic phenotype. Arterioscler Thromb Vasc Biol. 2016;36:1240–1246. doi: 10.1161/ATVBAHA.115.306958. [DOI] [PubMed] [Google Scholar]
- 24.Hamamdzic D., Wilensky R.L. Porcine models of accelerated coronary atherosclerosis: role of diabetes mellitus and hypercholesterolemia. J Diabetes Res. 2013;2013:761415. doi: 10.1155/2013/761415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borén J., Williams K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–483. doi: 10.1097/MOL.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 26.Gustafsson M., Levin M., Skålén K. Retention of low-density lipoprotein in atherosclerotic lesions of the mouse: evidence for a role of lipoprotein lipase. Circ Res. 2007;101:777–783. doi: 10.1161/CIRCRESAHA.107.149666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.