

Corresponding Author

Key Words: antidiabetic drugs, diabetic mellitus, heart failure, insulin
Summary
The heightened risk of heart failure in type 2 diabetes cannot be explained by the occurrence of clinically overt myocardial ischemic events or hyperglycemia. Experimentally, insulin exerts detrimental effects on the heart, vasculature, kidneys, and adipose tissue that can lead to heart failure. In both randomized clinical trials and observational studies, antihyperglycemic drugs that act through insulin signaling (i.e., sulfonylureas, thiazolidinediones, and incretins) increase the risk or worsen the clinical course of heart failure, whereas drugs that ameliorate hyperinsulinemia and do not signal through insulin (i.e., metformin and sodium-glucose cotransporter 2 inhibitors) reduce the risk of heart failure.
The primary aims of the treatment of patients with type 2 diabetes are control of hyperglycemia and reduction in the risk of macrovascular and microvascular events. Historically, the definition of macrovascular events has generally focused on the occurrence of occlusive or thrombotic events in atherosclerotic vessels (i.e., myocardial infarction, stroke, and major limb ischemia); however, heart failure is a common and serious complication of type 2 diabetes (1), and its occurrence cannot be explained by ischemic injury to the heart. Little is known about why heart failure is so common in patients with type 2 diabetes, even when cardiovascular risk factors are well-controlled (Supplemental Appendix A).
The most distinctive feature of type 2 diabetes is hyperinsulinemia. Insulin can exert important adverse effects on the heart, vasculature, kidneys, and adipose tissue, which can hasten the onset of heart failure or lead to worsening of its clinical course. Of interest, the phenotype of heart failure appears to differ in patients with types 1 and 2 diabetes, possibly because only the latter patients have sustained hyperinsulinemia. Furthermore, many antidiabetic drugs that work through the actions of insulin have been associated with an increased risk of heart failure and an increased mortality in patients with established symptoms (1). By contrast, antihyperglycemic drugs that do not depend on insulin signaling often reduce the risk of heart failure, and it is possible that their action to ameliorate hyperinsulinemia contributes to this benefit (Supplemental Appendix A).
Effect of Insulin on the Heart, Vasculature, Kidneys, and Adipose Tissue
Insulin receptors are ubiquitously expressed and are abundant in the heart and blood vessels. Interaction of insulin with its receptors leads to activation of 2 intracellular pathways: the Akt/mTOR signaling cascade and the mitogen-activated protein kinase (MAPK)/extracellular-regulated kinase pathway. Experimentally, overexpression of Akt leads to pathological cardiac hypertrophy, and if sustained for long periods, to heart failure. A reduction of insulin or Akt signaling in murine models of cardiac hypertrophy prevents heart failure, suggesting that this mechanism contributes to adverse cardiac remodeling. At the same time, activation of the MAPK/extracellular-regulated kinase pathway depresses cardiac contractility and enhances matrix remodeling, even in the absence of hypertrophy; it may also lead to the release of proinflammatory cytokines, leading to fibrosis. Either pathway (acting alone or in concert) might provide a molecular basis for an effect of insulin to cause heart failure (Supplemental Appendix B).
Hyperinsulinemia may also have important effects on the structure and function of the vasculature. Although insulin signaling through the Akt pathway might be impaired in endothelial cells in states of insulin resistance, activation of MAPK signaling pathways remains active, resulting in the induction and potentiation of various endogenous vasoconstrictors. Additionally, insulin can contribute directly to vascular smooth muscle cell hyperplasia (Supplemental Appendix B).
Insulin exerts a powerful antinatriuretic action, which may have evolved to counter the natriuresis caused by hyperglycemia. The infusion of insulin increases sodium reabsorption in the proximal and distal tubules, promotes the actions of angiotensin II, and inhibits the effects of endogenous natriuretic peptides. Insulin can directly increase activity of numerous ion transport mechanisms in the renal tubules, including the epithelial sodium channel, sodium-phosphate cotransporter, sodium-hydrogen exchanger type 3, and sodium-potassium adenosine triphosphatase (Supplemental Appendix B).
Additionally, insulin promotes adipogenesis by facilitating the transition of mesenchymal cells into preadipocytes and by enhancing their differentiation into mature fat cells (Supplemental Appendix C). Such an action may be particularly important if it occurs in epicardial adipose tissue, which is characteristically hypertrophied in type 2 diabetes (2). The accumulation and dysfunction of epicardial adipose tissue causes the release of numerous proinflammatory adipocytokines, which can adversely affect the structure and function of the adjoining myocardium, with which it shares an unobstructed microcirculation. This inflammatory response may lead to microvascular rarefaction and cardiac fibrosis, both of which can impair ventricular distensibility. Insulin can also promote fibroblast proliferation in the myocardium and the secretion of collagen, further impairing the ability of the cardiac chambers to enlarge (Supplemental Appendix C). If the antinatriuretic action of insulin leads to plasma volume expansion, the net result is ventricular overfilling, which leads to the syndrome of heart failure with a preserved ejection fraction. This phenotype is particularly common in patients with type 2 diabetes. Insulin can therefore exert adverse effects on the heart, vasculature, kidneys, and adipose tissue, which (acting in concert) can lead to heart failure or accelerate the clinical course of the disease (Figure 1).
Figure 1.
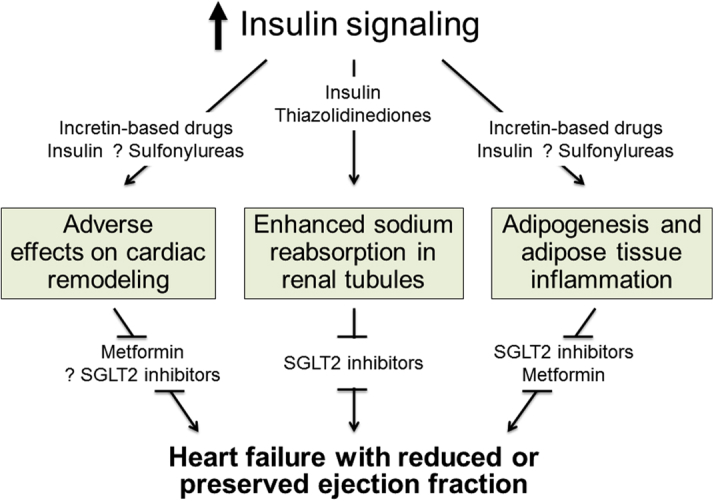
Pathophysiological Mechanisms by Which Enhanced Insulin Signaling May Lead to Heart Failure
Enhanced insulin signaling can lead to heart failure by exerting adverse effects on cardiac remodeling, promoting renal tubular sodium reabsorption, and stimulating accumulation and inflammation of epicardial adipose tissue, thereby aggravating its deleterious effects on the underlying myocardium. (Top) Drugs that potentiate insulin signaling (insulin, sulfonylureas, and thiazolidinediones). (Bottom) Drugs that attenuate insulin signaling (metformin and sodium-glucose cotransporter-2 [SGLT2] inhibitors).
Contrasting Effects of Types 1 and 2 Diabetes on Heart Failure
If insulin exerts adverse biological and pathophysiological effects that lead to heart failure, then the incidence of heart failure might be expected to differ in patients with type 1 versus type 2 diabetes because only the latter characteristically have sustained hyperinsulinemia (Supplemental Appendix D). In the only prospective study in type 1 diabetes that measured ventricular function and biomarkers of heart failure, the incidence of heart failure over a 7-year follow-up period was low, unless patients developed overt hypertension or coronary artery disease (3). In contrast, the incidence of heart failure is increased 2- to 3-fold in type 2 diabetes, and this risk cannot be readily attributed to known cardiovascular risk factors.
Differences in the cardiac effects of types 1 and 2 diabetes have also been observed in animal models of the 2 diseases (Supplemental Appendix D). Rodent models of type 2 diabetes, including db/db mice, ob/ob mice, and Zucker diabetic fatty rats, exhibit notable degrees of ventricular hypertrophy and fibrosis, which is accompanied by mitochondrial dysfunction, oxidative stress, abnormalities of calcium handling, and activation of neurohormonal systems. Echocardiographic studies reveal both systolic and diastolic dysfunction. In contrast, animal models of type 1 diabetes, streptozotocin-treated mice or Akita diabetic mice, do not exhibit cardiac hypertrophy, fibrosis, inflammation, or oxidative stress; instead, the hearts of diabetic animals are smaller than in control animals, possibly because of the absence of insulin’s effects on myocardial growth.
Contrasting Effects of Antidiabetic Drugs on the Risk of Heart Failure
It is commonly believed that inadequate glycemic control increases the risk of heart failure in the clinical setting. The likelihood of heart failure increases as the level of glycated hemoglobin rises; yet, it is not clear whether this observation is explained by an adverse effect of hyperglycemia on the heart or is attributable to a deleterious action of the hyperinsulinemia that invariably accompanies hyperglycemia in patients with type 2 diabetes (Supplemental Appendix E). Nonetheless, it is noteworthy that pre-diabetes is associated with a striking increase in the risk of heart failure, despite only modest elevations of blood glucose. Hyperinsulinemia is also associated with poor outcomes following an acute myocardial infarction in patients without diabetes (4).
Is the relationship between glycated hemoglobin and heart failure related to poor glycemic control and its adverse effects on cardiac energy utilization or metabolism? Alternatively, is the relationship between glycated hemoglobin and heart failure related to their shared association with hyperinsulinemia, which exerts direct detrimental effects on the circulation? These 2 possibilities can be distinguished by examining the effects of interventions that influence insulin levels and blood glucose in opposite directions (1).
Two classes of medications exert an antihyperglycemic effect that is not dependent on insulin (Figure 1). Biguanides (such as metformin) decrease hepatic glucose production, mostly through inhibition of the mitochondrial respiratory-chain complex 1 and activation of AMP kinase. Sodium-glucose cotransporter 2 (SGLT2) inhibitors promote glycosuria by an inhibitory effect on the proximal tubular reabsorption of glucose. The action of both drugs is accompanied by amelioration of hyperinsulinemia in patients with type 2 diabetes. It is therefore noteworthy that the use of metformin has been reported to decrease the risk of heart failure in observational studies, although evidence from clinical trials is lacking. Similarly, in large-scale randomized controlled trials, long-term treatment with SGLT2 inhibitors has been shown to reduce the risk of new-onset heart failure as well as hospitalization and death from heart failure (Supplemental Appendix F).
The mechanisms by which metformin and SGLT2 inhibitors may act on heart failure have not been clearly defined. Through its action on AMP kinase, metformin orchestrates a metabolic response that preserves myocardial adenosine triphosphate (ATP); it also has antiproliferative, antifibrotic, and antiapoptotic properties that may contribute to favorable effects on cardiac remodeling. The drug also inhibits adipocyte differentiation and adipose tissue inflammation, thereby potentially minimizing the ability of diabetes-related derangements of epicardial adipose tissue biology that cause microvascular rarefaction, cardiac fibrosis, and impaired ventricular distensibility. SGLT2 inhibitors have also been shown to inhibit the accumulation and inflammation of epicardial fat in patients with type 2 diabetes. Additionally, SGLT2 inhibitors may inhibit the deleterious effect of sodium-hydrogen exchanger activation in the myocardium and in the kidney, thus leading to cardioprotection and natriuresis (Supplemental Appendix F). Because insulin itself exerts detrimental effects of insulin on the heart, vasculature, and kidneys, however, it is possible that the benefits of both biguanides and SGLT2 inhibitors to reduce the risk of heart failure may (in part) be related to their shared ability to reduce hyperinsulinemia.
By contrast, the majority of antihyperglycemic medications act by stimulating the release of or potentiating the actions of insulin (Figure 1). Insulin use is independently associated with an increased risk of heart failure, despite improved glycemic control (5). Thiazolidinediones promote insulin signaling by increasing the sensitivity of tissues to its metabolic actions. Both pioglitazone and rosiglitazone have been shown to increase the risk of heart failure in randomized controlled clinical trials. Incretin-based drugs (i.e., glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors stimulate the release of insulin from pancreatic β cells by potentiating the actions of GLP-1). The GLP-1 receptor agonist liraglutide increases the clinical instability of patients with overt left ventricular systolic dysfunction, and treatment with the DPP-4 inhibitors saxagliptin, alogliptin, and vildagliptin have been associated with adverse remodeling or an increased risk of heart failure in patients with type 2 diabetes. Sulfonylureas are insulin secretagogues as a result of their action to inhibit the ATP-sensitive potassium channel, and their use has been associated with an increased risk of heart failure that is comparable to that seen with thiazolidinediones (Supplemental Appendix G).
The mechanisms by which these drugs exert their deleterious effects on the clinical course of heart failure are unknown. The action of sulfonylureas to inhibit the ATP-sensitive potassium channel may interfere with an important endogenous compensatory mechanism that serves to reduce calcium overload during periods of stress. Both GLP-1 receptor agonists and DPP-4 inhibitors have been shown to augment cyclic AMP and potentiate adrenergic mechanisms, both of which may exacerbate heart failure. Furthermore, signaling through the GLP-1 receptor can promote inflammation in adipose tissue, which may explain why the use of DPP4 inhibitors promotes cardiac fibrosis in diabetes, predisposing to the development of heart failure with a preserved ejection fraction. Although thiazolidinediones can ameliorate adipose tissue inflammation, both pioglitazone and rosiglitazone exert deleterious effects on the clinical course of heart failure because of their action to promote sodium reabsorption in the renal tubules. In experimental studies, thiazolidinediones exert their sodium-retentive effects (in part) by enhancing insulin signaling in the renal tubules; in clinical studies, the risk of heart failure produced by these drugs is increased by concomitant treatment with insulin. For many antihyperglycemic drugs, therefore, an increase in insulin signaling may exacerbate other deleterious effects of these drugs or may supersede potentially favorable effects on the heart. Interestingly, drugs that signal through insulin are often associated with weight gain; in clinical trials, such weight gain is accompanied by an increased risk of heart failure, even if glycemic control is improved (Supplemental Appendix G).
If insulin signaling exerts adverse effects to increase the risk of heart failure in patients with diabetes without overt symptoms of left ventricular dysfunction, then the same mechanism would also be expected to worsen the clinical course of patients with diabetes who have established heart failure. It is therefore noteworthy that, in several observational studies, patients with chronic heart failure who had levels of glycated hemoglobin ≤7.0% had a worse survival than those with levels of 7.0% to 8.0%, even after adjustment for other predictors of a poor outcome. Importantly, that this relationship was observed only in patients whose diabetes was treated with drugs, not in those who were managed only with diet. This finding suggests that the poor survival of patients with heart failure with a glycated hemoglobin ≤7.0% may have been related to the use of antihyperglycemic medications. The drugs used to intensify glycemic control in these studies (insulin, sulfonylureas, and thiazolidinediones) acted through insulin signaling (Supplemental Appendix H). A relationship between mortality and intensive glycemic control has not been seen in patients with heart failure whose diabetes has been managed using non–insulin-dependent therapeutic regimens.
Summary and Conclusions
The development of heart failure is common in states of hyperinsulinemia, but not in states of glucose intolerance that is accompanied by a deficiency of insulin. This finding has been supported by the findings in both experimental models of types 1 and 2 diabetes as well as in cohort studies of patients with diabetes in the community. Insulin can exert a wide range of detrimental effects on the heart, vasculature, kidneys, and adipose tissue that can lead to heart failure. Of importance, antihyperglycemic drugs that act through insulin signaling appear to consistently increase the risk of heart failure, whereas drugs that ameliorate hyperinsulinemia have been shown to reduce the onset and progression of heart failure. In patients with diabetes and established heart failure, intensive glycemic control with insulin-dependent therapeutic regimens is associated with an increased risk of death. The totality of evidence therefore supports the hypothesis that heightened insulin signaling, rather than hyperglycemia, may be the primary determinant of the development and progression of heart failure in type 2 diabetes. This conclusion suggests that the risk and course of heart failure are meaningfully influenced by the choices physicians make to lower blood glucose.
Footnotes
Dr. Packer has recently consulted for Admittance, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cardiorentis, Celyad, Daiichi Sankyo, Gilead, Novartis, Novo Nordisk, Relypsa, Sanofi, Takeda, and ZS Pharma.
The author attests he is in compliance with human studies committees and animal welfare regulations of the authors' institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
References
- 1.Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41:11–13. doi: 10.2337/dci17-0052. [DOI] [PubMed] [Google Scholar]
- 2.Greulich S., Maxhera B., Vandenplas G. Secretory products from epicardial adipose tissue of patients with type 2 diabetes mellitus induce cardiomyocyte dysfunction. Circulation. 2012;126:2324–2334. doi: 10.1161/CIRCULATIONAHA.111.039586. [DOI] [PubMed] [Google Scholar]
- 3.Konduracka E., Cieslik G., Galicka-Latala D. Myocardial dysfunction and chronic heart failure in patients with long-lasting type 1 diabetes: a 7-year prospective cohort study. Acta Diabetol. 2013;50:597–606. doi: 10.1007/s00592-013-0455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols G.A., Koro C.E., Gullion C.M., Ephross S.A., Brown J.B. The incidence of congestive heart failure associated with antidiabetic therapies. Diabetes Metab Res Rev. 2005;21:51–57. doi: 10.1002/dmrr.480. [DOI] [PubMed] [Google Scholar]
- 5.Kragelund C., Snorgaard O., Køber L. Hyperinsulinaemia is associated with increased long-term mortality following acute myocardial infarction in non-diabetic patients. Eur Heart J. 2004;25:1891–1897. doi: 10.1016/j.ehj.2004.07.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.