

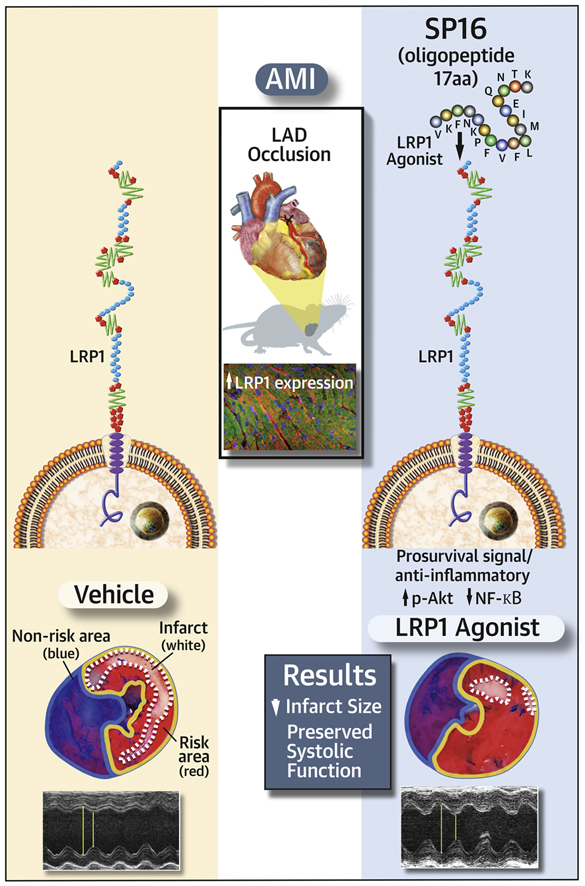
Visual Abstract
Key Words: ischemia reperfusion, low-density lipoprotein receptor-related protein-1, serine protease inhibitor, SERPINs
Abbreviations and Acronyms: A2MG, alpha-2 macroglobulin; AAT, alpha-1 antitrypsin; AMI, acute myocardial infarction; ATIII, antithrombin III; HRP, horseradish peroxidase; IL, interleukin; IV, intravenous; LPS, lipopolysaccharide; LRP1, low-density lipoprotein receptor–related protein-1; LV, left ventricular; LVFS, left ventricular fractional shortening; PBS, phosphate-buffered saline; SEC, serine protease inhibitor–enzyme complex; SERPIN, serine protease inhibitor; TBS, tris-buffered saline; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling
Highlights
-
•
LRP1 is a scavenger receptor with regulatory function, that also transduces a cytoprotective and anti-inflammatory signal.
-
•
The expression of LRP1 in the heart is increased in a mouse model of acute ischemia and in patients with ischemic heart failure.
-
•
The binding of the serine protease inhibitor–enzyme complex to the LRP1 receptor induces a protective signal, that can be leveraged by the use of a small peptide functioning as LRP1 agonist.
-
•
The LRP1 agonist recapitulates the cytoprotective effect of serine protease inhibitor–enzyme complex, and reduces the myocardial ischemia–reperfusion injury in the mouse model.
Summary
Low-density lipoprotein receptor–related protein-1 (LRP1) is a ubiquitous membrane receptor functioning as a scavenger and regulatory receptor, inducing anti-inflammatory and prosurvival signals. Based on the known structure–activity of the LRP1 receptor binding site, the authors synthesized a small peptide (SP16). SP16 induced a >50% reduction in infarct size (p < 0.001) and preservation of left ventricular systolic function (p < 0.001), and treatment with an LRP1 blocking antibody eliminated the protective effects of SP16. In conclusion, LRP1 activation with SP16 given within 30 min of reperfusion during experimental acute myocardial infarction leads to a cardioprotective signal reducing infarct size and preservation of cardiac systolic function.
The inflammatory response as a result of tissue injury is necessary for the clearance of cellular debris 1, 2. Low-density lipoprotein receptor–related protein-1 (LRP1) is a ubiquitous membrane receptor functioning as a scavenger and regulatory receptor 3, 4, 5. Its expression increases during hypoxia, ischemia, and tissue injury 3, 4, 5, 6. LRP1 is a nonselective receptor, binding several plasma proteins 3, 4, 5, 6. Also known as an alpha-2-macroglobulin (A2MG) receptor, LRP1 has the ability to bind the complex of A2MG and plasma proteases 7, 8. The binding of the protease–inhibitor complex to LRP1 is seen across the entire spectrum of serine protease inhibitors (SERPINs) such as alpha-1 antitrypsin (AAT) and antithrombin III (ATIII), and referred to as a SERPIN-enzyme complex (SEC) receptor 3, 4, 5 The binding of protease–inhibitor complexes to LRP1 inhibits the inflammatory response (5) and induces a prosurvival signal through phosphorylation of protein kinase Akt (9). In preclinical animal studies, the administration of plasma-derived AAT or ATIII led to a significant reduction in myocardial injury in experimental acute myocardial infarction (AMI) 10, 11. Therefore, we hypothesized that the synthesis of an LRP1 agonist in the form of a small peptide would provide cardioprotection in AMI.
Methods
Synthesis of the LRP1 agonist
SERPINs are a family of proteins characterized by the ability to inhibit plasma serine proteases such as trypsin, elastase, thrombin, and others (12). When SERPINs bind the serine proteases and consequently inactivate them, a conformational change occurs by which a short peptide containing a unique motif (5 to 11 amino acids) is exposed (13). This unique motif is responsible for the binding to LRP1 3, 14. Based on the known structure–activity of the LRP1 receptor binding site, we synthesized a series of short peptides ranging from 11 to 38 amino acids in length. Based on the anti-inflammatory activity in several in vitro assays we chose a 17 amino acid sequence (VKFNKPFVFLMIEQNTK). This peptide was then modified via replacement of a methionine (M) with norleucin (Nle) to improve stability and binding to LRP1 leading to an oligopeptide, named SP16 (Ac-VKFNKPFVFLNleIEQNTK-NH2). We also created a scrambled control peptide, SP34, containing all the amino acids of the SP16 peptide, including the core motif responsible for binding LRP1, in a randomly scrambled sequence order (FPKMVPQFNTELKIFPEVNIK). Peptides were synthesized by CPC Scientific Inc. (Sunnyvale, California) with purity >90% as verified by high-performance liquid chromatography and mass spectrometry. The pharmacokinetic profile of SP16 in rats was evaluated following single administration of 3 different intravenous (IV) doses (5 mg/kg bolus, 25 mg/kg bolus, 25 mg/kg infusion). Each dose was administered to 2 healthy adult rats (1 male and 1 female) for a total of 6 rats, and blood samples were obtained at baseline, 5, 15, 30, 60, 120, and 240 min, and at 8 and 24 h after IV administration. SP16 concentration was measured using high-performance liquid chromatography–mass spectrometry analysis (Shimadzu CBM20A; Applied Biosystems API 5500 LC/MS/MS instrument) and a Waters XSelect CSH C18 2.5 μm (2.1 mm × 50 mm) column. Pharmacokinetic parameters were calculated based on a noncompartmental model (WinNonlin 6.4).
Cell culture experiments
Murine macrophage J774A.1 cells were maintained in Dulbecco’s modified minimum essential medium (Dulbecco’s modified Eagle medium) (Hyclone, Thermo Fisher Scientific, Waltham, Massachusetts) supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin–streptomycin. THP1-XBlue-MD2-CD14 Cells (InvivoGen, San Diego, California) were maintained in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% heat inactivated fetal bovine serum, 1% penicillin–streptomycin, 100 μg/ml Normocin, 200 μg/ml Zeocin, and 250 μg/ml of G418. All cell lines were maintained in 5% CO2 at 37°C and in accordance with the distributor’s guidelines.
LRP1 binding assay in vitro
Recombinant human LRP1 protein (1 μg/ml) (R&D Systems, Minneapolis, Minnesota) was incubated on ice in the presence of biotin-SP16 (1 to 5 μg/ml) at 4°C for 1 h. The mixture was then incubated with 1 mg/ml M280 Streptavidin Dynabeads (Invitrogen, Carlsbad, California) for 1.5 h at 4°C. Beads were washed in phosphate-buffered saline (PBS) + 0.1% Tween 20 and boiled in sodium dodecyl sulfate loading buffer for 5 min. The supernatants were separated on NuPAGE 4% to 12% Bis-Tris gels (Invitrogen, Carlsbad, California) and transferred to nitrocellulose membranes. The membranes were probed with anti-LRP1 (LRP1 Cluster II Affinity Purified Polyclonal Ab, R&D Systems) and incubated overnight at 4°C. The membranes were then washed 3 times with tris-buffered saline (TBS) and Tween 20 (TBS-T) and incubated with secondary horseradish peroxidase (HRP)–coupled anti-goat diluted 1:10,000 in 5% milk. The Western blots were visualized by chemiluminescence using SuperSignal West Femto Maximum Sensitivity Substrate kit (Thermo Fisher Scientific) and a Bio-Rad Molecular Imager ChemiDoc XRS system (Bio-Rad, Hercules, California).
Induction of the inflammasome in vitro
The inflammasome is a macromolecular intracellular structure responsible for sensing injury and amplifying the inflammatory response, while inducing more cell death (15). To study the effects of SP16 on the inflammasome, J774A.1 cells were plated at 5 × 104 cells/well in a 96 multiwell plate and cultured overnight in Dulbecco’s modified Eagle medium. The cells were primed with either Escherichia coli 0111:B4 lipopolysaccharide (LPS) (25 ng/ml) (Ultrapure, InvivoGen, San Diego, California) or Gp96 (25 nM) (Protein Tech, Chicago, Illinois) for 4 h and then nigericin (20 μM) (Sigma-Aldrich, St. Louis, Missouri) or adenosine triphosphate (5 mM) (Sigma-Aldrich) for 2 h to induce the inflammasome formation, as previously described (10). The cells were treated with SP16 peptide (100 μg/ml), scrambled control peptide (100 μg/ml), or matching volume of vehicle at the time of priming with LPS. Mature interleukin (IL)-1β was used as the readout for the inhibitory effect of the peptide on inflammasome activation. The supernatants were collected and levels of IL-1β were measured with a mouse IL-1β enzyme-linked immunosorbent assay kit (Invitrogen, Grand Island, New York).
NFκB reporter assay
THP1-XBlue-MD2-CD14 cells were seeded at 1 × 105 cells/well in a 96-well cell culture plate. The cells were treated with peptide or a scrambled control peptide (100 μg/ml), for 30 min before the addition of LPS (5 ng/ml) or Gp96 (100 pM). After an 18- to 24-h incubation at 37°C, 20 μl of supernatant was transferred to 180 μl of QUANTI-Blue SEAP (Invivogen, San Diego, California) detection medium. NF-κB–inducible SEAP levels were detected by measuring the absorbance. In experiments blocking LRP1, the anti-LRP1 antibody (125 μg/ml) (clone 5A6, Molecular Innovations, Novi, Michigan) or a nontargeted murine immunoglobulin G class (125 μg/ml) (IgG2b, C-0008 clone, endotoxin-free, CrownBio, Santa Clara, California) were used 30 min before the addition of the peptide.
Endotoxemia in mice
We used a mouse model of endotoxemia to test the anti-inflammatory effects of SP16 in vivo. After acclimation, female BALB/c mice were treated with SP16 peptide (12 μg) by intraperitoneal injection at –2 h, 0 h, and +0.5 h before high-dose of E. coli 0111:B4 LPS (15 mg/kg) (Sigma-Aldrich) given intraperitoneally to induce lethal endotoxemia. The animals were monitored for 72 h and survival was recorded. The study was conducted in accordance with the standards of the American Association for Accreditation of Laboratory Animal Care and with prior approval of the Institutional Animal Care and Use Committee at BioQual, Inc. (Rockville, Maryland).
AMI in mice
Adult outbred male CD1 mice (10 weeks of age) were supplied by Harlan Sprague Dawley (Indianapolis, Indiana). A single operator (S.T.) skilled in coronary artery surgical ligation in mice performed all surgeries. Experimental AMI was induced by transient coronary artery ligation for 30 min to induce ischemia of the anterior wall and the apex (visible as pallor) followed by reperfusion, and leading to an infarct involving approximately 15% of the left ventricle (10). Mice were deeply sedated with sodium pentobarbital (50 to 70 mg/kg), intubated and placed in the right lateral decubitus. A left thoracotomy was performed followed by pericardiectomy and ligation of the proximal left coronary artery, followed by reperfusion. After closure of the thoracic access, the mice were left to recover for up to 1 week with unlimited access to food and water. Only the mice that showed evidence of ischemia at visual inspection during surgery and involving the whole apex were randomly assigned to the different treatments by an investigator (A.G.M.) not involved in the adjudication of the endpoints of interest. The experiments were conducted under the guidelines of laboratory animals for biomedical research published by the National Institutes of Health (revised 2011). The study protocol was approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Assessment of LRP1 expression in the heart
We investigated the expression of LRP1 in the hearts of 6 patients with ischemic heart failure undergoing heart transplant using western blot, and 6 subjects who had died of traumatic death and free of cardiac disease. A portion of the left ventricle, close to the apex, was taken and snap frozen with liquid nitrogen and then stored at –80°C. The samples were then thawed and processed for western blot using a rabbit monoclonal LRP1 antibody (Abcam, ab-92544 Cambridge, United Kingdom). We then studied LRP1 expression at western blot and immunofluorescence in the heart of 8 mice that had had myocardial ischemia surgery 24 h previously and 4 mice that underwent sham surgery. The western blot was performed using a similar approach as outlined for the human samples using glyceraldehyde 3-phosphate dehydrogenase as loading control protein. Immunofluorescence was performed on tissue slides derived from formalin-fixed paraffin-embedded myocardial samples, using the anti-LRP1 antibody (Abcam, ab-92544) and anticardiac myosin heavy chain (Sigma-Aldrich) to stain for cardiomyocytes.
Treatment groups
We randomly assigned 5 to 8 mice to the different groups of treatment given as a single intraperitoneal administration immediately after reperfusion: SP16 10 μg, SP16 100 μg, SP34 100 μg, matching volume of vehicle to SP16, LRP1 blocking antibody clone 5A6 (Molecular Innovations) 3 mg/kg, LRP1 blocking antibody (3 mg/kg) and SP16 100 μg, plasma-derived AAT 60 mg/kg (Aralast NP, Baxter, Deerfield, Illinois), and LRP1 antibody and AAT.
To simulate a clinical scenario in which treatment may not occur immediately at reperfusion, we added another group of 5 to 8 mice in which SP16 was administered intraperitoneally with a delay of 30 min after reperfusion. Each group included 5 to 8 mice per experiment.
In a final set of experiments we treated 4 to 6 mice with SP16 administered a single dose of 3, 10, 100, or 500 μg given subcutaneously after reperfusion, to test the clinical value of this delivery route.
Assessment of survival and death signals
To assess Akt signaling by western blot, we took the heart of 9 mice that had underwent surgery for myocardial ischemia (30 min) followed by reperfusion (180 min). Thirty to 50 mg of tissue was homogenized in radioimmunoprecipitation assay buffer (Sigma-Aldrich) supplemented with protease (Sigma-Aldrich) and phosphatase (Thermo Fisher Scientific) inhibitor cocktails. The samples were then spun down (10,000 g) and the soluble proteins were collected. Protein quantification was performed and 20 μg of each sample was separated on a NuPAGE 4% to 12% Bis-Tris gel (Invitrogen, Carlsbad, California) and transferred to a nitrocellulose membrane. The membranes were then probed with anti-Akt-(Ser(P)473) antibody (Cell Signaling Technology, Beverly, Massachusetts) overnight at 4°C. The membranes were then washed 3 times with TBS-Tween 20 (0.1%) and incubated with secondary HRP-coupled anti-rabbit antibody (Cell Signaling Technology). After washing the blots 3 times with TBS-Tween 20, The blots were visualized by chemiluminescence using SuperSignal West Femto Maximum Sensitivity Substrate kit (Thermo Fisher Scientific) and a Bio-Rad Molecular Imager ChemiDoc XRS system (Bio-Rad). The membrane was then stripped with a mild stripping buffer and reprobed for total Akt (Cell Signaling Technology), incubated overnight at 4°C. The membranes were then washed with TBS-Tween 20 as before, incubated with HRP-coupled anti-rabbit antibody (Cell Signaling Technology), and visualized as previously described.
The same samples were reprobed against the anti-Bax (Cell Signaling Technology) and then the anti-Bcl2 (Santa Cruz Biotechnology, Dallas, Texas) antibodies overnight at 4°C. After chemiluminescence and development, the Bax and Bcl2 bands were measured and the Bax/Bcl2 ratio was calculated.
Finally, caspase-3 activity was measured in the same samples using a fluorogenic enzymatic assay, as previously performed (10). Briefly, 100 mg of protein samples were diluted with caspase assay buffer (31.25% sucrose, 0.3125% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate, and 312.5 mM 4-[2-hydroxyethyl]piperazine-1-ethanesulfonic acid, N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid], pH 7.4) to reach 42 ml of final volume. Duplicates of each samples were then incubated with 56 ml of assay diluent (18 mM dithiotreitol, 3.6% dimethyl sulfoxide) with or without the caspase-3 inhibitor Ac-DEVD-CHO (2 mM final concentration, Promega, Fitchburg, Wisconsin) at 30°C for 30 min. Following this first incubation, 2 ml of caspase-3 fluorogenic substrate Ac-DEVD-AMC (2 mM final concentration, Enzo Life Sciences, Farmingdale, New York) were added to the mix for additional 70 min. The fluorescence was measured using a GloMax multiplate reader (Promega) and the specific enzymatic activity was calculated by subtracting the sample fluorescence without inhibitor to the sample fluorescence with inhibitor. Results were expressed as unit of fluorescence per minute per milligram.
Infarct size assessment
Infarct size was measured as previously described (10). A subgroup of mice were sacrificed 24 h after surgery and the hearts were quickly removed and mounted on a Langendorff apparatus, where the coronary arteries were antegradely perfused with PBS 1X pH 7.4, containing heparin (40 U/ml). After the blood was washed out, 10% triphenyltetrazolium chloride (Sigma-Aldrich) in PBS 1X was perfused, followed by the tying of the ligature and infusion of 1% Phthalo blue dye (Quantum Ink, Louisville, Kentucky); 5 mM adenosine in PBS 1X was injected as a bolus into the aorta until most of the heart turned blue. Afterward, the hearts were then removed from the Langendorff apparatus, frozen, and cut into 6 transverse slices of equal thickness, about 1 mm, from apex to base. The infarcted tissue (appearing white) and the viable tissue (bright red) were measured by computer morphometry using Image Pro Plus 6.0 software (Media Cybernetics, Silver Spring, Maryland). Infarct size is reported as percentage of the area at risk in the left ventricle.
In a subgroup of mice treated with SP16 and vehicle, immediately before sacrifice at 24 h, blood was drawn from the inferior vena cava and collected in BD Vacutainer tubes (BD Vacutainer, Franklin Lakes, New Jersey) for serum isolation. Mouse cardiac troponin I level was determined by an enzyme-linked immunosorbent assay (Life Diagnostic Inc., West Chester, Pennsylvania), as a surrogate for infarct size.
Determination of myocardial fibrosis, myocardial inflammatory cells infiltration, and tunel assay
Seven days following ischemia–reperfusion injury, the hearts were collected in formalin, embedded in paraffin, and sliced in 5-mM sections. Masson’s trichrome staining (Richard-Allan Scientific Masson’s trichrome kit, Thermo Fisher Scientific) was performed following the supplier’s instructions. The fibrosis was calculated as percentage of the left ventricle. Infiltrating leukocytes were measured by probing the sections at 7 days after ischemia–reperfusion with a rat anti-mouse CD-45 (Millipore, Temecula, California) for 3 h at room temperature followed by a goat anti-rat HRP for 2 h and reaction with Novared staining (Vector Laboratories, Burlingame, California).
Deoxyribonucleic acid fragmentation was determined in the infarct border zone, defined as the area of the myocardium bordering the scar where the cardiomyocytes represents >75% of the area, by using a fluorescence-based terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, ApoAlert (Clontech, Mountain View, California), following the manufacturer instruction. An antibody against mouse cardiac actin (Sigma-Aldrich) was used to stain the cardiomyocytes and 4′,6-diamidine-2′-phenylindole dihydrochloride (Sigma-Aldrich) was used to counterstain the nuclei. TUNEL specificity was verified by the costaining of the TUNEL product and 4′,6-diamidine-2′-phenylindole dihydrochloride. Only TUNEL-positive cardiomyocytes were counted and the results were expressed as percentage.
Echocardiography
The mice underwent transthoracic echocardiography at baseline (before surgery) under mild anesthesia with sodium pentobarbital (30 to 50 mg/kg) and at 24 h after surgery. Echocardiography was performed with the Vevo770 imaging system (VisualSonics Inc., Toronto, Ontario, Canada) and a 30-MHz probe 10, 16. The heart was visualized in B-mode from parasternal short axis and apical views. We measured the left ventricular (LV) end-diastolic and end-systolic areas at B-Mode and the LV end-diastolic and LV end-systolic diameters at M-mode. LV fractional shortening (FS) and LV ejection fraction were calculated. A subgroup of mice underwent a repeated echocardiography at 7 days to measure the infarct size (number of segments with akinesis) (16) and the LV systolic function.
As an additional experiment, a group of mice were treated with SP16 100 μg, SP34 100 μg, or matching vehicle (sterile water), and after 180 min they underwent echocardiography to measure LV systolic function at rest and 3 min after challenge with isoproterenol (Sigma-Aldrich) 10 ng to measure contractile reserve (17). The investigators performing and reading the echocardiogram were blinded to the treatment allocation.
Statistical analysis
All results are expressed as mean ± SE. Comparisons among 3 or more groups were performed using analysis of variance, followed by a t test for unpaired data to compare each treatment group with the respective control group. Being a controlled experimental setting, we expected small variability within groups and for the main variable of interest (infarct size) we expected to see a difference or effect (δ) (i.e., 30% reduction) that was, at least, twice as large the SD (σ), so we chose a sample size of ≥6 in each group for the key experiments to provide a >80% power to detect an effect on infarct size of δ = |μ1 – μ2|/σ = 2 using an estimate (2-sided, α = 0.05).
Kaplan-Meier survival curves for SP16 and vehicle after LPS challenge were compared using the log-rank test. We used SPSS version 22.0 (IBM Corporation, New York, New York) for all analysis. A p value <0.05 was considered significant.
Results
Synthesis of the LRP1 agonist
Based on the known structure–activity of the LRP1 receptor binding site, we synthesized a small peptide designated SP16. The structure for SP16 is depicted in Figure 1. SP16 was shown to bind to LRP1 in vitro, whereas SP34 did not (Figure 1).
Figure 1.

SP16 Is an LRP1 Agonist
When serine protease inhibitors (SERPINs) bind the preoteases inactivating them, a conformational change occurs by which a short peptide containing a unique motif (5 to 11 amino acids) is exposed. SP16 contains alpha-1 antitrypsins (AATs) pentapeptide FVFLM responsible for binding to low-density lipoprotein receptor–related protein-1(LRP1). (A) Scheme modified from Joslin et al. (13) and Lillis et al. (3). (B) SP16 binds LRP1 in vitro, whereas SP34 does not. (C,D) SP16 inhibits NF-κB signaling induced by lipopolysaccharide (LPS) or Gp96 in THP1-XBlue-MD2-CD14 cells, and that treatment with LRP1 blocking antibody limits SP16-related inhibition. Ab = antibody; IgG = immunoglobulin G.
Pharmacokinetics of SP16 obtained in rats revealed maximum plasma concentrations of 1.5 μg/ml, 16.4 μg/ml, and 5.8 μg/ml when measured after 5 mg/kg IV bolus, 25 mg/kg IV bolus, and 25 mg/kg IV infusion, respectively. SP16 pharmacokinetics were characterized by rapid initial clearance (216 ml·min–1·kg–1) with a mean residence time of 15 min, accompanied by terminal elimination half-life of 3.3 h, such that detectable concentrations of SP16 (>0.5 ng/ml) were observed for 8 to 24 h following a 25-mg/kg doses. Steady-state volume of distribution (Vdss) was 3.4 l/kg. According to standard extrapolation formulas across species, these parameters predict peak concentrations in the range of 0.5 to 50 μg/ml following a single dose of 3, 10, 100, and 500 μg in mice.
SP16 functions as a LRP1 agonist
SP16 significantly inhibited NF-κB activation induced by LPS or Gp96 in monocytes in comparison with SP34 or a vehicle solution (Figure 1). Treatment with an LRP1 blocking antibody, interfering with receptor binding, significantly reduced the anti-inflammatory signal induced by SP16 (Figure 1). These data indicate that SP16 functions as a LRP1 agonist. Moreover, SP16, and not SP34, significantly inhibited (>50%) the formation of the NLRP3 inflammasome in macrophages in vitro, measured by mature IL-1β levels following stimulation with LPS and nigericin or Gp96 and adenosine triphosphate in J774A.1 cells in vitro (Supplemental Figure 1), confirming the anti-inflammatory effects of LRP1 agonist, SP16, on the inflammasome induced by distinct stimuli.
Anti-inflammatory effects of SP16 in vivo
We tested the effects of SP16 in a mouse model of endotoxemia. SP16 peptide was given in 3 doses of 4 μg intraperitoneally at –2, 0, and +0.5 h before high-dose LPS challenge (15 mg/kg) significantly decreased mortality at 72 h from 100% to 40% (p < 0.001) (Supplemental Figure 2), thus confirming preservation of the LRP1 agonist activity in vivo.
LRP1 expression is increased in the heart after AMI
We measured expression of LRP1 in the heart of 6 male patients with ischemic heart disease undergoing heart transplantation (median 61 [range 47 to 67] years of age, 3 patients were supported by a LV assist device before transplant, 1 patient had a total artificial heart, and 2 had direct transplantation). Figure 2 shows a >2-fold increase in LRP1 expression in the hearts of patients versus control patients (p < 0.001). We also measured LRP1 expression in the heart of mice 24 h after ischemia–reperfusion injury. Figure 2 shows >3-fold increase in mice with ischemia–reperfusion versus sham-operated mice (p = 0.003). Immunofluorescence images show intense LRP1 staining in the heart of ischemia–reperfusion mice and not in sham-operated mice.
Figure 2.

LRP1 Expression Is Increased in Heart During Acute Myocardial Infarction
(A) Low-density lipoprotein receptor–related protein-1 (LRP1) expression is significantly increased in the heart of patients with end-stage heart failure due to prior myocardial infarction undergoing heart transplantation, compared with subjects who died of noncardiac causes (p < 0.001; n = 6 per group). (B) Increased LRP1 expression is seen in the heart of mice subject to myocardial ischemia (30 min) and reperfusion (24 h) (top right panel) (p = 0.003, n = 4 per group). The immunofluorescence for LRP1 (red) and cardiac muscle actin (green) showing (C) increased LRP1 expression in areas bordering the infarct zone, (D) compared with sham-operated mice. GAPDH = glyceraldehyde 3-phosphate dehydrogenase.
SP16 induces rapid phosphorylation of Akt during ischemia–reperfusion
We measured phosphorylation of Akt (Ser473) during ischemia–reperfusion as a measure of the prosurvival effect of SP16: compared with vehicle-treated mice, mice receiving 100 μg intraperitoneally at time of reperfusion had a significant increase in pAkt/total Akt ratio (p = 0.029), associated with a reduction in the ratio of the proapoptotic to antiapoptotic Bax/Bcl2 ratio (p = 0.010), and a reduction in the caspase-3 activity (p = 0.034) in the heart tissue (Figure 3).
Figure 3.

SP16 Induces Rapid Phosphorylation of Akt During Ischemia–Reperfusion
We measured phosphorylation at Ser473 of Akt in myocardial samples during ischemia–reperfusion as a measure of the prosurvival effect of SP16. As compared with vehicle-treated mice, mice receiving SP16 100 μg intraperitoneally at time of reperfusion had a significant increase in pAkt/total Akt ratio, associated with a reduction in the ratio of the proapoptotic to antiapoptotic Bax/Bcl2 ratio and of caspase-3 myocardial tissue activity. i.p. = intraperitoneal; p-Akt = phosphorylated Akt.
Infarct-sparing effects of SP16
We induced AMI in adult male mice by coronary ligation and transient ischemia (30 min) followed by reperfusion. When compared with vehicle solution, SP16, but not SP34, given intraperitoneally at reperfusion significantly reduced in a dose-dependent fashion infarct size, measured as percent of area at risk at triphenyl tetrazolium chloride staining at pathology and preserved LV systolic function measured as LVFS % (Figure 4, Supplemental Table 1). The protective effects of SP16 were seen also in a significantly lower cardiac troponin I levels at 24 h in the mice treated with SP16 (1.0 ± 0.7 ng/ml) versus vehicle (10.0 ± 1.1 ng/ml; p < 0.001).
Figure 4.

Infarct-Sparing Effect of SP16
We induced acute myocardial infarction (AMI) in adult male mice by coronary ligation and transient ischemia (30 min) followed by reperfusion. When compared with vehicle solution, SP16, but not SP34, given intraperitoneally at reperfusion significantly reduced in a dose-dependent fashion infarct size, measured as percent of area at risk at tetrazolium chloride (TTC) staining at pathology and preserved left ventricular systolic function measured as left ventricular (LV) fractional shortening (LVFS) %. n = 5 to 8 mice/group. c-Trop I = cardiac troponin I; LVEDD = left ventricular end-diastolic diameter; LVESD = left ventricular end-systolic diameter.
To simulate a clinical scenario in which treatment may not occur immediately at reperfusion, we tested whether a delay of 30 min in treatment with SP16 would affect the results, and found that the cardioprotective effects were maintained (Supplemental Figure 3). These data support a translational therapeutic value of this strategy. SP16 given subcutaneously at time of reperfusion showed a similar reduction in infarct size (all p = NS compared to corresponding intraperitoneal dose) (Supplemental Figure 4).
Effects of SP16 on cardiac systolic function
LRP1 has been shown to affect intracellular calcium content by regulating the expression of SERCA2 in vitro in HL-1 cardiomyocytes (18). To determine the acute effects of LRP1 agonist SP16 on cardiac systolic function, we measured LV systolic function with SP16 at rest and after challenge with isoproterenol to evaluate also contractile reserve. As shown in Figure 5, SP16 had no significant effect on LVFS % at rest or after isoproterenol challenge. These data show that, as tested, SP16 has no appreciable inotropic activity in vivo.
Figure 5.

Effects of SP16 on LV Systolic Function
We measured left ventricular (LV) systolic function with SP16 at rest and after challenge with isoproterenol to evaluate also contractile reserve. SP16 had no significant effect on LV fractional shortening (LVFS) (A) at rest or (B) after isoproterenol challenge. These data show that, as tested, SP16 has no appreciable inotropic activity in vivo. We also measured LVFS at 7 days after acute myocardial infarction (AMI) in mice. (C) Mice treated with SP16 at reperfusion had significantly greater LVFS than did vehicle-treated mice. (D) This was associated with a significantly smaller area of wall motion abnormality, a surrogate for infarct size. These data show that the early cardioprotective effects of SP16 are maintained at 7 days after surgery. n = 5 to 8 mice/group. d = days.
We also measured LVFS % at 7 days after AMI in mice. Mice treated with SP16 at reperfusion had significantly greater LVFS% than did vehicle-treated mice (Figure 5). This was associated with a significantly smaller area of wall motion abnormality, a surrogate for infarct size (16) (Figure 5). These data show that the early cardioprotective effects of SP16 are maintained at 7 days after surgery.
The use if pentobarbital as sedative led to a reduction in contractility across all groups but none of the effects on systolic function between groups can be attributed to the use of pentobarbital given the use of the same protocol in all cases.
Effects for SP16 on cardiomyocytes apoptosis, myocardial inflammation, and fibrosis 7 days after ischemia–reperfusion injury
The protective effects in terms of preserved regional and global systolic function seen with SP16 treatment were associated with a reduction in fibrotic scar size, a reduction in cardiomyocytes in border zone, and a reduced leukocyte infiltration, compared with vehicle-treated mice (Figure 6).
Figure 6.

Effects of SP16 on Cardiomyocyte Apoptosis and Myocardial Inflammation and Fibrosis
We measured the percentage of (A) apoptotic cardiomyocytes by detecting nuclei showing in situ end labeling of deoxyribonucleic acid fragmentation (terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL]) in the peri-infarct border zone, (B) the number of leukocytes (positive for cluster of differentiation 45) per high-power field (HPF) ×40, and (C) the extent of the fibrotic infarct scar (at Masson trichrome stain) in the mouse hearts 7 days after 30 min of ischemia. Treatment with SP16 led to a significant reduction in infarct scar size and a reduction in cardiomyocyte apoptosis and myocardial inflammation in the peri-infarct border zone. n = 4 mice/group. LV = left ventricle.
LRP1 mediates the cardioprotective signaling of SP16
To test whether LRP1 mediated the cardioprotective signal of SERPINs and of SP16, we pretreated mice with a LRP1 blocking antibody. Treatment with the LRP1 blocking antibody had no effects on the infarct size when given by itself, but eliminated the protective effects of both plasma-derived AAT and of SP16, indicating that a functional LRP1 receptor is necessary for the protective effect of AAT and of SP16 (Figure 7). Moreover, when AAT was given with the LRP1 blocking antibody it led to a significant increase in infarct size (Figure 7).
Figure 7.

LRP1 Mediates SP16-Induced Cardioprotection
We pretreated mice with a Low-density lipoprotein receptor–related protein-1 (LRP1) blocking antibody to test whether LRP1 mediated the cardioprotective signal of SP16 and alpha-1 antitrypsins (AAT). Treatment with the blocking antibody eliminated the protective effects of both SP16 and plasma-derived AAT. n = 5 to 8 mice/group. Ab = antibody; LV = left ventricular.
Discussion
The data presented herein show that SP16, a novel LRP1 agonist, leads to cardioprotective signaling, significant reduction in infarct size, and preservation of cardiac function in myocardial ischemia–reperfusion injury.
LRP1 is a membrane receptor mostly known for being part of the low-density lipoprotein receptor family and responsible for lipoprotein scavenging 3, 4, 5. LRP1 is, however, a multifunctional protein 6, 7, 8. LRP1, also known as A2MG receptor, is known to bind A2MG-protease complexes, and more in general binds SECs, acting as an SEC receptor 4, 13, 14. The binding of LRP1 to the enzyme–inhibitor complex acts not only as a scavenger but also as impacting anti-inflammatory and prosurvival signaling 3, 4, 5, 6, 7, 8. In leukocytes, LRP1, also known as CD91, functions as scavenger receptor for apoptotic cells leading to an anti-inflammatory signal (19). LRP1/CD91 is also involved in antigen processing and presentation to T cells by antigen presenting cells through the major histocompatibility complex (20).
LRP1 expression is increased during hypoxia, ischemia, and injury, and as the inflammatory process takes place in the tissue, the generation of enzyme-inhibitor complexes initiates, through LRP1, a down-regulation of the same inflammatory response leading to modulation and resolution of inflammation. The inflammatory response following tissue injury is necessary for the clearance of cellular debris and pathogen load, in case of microbial infections, preventing an exaggerated inflammatory response and thereby abrogating further injury 3, 4, 5, 21. Uncontrolled rampant inflammation, as it occurs during AMI, however, leads to a greater injury 2, 22. The peak of the inflammatory response in AMI occurs within hours to days after the initial insult, in correspondence of leukocytes infiltration and in conjunction with increased expression of plasma proteases and inhibitors 1, 2, 15. As such the role of endogenous LRP1 signaling may be negligible during the first few hours of AMI due to the limited presence of enzyme–inhibitor complexes triggering the signal. Such a time lag in LRP1-mediated signaling in AMI correlates with the initial overly aggressive inflammatory response in AMI, and it may potentially have a negative impact on the degree of injury and the recovery. We therefore hypothesized that an LRP1 agonist, given early during AMI, could provide cardioprotection by providing a survival signal and limiting the inflammatory injury. Based on the known structure–activity of the LRP1 receptor, we designed a small peptide, SP16, containing the unique 5 amino acids motif responsible for the binding of the enzyme/inhibitor complex to LRP1 13, 14. SP16 has the advantage of being constitutively active and does not require activation by the plasma proteases. We have shown that SP16 leads to down-regulation NF-κB signaling and prevention of NLRP3 inflammasome activation following diverse stimuli in a leukocyte assay in vitro. During in vivo experiments, SP16 induces anti-inflammatory and cytoprotective signals by binding to LRP1 early in the course of the injury, largely in advance of the predicted activation of LRP1 by the endogenous enzyme–inhibitor complexes. Accordingly, we observed that SP16 provides rapid phosphorylation of Akt in the heart following ischemia–reperfusion providing powerful cardioprotection when administered within 30 min of reperfusion during experimental AMI, associated with inhibition of programmed cell death in the myocardium. SP16 also protected the viable myocardium bordering the infarct scar and preserved cardiac function at 7 days. Treatment with LRP1 blocking antibody eliminates the benefits of SP16, confirming the central role of LRP1 in the cardioprotective mechanism. These effects are likely not only attributable to an effect on LRP1 in leukocytes, as cardiomyocyte death is the major determinant of infarct size. LRP1 is indeed expressed ubiquitously and hence the activity of SP16 is likely to have affected multiple cell types. Future studies using conditional knockout restricted to cardiomyocytes are being planned to determine the specific role of cardiomyocyte LRP1 signaling in vivo.
The cardioprotective effects of SP16 are consistent with the effects seen with plasma-derived AAT and ATIII, and with recombinant human AAT 10, 11, 23. We showed in 2011 that plasma-derived AAT inhibited induction of the NLRP3 inflammasome and ischemia–reperfusion cardiomyocyte death in vitro and in vivo (10). In 2013, Wang et al. (11) showed that ATIII, another member of the SERPIN family, markedly reduced infarct size, independent of its anticoagulant activity. More recently, using different recombinant fusion proteins of human AAT and fragment crystallizable region of immunoglobulin G, we showed that the cardioprotective effects of AAT were independent of its ability to inhibit elastase (23). Collectively, the data show that distinct LRP1 agonists derived from SERPINs induce a similar cardioprotective effect, independent of their ability to inhibit the proteases. Of interest when AAT was given with the LRP1 blocking antibody, it led to a significant increase in infarct size, showing that when LRP1-mediated protective effect is blocked, AAT may exert a toxic rather than a cardioprotective effect, through a different mechanism. In the current study, we also show that SP16 is effective when tested in a clinically relevant setting. In fact, the treatment was administered not before the onset of ischemia, which would not be feasible in patients, but at reperfusion or even within 30 min of reperfusion in the mouse, providing, in essence, a large therapeutic window for treatment of patients with AMI.
Despite the progress in the treatment of AMI, further improvement to prevent heart failure and death after AMI remains an urgent unmet medical need. Each year about 935,000 Americans have an AMI (24). A significant number of patients continue to die of AMI, and survivors are at significantly higher risk of developing heart failure (25). Heart failure after AMI presents a very unfavorable prognosis (>30% of patients die within 5 years of diagnosis). With a rising prevalence (approximately 2% of the population), and high incidence (>500,000 new cases each year in the United States alone), AMI and heart failure represent important public enemies. Prompt reperfusion has revolutionized the treatment of AMI. The restoration of antegrade flow in the occluded coronary artery, by means of fibrinolysis or angioplasty with balloons and stents, salvages a large amount of myocardium at risk, thus reducing the infarct size and its consequences on global cardiac function and dimensions. Reimer et al. (26) had shown that, in the dog, a wavefront phenomenon of ischemic cell death would occur by which the final size of the infarct would be correlated with the duration of ischemia. Without reperfusion, the infarct size is predicted to represent approximately 70% of the myocardium at ischemic risk (27). Reperfusion alone is predicted to double the amount of salvaged myocardium and reduce infarct size in half (27). However, Kloner et al. (28) had already shown in 1974 that reperfusion could be associated with incomplete salvage due to failure to achieve uniform reperfusion and had noted that the death of severely ischemic myocardial cells occurred before the so-called no-reflow phenomenon. The description of the ischemic preconditioning by Murry et al. (29) in 1986 revealed that not all the events occurring with reperfusion were beneficial, and that treatment with intermittent short bouts of ischemia before prolonged ischemia could modulate the myocardial response during reperfusion and further reduce infarct size. This led to development of the concept of reperfusion injury in AMI 27, 30, a phenomenon by which only part of the potentially salvageable myocardium is actually salvaged by reperfusion. Based on experimental data, it is expected that a strategy aimed at limiting reperfusion injury in AMI “would reduce infarct size by a further 25%, realizing the full benefit of reperfusion” (27). Reperfusion strategies have been successful in shortening the duration of ischemia, thus reducing infarct size. Prevention of mitochondria-mediated events have only partially and inconsistently translated into reduction in infarct size in preclinical and clinical studies, possibly because treatment is only effective if administered before ischemia or reperfusion (31), limiting the clinical applicability of these strategies due to their narrow therapeutic window. There are no clinically available strategies to limit the inflammatory injury 22, 31. Treatment with SP16 may therefore represent a significant step forward in the treatment of AMI by providing an effective strategy limiting inflammatory injury following reperfusion.
SP16 also provides several clear advantages over the use of plasma-derived formulations of SERPINs. SP16 contains the SERPINs’ highly conserved motif required for LRP1 binding and shown to exert an anti-inflammatory action, devoid of any inhibition of plasma proteases. Unlike SERPINs, SP16 is already active and does not require interaction with serine proteases to become active. SP16 does not require isolation from the plasma, limiting the inherent infectious or anaphylaxis risks. On the other hand, being highly homologous to the C-terminus of SERPINs, SP16 is expected to have a very high tolerability and safety profile, as shown in the preliminary toxicology profile. In patients with ST-segment elevation AMI, plasma-derived AAT was safe and associated with a blunted inflammatory response (32). Moreover, the synthesis of SP16 is rather inexpensive, and it not affected by potential limitations in plasma supply. Given the smaller molecular weight and efficacy at low dose, SP16 can be given subcutaneously, thus facilitating administration.
Study limitations
First, we only studied young adult male mice. Future studies should examine the impact of LRP1 agonism in mice of both sexes and of different ages. In addition, the mice used in the present study where without comorbidities. Myocardial LRP1 expression may change with hypertension, diabetes, or obesity. Therefore, future studies are warranted to determine the effectiveness of LRP1 agonism in reducing ischemic myocardial damage in the presence of these diseases.
Conclusions
LRP1 activation with SP16 during experimental AMI leads to a cardioprotective signal, reducing infarct size and preserving cardiac systolic function in mice. SP16 represents a first-in-class pharmacologic agent exploring an entirely novel approach of cardioprotection in AMI.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Coronary artery reperfusion is a critical therapeutic strategy to rescue the ischemic myocardium during AMI. Despite reperfusion, however, a significant amount of myocardium is lost and patients are at risk for the development of heart failure. Additional therapies are therefore warranted to reduce damage associated with reperfusion and prevent long-term adverse effects. The findings of this study demonstrate that the LRP1 receptor is a valuable target for intervention in AMI, and proposes a small peptide LRP1 agonist as a therapeutic strategy in AMI.
TRANSLATIONAL OUTLOOK 1: There is an increased expression of LRP1 in the heart following ischemia and reperfusion. Selective agonism of LRP1 increases the activation of Akt, a prosurvival factor, decreases the Bax/Bcl2 ratio and reduces caspase-3 activity, resulting in a smaller infarct size and preserved cardiac function.
TRANSLATIONAL OUTLOOK 2: LRP1 agonists can be developed to reduce myocardial ischemic injury. Synthetic LRP1 agonists may be advantageous, as they can be synthesized at low cost, and they lack the structural complexity to induce immunogenicity. Moreover, given the small molecular weight, they can be delivered subcutaneously.
Footnotes
The study was completed as part of a collaboration between Virginia Commonwealth University and Serpin Pharma. Ms. Austin and Dr. Gelber are employees of Serpin Pharma. Dr. Abbate is supported by National Heart, Lung, and Blood Institute grants HL121402 and HL136816. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Toldo and Austin contributed equally to this work.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Frangogiannis N.G. The inflammatory response in myocardial injury, repair, and remodeling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christia P., Bujak M., Gonzalez-Quesada C. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J Histochem Cytochem. 2013;61:555–570. doi: 10.1369/0022155413493912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lillis A.P., Van Duyn L.B., Murphy-Ullrich J.E., Strickland D.K. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strickland D.K., Muratoglu S.C., Antalis T.M. Serpin-enzyme receptors LDL receptor-related protein 1. Methods Enzymol. 2011;499:17–31. doi: 10.1016/B978-0-12-386471-0.00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.May P. The low-density lipoprotein receptor-related protein 1 in inflammation. Curr Opin Lipidol. 2013;24:134–137. doi: 10.1097/MOL.0b013e32835e809c. [DOI] [PubMed] [Google Scholar]
- 6.Cal R., Castellano J., Revuelta-López E. Low-density lipoprotein receptor-related protein 1 mediates hypoxia-induced very low density lipoprotein-cholesteryl ester uptake and accumulation in cardiomyocytes. Cardiovasc Res. 2012;94:469–479. doi: 10.1093/cvr/cvs136. [DOI] [PubMed] [Google Scholar]
- 7.Williams S.E., Kounnas M.Z., Argraves K.M., Argraves W.S., Strickland D.K. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein and the receptor-associated protein. An overview. Ann N Y Acad Sci. 1994;737:1–13. doi: 10.1111/j.1749-6632.1994.tb44297.x. [DOI] [PubMed] [Google Scholar]
- 8.Franchini M., Montagnana M. Low-density lipoprotein receptor-related protein 1: new functions for an old molecule. Clin Chem Lab Med. 2011;49:967–970. doi: 10.1515/CCLM.2011.154. [DOI] [PubMed] [Google Scholar]
- 9.Fuentealba R.A., Liu Q., Kanekiyo T., Zhang J., Bu G. Low density lipoprotein receptor-related protein 1 promotes anti-apoptotic signaling in neurons by activating Akt survival pathway. J Biol Chem. 2009;284:34045–34053. doi: 10.1074/jbc.M109.021030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toldo S., Seropian I.M., Mezzaroma E. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia–reperfusion injury. J Mol Cell Cardiol. 2011;51:244–251. doi: 10.1016/j.yjmcc.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Wang Y., Wang J. Antithrombin is protective against myocardial ischemia and reperfusion injury. J Thromb Haemost. 2013;11:1020–1028. doi: 10.1111/jth.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Potempa J., Korzus E., Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem. 1994;23:15957–15960. [PubMed] [Google Scholar]
- 13.Joslin G., Fallon R.J., Bullock J., Adams S.P., Perlmutter D.H. The SEC receptor recognizes a pentapeptide neodomain of alpha 1-antitrypsin-protease complexes. J Biol Chem. 1991;266:11282–11288. [PubMed] [Google Scholar]
- 14.Joslin G., Griffin G.L., August A.M. The serpin-enzyme complex (SEC) receptor mediates the neutrophil chemotactic effect of alpha-1 antitrypsin-elastase complexes and amyloid-beta peptide. J Clin Invest. 1992;90:1150–1154. doi: 10.1172/JCI115934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toldo S., Mezzaroma E., Mauro A.G., Salloum F., Van Tassell B.W., Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22:1146–1161. doi: 10.1089/ars.2014.5989. [DOI] [PubMed] [Google Scholar]
- 16.Seropian I.M., Abbate A., Toldo S. Pharmacologic inhibition of phosphoinositide 3-kinase gamma (PI3Kγ) promotes infarct resorption and prevents adverse cardiac remodeling after myocardial infarction in mice. J Cardiovasc Pharmacol. 2010;56:651–658. doi: 10.1097/FJC.0b013e3181f9a905. [DOI] [PubMed] [Google Scholar]
- 17.Toldo S., Mezzaroma E., Bressi E. Interleukin-1β blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J Cardiovasc Pharmacol. 2014;64:1–6. doi: 10.1097/FJC.0000000000000106. [DOI] [PubMed] [Google Scholar]
- 18.Revuelta-López E., Cal R., Herraiz-Martínez A. Hypoxia-driven sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) downregulation depends on low-density lipoprotein receptor-related protein 1 (LRP1)-signalling in cardiomyocytes. J Mol Cell Cardiol. 2015;85:25–36. doi: 10.1016/j.yjmcc.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 19.Vandivier R.W., Ogden C.A., Fadok V.A. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol. 2002;169:3978–3986. doi: 10.4049/jimmunol.169.7.3978. [DOI] [PubMed] [Google Scholar]
- 20.Binder R.J., Srivastava P.K. Essential role of CD91 in re-presentation of Gp96-chaperoned peptides. Proc Natl Acad Sci U S A. 2004;101:6128–6133. doi: 10.1073/pnas.0308180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walley K.R., Thain K.R., Russell J.A. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med. 2014;258:258ra143. doi: 10.1126/scitranslmed.3008782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seropian I.M., Toldo S., Van Tassell B.W., Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014;63:1593–1603. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Mauro A.G., Mezzaroma E., Marchetti C. A preclinical translational study of the cardioprotective effects of plasma derived alpha-1 anti-trypsin in acute myocardial infarction. J Cardiovasc Pharmacol. 2017;69:273–278. doi: 10.1097/FJC.0000000000000474. [DOI] [PubMed] [Google Scholar]
- 24.Mozaffarian D., Benjamin E.J., Go A.S. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–e322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 25.Velagaleti R.S., Pencina M.J., Murabito J.M. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–2062. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reimer K.A., Lowe J.E., Rasmussen M.M., Jennings R.B. The wavefront phenomenon of ischemic cell death. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation. 1977;56:786–794. doi: 10.1161/01.cir.56.5.786. [DOI] [PubMed] [Google Scholar]
- 27.Yellon D.M., Hausenloy D.J. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 28.Kloner R.A., Ganote C.E., Jennings R.B. The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974;54:1496–1508. doi: 10.1172/JCI107898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murry C.E., Jennings R.B., Reimer K.A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 30.Ibáñez B., Heusch G., Ovize M., Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 31.Trankle C., Thurber C.J., Toldo S., Abbate A. Mitochondrial membrane permeability inhibitors in acute myocardial infarction. Still awaiting translation. J Am Coll Cardiol Basic Trans Science. 2016;1:524–535. doi: 10.1016/j.jacbts.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbate A., Van Tassell B.W., Christopher S. Effects of prolastin C (plasma-derived alpha-1 antitrypsin) on the acute inflammatory response in patients with ST-segment elevation myocardial infarction (from the VCU-alpha 1-RT pilot study) Am J Cardiol. 2015;115:8–12. doi: 10.1016/j.amjcard.2014.09.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.