

Abstract
Crystal structures of the human angiotensin II type 1 receptor (AT1R) complex with the antihypertensive agent ZD7155 (PDB id: 4YAY) and the blood pressure medication Benicar (PDB id: 4ZUD) showed that binding poses of both antagonists are similar. This finding implies that clinically used angiotensin receptor blocking (ARB) drugs may interact in a similar fashion. However, clinically observed differences in pharmacological and therapeutic efficacies of ARBs lead to the question of whether the dynamic interactions of AT1R with ARBs vary. To address this, we performed induced-fit docking (IFD) of eight clinically used ARBs to AT1R followed by 200 ns molecular dynamic (MD) simulation. The experimental Ki values for ARBs correlated remarkably well with calculated free energy with R2 = 0.95 and 0.70 for AT1R-ARB models generated respectively by IFD and MD simulation. The eight ARB–AT1R complexes share a common set of binding residues. In addition, MD simulation results validated by mutagenesis data discovered distinctive spatiotemporal interactions that display unique bonding between an individual ARB and AT1R. These findings provide a reasonably broader picture reconciling the structure-based observations with clinical studies reporting efficacy variations for ARBs. The unique differences unraveled for ARBs in this study will be useful for structure-based design of the next generation of more potent and selective ARBs.
INTRODUCTION
The angiotensin receptor blockers (ARBs) are lifesaving blood pressure (BP) medications created for treatment of hypertensive patients. Eight ARBs are approved by the Food and Drug Administration. They are receptor-selective competitive nonpeptide antagonists, and the most prescribed therapeutic antihypertensive class with global sales exceeding US $15 billion in 2014. Large-scale clinical trial results (>43 trials) suggested that BP control at the maximal doses of different ARBs statistically differ.1–7 In addition, the clinical trials reported that therapeutic efficacy of ARBs widely differs for BP-independent treatment of several cardiovascular diseases (CVD) such as congestive heart failure, renal failure, diabetic retinopathy, and degenerative vascular diseases.8,9 Differences in chemical structures of ARBs may influence some of the clinical outcomes, but the molecular mechanism is unknown.
In humans, the target of ARBs is the angiotensin type 1 receptor (AT1R), which in vivo mediates responses to the renin–angiotensin system (RAS) hormone, angiotensin II (Angll). In the pathogenesis of CVDs, blood pressure and water-electrolyte control by RAS fail and they are treated with ARBs.8,9 Of two G protein coupled receptors (GPCRs, i.e. AT1R and AT2R) for AngII, AT1R is the predominant AngII receptor in vasculature, heart, adrenal glands, brain, liver, and kidneys in adult life where it also stipulates efficacy of ARBs under therapy. The competitive behavior of AngII and ARBs suggests that they bind at overlapping if not identical ligand pocket of AT1R.9
Generally, ARB interaction with AT1R was mapped in three major phases. First, during the development of losartan10 and eprosartan,11 structure–activity analysis of a large array of intermediary candidates revealed three common pharmacophores necessary to achieve selective binding to AT1R. As a result, common features of ARBs include a biphenyl scaffold attached to acidic/tetrazole group at one end and an imidazole/H-bond acceptor on the opposite end (see Figure 1A).12–14 Second, site-directed mutagenesis experiments and modeling analyses (Figure 1B) suggested that the AT1R residue Asn294TM7 may contact the imidazole/H-bond acceptor, the residues Asp74-Phe77TM2, Ser105-Tyr113TM3, and Trp253-His256TM6 may bind the hydrophobic biphenyl scaffold, and the residues Arg167ECL2 and Lys199TM5 may interact with the acidic tetrazole group in ARBs.14–19 Third, high-resolution X-ray crystallographic analysis correctly identified the interactions of three critical residues of AT1R, Tyr35TM1, Trp84TM2, and Arg167ECL2 for binding olmesartan and ZD7155.20,21 Modeling of additional antagonist docking highlighted conservation of interaction with Tyr35TM1, Trp84TM2, and Arg167ECL2.20 Further, analyses of olmesartan analogs indicated that alterations of ARB structures extend interactions to additional subpockets in AT1R.21
Figure 1.
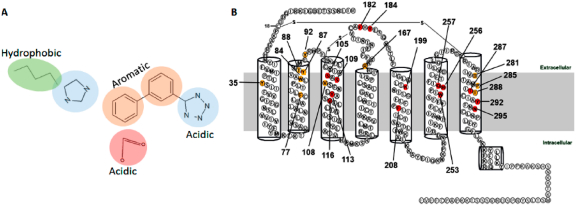
(A) Canonical structure of ARBs. (B) Two-dimensional annotation of AT1R structure showing red colored residues identified as important for binding ARBs by mutagenesis. The yellow colored residues were subsequently identified in the crystal structure of AT1R and confirmed by mutagenesis.
Binding pockets elucidated for ZD7155 (a drug precursor of Atacand, i.e. candesartan) and olmesartan (Benicar) raise questions regarding interactions of clinical ARBs such as Cozaar (losartan), Avapro (irbesartan), Teveten (eprosartan), Diovan (valsartan), Micardis (telmisartan), and Edarbi (azilsaratn) which failed in crystallization attempts. Clinical trials show that efficacy of losartan is lower in reducing the risk for various CVDs than most ARBs.22 While losartan is a weaker inverse agonist of AT1R, olmesartan, candesartan, irbesartan, and eprosartan are experimentally confirmed inverse agonists.13,23,24 As shown in Figure S1, losartan, irbesartan, and valsartan contain a biphenyl tetrazole scafold, whereas azilsartan and telmisartan contain an acidic group in place of tetrazole. Eprosartan has the most differentiated structure in which the biphenyl tetrazole is substituted with benzoic acid.13,14,25 Since all ARBs block AngII binding to AT1R, the mechanism responsible for differences in clinical outcomes remain elusive.3,6,26,27 Clinical efficacy data for CVD end points are not available for all ARBs.15,28 Further, the crystal structures of AT1R are limited to showing the binding poses of olmesartan and ZD7155. Although structure–function studies validated these crystal structures, some of the mutagenesis observations have not been satisfactorily explained. We hypothesize that divergent spatiotemporal interactions of ARBs with different binding residues lead to efficacy differences. Several studies compared the interactions of different ARBs with AT1R but none involving all ARBs in the same experimental set up. In this study, therefore, we used the same experimental setup to compare binding affinities, the docking poses and molecular dynamics of AT1R with all the ARBs to identify differential binding pattern of biphenyl tetrazole and non- biphenyl tetrazole ARBs which may influence the clinical outcome.
Development of next generation ARBs remains an active research area based on the anticipation of better treatment of blood pressure-independent CVD pathologies. Hence, dynamic modeling of specific interactions of individual ARBs with AT1R represented an important benchmark necessary for computer-aided design of next generation ARBs. We used induced-fit docking followed by 200 ns of molecular dynamics (MD) simulation for the drug Benicar. The simulated model was compared to crystal structure as a training exercise. This was followed by MD simulation modeling of all clinically used ARBs complexed with AT1R to decipher spatiotemporal interactions of different ARBs with divergent binding residues. We integrate MD simulation modeling results with competition binding and mutagenesis experimental data. Finally, we provide the details of differential binding mode of all the ARBs including their binding orientation, i.e., sys- and anti- conformation (Figure S2). We will discuss the usefulness of our analysis to guide design of next generation ARBs.
MATERIAL AND METHODS
Protein Preparation.
The starting coordinates of the AT1R [PDB ID: 4ZUD] were retrieved from Protein Data Bank (www.rcsb.org) and further modified for Glide docking calculations.29 For these calculations, the protein was minimized using the Protein Preparation Wizard by applying an OPLS3 (Optimized Potentials for Liquid Simulations) force field.30 Only the non-hydrogen atoms were applied for progressively weaker restraints and the refinement procedure was done based on the default protocol of Schrodinger, LLC, NY, USA, as the Glide uses the full OPLS3 force field at an intermediate docking stage and is claimed to be more sensitive to geometrical details than other docking tools. The most likely positions of hydroxyl and thiol hydrogen atoms, protonation states, and tautomers of His residues, and Chi “flip” assignments for Asn, Gln, and His residues were selected. Finally, the minimizations of protein were performed until the average root-mean-square deviation of the non-hydrogen atoms reached 0.3 Å.
Ligand Preparation.
All the eight clinically used ARBs were drawn in Maestro (Schrodinger, LLC, New York, NY, USA). Each structure was assigned an appropriate bond order using the LigPrep package from Schrodinger, LLC, NY, USA.31 The ligands were converted to .mae format (Maestro, Schrodinger, LLC, NY, USA) and geometrically optimize the ligand and to compute partial atomic charges. Then, at most, 32 poses per ligand were generated with different steric features for the subsequent docking study.
Induced Fit Docking (IFD).
Induced fit docking (IFD) (Schrodinger, LLC, NY, USA) was used to dock the ARBs inside the ligand binding pocket of AT1R. First, the ligand was docked into a rigid receptor model with scaled-down vdW radii. A vdW scaling of 0.5 was used for both the protein and ligand nonpolar atoms. A constrained energy minimization was carried out on the protein structure, keeping it close to the original crystal structure while removing bad steric contacts. Energy minimization was carried out using the OPLS3 force field with implicit solvation model until default criteria were met. The Glide XP mode was used for the initial docking, and ligand poses were retained for protein structural refinements, and then, Prime (Schrodinger, LLC, NY, USA) was used to generate the induced-fit protein–ligand complexes. Each of the structures from the previous step was subjected to side-chain and backbone refinements.32 All residues with at least one atom located within 4.0 Å of each corresponding ligand pose were included in the Prime refinement. The refined complexes were ranked by Prime energy, and the 20 receptor structures within 30 kcal/mol of the minimum energy structure were passed through for a final round of Glide docking and scoring. In the final step, each ligand was redocked into top 20 refined structure using Glide XP.
MM/GBSA.
Prime/MM-GBSA was used to predict the free energy of binding between the receptor and the set of ligands. The binding free energy (ΔGbind) was calculated using the default parameters of Prime, Schrodinger, LLC, NY, USA.33,34 The binding free energy (Gbind) is then estimated using the following equation:
where ER:L is energy of the complex, ER + EL is sum of the energies of the ligand and the apo protein, using the OPLS3 force field, Gsolv (GSA) is the difference between GBSA solvation energy (surface area energy) of complex and sum of the corresponding energies for the ligand and apoprotein.35–37
MD Simulation.
Molecular dynamics (MD) simulations were carried out for all the clinically used ARBs–AT1R complexes using Desmond MD code and the OPLS3 force field30 for minimization of the system. Using the Desmond system builder, a 10 Å buffered orthorhombic system with periodic boundary conditions was constructed using a POPC lipid membrane38–41 and an SPC explicit water solvent.34 The overall charge was neutralized by 0.15 mol/L NaCl. The simulations were performed in the NPT ensemble with number of elements, pressure and temperature are controlled. The temperature of 300 K and pressure of 1.013 bar were kept constant by coupling the system to a Berendsen thermostat and barostat; which is normal temperature and pressure mimicking the real life environment. An integration step of 2.0 was used, Coulombic interactions were calculated using a cutoff radius of 9.0 Å, and long-range electrostatic interactions were calculated using the smooth particle mesh Ewald method.42 Before each MD simulation, a default Desmond membrane protein relaxation protocol was applied.43 To monitor protein stability and conformational fluctuations throughout the simulations, we computed backbone RMSDs after least-squares fitting to the starting structure. This was done for the entire protein and transmembrane α-helices.
ARB Binding and Site-Directed Mutation Effects.
Competitive binding kinetics of all eight ARBs in clinical use were performed in a single platform using methods as described in previous reports from our laboratory.20,21 COS1 cells transfected with AT1R were used to prepare total membrane and used in 125I-AngII competitive equilibrium binding assay. IC50 values were estimated from four replicate analyses. Mutation effect on the IC50 for ARBs was obtained from previously published reports and presented as fold-change compared to the wild-type control in the same study as detailed in the Supporting Information Table S1.
RESULTS AND DISCUSSION
Induced Fit Docking of ARBs and MM/GBSA Free Energy Calculation.
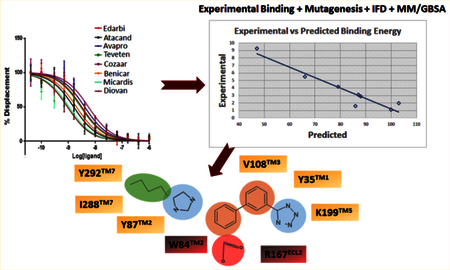
Each docking simulation began with the protein conformation based on the 2.4-Å resolution crystal structure (PDB entry 4ZUD) of Zhang et al.21 Induced fit docking protocol of Schrodinger, LLC, positioned all clinically used ARBs at the same space within the orthosteric pocket (see Figure 2). Critical contact residues, Tyr35TM1, Trp84TM2, and Arg167ECL2 appear to project toward all ARBs. Our mutagenesis studies have shown that these three residues are essential for binding AngII, which validates competitive antagonism of clinically used ARBs. However, binding conformation and the nature of bonding interactions of each ARB with critical contact residues significantly differed in IFD simulation (Figure 3). Arg167ECL2 strongly interacted with all ARBs. While Tyr35TM1 interacted with seven ARBs but not with Valsartan, Trp84TM2 did not appear to interact with all of the ARBs, an observation which contradicted mutagenesis results. Instead Tyr87TM2 appears to interact with six ARBs but not with irbesartan and telmisartan. Further, Ser105TM3, Ser109TM3, and Asp281TM7 interacted with more than one ARBs, but not all. Telmisartan interacted strongly with several additional residues.
Figure 2.
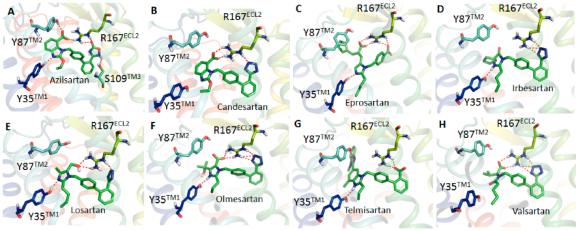
Binding mode of AT1R with azilsartan (A), candesartan (B), eprosartan (C), irbesartan (D), losartan (E), olmesartan (F), telmisartan (G), and valsartan (H). Biphenyl tetrazole imidazole ARBs bind in a similar binding mode and other ARBs are in different conformations. The acid moieties of all ARBs are projecting toward Arg167ECL2, but the extent of interaction varies between different ARBs.
Figure 3.
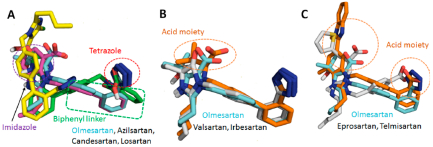
Binding conformation of eight clinically used ARBs in the binding pocket of AT1R. ARBs which have the biphenyl tetrazole imidazole group (A) have a similar conformational orientation. Biphenyl tetrazole group superimposed with Olmesartan (B). Nonbiphenyl tetrazole ARBs superimposed with olmesartan (C).
Experimental and MM/GBSA Binding Energies.
125I-AngII competitive binding experiments were performed for all clinically used ARBs and found that telmisartan has high Ki followed by candesartan, olmesartan, irbesartan, azilsartan, eprosartan, losartan, and valsartan (Figure 4A). IFD followed by MM/GBSA free energy calculation result was in agreement with the experimental binding data for ARBs (Table 1). Therefore, we believe that the binding poses generated by IFD are reliable. In order to further refine individual ARB poses we performed 200 ns MD simulation study.
Figure 4.
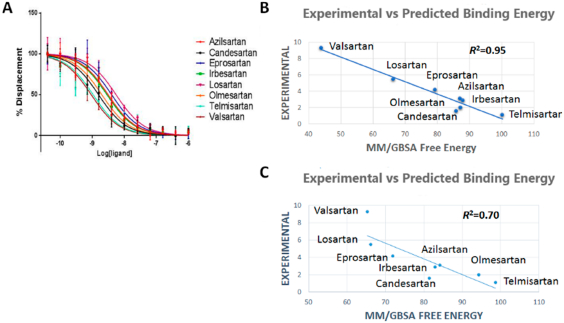
(A) Competitive binding experiment showing the binding affinity of ARBs. (B) Experimental vs predicted binding energy of all ARBs from IFD pose. (C) Experimental vs predicted binding energy of all ARBs from MD simulation pose.
Table 1.
Average MM/GBSA Binding Free Energy from Induced Fit Docking (IFD) Pose and Average MM/GBSA Binding Free Energy from Representative Structures Generated from the 200 ns MD Simulation Trajectoriesa
| compound name | experimental binding | induced fit docking (IFD) | representative structure from MD simulation trajectories | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (ki nM) | ΔGBind (kcal/mol) | ΔGBind Coulomb (kcal/mol) | ΔGBind H-bond (kcal/mol) | ΔGBind vdw (kcal/mol) | ΔGBind (kcal/mol) | ΔGBind Coulomb (kcal/mol) | ΔGBind H-bond (kcal/mol) | ΔGBind vdw (kcal/mol) | |
| azilsartan | 3.1 | −87.034 ± 3.447 | −29.555 ± 2.624 | −3.200 ± 0.277 | −30.923 ± 48.095 | −84.193 ± 7.969 | −64.446 ± 12.191 | −4.251 ± 0.992 | −55.266 ± 5.543 |
| candesartan | 1.6 | −85.863 ± 1.728 | −169.006 ± 5.042 | −6.751 ± 0.184 | −51.735 ± 3.003 | −81.398 ± 7.143 | −160.073 ± 9.700 | −8.689 ± 1.124 | −52.265 ± 2.713 |
| eprosartan | 4.2 | −79.323 ± 9.809 | −109.319 ± 4.227 | −5.127 ± 0.231 | −44.810 ± 5.602 | −65.302 ± 10.884 | −149.366 ± 12.807 | −5.825 ± 1.078 | −59.156 ± 2.159 |
| irbesartan | 2.9 | −87.932 ± 2.404 | −51.603 ± 2.648 | −3.342 ± 0.289 | −56.780 ± 1.733 | −82.890 ± 9.032 | −65.385 ± 6.386 | −5.736 ± 0.312 | −52.432 ± 1.812 |
| losartan | 5.5 | −66.308 ± 7.619 | −95.575 ± 11.843 | −3.023 ± 1.174 | −45.282 ± 5.694 | −66.107 ± 6.132 | −83.837 ± 6.428 | −1.905 ± 0.894 | −49.037 ± 4.586 |
| olmesartan | 2.0 | −87.212 ± 9.969 | −87.661 ± 8.315 | −5.94 ± 0.410 | −53.889 ± 2.847 | −94.336 ± 8.593 | −92.139 ± 8.017 | −6.390 ± 0.582 | −64.207 ± 4.344 |
| telmisartan | 1.1 | −100.237 ± 16.677 | −5.065 ± 10.538 | −3.168 ± 0.426 | −60.118 ± 4.895 | −98.627 ± 9.030 | −17.306 ± 2.974 | −3.206 ± 0.454 | −62.000 ± 8.700 |
| valsartan | 9.3 | −43.943 ± 10.083 | −146.492 ± 6.627 | −5.149 ± 0.940 | −46.370 ± 3.492 | −65.302 ± 10.884 | −149.366 ± 12.807 | −5.825 ± 1.078 | −59.156 ± 2.159 |
Mean ± SD.
Binding and MM/GBSA results reveal that Ki of losartan and valsartan are lowest among the ARBs. Losartan does not have a carboxylic group among the biphenyl tetrazole ARBs. IFD indicated that the hydroxyl group attached to the imidazole in losartan could not form H-bond with Tyr87TM2 while other biphenyl tetrazole ARBs could form multiple H-bonds as seen in Figure 2. In the case of valsartan, due to replacement of imidazole group by methylamino-butanoic acid, the carboxylic group attached in N atom displayed anti conformation (i.e., orientation of groups at the ends of the biphenyl spacer) with respect to the tetrazole group (Figures 3 and S2). Hence the carboxylic group could not form H-bond with Arg167ECL2 or Tyr87TM2; instead it forms H-bond with Asp281TM7. These observations reveal that syn and anti conformations of ARBs may play a paramount role in bonding with Arg167ECL2 or Tyr87TM2 to achieve better affinity.
The five best IFD poses for each of the eight ARBs were rescored to obtain MM/GSBA ensemble-average free energy which was in agreement (R2 = 0.95) with the experimental binding energy (Figure 4B). We further calculated the MM/GBSA free energy from the 200 ns MD simulation trajectory (Table 1). Five representative structures generated by clustering the whole trajectories using Desmond_RMSD_Clustering application and MM/GBSA binding free energy were calculated based on the five representative structures. We observed an R2 = 0.70 correlation between the experimental binding and predicted binding energy (Figure 4C). This result further reveals that the MD simulation result is in agreement with the experimental binding experiment.
The binding energies of ARBs varied from −43 to −102 kcal/mol. The decomposition of calculated binding energies reveals that electrostatic, H-bond, and van der Waals interactions contributed in the binding of all ARBs. Contribution of electrostatic interaction is higher in binding candesartan, valsartan, olmesartan, eprosartan and losartan. The contribution of H-bond interactions and the van der Waals interactions are in the range from −30 to −60 kcal/mol. Telmisartan shows the highest van der Waals interactions due to its bigger size and higher surface area contact.
Molecular Dynamics Simulation.
The flexible docking procedure we adapted evaluates both syn and anti conformations of ARBs in the active site of AT1R. Our MD simulation favors anti conformation probability for seven clinically used ARBs (see Figure S2), which is also the orientation found in two crystallographic structures (4YAY and 4ZUD). All simulations were carried out using the IFD pose. The backbone RMSD of AT1R bound with olmesartan reach a plateau after 50 ns of MD simulation (Figure 5). The RMSD of olmesartan is quite stable in the orthosteric site of AT1R during 200 ns MD simulation (Figure 5). The backbone RMSD of other seven ARBs are shown in Figure S3. Average structures of all AT1R bound ARBs observed from the MD simulation trajectory are similar to the IFD pose as shown in Figure S4.
Figure 5.
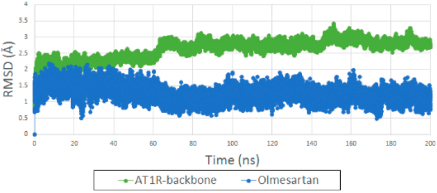
(green) RMSD backbone of AT1R bound with olmesartan generated from IFD. (blue) RMSD of olmesartan in the orthosteric site of AT1R in 200 ns MD simulation.
MD Simulation of Benicar (Olmesartan) Binding.
The starting point of our MD simulation was the IFD model (Figure 4) in which H-bonding is observed between Arg167ECL2, tetrazole group and imidazole ring of olmesartan, Tyr35TM1 hydroxyl with the imidazole ring and Tyr87TM2 hydroxyl group with imidazole hydroxyl group. Interactions that occur between a residue and the ligand more than 30% of MD simulation time in the selected trajectory (0–200 ns) are detailed in Figure 6. Crystal structure and MD simulation results are in agreement regarding olmesartan interactions with Tyr35TM1, Phe77TM2, Trp84TM2, Tyr87TM2, Tyr92ECL1, Val108TM3, Ser109TM3, Arg167ECL2, Cys180ECL2, Lys199TM5, and Ile288TM7.
Figure 6.
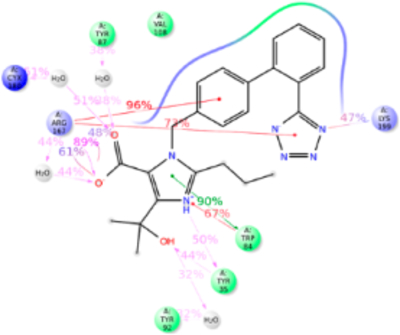
Schematic detailing olmesartan interactions with AT1R residues. Interactions that occur more than 30.0% of the simulation time in the selected trajectory (0.00–200.00 ns) are shown. It is possible to have interactions with >100% as some residues may have multiple interactions of a single type with the same ligand atom.
The Tyr35TM1 side-chain formed H-bond with tetrazole ring, hydrophobic interaction with the alkyl tail and water bridge interactions. Crystal structural analysis reveals that the N atom of imidazole group of olmesartan forms H-bond with phenolic side chain. The alkyl tail attached to imidazole ring forms a weak hydrophobic interaction projected phenyl ring of Tyr35TM1. Observed interactions of Tyr35TM1 with olmesartan are supported by mutagenesis analyses.20,21 The Tyr35AlaTM1 mutant significantly reduces the olmesartan binding affinity, but the mutations to Phe and Ile do not cause significant changes. Mutations of nonpolar residues Phe or Ile cause loss of H-bond formation with olmesartan, but it can form a stronger hydrophobic interaction with the alkyl tail which compensates the loss of H-bond. This hydrophobic interaction may be significantly reduced when mutated with smaller residue Ala due to increase in distance between Ala side chain and alkyl tail. But mutation with electrically positive charged residue Lys will prevent the formation of h-bond with imidazole ring as well as hydrophobic interaction with alkyl tail. This could be the reason why Tyr35LysTM5 mutation abolished the olmesartan binding (Table S1).
The aromatic side chain of Trp84TM2 forms π–π stacking and hydrophobic interaction with the phenyl ring of olmesartan during 50% of the simulation time. Mutation with another aromatic residue, Phe, showed no marked change in the binding affinity because Phe still retains the π–π stacking and hydrophobic interactions. However, mutations to Ala and Ile completely abolished the binding of olmesartan, since side chains of Ala or Ile could not form π–π stacking (Figure S5).
MD simulation result recapitulates the crystallographic observation that Arg167ECL2, contributed most interactions in olmesartan binding, with 250% contacts throughout the simulation (Figure 6). Hydrophobic residue Ala or polar residue Gln mutations, abolish binding as these side chains could not interact with imidazole-carboxylic and the tetrazole ring on two ends of the biphenyl linker. Substitution of electrically positive charged residue, Lys has a single -NH2 group which could partially interact with either tetrazole group or imidazole-carboxylic group and hence reduces it binding affinity.
In the current MD simulation study we observed the ε-NH2 group of Lys199TM5 side chain forming an ionic bond with the phenyl group and tetrazole ring 88% of simulation time. Crystal structures could not detect Lys199TM5 role due to poor electron density. However, Lys199TM5 → Arg mutation slightly enhanced the binding affinity because the Arg side chain could interact with the tetrazole and phenyl groups of olmesartan. This result suggests that Lys199TM5 replacement with other residues should abolish olmesartan binding. As expected Lys199TM5 → Gln and Ala decreased the olmesartan binding affinity. Structural analysis reveals that a shorter Ala199TM5 side chain does not disturb the binding pocket of AT1R. But a larger polar Gln199TM5 mutant side chain projected toward the binding pocket and hindered other pocket residues and hence impeded olmesartan binding. Extensive mutagenesis studies have shown that Lys199TM5 is important for ARBs/AngII binding (Table S1).
Furthermore, residues Phe77TM2, Tyr87™2 Tyr92ECL1, Val108TM3, Ser109TM3, Cys180ECL2, and Ile288TM7 were involved in interaction with olmesartan about 50% of MD simulation time, primarily forming hydrophobic interactions. Ala substitution mutations at these positions significantly alter the binding affinity of olmesartan,21 suggesting functionally specific interactions with these residues in AT1R.
MD Simulation of Biphenyl Imidazole ARBs Binding.
Five currently used ARBs in clinical practice—osartan, candesartan, azilsartan, valsartan, and irbesartan—are variants of the biphenyl scaffold structure in olmesartan. Their binding to AT1R is anticipated to recapitulate interactions with residues that were found to interact with olmesartan in the crystal structure.
Interactions of Cozaar (Losartan).
Losartan is considered founder ARB and structurally closest to olmesartan; with hydroxyl and chloride group attached to imidazole instead of carboxylic group (mimicking the losartan metabolite, EXP3174) and propanol in olmesartan (see Figure 7). The anti conformation of losartan-salt was reported in the crystal structure44 and the same result was obtained in quantum mechanics45 and molecular modeling studies46 by different groups. Our MD simulation recapitulated anti conformation for losartan in the AT1R pocket interacting with Tyr35TM1, Trp84TM2, Tyr87TM2, Val108TM3, Arg167ECL2, and Lys199TM5. The binding affinity of losartan is lower than that of olmesartan. The losartan imidazole ring does not interact with Arg167ECL2; however the hydroxyl group forms a strong H-bond with Tyr87TM2 and Arg167ECL2 forms a strong ionic bond with tetrazole and phenyl ring. In addition, the tetrazole forms a strong ionic bond with Lys199TM5. MD simulation further reveals that Arg167ECL2 and Lys199TM5 contribute 300% interactions with strong ionic and hydrophobic interactions.
Figure 7.
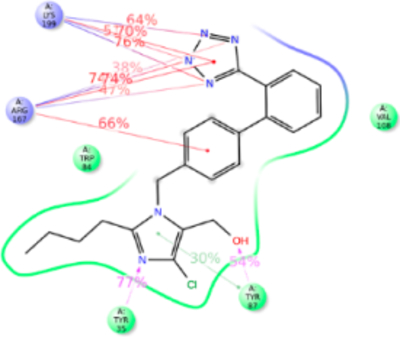
Schematic of detailed losartan interactions with AT1R residues.
The Tyr35PheTM1 mutation resulted in ~4-fold decreased binding affinity, whereas affinity for olmesartan in the Tyr35PheTM1 mutant was unaffected (Table S1). Phe35TM1 could not compensate the loss of H-bond in losartan binding while imidazole ring in olmesartan forms π–π stacking. This difference may help the imidazole moiety of losartan pull toward the Tyr87TM2 in the Tyr35Phe mutant. Trp84TM2 forms a weak π–π stacking with phenyl ring attached to the imidazole group. However, the Trp84PheTM2 mutation reduces the binding 20-fold since a Phe84TM2 could not form π–π stacking with the phenyl group of losartan because the losartan phenyl group also has a strong ionic interaction with Arg167ECL2 which pulls this phenyl ring away. As expected the Arg167LysECL2 mutant binding affinity for losartan reduced 3-fold and no significant change was present in the Lys199AlaTM5 mutant which was similar to olmesartan binding. The role of Lys199TM5 ionic bond with tetrazole group of losartan is compensated by a stronger ionic bond with Arg167ECL2 (Figure S6).
Interactions of Atacand (Candesartan).
Structure of candesartan includes biphenyl tetrazole scaffold similar to olmesartan and losartan, but the imidazole moiety in candesartan is substituted with alkyl ether tail and benzoic acid. MD simulation displayed candesartan in the anti conformation interacting with Tyr35TM1, Trp84TM2, Tyr87TM2, Arg167ECL2, and Lys199TM5 (Figure 8). Of these, a significant functional role of Tyr87TM2 and Arg167ECL2 in candesartan binding is revealed by mutagenesis while mutations of Tyr35TM1, Trp84TM2, and Lys199TM5 do not significantly alter the affinity. The H-bond between Tyr35TM1 and imidazole ring is replaced by stronger hydrophobic interaction Phe35TM1 with the alkyl ether tail and the phenyl group in the Tyr35Phe mutation which does not change candesartan affinity. Similarly, weak hydrophobic interaction with Trp84TM2 is unaffected in the Trp84Phe mutation due to hydrophobic interaction of Phe84TM2 with the ether group substituted in imidazole ring in candesartan. Tyr87TM2 forms a stronger H-bond and hydrophobic interaction with a carboxylic group of benzoic acid ring, contributing about ~100% interaction with candesartan in simulation. Arg167ECL2 contributed about 600% of interactions through H-bond, ionic bond, and hydrophobic interactions with the benzyl carboxylic group, tetrazole group and strong ionic interaction with the phenyl group. Mutagenesis reveals that Arg167Lys mutation reduces candesartan affinity 4-fold, due to loss of H-bond interaction with benzoic acid and tetrazole group but retains only the ionic interaction (Table S1). Lys199TM5 contributed about 100% interaction and forms H-bond and ionic bond with the tetrazole group, plays a less critical role in binding candesartan as indicated by the Lys199Ala mutant which does not change the binding affinity of candesartan (Table S1). The Ala199TM5side chain of mutant projected toward the phenyl ring of candesartan establishing a strong hydrophobic interaction (Figure S7).
Figure 8.
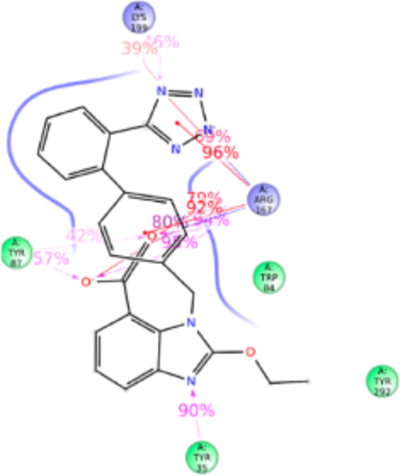
Schematic of detailed candesartan interactions with AT1R residues.
Interactions of Edarbi (Azilsartan).
Azilsartan structure is similar to candesartan but tetrazole ring is substituted with keto-oxadiazol. MD simulation displayed azilsartan in anti conformation interacting with Tyr35TM1, Trp84TM2, Tyr87TM2, Tyr92ECL1, Val108TM3, Ser109TM3, Leu112TM3, Tyr113TM3, Arg167ECL2, Phe182ECL2, Lys199TM5, Pro285TM7, Ile288TM7, and Tyr292TM7 (Figure 9). Mutagenesis indicated that Tyr113AlaTM3, Lys199AlaTM5, and Gln257AlaTM6 mutations significantly reduce azilsartan binding affinity. The Tyr113TM3 forms H-bond and hydrophobic interaction with the keto-oxadiazol and phenyl group, and in the Ala113TM3 mutant loss of H-bond and weakened hydrophobic interaction reduce the binding affinity. Lys199TM5 forms an ionic bond followed by weak h-bond and hydrophobic interaction with keto-oxadiazole moiety of azilsartan. Lys199AlaTM5 or Lys199GlnTM5 mutations significantly reduce azilsartan affinity (Table S1). Lys199TM5 mutation to a polar residue, Gln or a hydrophobic residue Ala could not form an ionic bond as well as could not compensate hydrophobic interaction. The Gln257Ala mutation significantly reduces azilsartan affinity, but His256TM6 does not dynamically participate in azilsartan binding although it is a part of azilsartan binding pocket. The polar Gln256TM6 side chain of His256GlnTM6 mutant projects toward azilsartan in the binding pocket and helps maintain the charge-state of the binding pocket. Arg167ECL2 interacts 200% of simulation period, through H-bonds to the acid moiety. We do not have the mutagenesis data for azilsartan binding in Arg167ECL2 mutants, although it interacts 200% with predominantly H-bonding (Figure S8).
Figure 9.
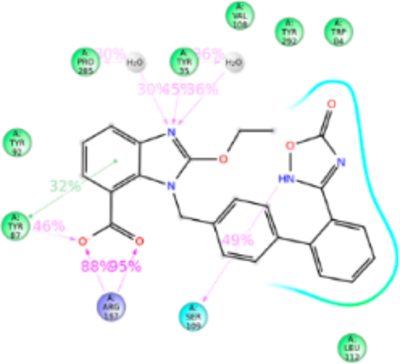
Schematic of detailed Azilsartan interactions with AT1R residues.
Interactions of Diovan (Valsartan).
Valsartan lacks the imidazole group which is substituted with methylamino-butanoic acid. The alkyl tail orientation in valsartan is anomalous compared with other ARBs, in the same plane as the tetrazole group. As a consequence valsartan adopted a slightly different conformation which resulted in strong bonding with Trp84TM2, Tyr87TM2, Tyr92ECL1, Arg167ECL2, Asp281TM7, Ile288TM7, and Tyr292TM7 (Figure 10). Valsartan interaction (tetrazole) with Lys199TM5 is water-mediated and not very strong. Most of the orthosteric site residue mutations which exist in our library do not significantly alter valsartan binding. The Tyr113AlaTM3 mutation reduced valsartan binding (Table S1). Simulation result suggests that valsartan binds far away from the Tyr133TM3. It is possible that side chain of Tyr113TM3 could maintain the helix orientation while binding valsartan (Figure S9).
Figure 10.
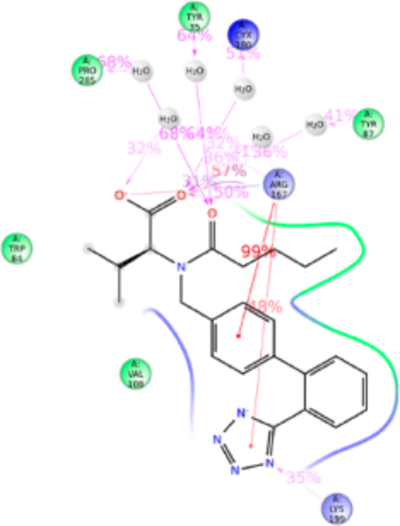
Schematic of detailed valsartan interactions with AT1R residues.
Interactions of Avapro (Irbesartan).
Imidazole group is substituted with a keto cyclopentane structure in irbesartan. MD simulation suggests binding of this substituent in syn conformation akin to methylamino-butanoic acid group in valsartan observed in IFD. Alkyl tail in other ARBs are in a different plane and consequently irbesartan forms a strong H-bond with Asp281TM7. MD simulation indicates that Trp84TM2, Arg167ECL2, Lys199TM5, Asp281TM7, and Tyr292TM7 are important for irbesartan binding (Figure 11). The tetrazole ring of irbesartan forms ionic bond and H-bonding with Arg167ECL2 and Lys199TM5. During the course of MD simulation, the oxygen atom of the keto group attached to imidazole ring forms H-bond with Arg167ECL2. The biphenyl ring forms π–π stacking with Trp84TM2 throughout the MD simulation. Asp281TM7 forms strong H-bond with N atom of imidazole ring and contributed ~100% interaction. Tyr292TM7 forms π–π stacking with phenyl ring and H-bonding with tetrazole ring. We observed that Tyr113Ala mutation causes a significant reduction of irbesartan binding affinity (Table S1), although MD simulation does not indicate interaction with Tyr113TM3 and irbesartan. Ala113TM3 may change helical conformation altering projection of Ser105TM3 and Val108TM3 side chains which are involved in weak interactions with irbesartan (Figure S10).
Figure 11.
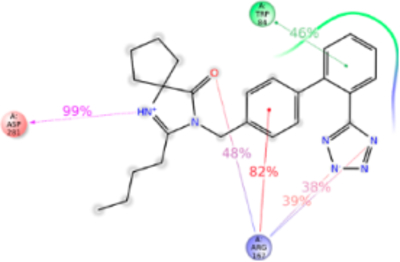
Schematic of detailed irbesartan interactions with AT1R residues.
MD Simulation of Nonbiphenyl Scaffold ARBs Binding.
Eprosartan and telmisartan lack the biphenyl scaffold linking the tetrazole and imidazole moieties present in majority of ARBs. Their interactions with AT1R are anticipated to reveal nonredundant contacts, potentially unravelling subpockets that may be further explored in future drug design.
Interactions of Teveten (Eprosartan).
Eprosartan adopted a scaffold consisting of thiophene connected to benzylimmidazole carboxylate by an alkyl chain. A carboxylic group is substituted on the alkyl chain. MD simulation study observed that Tyr35TM1, Phe77TM2, Trp84TM2, Tyr92ECL1, Val108TM3, Ser109TM3, Arg167ECL2, Phe182ECL2, Lys199TM5, Ile288TM7, and Tyr292TM7 are involved in eprosartan binding (Figure 12). Mutagenesis validated that the residues Tyr35TM1, Trp84TM2, Arg167ECL2, Phe182ECL2, Lys199TM5, Pro285TM7, Ile288TM7, and Tyr292TM7 are important for eprosartan binding (Table S1). Tyr35TM1 contributes H-bond, hydrophobic interaction and water bridges interaction with imidazole ring. Tyr35Ala mutation significantly reduces the binding affinity of eprosartan. In Ala35TM1 mutation, the H-bond and water bridges interaction with eprosartan is lost resulting in 1000-fold reduction of binding affinity. Trp84TM2 contributed 90% hydrophobic interaction in eprosartan binding. Trp84Phe mutation reduces eprosartan binding by ~8-fold; the indole side chain of Trp84TM2 could form a stronger hydrophobic interaction than with Phe. The two carboxylic groups of eprosartan form strong H-bond and ionic bond with Arg167ECL2, and Lys199TM5 throughout our simulation. Mutating either of these important residues does not affect the binding affinity of eprosartan since either of these residues compensates each other. The Pro285Ala mutation reduces the binding affinity ~4-fold. The substituted thiophene group of eprosartan forms a hydrophobic interaction with Pro285TM7 and mutating to smaller side chain residue Ala disrupts the interaction. Ile288Ala mutation reduced eprosartan binding significantly, validating strong hydrophobic interaction of Ile288TM7 with alkyl tail and thiophene group of eprosartan. Tyr292TM7 forms hydrophobic interaction and water-bridge with alkyl tail and imidazole group respectively and contributed about 70% of the interaction. The Tyr292Ala mutant reduces the binding affinity by ~12-fold, since the shorter side chain of Ala could only form a weak hydrophobic interaction (Figure S11).
Figure 12.
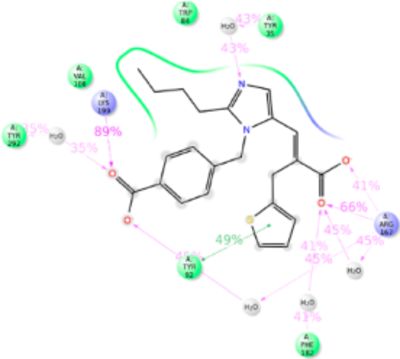
Schematic of detailed eprosartan interactions with AT1R residues.
Interactions of Micardis (Telmisartan).
Telmisartan is the biggest ARB molecule. It is substituted with two benzimidazoles connected to each other and lacks tetrazole ring which is replaced by a carboxylic group. MD simulation suggested that residues Arg167ECL2, Phe182ECL2, Lys199TM5, His256TM6, Asp263TM6, and Ile288TM7 are important in telmisartan binding (Figure 13). Interactions with Tyr35TM1, Trp84TM2, and Lys199TM5 are comparatively reduced. Mutagenesis showed that Trp84TM2, Tyr92TM2, His256TM6, Gln257TM6, and Ile288TM7 are the important residues for telmisartan binding (Table S1). Mutation of Trp84Phe reduced the binding affinity of telmisartan by ~5-fold. Hydrophobic indole side chain of Trp84TM2 forms a hydrophobic interaction with benzimidazole of telmisartan but mutation with Phe which has shorter phenyl chain could weaken the hydrophobic interaction and hence reduce in binding affinity. Tyr92Ala mutation resulted in ~8 fold reduction of binding affinity. MD simulation result shows that it has a weak hydrophobic interaction with telmisartan but the phenol side chain of Tyr is projected toward the Arg167ECL2 and could help in balancing the interaction between Arg167ECL2 and telmisartan. Arg167ECL2 is predicted as the major residue contributing 140% of interactions for telmisartan binding. It forms h-bond with the carboxylic group and ionic bond with the phenyl ring of benzimidazole. Simultaneously these functional groups of telmisartan also interact with Asp263TM6, Phe182ECL2, and His256TM6. Therefore, mutation of Arg167ECL2 does not significantly affect telmisartan binding. Similarly, Lys199TM5 also has a weak H-bond with N atom of benzimidazole group but Asn200TM5 has also formed the h-bond in the same site in the course of MD simulation, thus compensating for the Lys199TM5 binding. Hence, the Lys199TM5 mutation does not affect the binding affinity of telmisartan. Mutagenesis result further demonstrated that Ile288Ala mutation reduces the binding affinity of telmisartan by ~12-fold (Table S1). MD simulation observed that bigger and flanking alkyl side chain of Ile288TM7 is projected in between the two benzimidazole groups and forms strong hydrophobic interactions. But mutation to smaller hydrophobic side chain residue Ala could only form a weak hydrophobic interaction and hence resulted in reducing the binding affinity. The anomaly observed in telmisartan binding in comparison with other ARB binding is the π–π stacking with benzimidazole and aromatic side chains of Trp253TM5 and His256TM5. Our mutagenesis result observed that H256A mutation resulted in ~6 fold decreased in binding affinity. The earlier report suggests that Trp253TM5 and His256TM5 are the important residues for AT1R activation. Therefore, we suspect the difference in the clinical outcome of telmisartan is mainly due to these interactions (Figure S12).
Figure 13.
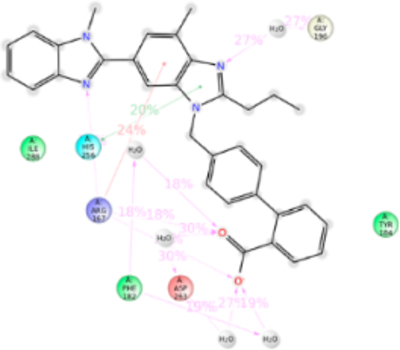
Schematic of detailed telmisartan interactions with AT1R residues.
Modules from Comparison of AT1R Interaction with AT1R ARBs.
Table S2 provides a comparative view of interaction of clinical ARBs with AT1R. IFD combined with MD simulation analyses of biphenyl tetrazole ARBs show that the nature and importance of bonding significantly differ even when the same residue is involved in binding all ARBs. This principle is best illustrated when Arg167ECL2 interaction with six biphenyl ARBs is compared. Arg167ECL2 interacted with both imidazole and tetrazole groups in olmesartan. However, its interaction differs in other ARBs particularly in losartan, valsartan, and irbesartan. MD simulation also revealed unequivocal interactions with Lys199TM5 in the case of losartan and irbesartan. Valsartan interactions with Lys199TM5 is a weak water-mediated interaction. Irbesartan forms ionic and H-bonding with Arg167ECL2 and Lys199TM5. Arg167ECL2 contributed strong interactions for candesartan binding, but not Lys199TM5. Both Arg167ECL2 and Lys199TM5 bond with two carboxylic groups of eprosartan but mutating either of these residues does not affect eprosartan binding. Arg167ECL2 and Lys199TM5 do not significantly affect telmisartan binding.
Strength of binding of different ARBs involves interplay of π–π stacking, hydrogen-bond, ionic, and hydrophobic interactions that clearly differ. We also note that leaning of ARBs within orthosteric pocket show differences. For instance, olmesartan, azilsartan and losartan lean relatively more on TM1–TM3 residues, whereas valsartan, eprosartan, and telmisartan shift toward residues in TM6–TM7. That such differences are important is independently demonstrated by mutation effects that significantly differ for different ARBs. Mutagenesis showed that common residues, Tyr35TM1, Trp84TM2, Arg167ECL2, Lys199TM5, and Ile288TM7 interact with both AngII and ARBs accounting for competitive interaction with AngII. Observed interaction of ARBs with residues such as Lys199TM5 and Asp281TM7 which eluded crystal structures indicates alternative contacts that could be targeted in designing ARBs to modulate effects, such as inverse agonism and mechanical activation of the receptor. In a recent study, Takezako et al.24 suggested that Ser109TM3, Phe182ECL2, Gln257TM6, Tyr292TM7, and Asn295TM7 are important residues for strong inverse agonism. We observed that all ARBs interact with more than one of these residues (Figures 6–13 and S5–S12). However, losartan shows weak interactions with these residues which is consistent with the reports that losartan is a weak inverse agonist. MD simulation results indicate that eprosartan and telmisartan leaned toward the ECL2 region (Table S2) more than the biphenyl ARBs. Conformational mapping studies previously reported from our lab indicated the formation of a ligand-sensitive lid by the ECL2 in AT1R.47 Recently LSD binding to serotonin receptor (5-HT2B) has been found to induce a similar lid in the ECL2 of 5-HT2B.48 Closing the lid makes the LSD stay longer in the orthosteric pocket. Telmisartan is already reported to be a long-acting ARB. Eprosartan, candesartan and azilsartan which demonstrate interactions with residues in ECL2 in modeling may also be expected to display long-acting pharmacology.
The relationship between the binding modes of ARBs with differences in efficacy is complex. In general the ARBs interfere with activation mechanisms of AT1R. The mechanism of AT1R activation by AngII studied by mutagenesis and molecular modeling techniques19,49–52 indicate that C-terminal tail of AngII binds with Lys199TM5. The hydrophobic phenyl group of Phe8AngII interacts with Trp84TM2, Val108TM3, Pro284TM7, Ile288TM7 and Tyr292TM7. Tyr4 of AngII interacts with R167ECL2 which is an important interaction shown by recent studies.20,21 Thus, interactions with aromatic residues of AngII leads to activation of AT1R.51 In order to compete with the AngII binding, the ARBs should interact with these residues. Indeed, current study reveals that acid moiety i.e. tetrazole and carboxylic groups of all ARBs bind with Arg167ECL2, Ile288TM7, and Tyr288TM7. The interaction with biphenyl rings of ARBs with Trp84TM2, Val108TM3, Pro284TM7, Ile288TM7, and Tyr292TM7could block the activation of AT1R by aromatic residues of AngII. Difference in the inverse agnostic property of ARBs has been shown in our previous study,24 which is relevant to AngII independent activation of AT1R. The residues Phe77TM2, Asn111™3, Asn294TM7, Ile288TM7, and Tyr292TM7 that control AngII independent activation of AT1R interact with aromatic rings in ARBs, presumably stabilizing distinct inactive states of the receptor.
Some stereochemical features that would be useful to design next generation ARBs are revealed. Functional group/pharmacophores necessary in an ARB include two acid moieties (tetrazole and carboxylate) at either end of an aromatic-hydrophobic core and a hydrophobic tail. The tetrazole and carboxylic acid moieties in syn conformation project toward the Arg167ECL2. If three acid moieties are present an anticonformation will result projecting toward the Tyr35TM1 and will form a stronger interaction. Thus, targeting the acidic pharmacophore sites may enhance the potency. In addition, it could enhance interaction with additional important residues such as Lys199TM5 and Asp281TM7 to modulate effects such as inverse agonism. The aromatic hydrophobic core structure could be targeted for modification to alter the ensemble effect of π–π stacking, hydrogen-bond, ionic, and hydrophobic interactions. It is important to note that new generation ARBs leaning more toward either TM1–TM3 residues or toward residues in TM6–TM7 within the orthosteric pocket could result in functional differences. Therefore, this IFD combined with MD simulation study will help the medicinal chemist to design novel and potent ARBs.
General GPCR activation models suggest that full, partial, and biased agonists harbor the ability to activate different levels of receptor signaling, which is primarily related to ligand-induced receptor conformational changes. Differences in ARB interactions we demonstrated here provides important insight for the speculation that extent of inhibition of basal function of AT1R by ARBs likely differ. Differences observed in nature of interactions and/or leaning of ligands could be initiation sites for propagation of ARB-bound AT1R conformation that ultimately locks the receptor in an inhibited functional state. Stability and half-life of this state likely differ depending on the ARB structure and its interactions with AT1R.
CONCLUSION
A combination of IFD, MM/GBSA, and MD simulation reveals distinctive features of binding sites for the eight clinically used ARBs that fully integrate the experimentally determined crystal structure of AT1R and archival mutagenesis data. For instance, Tyr87 TM2, Phe182ECL2, Lys199TM5, and Asp281TM7 observed as ancillary residues in the ligand binding pocket in the crystal structure of AT1R are found to be critical spatiotemporal contacts for ARBs by MD simulation, which is in agreement with mutagenesis results. The biphenyl tetrazole-imidazole ARBs (losartan, olmesartan, valsartan, candesartan) bind in anti conformation. However, azilsartan and irbesartan differ in structure and prefer anti conformation that may weaken interaction with Arg167ECL2 and Lys199TM5 and form a strong H-bond with Asp281TM7. The critical residues in binding all ARBs are Arg167ECL2 and Trp84TM2. Residues Tyr35TM1, Tyr87TM2, and Lys199TM5 interacted with most of the ARBs. The nonbiphenyl ARBs, eprosartan and telmisartan, display less interaction with Tyr35TM1, Trp84TM2, and Trp87TM2 owing to structural difference. In designing a novel ARB, incorporating the prospect for interaction with an ECL2-lid may produce a longer acting class of ARBs.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health Grants, HL115964 and HL132351 to S.S.K.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00424.
2D structures of clinically used ARBs, predicted conformation of ARBs binding in the orthosteric site of AT1R, RMSD of AT1R backbone and ARBs in 200 ns MD simulation study, average structures of ARBs bound to AT1R generated from MD simulation trajectory, histogram showing the percentage of interaction of each residue with the ARBs, and tables showing the effect of amino acid mutation in ARB binding and occupancy of each residue with the ligand in MD simulation (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Barreras A; Gurk-Turner C Angiotensin II Receptor Blockers. Proc. (Bayl Univ Med. Cent) 2003, 16, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Abraham HM; White CM; White WB The Comparative Efficacy and Safety of the Angiotensin Receptor Blockers in the Management of Hypertension and other Cardiovascular Diseases. Drug Saf 2015, 38, 33–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Verdecchia P; Angeli F; Repaci S; Mazzotta G; Gentile G; Reboldi G Comparative Assessment of Angiotensin Receptor Blockers in Different Clinical Settings. Vasc. Health Risk Manage 2009, 5, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Baumhakel M; Bohm M Cardiovascular Outcomes with Angiotensin II Receptor Blockers: Clinical Implications of Recent Trials. Vasc. Health Risk Manage 2011, 7, 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kurtz TW; Kajiya T Differential Pharmacology and Benefit/Risk of Azilsartan Compared to other Sartans. Vasc. Health Risk Manage 2012, 8, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Miura S; Karnik SS; Saku K Review: Angiotensin II Type 1 Receptor Blockers: Class Effects versus Molecular Effects. JRAAS 2011, 12, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burnier M Angiotensin II Type 1 Receptor Blockers. Circulation 2001, 103, 904–912. [DOI] [PubMed] [Google Scholar]
- (8).Frohlich ED Local Hemodynamic Changes in Hypertension: Insights for Therapeutic Preservation of Target Organs. Hypertension 2001, 38, 1388–1394. [PubMed] [Google Scholar]
- (9).Karnik SS; Unal H; Kemp JR; Tirupula KC; Eguchi S; Vanderheyden PM; Thomas WG International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharmacol. Rev 2015, 67, 754–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wexler RR; Carini DJ; Duncia JV; Johnson AL; Wells GJ; Chiu AT; Wong PC; Timmermans PB Rationale for the Chemical Development of Angiotensin II Receptor Antagonists. Am. J. Hypertens 1992, 5, 209S–220S. [DOI] [PubMed] [Google Scholar]
- (11).Brooks DP; Ohlstein EH; Ruffolo RR Jr. Pharmacology of Eprosartan, an Angiotensin II Receptor Antagonist: Exploring Hypotheses from Clinical Data. Am. Heart J 1999, 138, 246–251. [DOI] [PubMed] [Google Scholar]
- (12).Kohara Y; Kubo K; Imamiya E; Wada T; Inada Y; Naka T Synthesis and Angiotensin II Receptor Antagonistic Activities of Benzimidazole Derivatives Bearing Acidic Heterocycles as Novel Tetrazole Bioisosteres. J. Med. Chem 1996, 39, 5228–5235. [DOI] [PubMed] [Google Scholar]
- (13).Michel MC; Foster C; Brunner HR; Liu L A Systematic Comparison of the Properties of Clinically Used Angiotensin II Type 1 Receptor Antagonists. Pharmacol. Rev 2013, 65, 809–848. [DOI] [PubMed] [Google Scholar]
- (14).Miura S; Nakao N; Hanzawa H; Matsuo Y; Saku K; Karnik SS Reassessment of the Unique Mode of Binding Between Angiotensin II Type 1 Receptor and Their Blockers. PLoS One 2013, 8, e79914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yamano Y; Ohyama K; Chaki S; Guo DF; Inagami T Identification of Amino Acid Residues of Rat Angiotensin II Receptor for Ligand Binding by Site Directed Mutagenesis. Biochem. Biophys. Res. Commun 1992, 187, 1426–1431. [DOI] [PubMed] [Google Scholar]
- (16).Yamano Y; Ohyama K; Kikyo M; Sano T; Nakagomi Y; Inoue Y; Nakamura N; Morishima I; Guo DF; Hamakubo T; Inagami T Mutagenesis and the Molecular Modeling of the Rat Angiotensin II Receptor (AT1). J. Biol. Chem 1995, 270, 14024–14030. [DOI] [PubMed] [Google Scholar]
- (17).Noda K; Saad Y; Karnik SS Interaction of Phe8 of Angiotensin II with Lys199 and His256 of AT1 Receptor in Agonist Activation. J. Biol. Chem 1995, 270, 28511–28514. [DOI] [PubMed] [Google Scholar]
- (18).Noda K; Saad Y; Kinoshita A; Boyle TP; Graham RM; Husain A; Karnik SS Tetrazole and Carboxylate Groups of Angiotensin Receptor Antagonists Bind to the Same Subsite by Different Mechanisms. J. Biol. Chem 1995, 270, 2284–2289. [DOI] [PubMed] [Google Scholar]
- (19).Matsoukas MT; Cordomi A; Rios S; Pardo L; Tselios T Ligand Binding Determinants for Angiotensin II Type 1 Receptor from Computer Simulations. J. Chem. Inf. Model 2013, 53, 2874–2883. [DOI] [PubMed] [Google Scholar]
- (20).Zhang H; Unal H; Gati C; Han GW; Liu W; Zatsepin NA; James D; Wang D; Nelson G; Weierstall U; Sawaya MR; Xu Q; Messerschmidt M; Williams GJ; Boutet S; Yefanov OM; White TA; Wang C; Ishchenko A; Tirupula KC; Desnoyer R; Coe J; Conrad CE; Fromme P; Stevens RC; Katritch V; Karnik SS; Cherezov V Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang H; Unal H; Desnoyer R; Han GW; Patel N; Katritch V; Karnik SS; Cherezov V; Stevens RC Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem 2015, 290, 29127–29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kjeldsen SE; Stalhammar J; Hasvold P; Bodegard J; Olsson U; Russell D Effects of Losartan vs Candesartan in Reducing Cardiovascular Events in the Primary Treatment of Hypertension. J. Hum. Hypertens 2010, 24, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Aulakh GK; Sodhi RK; Singh M An Update on Non-Peptide Angiotensin Receptor Antagonists and Related RAAS Modulators. Life Sci 2007, 81, 615–639. [DOI] [PubMed] [Google Scholar]
- (24).Takezako T; Unal H; Karnik SS; Node K Structure-Function Basis of Attenuated Inverse Agonism of Angiotensin II Type 1 Receptor Blockers for Active-State Angiotensin II Type 1 Receptor. Mol. Pharmacol 2015, 88, 488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kohara Y; Kubo K; Imamiya E; Wada T; Inada Y; Naka T Synthesis and Angiotensin II Receptor Antagonistic Activities of Benzimidazole Derivatives Bearing Acidic Heterocycles as Novel Tetrazole Bioisosteres. J. Med. Chem 1996, 39, 5228–5235. [DOI] [PubMed] [Google Scholar]
- (26).Arumugam S; Sreedhar R; Thandavarayan RA; Karuppagounder V; Krishnamurthy P; Suzuki K; Nakamura M; Watanabe K Angiotensin Receptor Blockers: Focus on Cardiac and Renal Injury. Trends Cardiovasc. Med 2016, 26, 221–228. [DOI] [PubMed] [Google Scholar]
- (27).Abraham HM; White CM; White WB The Comparative Efficacy and Safety of the Angiotensin Receptor Blockers in the Management of Hypertension and other Cardiovascular Diseases. Drug Saf 2015, 38, 33–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Arumugam S; Sreedhar R; Thandavarayan RA; Karuppagounder V; Krishnamurthy P; Suzuki K; Nakamura M; Watanabe K Angiotensin Receptor Blockers: Focus on Cardiac and Renal Injury. Trends Cardiovasc. Med 2016, 26, 221–228. [DOI] [PubMed] [Google Scholar]
- (29).Glide; Schrödinger Release 2017–2, Schrödinger, LLC: New York, NY, 2017. [Google Scholar]
- (30).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (31).LigPrep; Schrödinger Release 2017–2, Schrödinger, LLC: New York, NY, 2017. [Google Scholar]
- (32).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (33).Prime; Schrödinger Release 2017–2, Schrödinger, LLC: New York, NY, 2017. [Google Scholar]
- (34).Dhanachandra Singh K; Karthikeyan M; Kirubakaran P; Nagamani S Pharmacophore Filtering and 3D-QSAR in the Discovery of New Jak2 Inhibitors. J. Mol. Graphics Modell 2011, 30, 186–197. [DOI] [PubMed] [Google Scholar]
- (35).Osajima T; Suzuki M; Neya S; Hoshino T Computational and Statistical Study on the Molecular Interaction between Antigen and Antibody. J. Mol. Graphics Modell 2014, 53, 128–139. [DOI] [PubMed] [Google Scholar]
- (36).Sun H; Tian S; Zhou S; Li Y; Li D; Xu L; Shen M; Pan P; Hou T Revealing the Favorable Dissociation Pathway of Type II Kinase Inhibitors via Enhanced Sampling Simulations and Two-End-State Calculations. Sci. Rep 2015, 5, 8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhang X; Perez-Sanchez H; Lightstone FC A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem 2017, 17, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Braganza LF; Worcester DL Hydrostatic Pressure Induces Hydrocarbon Chain Interdigitation in Single-Component Phospholipid Bilayers. Biochemistry 1986, 25, 2591–2596. [DOI] [PubMed] [Google Scholar]
- (39).Faraldo-Gomez JD; Smith GR; Sansom MS Setting up and Optimization of Membrane Protein Simulations. Eur. Biophys. J 2002, 31, 217–227. [DOI] [PubMed] [Google Scholar]
- (40).Schlegel B; Sippl W; Holtje HD Molecular Dynamics Simulations of Bovine Rhodopsin: Influence of Protonation States and Different Membrane-Mimicking Environments. J. Mol. Model 2005, 12, 49–64. [DOI] [PubMed] [Google Scholar]
- (41).Nath Chakraborty S; Gelb LD A Monte Carlo Simulation Study of Methane Clathrate Hydrates Confined in Slit-Shaped Pores. J. Phys. Chem. B 2012, 116, 2183–2197. [DOI] [PubMed] [Google Scholar]
- (42).Darden T; York D; Pedersen L Particle Mesh Ewald: an N log (N) method for Ewald sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (43).Isberg V; Balle T; Sander T; Jorgensen FS; Gloriam DE G protein- and Agonist-bound Serotonin 5-HT2A Receptor Model Activated by Steered Molecular Dynamics Simulations. J. Chem. Inf. Model 2011, 51, 315–325. [DOI] [PubMed] [Google Scholar]
- (44).Hu X–R; Wang Y-W; Gu J-M Losartan Potassium 3.5-hydrate, A New Crystalline form. Acta Crystallogr., Sect. E: Struct. Rep. Online 2005, E61, m1686–m1688. [Google Scholar]
- (45).Kujawski J; Czaja K; Ratajczak T; Jodlowska E; Chmielewski MK Investigations on Synperiplanar and Antiperiplanar Isomers of Losartan: Theoretical and Experimental NMR Studies. Molecules 2015, 20, 11875–11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wilkes BC; Masaro L; Schiller PW; Carpenter KA Angiotensin II vs its Type I Antagonists: Conformational Requirements for Receptor Binding Assessed from NMR Spectroscopic and Receptor Rocking Experiments. J. Med. Chem 2002, 45, 4410–4418. [DOI] [PubMed] [Google Scholar]
- (47).Unal H; Jagannathan R; Bhat MB; Karnik SS Ligand-specific Conformation of Extracellular Loop-2 in the Angiotensin II Type 1 Receptor. J. Biol. Chem 2010, 285, 16341–16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wacker D; Wang S; McCorvy JD; Betz RM; Venkatakrishnan AJ; Levit A; Lansu K; Schools ZL; Che T; Nichols DE; Shoichet BK; Dror RO; Roth BL Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Fillion D; Cabana J; Guillemette G; Leduc R; Lavigne P; Escher E Structure Of The Human Angiotensin II Type 1 (AT1) Receptor Bound to Angiotensin II from Multiple Chemoselective Photoprobe Contacts Reveals A Unique Peptide Binding Mode. J. Biol. Chem 2013, 288, 8187–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Clement M; Martin SS; Beaulieu ME; Chamberland C; Lavigne P; Leduc R; Guillemette G; Escher E Determining the Environment of the Ligand Binding Pocket of The Human Angiotensin II Type I (Hat1) Receptor Using the Methionine Proximity Assay. J. Biol. Chem 2005, 280, 27121–27129. [DOI] [PubMed] [Google Scholar]
- (51).Miura S; Feng YH; Husain A; Karnik SS Role of Aromaticity of Agonist Switches of Angiotensin II in the Activation of The AT1 Receptor. J. Biol. Chem 1999, 274, 7103–7110. [DOI] [PubMed] [Google Scholar]
- (52).Balakumar P; Jagadeesh G Structural Determinants for Binding, Activation, and Functional Selectivity of the Angiotensin AT1 Receptor. J. Mol. Endocrinol 2014, 53, R71–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.