

Abstract
Mantle cell lymphoma (MCL) comprises around 6% of all non-Hodgkin’s lymphoma (NHL) diagnoses. In younger patients, age less than 60 to 65 years, aggressive induction often followed by consolidation with autologous stem cell transplant has suggested improved outcomes in this population. Less intensive therapies in older patients often followed by maintenance have been studied or are under active investigation. However, despite recent advances, MCL remains incurable, with a median overall survival of around five years. Patients with high-risk disease have particularly poor outcomes. Treatment varies widely across institutions, and to date no randomized trials comparing intensive vs less intensive approaches have been reported. Although recent data have highlighted the heterogeneity of MCL outcomes, patient assessment for treatment selection has largely been driven by patient age with little regard to fitness, disease biology, or disease risk. One critical advance is the finding that minimal residual disease status (MRD) after induction correlates with long-term outcomes. As such, its use as a potential end point could inform clinical trial design. In order to more rapidly improve the outcomes of MCL patients, clinical trials are needed that prospectively stratify patients on the basis of MCL biology and disease risk, incorporate novel agents, and use MRD to guide the need for additional therapy.
Mantle cell lymphoma (MCL), which accounts for approximately 6% of non-Hodgkin lymphoma (NHL), was recognized internationally as a distinct diagnosis in 1994 (1,2). In the 1990s, cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or purine analog-based chemotherapy regimens were largely accepted as the standard first-line treatment. Since that time, efforts to improve MCL outcomes have incorporated rituximab as well as more aggressive therapeutic strategies, including autologous transplant and high-dose cytarabine-containing regimens. These strategies appear to have had a substantial impact on disease outcomes, with several studies suggesting improved overall survival (OS). Despite this therapeutic progress, median OS remains around five years, and it is not yet entirely clear how the improved outcomes observed in clinical trial settings have translated to the general US population (3–6). In an attempt to improve outcomes, studies have incorporated different induction and consolidation strategies, maintenance, and agents with unique mechanisms of action including proteasome inhibitors, immunomodulatory drugs/cereblon inhibitors, and kinase inhibitors. Despite these therapeutic advances, the majority of patients will relapse and die from their disease. Therefore, relapsed and refractory disease also remains an area where novel agents are needed.
Only recently has the heterogeneity of MCL been better defined. Several studies have identified a subset of patients with a more indolent lymphoma that may be safely observed for a period of time prior to initiation of treatment (7–9). Conversely, certain clinicopathological features such as advanced age, male sex, blastoid variant morphology, elevated lactate dehydrogenase (LDH) or white blood cell (WBC) count, high proliferative index (Ki-67), complex karyotype, unmutated (<5%) immunoglobulin variable region heavy chain (IgVh), and p53 abnormalities have been associated with inferior outcomes (6,10–14). However, few prospective data are available to inform optimal therapeutic approaches based on disease risk.
The National Clinical Trials Network member groups have engaged in a number of informative therapeutic studies in MCL (Table 1) that have helped shape the current treatment landscape. This landscape is rapidly evolving as novel targets are identified, clinically effective oral agents are developed, prognostic subgroups based on disease biology are defined, more sensitive indicators of remission are employed, and better consolidation and maintenance strategies are incorporated into treatment algorithms. These advances afford a noteworthy opportunity to impact the standard of care for this disease. In trying to foresee where we want the field to be in the future, we recognize that certain studies may not be feasible, and results from current ongoing trials will likely have a major influence on treatment standards and inform future clinical trial priorities.
Table 1.
Recent NCTN clinical trials in mantle cell lymphoma*
| Protocol title | Treatment regimen | Date reported/published |
|---|---|---|
| A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213 (17) | R-HyperCVAD | 2013 |
| R-CHOP/MTX plus ara-C/etoposide →carmustine/etoposide/cyclophosphamide followed by ASCT |
|
|
| Phase II study of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone immunochemotherapy followed by yttrium-90-ibritumomab tiuxetan in untreated mantle cell lymphoma: E1499 (42) | R-CHOP x 4 followed by (90)Y-ibritumomab tiuxetan | 2012 |
| Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: An Eastern Cooperative Oncology Group study (E1405) (31) | VcR-CVAD (∼1/3 received consolidation with ASCT) | 2014 |
| Phase II trial of R-CHOP plus bortezomib induction therapy followed by bortezomib maintenance for previously untreated mantle cell lymphoma: SWOG 0601 (29) | R-CHOP plus bortezemib followed by bortezemib maintenance | 2014 |
| Therapy with bortezomib plus lenalidomide for relapsed/refractory mantle cell lymphoma: Final results of a phase II trial: CALGB 50501 (43) | Lenalidomide plus bortezomib followed by maintenance lenalidomide and bortezomib | 2015 |
ASCT = autologous stem cell transplantation; hyperCVAD = hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone; MTX/Ara-C = methotrexate/cytarabine; NCTN = National Cancer Institute’s National Clinical Trials Network; R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; VcR CVAD = bortezomib, rituximab, cyclophosphamide, vincristine, doxorubicin, and dexamethasone.
Current Landscape of Clinical Trials in Mantle Cell Lymphoma
There are currently many different treatment approaches for MCL, often influenced by age and comorbidities, as well as patient and physician preference. This is reflected by the initial treatment guidelines outlined by the National Comprehensive Cancer Network (NCCN), which lists 10 potential regimens for the initial treatment of MCL. Treatment strategies are typically divided into “intensive” and “less intensive” approaches. Intensive approaches typically include more aggressive induction regimens as well as consolidative autologous stem cell transplantation (ASCT). Less intensive approaches include less aggressive induction regimens frequently followed by longer-term consolidation/maintenance therapy rather than ASCT (15). Most trials published to date have selected patients solely based on age, with limited consideration for fitness/comorbidities or disease biology. Notably, there have been no phase III trials comparing intensive vs less intensive strategies in the rituximab and targeted agent era. A number of recently reported or ongoing trials, listed below (Table 2), will help define potential standards moving forward.
Table 2.
Recently reported and ongoing clinical trials in MCL*
| Study | Intensity | Treatment | Status/results |
|---|---|---|---|
| TRIANGLE (EMCL) | Intensive | Induction +/− consolidative auto vs ibrutinib; R-CHOP/DHAP → ASCT vs R-CHOP/DHAP + ibrutinib → ASCT → ibrutinib maintenance vs R-CHOP/DHAP + ibrutinib → ibrutinib maintenance | Ongoing |
| S1106 | Intensive | A randomized phase II trial of R-HCVAD-MTX/ARA-C induction followed by consolidation with an autologous stem cell transplant vs R-bendamustine induction followed by consolidation with an autologous stem cell transplant for patients age ≤65 y with previously untreated mantle cell lymphoma | Terminated |
| LYM-3002 | Less intensive | Randomized phase III study of R-CHOP vs bortezomib, rituximab, cyclophosphamide, doxorubicin, prednisone (VR-CAP), which showed significantly improved 2-year PFS in the VR-CAP arm | Reported |
| S0601 | Less intensive | Single-arm phase II study of RCHOP plus bortezomib, improved PFS as compared with R-CHOP historical controls | Reported |
| SHINE | Less intensive | Bendamustine-rituximab +/− ibrutinib or placebo followed by rituximab and ibrutinib maintenance in patients age > 65 y (Janssen sponsored, phase III, international study) | Enrollment completed |
| E1411 (CTSU)/Intergroup | Less intensive |
|
Ongoing: expected to reach target accrual in third quarter of 2016 |
| MCL elderly R2 (EMCL) | Less intensive | 8x R-CHOP vs 6x R-CHOP/R-DHA -> rituximab maintenance vs rituximab/lenalidomide (2 y) | Ongoing |
ASCT = autologous stem cell transplantation; BR = bendamustine plus rituximab; BVR = BR plus bortezomib; LR = lenalidomide plus R consolidation; MCL = mantle cell lymphoma; PFS = progression-free survival; R-CHOP/DHA = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone/dexamethasone, high-dose cytarabine; R-CHOP/DHAP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone, high-dose cytarabine, cisplatin.
Standard Therapy for Treatment-Naïve MCL: Defining the Appropriate Comparator Arm for Randomized Studies
Several active regimens are commonly used, but have not been directly compared, thus a comparator arm is not yet completely defined. Furthermore, any standard selected may become obsolete as ongoing studies may define new standards of care.
Induction and Consolidation
One potential treatment paradigm classifies therapeutic approaches as intensive and less intensive. Although this may be appropriate for certain studies, in other instances subjects should be selected or stratified by disease risk, clinical features, and comorbidities.
Intensive
There are a number of active regimens in this setting, many of which have been studied in combination with postinduction consolidation, that is, ASCT. The most active regimens have included rituximab (R) and cytarabine, and therefore these agents should be incorporated as part of any standard intensive induction arm. Although rituximab-hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (R-HyperCVAD) alternating with high dose methotrexate plus cytarabine (HDMTX/Ara-C) was active in a single-center experience (16), it has not performed as well in cooperative group settings (17). Furthermore, this regimen has been associated with substantial toxicity, resulting in the inability to deliver full doses in many patients (17,18). Additionally, HyperCVAD/HDMTX/Ara-C was studied as a pre-ASCT induction regimen in a US intergroup trial (S1106), and the study was closed early because of toxicity and an unacceptably high rate of stem cell collection failures in the R-HyperCVAD arm (19). Therefore, we recommend that R-HyperCVAD alternating with high-dose MTX/Ara-C not be used as a standard comparator.
Four intensive induction regimens containing high-dose cytarabine have been studied in Europe, each followed by ASCT: rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) with augmented doses alternating with R + high-dose cytarabine (20,21), CHOP x 2 and R-CHOP x 1, followed by three cycles of rituximab-dexamethasone, high-dose cytarabine, and cisplatin (R-DHAP) (22), R-CHOP x 3 alternating with R-DHAP x 3 (23), and R-DHAP x 4 followed by R-CHOP x 4 for patients not achieving at least a partial response (PR) +/− R maintenance in responding patients (24). These regimens all produced similarly high complete response (CR) rates and relatively prolonged progression-free survival (PFS), and it is not clear that one regimen is superior as they have not been directly compared. The randomized study by Hermine et al. (23), however, did establish the superiority of a high-dose cytarabine–containing induction regimen over R-CHOP alone.
Furthermore, the optimal ASCT conditioning regimen is not established and has largely depended on institutional preferences, with the most commonly used regimens being total body irradiation (TBI), Ara-C, and melphalan (TAM), TBI plus cyclophosphamide, or carmustine, etoposide, Ara-C, and melphalan or cyclophosphamide (BEAM/BEAC), all of which are acceptable standards.
Less Intensive
A number of regimens have shown promising activity. While rituximab is generally included, otherwise the standard is yet to be completely defined and may be informed by ongoing studies. To this end, regimens that may be considered include bendamustine plus rituximab (25,26), rituximab plus cladribine (27,28), or bortezomib-containing regimens (29–32). Given the relatively poor results with R-CHOP alone (without maintenance) (33,34), R-CHOP alone should not be used as a potential standard comparator. In SWOG S0601 and the LYM-3002 study, R-CHOP in combination with bortezomib or VR-CAP (R-CHOP, replacing vincristine with bortezomib), respectively, have established bortezomib plus chemo-immunotherapy as potential comparators. The LYM-3002 study reported a statistically significant improvement in PFS (24.7 months, 95% confidence interval [CI] = 19.8 to 31.8 months, vs 14.4 months, 95% CI = 12.0 to 16.9 months, P < .001, two-sided) with VR-CAP compared with R-CHOP (29,30). These results led to US Food and Drug Administration (FDA) approval of bortezomib in combination with R-CAP for the initial treatment of MCL.
Recently, the bendamustine + rituximab (B-R) regimen has been adopted by many as a standard first-line regimen because of superior outcomes and somewhat less toxicity than R-CHOP in the MCL subset of patients in a randomized trial (25), as well as improved CR rates in the MCL subset within the BRIGHT trial (26). Because B-R and VR-CAP have not been compared directly and both are superior to R-CHOP, we consider either to be an acceptable standard regimen. Ultimately, E1411, which utilizes the B-R backbone and compares the addition of bortezomib to induction, may define the optimal standard induction treatment arm for future trials. The addition of cytarabine to B-R has shown efficacy in an older patient population (35), and studies are underway to better define treatment sequencing. As such, incorporation of cytarabine into the B-R backbone is another potential induction strategy.
Maintenance
Rituximab maintenance until disease progression provided PFS and overall survival benefit compared with interferon maintenance after R-CHOP chemotherapy in patients age 60 years or older (36). Rituximab maintenance is also effective following ASCT, with statistically significant improvement in PFS (hazard ratio = 0.48, 95% CI = 0.29 to 0.82, P = .007, two-sided) (24,37). Prospective data showing an OS benefit are lacking; however, recently published retrospective data do show potential improvement in OS with rituximab maintenance after ASCT (37). Maintenance in other patient groups and settings remains largely unexplored, although the potential benefits of rituximab maintenance and/or alternative maintenance strategy standards will be further informed by ongoing studies. The efficacy of rituximab maintenance after other induction regimens has yet to be established, but given the benefit in follicular lymphoma, another incurable B-cell lymphoma with a pattern of continuous relapse, as well as the low toxicity burden of rituximab, it is reasonable to consider rituximab maintenance a standard option after induction chemotherapy. The optimal timing and duration of maintenance therapy have also not yet been defined. The role of bortezomib consolidation or maintenance after ASCT is still evolving, with recently presented data suggesting a possible PFS benefit (38) and randomized data on a relatively small number of patients where no benefit was seen (39). No randomized data are available yet to define maintenance strategies using bortezomib after less intense induction, lenalidomide, or novel oral agents such as ibrutinib. E1411 and the European Triangle study will inform some of these approaches.
Recommended Clinical Trial Questions
After considering a wide range of clinical and scientific questions of interest, as well as feasibility of trials, we recommend that therapeutic trials focus on: 1) the initial therapy of treatment-naïve patients including: a) induction and b) consolidation/maintenance; and 2) relapsed/refractory patients with a special interest in those whose disease has progressed after ibrutinib treatment. For initial therapy, trials should select and/or stratify patients for specific trials based on a) disease risk/biology and/or b) fitness/candidacy for intensive vs. less intensive treatment strategies. As even intensive approaches are not curative, it is imperative that novel agents be incorporated earlier in the course of disease treatment. Trials that include patients following ibrutinib therapy should focus on emerging mechanisms of resistance to potentially individualize subsequent use of novel agents. Lastly, although not a therapeutic intervention, in an effort to better understand and learn from MCL treatment in the United States, the committee also proposes establishment of a multicenter and/or population-based MCL registry, preferably with sample collection.
Given the potential for earlier application of novel agents and to enhance patient accrual, we propose that novel agents be incorporated into both intense regimens (including ASCT) and less intense regimens. To date, the majority of studies have used age as the primary eligibility criterion for evaluating a given treatment regimen. We caution that this may interfere with trial feasibility and applicability, limiting timely accrual and consequently hampering the ability to measure efficacy of the investigational therapy in subsets of disease risk/biology.
Induction
Fit patients: A potential research question of interest is to investigate novel induction strategies, incorporating targeted agents (kinase inhibitors, CDK inhibitors, proteasome inhibitors, bcl-2 inhibitors) into standard immunochemotherapy backbones with a goal of increasing the proportion of patients achieving MRD negativity with induction therapy.
Less fit/unfit patients: With the increasing availability, tolerability, and effectiveness of therapeutics deemed “biologics” or “non-DNA-damaging,” the committee recommends that combinations of novel therapies with or without standard cytotoxic agents should be studied. We specify combinations as current data in the relapsed setting suggest that approved single agents (bortezomib, ibrutinib, or lenalidomide) are likely not adequate therapy.
High-risk patients: Despite the incorporation of intensive therapies, patients with poor risk features including complex karyotype, p53 abnormalities, high Ki-67, or blastoid histology continue to have extremely poor outcomes. As such, this population represents an area of unmet need and an opportunity to use novel treatment approaches, although the accrual challenges in this rare population are recognized to be substantial.
Consolidation
Consolidation is defined as any therapy utilized after completion of induction. This may include approaches currently referred to as “maintenance.” Consolidation strategies should focus on high-risk subgroups, although precisely defining such groups remains an open question. Study designs that prospectively stratify consolidation using pretreatment disease risk factors such as proliferation index (as defined by Ki67 staining), Mantle Cell Lymphoma International Prognostic Index-biologic (MIPI-b), and/or p53 status are deemed high-priority questions. Of equal potential importance, however, are studies utilizing a response-adapted approach, such as minimal residual disease (MRD) status or occurrence of molecular relapse. Given the potential expense and toxicities of ongoing therapy with novel agents, as well as the general favorable outcomes seen in low-risk patients, “one size fits all” studies that do not risk-stratify patients should be avoided.
A potential research question of interest is to investigate consolidation treatment duration based on molecular response. Our consensus is that, although a trial investigating ASCT vs no ASCT in all young, fit patients is of clinical interest, this is not likely to be feasible. Instead, given the promising outcomes of patients who are MRD negative after induction, a more refined approach that incorporates MRD response as a tool to risk-stratify and/or select patients for ASCT is warranted. For example, patients who are MRD negative after appropriate induction therapy (using MRD as a surrogate for a “deep” remission) may be appropriate to consider for random assignment to ASCT vs no ASCT.
Relapsed/Refractory Patients Including Prior Ibrutinib Exposure
Relapsed and refractory MCL remains a major therapeutic challenge even in the era of novel agents. Although the BTK inhibitor ibrutinib has shown promising activity in relapsed and refractory MCL, only one-third of patients remain progression free two years after starting therapy (46,47). Patients who develop ibrutinib resistance appear to have extremely poor outcomes, especially those with primary drug resistance (66,67). Emerging data may further define the biology of resistance and potentially uncover other targetable pathways (68–70). Therefore, we recognize ibrutinib failure as an area of unmet need. Efforts to define “resistance signatures” will require obtaining biopsies of involved sites both prior to treatment and upon progression. Ultimately, this has the potential to allow for the development of biomarker-targeted study designs, which could include simultaneous phase II studies that rationally select therapeutic combinations based on novel biomarkers. It is recognized that, although scientifically and clinically important, such studies may be limited by small patient numbers. Nonetheless, in-depth molecular studies of resistance even in small patient populations may be productive.
Registry
As novel therapies become more widely available—and more expensive—studies that can evaluate therapeutic approaches as well as outcomes and cost-effectiveness across broader populations are needed. Although the National Cancer Institute’s National Clinical Trials Network (NCTN) may not provide an optimal venue for MCL registry studies, the committee recommends establishing a MCL registry, perhaps using the Cancer Outcome Tracking and Analysis (COTA) database. In addition to better defining actual treatment patterns, outcomes, and cost, such a database could ultimately also serve as a platform for evaluation of biomarkers.
Trial End Points
Appropriate trial end points will differ by study design. We support the following trial end points for frontline studies:
Randomized phase III: OS
Randomized phase II: PFS and/or MRD-negative CR as an alternative to PFS
Single-arm phase II: MRD-negative CR
Alternative secondary end points: OS, duration of response (DOR), MRD, positron emission tomography (PET), gene expression profiling (GEP), sequencing
Although definitive data correlating response or PFS with OS are limited, for many current novel treatments OS as a primary end point may not be feasible at this time. The current concern for using OS as an end point is based on the likelihood that improved survival outcomes and rapid rate of discovery and development of better therapeutics will frequently render OS end points impractical, especially as novel agents progressively prolong survival after disease relapse. In the situation where an impact on OS is feasible and there is high likelihood that a difference in PFS (or an alternative early end point such as MRD status) would be required to see an overall survival impact by treatment, then a phase II/III design could be undertaken. There would be a phase II determination based on PFS to proceed to the definitive comparison, but with the efficiency of utilizing the phase II patients for the phase III comparison.
OS should be evaluated as a secondary end point in phase II studies. Potential phase II end points include PFS, PET CR, and MRD status. However, it is also recognized that PFS may be confounded by maintenance and consolidation strategies and, as such, may interfere with interpreting PFS results in single-arm studies.
Special discussion on the definition of the depth of response including CR and MRD is warranted. The emergence of PET imaging and MRD techniques is redefining the ability to identify high-quality remissions. PET has been evaluated in a number of MCL studies, but the interpretation of some of these studies is limited by small sample size. To date, studies in the setting of aggressive therapeutic approaches evaluating the prognostic significance of a negative PET scan after induction therapy suggest that PET negativity after initial therapy correlates with improved outcomes; however, results have not been uniform across studies (71–77). We recognize that additional studies are necessary to further validate these findings. The E1411 trial is evaluating PET response at the end of induction and should help further define the role of PET in MCL. It is imperative that all imaging studies be reviewed centrally and with a uniform definition of response, for example, Deauville criteria. Although potentially interesting as an exploratory end point, current data do not support the use of interim or surveillance PET.
That MCL patients almost universally relapse despite achieving a clinical CR suggests the existence of disease below the limit of detection by imaging and pathology. More sensitive methods of detecting and quantifying minimal levels of residual disease could therefore potentially predict clinical relapse. This concept when first examined almost 20 years ago (78,79) showed that polymerase chain reaction (PCR) could detect either clonal rearranged immunoglobulin H (IgH) or the Bcl-1/IgH translocation in the peripheral blood or marrow, correlating with a PCR product from diagnostic material and demonstrating the presence of MRD in patients in clinical remission. The number of patients in these studies was too small to draw statistical conclusions. The first large study to show a correlation between MRD status and outcome was reported by Pott et al. in an analysis of a subset of patients from the European MCL Younger and MCL Elderly studies (80). In these trials, the presence of MRD as determined by real-time quantitative PCR (RQ-PCR) for clonal IgH or Bcl-1/IgH was associated with inferior two-year remission duration of both young and elderly patients. Longer-term follow-up of these patients demonstrated superior PFS for patients with undetectable MRD compared with patients who developed MRD positivity within one year (81).
The impact of MRD status, using standard nested PCR to detect either Bcl-1/IgH translocation or a clonal IgH band following therapy, was also evaluated by the Nordic Group in a population of younger patients receiving intensive induction therapy and ASCT (MCL-2 study) (20,21). The timing of PCR-detectable relapse correlated with PFS, with patients remaining continuously MRD-negative having the longest PFS, followed by patients who became MRD positive after one year. Patients who developed detectable MRD within the first year had the shortest PFS. Similar findings were reported in the Nordic MCL-3 study, with statistically significantly better five-year PFS of patients who were MRD negative post-ASCT compared with patients who still had measurable disease by PCR (P < .01, two-sided) (71).
Recent data from the French LyMa trial confirmed the importance of pre-ASCT MRD status. In this study, patients received four cycles of R-DHAP induction followed by ASCT, and were then randomly assigned to three years of rituximab maintenance or observation (82). Following induction, the rate of MRD negativity was 66% in the marrow and 80% in the peripheral blood. Patients who achieved an MRD-negative status after induction in either the marrow or blood had a longer PFS, whether they received maintenance rituximab or observation post-ASCT. In the CALGB 50403 study, which added post-auto-HCT bortezomib in two different extended dosing schedules to the CALGB 59909 induction backbone, the five-year PFS was 93% for patients who were MRD negative after induction and 51% for patients who were MRD positive (38). In a subset analysis of patients in the S1106 trial who were treated with BR prior to auto-HCT, eight of nine evaluable patients achieved an MRD-negative CR following induction therapy (19).
The Nordic group also tested the hypothesis that molecular relapse heralds clinical relapse and that preemptive therapy upon conversion from MRD negativity to MRD positivity could postpone clinical relapse while reducing the overall number of cycles of therapy compared with treating all patients with maintenance therapy. In the MCL-2 study, upon conversion to PCR-positive status post-ASCT, patients were treated with four weekly doses of rituximab. There was no untreated control group for comparison, but patients who received rituximab had a median relapse-free survival of 3.7 years from the time of preemptive therapy and a median molecular complete remission of 1.5 years (83). As 10% of the patients in this study (and 13% in the German/French study) relapsed clinically while remaining MRD negative, molecular relapse using current technologies is not a universal predictor or a surrogate for clinical relapse. Another limitation of PCR-based molecular monitoring is absence of a suitable MRD marker in approximately 10% of cases. Therefore, it will be important to prospectively evaluate more sensitive sequencing approaches as they continue to emerge.
Flow cytometry has been evaluated in MCL (84) and may be an effective tool; however, it has not yet been correlated with outcomes. More recently, efforts have been made to use other techniques for MRD testing including next-generation sequencing (NGS) and evaluation of MRD on formalin-fixed, paraffin-embedded samples (85,86). NGS seems to be more sensitive and is commercially available. The E1411 trial will compare MRD assessments by NGS and flow cytometry relative to outcome, which may further define the roles of these assays in MCL.
In summary, MRD is emerging as a strong predictor of clinical outcome, and we believe that there are adequate data to support MRD negativity as a useful phase II end point at this time. Prospective studies should continue to incorporate MRD techniques that are feasible, can be standardized, are broadly applicable across all treatment settings, can be correlated with PFS and OS, and can be used experimentally to risk stratify and/or select patients for additional therapy. Additionally, we believe that there is an opportunity to include MRD assessment as an integral or integrated biomarker in future cooperative group studies to address several questions, including 1) whether patients can be risk-stratified by MRD status following induction or ASCT to allow treatment of only MRD-positive patients with consolidation therapy and 2) whether ongoing MRD monitoring post-therapy provides a means to allocate additional therapy in a rational and cost-effective manner, for example, by intervening only after the detection of MRD or stopping maintenance after achieving an MRD-negative status.
Biomarkers
As technology capable of discerning biologically distinct subsets of MCL evolves and becomes more readily available and our understanding of these biological differences improves, stratification by biology will become more important. It is likely that the optimal therapy for each biological subtype of MCL will be different, and it will be important for cooperative group trials to study these differences prospectively. Initially, this may be possible without changes in trial design, but eventually, as further data are generated using existing and emerging technologies, it will be imperative that studies incorporate commercially available sequencing and gene expression platforms, as well as MRD testing into trial designs. The ultimate goal is to utilize these data to 1) prospectively stratify patients into high- and/or low-risk disease, 2) prospectively use biologic or therapeutic end points to select patients for additional therapy including consolidation, and 3) identify biomarkers and/or unique “signatures” that can be specifically targeted with novel agents. Unfortunately, to date few validated biomarkers exist to help inform therapeutic approaches. Therefore, defining molecular and genetic markers of response to specific therapies remains an area of high clinical relevance.
Clinical prognostic tools have typically incorporated age, performance status, and laboratory values to stratify disease risk groups. The most extensively studied prognostic score is the mantle cell international prognostic index (MIPI), which includes age, ECOG performance status, LDH, and peripheral blood leukocytes. The MIPI appears to best stratify low-risk vs high-risk populations and has been validated when applied to aggressive and less aggressive treatment approaches. Specifically, the overwhelming majority of current studies show shorter PFS, time to treatment failure (TTF), and OS in patients with high-risk MIPI scores (14,18). Other clinical features, such as elevated beta-2 microglobulin (β2M) and absolute monocyte count at presentation, have also been found to correlate with inferior outcomes (87). Although MIPI has been validated across MCL populations (88), its predictive value for response to a given regimen may be suboptimal as a high MIPI score may be for one patient driven by patient frailty (greater age and poor performance status) and for a younger patient with good performance status by high WBC and LDH. In this scenario, despite similar MIPI scores, a standard biology-driven treatment approach may not be applicable. Therefore, stratification by MIPI alone is not adequate and patients should therefore be evaluated in the context of biologic factors.
To date, the most informative predictive biologic factors have measured cell proliferation. Histology, especially the blastoid variant, has also generally been associated with inferior outcomes; however, it is not clear if this is the result of increased proliferation or other biologic influences. Furthermore, stratifying solely on histologic subtype may be limited by the relatively low prevalence (∼10%) of blastoid variants. Further elucidation of the effect of histology should be explored, but is not sufficient for prognostic stratification at this time.
The most widely studied clinically relevant measurement of MCL cell proliferation is the percentage of MCL cells that stain positively for Ki-67. Ki-67 has been prospectively validated as a powerful prognostic tool across a number of treatment settings (11,13,89,90). The “biologic MIPI” (MIPI-b) combines Ki-67 score with the MIPI to discriminate high-risk from low-risk patients, and current data indicate MIPI-b may be applicable across all treatment settings. Using MIPI-b that incorporates a Ki-67 cutoff of greater than 30% to define poor-risk patients, it appears that low-risk MIPI-b patients may have superior outcomes irrespective of age and treatment intensity (Figure 1) (88). Thus, the use of MIPI-b allows for better discrimination of disease risk and supports the idea that age alone should not be used to select treatment. Further analysis of using the Ki-67 score plus MIPI (“MIPI-c”) has been presented. The combination of Ki67 score (cutoff > 30%) and MIPI stratified patients into four groups with statistically significantly different outcomes (P < .001) irrespective of age or treatment (91). As such, incorporation of MIPI-b or MIPI-c has important ramifications for future trial design.
Figure 1.
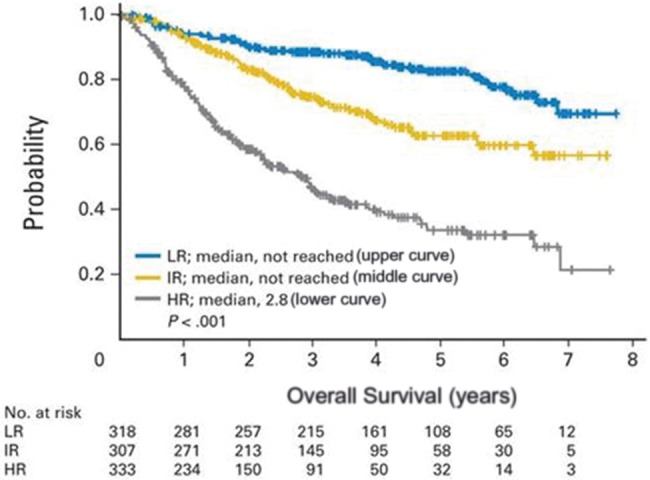
Overall survival by risk group in mantle cell lymphoma (MCL). Kaplan-Meier analysis of overall survival in mantle cell lymphoma by risk group. Reprinted from (88) with permission from the copyright holder. HR = high risk; IR = intermediate risk; LR = low risk.
It is less clear how much MIPI-b or MIPI-c improves upon the value of the Ki-67 score alone. Depending on the study, Ki-67 cutoffs below 10% (low-risk disease), between 10% and 30% (intermediate risk), and greater than 30% (poor risk) have been studied (14). However, for the purpose of risk stratification, the most compelling data available support defining groups as high-risk and low-risk disease using a Ki-67 cutoff of 30% (18,88,91). Accordingly, this is the cutoff that should be used to prospectively stratify patients in future trials.
Ki-67 staining may in some cases be limited by poor specimen quality, challenges of adequately defining Ki-67 on marrow samples, and variable reproducibility/consistency across institutions. Therefore, for future studies, it is imperative that quantification methods are standardized and that all specimens are gathered prospectively and reviewed centrally. If a Ki-67 value cannot be obtained, efforts to obtain additional tissue should be made.
Gene expression profiling (GEP) also defined a proliferation gene signature as an important prognostic indicator (92–94); however, this has not yet been validated across institutions or in prospective treatment trials and technically has remained a challenge. However, recently more widely accessible and more broadly applicable GEP platforms such as NanoString (Seattle, WA) have successfully been used to define cells of diffuse large B-cell lymphoma (DLBCL) origin (95) and evaluate key MCL genes. In addition to defining proliferation, these assays have the ability to further define pathway activation as well as other markers that have been shown to correlate with outcome such as p53 and SOX-11 (96).
Thus, we recommend that NanoString and/or similar GEP platforms be incorporated into all trials and the results compared with already validated prognostic indicators. We also encourage retrospective study of existing data sets for which archival tissue is available to better define prognostic groups and predictive markers that may define responses to a given therapy. As patients relapse after treatment, study designs should include repeat biopsies for analysis aimed at better understanding mechanisms of resistance, particularly in the setting of novel targeted agents. Furthermore, as reviewed above, MRD status postinduction may be used to define disease risk and has the potential to be a powerful biomarker in MCL.
Novel Agents
Including the currently approved drugs, bortezomib, ibrutinib, and lenalidomide, multiple agents have demonstrated single-agent activity in mantle cell lymphoma (Table 3). Cross-trial comparisons cannot be made as virtually every published trial has been a phase I or phase II design with divergent patient sample sizes, characteristics, treatment histories, and disease biology. Several other agents have demonstrated activity in combination with rituximab or chemotherapy, and a multitude of drugs is currently being evaluated. Identifying and studying novel agents with unique mechanisms of action, especially as further insight into disease biology emerges, is of particular importance. Specifically, early studies of compounds that target BTK, cyclin dependent kinases (CDK), Bcl-2, and PI3K appear to be especially promising. For example, ONO-4059, a highly selective next-generation BTK inhibitor has shown activity in a phase I study of relapsed/refractory NHL with 11 of 12 evaluable patients responding to therapy, including five (46%) who achieved CR (48). CDK4 as a therapeutic target in MCL is of particular interest given its interaction with cyclin D1 and enhanced CDK4-mediated cell cycle progression. Palbociclib and abemaciclib are selective inhibitors of CDK4 and CDK6 that have been evaluated in early phase trials. Single-agent activity has been relatively modest; however, durable responses have been noted. For example, in a phase I study of 17 relapsed/refractory MCLs, the ORR was 18%, with five of the 17 (29%) remaining progression free after one year of treatment (range = 14.9–30.1 months) (62). Bcl-2 as a unique target has also been evaluated. The phase I results in 44 relapsed/refractory NHLs using the bcl-2 inhibitor venetoclax, which was recently FDA approved for chronic lymphocytic leukemia, have been reported, with nine of 12 (1 CR) MCL patients responding to treatment (57). Phosphoinositide 3-kinase (PI3K) inhibitors have also shown notable activity in MCL. Initial results with idelalisib, a PI3K-delta isoform–specific inhibitor, in 40 relapsed/refractory MCL patients demonstrated a 40% response rate; however, response duration and was only 2.7 months (44). Accordingly, in an effort to potentially prolong response via broader anti-PI3K inhibition, agents that inhibit multiple PI3K isoforms are under investigation. For example, copanlisib, a PI3K inhibitor that targets both the alpha and delta isoforms has shown preliminary evidence of activity (45).
Table 3.
Selected trials of novel single agents in relapsed/refractory MCL*
| Drug class | Agent | No. | ORR, % | CR, % | Reference |
|---|---|---|---|---|---|
| PI3Ki | Idelalisib | 40 | 40 | 5 | 44 |
| Copanlisib | 6 | 83 | 17 | 45 | |
| BTKi | Ibrutinib | 111 | 68 | 21 | 46 |
| Ibrutinib | 120 | 63 | 21 | 47 | |
| ONO-4059 | 16 | 92 | 46 | 48 | |
| mTORi | Everolimus | 58 | 9 | 0 | 49 |
| Temsirolimus | 54 | 22 | 2 | 50,51 | |
| PKCi | Enzastaurin | 60 | 0 | 0 | 52 |
| SINE | Selinexor | 2 | 50 | 0 | 53 |
| Proteasome inhibitor | Bortezomib | 155 | 33 | 8 | 54 |
| Ixazomib | 2 | 0 | 0 | 55 | |
| IMID | Lenalidomide | 134 | 28 | 8 | 56 |
| Bcl2 | Venetoclax | 12 | 80 | 8 | 57 |
| VEGFi | Bevacizumab | 15 | 0 | 0 | 58 |
| Aurora A kinase inhibitor | Alisertib | 13 | 23 | 0 | 59 |
| HDACi | Vorinostat | 9 | 0 | 0 | 60 |
| Vorinostat | 4 | 0 | 0 | 61 | |
| CDK4/6i | Palbociclib | 17 | 18 | 6 | 62 |
| Abemaciclib | 28 | 36 | 0 | 63 | |
| Other CDKi | P276-00 | 13 | 0 | 0 | 64 |
| Flavopiridol | 30 | 11 | 0 | 65 |
Bcl2 = B-cell lymphoma 2; BTKi = bruton’s tyrosine kinase inhibitor; CDK4/6i = cyclin-dependent kinase 4/6 inhibitor IMiD = immunomodulator; HDACi = histone deacetylase inhibitor; mTORi = mammalian target of rapamycin inhibitor; PI3Ki = phosphatidyl inositol 3-kinase inhibitor; PKCi = protein kinase C inhibitor; SINE = selective inhibitor of nuclear export; VEGFi = vascular endothelial growth factor inhibitor.
These studies highlight the probability that new drugs and combinations will lead to improved patient care and outcomes but also, given the remarkable number of agents with pleotropic effects and uncertain mechanisms of resistance, highlight the challenges that lie ahead. In order to optimize rapid and appropriate drug/combination development, future trials will require coordinated efforts throughout the MCL community, in both basic and clinical sciences and across institutions and countries.
Conclusions
Clinical trial priorities include optimizing induction therapy and use of consolidation, as well as seeking effective therapy for patients with relapsed/refractory MCL following development of ibrutinib resistance. Key considerations for trial design, patient selection, and implementation include: basing selection of intensity of therapy on clinically assessed fitness as well as age cutoffs; stratifying patients based on assessment of disease risk that includes biologic factors such as proliferation markers; incorporating novel agents into induction, including “biologic”-based therapies in less fit patients; investigating consolidation strategies based on pre-induction disease risk assessment and/or postinduction response (eg, MRD status, PET).
Practice-changing phase III trials are the goal of the NCTN. It should be recognized that phase III trials in MCL will be logistically challenging and must be designed to be accrual-friendly.
A number of standard comparator arms may be proposed, but common key considerations include: intensive therapy regimens should include high-dose cytarabine; a number of potential standard chemo-immunotherapy regimens exist and are under study for less intensive approaches; results from ongoing clinical trials will further inform future treatment standards and lay the foundation for future phase III studies; the current consolidation standard is rituximab maintenance in older MCL patients and ASCT in younger MCL patients.
Disease stratification by Ki-67 (using a cutoff of >30%), p53 mutations, and/or MIPI-b or MIPI-c should be used to risk-stratify patients until better biomarkers are defined. MRD should be used to risk-stratify patients to intensive vs nonintensive strategies appears ready for testing.
An unmet need is the identification of biomarkers that will enhance understanding of MCL pathobiology, better define biologic risk groups, and enhance prognostic ability. All MCL patients should have tumor tissue prospectively collected to facilitate study of biologic factors. In addition to ongoing evaluation of established prognostic factors such as Ki-67, studies should evaluate new technologies such as GEP and/or sequencing platforms to better define broadly applicable biologic subsets. Proposed biologic factors need to be properly validated.
Investigations of novel agents may be designed to add to or replace current treatment standards. Such agents may also be compared with each other in randomized phase II studies. In the case of postibrutinib disease progression, concurrent phase II studies with assignment based on biomarkers and resistance signatures would be reasonable.
Ongoing exploration of potential surrogate end points such as MRD negativity and PET CR is warranted.
Funding
Partial support was from the Office of the Director, Coordinating Center for Clinical Trials, National Cancer Institute/National Institutes of Health.
Notes
This manuscript resulted from a Clinical Trials Planning Meeting in Lymphoma, November 21 and 22, 2014.
References
- 1. Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer. 2008;113(4):791–798. [DOI] [PubMed] [Google Scholar]
- 2. Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms: A proposal from the international lymphoma study group. Blood. 1994;84(5):1361–1392. [PubMed] [Google Scholar]
- 3. Chandran R, Gardiner S, Simon M, Spurgeon SE. Survival trends in mantle cell lymphoma in the United States over 16 years 1992-2007. Leuk Lymph. 2012;53(8):1488–1493. [DOI] [PubMed] [Google Scholar]
- 4. Herrmann A, Hoster E, Zwingers T, et al. Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol. 2009;27(4):511–518. [DOI] [PubMed] [Google Scholar]
- 5. Schmidt C, Dreyling M. Therapy of mantle cell lymphoma: Current standards and future strategies. Hematol Oncol Clin North Am. 2008;22(5):953–963. [DOI] [PubMed] [Google Scholar]
- 6. Abrahamsson A, Albertsson-Lindblad A, Brown PN, et al. Real world data on primary treatment for mantle cell lymphoma. A Nordic Lymphoma Group observational study. Blood. 2014;124(8):1288–1295. [DOI] [PubMed] [Google Scholar]
- 7. Martin P, Chadburn A, Christos P, et al. Outcome of deferred therapy in mantle-cell lymphoma. J Clin Oncol. 2009;27(8):1209–1213. [DOI] [PubMed] [Google Scholar]
- 8. Martin P, Leonard J. Is there a role for “watch and wait” in patients with mantle cell lymphoma? Semin Hematol. 2011;48(3):189–193. [DOI] [PubMed] [Google Scholar]
- 9. Hsi ED, Martin P. Indolent mantle cell lymphoma. Leuk Lymph. 2014;55(4):761–767. [DOI] [PubMed] [Google Scholar]
- 10. Slotta-Huspenina J, Koch I, de Laval L, et al. The impact of cyclin D1 mRNA isoforms, morphology, and p53 in mantle cell lymphoma: p53 alterations and blastoid morphology are strong predictors of a high proliferation index. Haematologica. 2012;97(9):1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): A clinicopathological study from the European MCL Network. Br J Haematol. 2005;131(1):29–38. [DOI] [PubMed] [Google Scholar]
- 12. Hernandez L, Fest T, Cazorla M, et al. p53 mutations and protein overexpression are associated with aggressive variants of mantle cell lymphomas. Blood. 1996;87(8):3351–3359. [PubMed] [Google Scholar]
- 13. Klapper W, Hoster E, Deutermann O, et al. Ki-67 as a prognostic marker in mantle cell lymphoma-consensus guidelines of the pathology panel of the European MCL Network. J Hematop. 2009;2(2):103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558–565. [DOI] [PubMed] [Google Scholar]
- 15. Zelenetz AD, Gordon LI, Wierda WG, et al. Non-Hodgkin’s lymphomas, version 4.2014. J Natl Compr Canc Netw. 2014;12(9):1282–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol. 2015;172(1):80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol. 2013;24(6):1587–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Merli F, Luminari S, IIariucci F, et al. Rituximab plus HyperCVAD alternating with high dose cytarabine and methotrexate for the initial treatment of patients with mantle cell lymphoma, a multicentre trial from Gruppo Italiano Studio Linfomi. Br J Haematol. 2012;156(3):346–353. [DOI] [PubMed] [Google Scholar]
- 19. Chen R, Hongli L, Bernstein SH, et al. Pre-transplant R-bendamustine induces high rates of minimal residual disease in MCL patients: Updated results of S1106: US intergroup study of a randomized phase II trial of R-HCVAD vs R-bendamustine followed by autologous stem cell transplants for patients with mantle cell lymphoma. Blood. 2015;126(23):518. [Google Scholar]
- 20. Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: A nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood. 2008;112(7):2687–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: Six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma. Br J Haematol. 2012;158(3):355–362. [DOI] [PubMed] [Google Scholar]
- 22. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: A phase 2 study from the Groupe d'Etude des Lymphomes de l'Adulte. Blood. 2013;121:48–53. [DOI] [PubMed] [Google Scholar]
- 23. Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) is superior to 6 courses CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: Results of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). Blood .2012;120(21):110. [Google Scholar]
- 24. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab maintenance versus wait and watch after four courses of R-DHAP followed by autologous stem cell transplantation in previously untreated young patients with mantle cell lymphoma: First interim analysis of the phase III prospective LyMa trial, a Lysa study. Paper presented at the 56th meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 25. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. [DOI] [PubMed] [Google Scholar]
- 26. Flinn IW, van der Jagt R, Kahl BS. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: The BRIGHT Study. Blood. 2014;123(19):2944–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inwards DJ, Fishkin PA, Hillman DW, et al. Long-term results of the treatment of patients with mantle cell lymphoma with cladribine (2-CDA) alone (95-80-53) or 2-CDA and rituximab (N0189) in the North Central Cancer Treatment Group. Cancer. 2008;113 (1):108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spurgeon SE, Pindyck T, Okada C, et al. Cladribine plus rituximab is an effective therapy for newly diagnosed mantle cell lymphoma. Leuk Lymphoma. 2011;52(8):1488–1494. [DOI] [PubMed] [Google Scholar]
- 29. Till B, Li H, Bernstein SH, et al. A phase II trial of R-CHOP plus bortezomib induction therapy followed by bortezomib maintenance for previously untreated mantle cell lymphoma: SWOG 0601. Br J Haematol. 2016;172 (2):208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944–953. [DOI] [PubMed] [Google Scholar]
- 31. Chang JE, Smith MR, Gascoyne RD, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: An Eastern Cooperative Oncology Group study (E1405). Blood. 2014;123(11):1665–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang JE, Peterson C, Choi S, et al. VcR-CVAD induction chemotherapy followed by maintenance rituximab in mantle cell lymphoma: A Wisconsin Oncology Network study. Br J Haematol. 2011;155(2):190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: Molecular complete responses are not predictive of progression-free survival. J Clin Oncol. 2002;20(5):1288–1294. [DOI] [PubMed] [Google Scholar]
- 34. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: Results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23(9):1984–1992. [DOI] [PubMed] [Google Scholar]
- 35. Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol. 2013;31(11):1442–1449. [DOI] [PubMed] [Google Scholar]
- 36. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle cell-lymphoma. N Engl J Med. 2012;367(6):520–531. [DOI] [PubMed] [Google Scholar]
- 37. Graf SA, Stevenson PA, Holmberg LA, et al. Maintenance rituximab after autologous transplantation in patients with mantle cell lymphoma. Ann Oncol. 2015;26:2323–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaplan LD,, Jung SH, Stock W. et al. Bortezomib maintenance (BM) versus consolidation (BC) following aggressive immunochemotherapy and autologous stem cell transplant (ASCT) for untreated mantle cell lymphoma (MCL): CALGB (Alliance) 50403. Paper presented at the 57th meeting of the American Society for Hematology December 2015; Orlando, FL.
- 39. Doorduijn JK, Minnema MC, Kersten MJ. et al. Bortezomib maintenance therapy after induction with R-CHOP, ARA-C and autologous stem cell transplantation in newly diagnosed MCL patients, results of a multicenter phase II HOVON study. Paper presented at the 57th meeting of the American Society for Hematology; December 2015; Orlando, FL.
- 40. Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol. 2009;27(36):6101–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu H, Johnson JL, Koval G, et al. Detection of minimal residual disease following induction immunochemotherapy predicts progression free survival in mantle cell lymphoma: Final results of CALGB 59909. Haematologica. 2012;97:579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith MR, Li H, Gordon L, et al. Phase II study of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone immunochemotherapy followed by yttrium-90-ibritumomab tiuxetan in untreated mantle-cell lymphoma: Eastern Cooperative Oncology Group Study E1499. J Clin Oncol. 2012;30(25):3119–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morrison VA, Jung SH, Johnson J. Therapy with bortezomib plus lenalidomide for relapsed/refractory mantle cell lymphoma: Final results of a phase II trial (CALGB 50501). Leuk Lymphoma. 2015;56(4):958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dreyling M, Morschhauser F, Bron D. et al. Preliminary results of a phase II study of single agent bay 80-6946, a novel PI3K inhibitor, in patients with relapsed/refractory, indolent or aggressive lymphoma. Paper presented at the 55th meeting of the American Society for Hematology; December 2013; New Orleans, LA.
- 46. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang ML, Goy A, Martin P. et al. Efficacy and safety of single-agent ibrutinib in patients with mantle cell lymphoma who progressed after bortezomib therapy. Blood. Paper presented at the 56th annual meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 48. Walter HS, Rule SA, Dyer MJ, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory B-cell malignancies. Blood. 2016;127(4):411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang M, Popplewell LL, Collins RJ. et al. Everolimus for patients with mantle cell lymphoma refractory to or intolerant of bortezomib: Multicentre, single-arm, phase 2 study. Br J Haematol. 2014;165(4):510–518. [DOI] [PubMed] [Google Scholar]
- 50. Hess G, Herbrecht R, Romaguera J, et al. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27(23):3822–3829. [DOI] [PubMed] [Google Scholar]
- 51. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: An international, randomized, open-label, phase 3 study. Lancet 2016;387(10020):770–778. [DOI] [PubMed] [Google Scholar]
- 52. Morschhauser F, Seymour JF, Kluin-Nelemans HC, et al. A phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Ann Oncol. 2008;19(2):247–253. [DOI] [PubMed] [Google Scholar]
- 53. Kuruvilla J. The oral selective inhibitor of nuclear export (SINE) selinexor (KPT-330) demonstrates broad and durable clinical activity in relapsed/refractory non Hodgkin’s lymphoma (NHL). Paper presented at the 56th meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 54. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–4874. [DOI] [PubMed] [Google Scholar]
- 55. Assouline SE, Chang J, Cheson BD, et al. Phase 1 dose-escalation study of IV ixazomib, an investigational proteasome inhibitor, in patients with relapsed/refractory lymphoma. Blood Cancer J. 2014;17(4) e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: Phase II MCL-001 (EMERGE) study. J Clin Oncol. 2013;31(29):3688–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davids MS, Seymour JF, Gerecitano JF. et al. Phase I study of ABT-199 (GDC-0199) in patients with relapsed/refractory (R/R) non-Hodgkin lymphoma (NHL): Responses observed in diffuse large B-cell (DLBCL) and follicular lymphoma (FL) at higher cohort doses. Paper presented at the meeting of the American Society for Clinical Oncology; June 2014; Chicago, IL. [PubMed]
- 58. Stopek AT, Unger JM, Rimsza LM, et al. A phase II trial of single agent bevacizumab in patients with relapsed, aggressive non-Hodgkin lymphoma: Southwest Oncology Group Study S0108. Leuk Lymphoma. 2009;50(5):728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kirschbaum M, Frankel P, Popplewell L, et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin's lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(9):12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ogura M, Ando K, Suzuki T, et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2014;165(6):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leonard JP, LaCasce AS, Smith MR, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119(20):4597–4607. [DOI] [PubMed] [Google Scholar]
- 63. Morschhauser F, Bouabdallah K, Stilgenbauer S. et al. Clinical activity of abemaciclib (LY2835219), a cell cycle inhibitor selective for CDK4 and CDK6, in patients with relapsed or refractory mantle cell lymphoma. Paper presented at the 56th meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 64. Cassaday RD, Goy A, Advani S, et al. A phase II, single-arm, open-label, multicenter study to evaluate the efficacy and safety of P276-00, a cyclin-dependent kinase inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Clin Lymphoma Myeloma Leuk. 2015;15(7):392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kouroukis CT, Belch A, Crump M. Flavopiridol in untreated or relapsed mantle-cell lymphoma: Results of a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2003;21(9):1740–1745. [DOI] [PubMed] [Google Scholar]
- 66. Martin P, Maddocks K, Leonard JP, et al. Post-ibrutinib outcomes with mantle cell lymphoma. Blood. 2016;127(12)1559–1563. [DOI] [PubMed] [Google Scholar]
- 67. Cheah CY, Chihara D, Romaguera JE. et al. Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol. 2015;26(6):1175–1179. [DOI] [PubMed] [Google Scholar]
- 68. Chiron D, Di Liberto M, Martin P. et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-kappa B-targeted treatment strategies for mantle cell lymphoma. Nature Med. 2014;20(1):87–92. [DOI] [PubMed] [Google Scholar]
- 70. Balasubramanian S, Schaffer M, Deraedt W, et al. Mutational analysis of patients with primary resistance to single-agent ibrutinib in relapsed or refractory mantle cell lymphoma (MCL). Paper presented at the 56th meeting of the American Society for Hematology; December 2014; San Francisco, CA.
- 71. Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood. 2014;123(19):2953–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Magnusson E, Cao Q, Linden MA, et al. Hematopoietic cell transplantation for mantle cell lymphoma: Predictive value of pre-transplant positron emission tomography/computed tomography and bone marrow evaluations for outcomes. Clin Lymph Myeloma Leuk. 2014;14(2):2152–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cohen JB, Hall NC, Ruppert AS, et al. Association of pre-transplantation positron emission tomography/computed tomography and outcome in mantle cell lymphoma. Bone Marrow Transplant. 2013;48(9):1212–1217. [DOI] [PubMed] [Google Scholar]
- 74. Brepoels L, Stroobants S, De Wever W, et al. Positron emission tomography in mantle cell lymphoma. Leuk Lymph. 2008;49(9):1693–1701. [DOI] [PubMed] [Google Scholar]
- 75. Mato AR, Svoboda J, Feldman T, et al. Post-treatment (not interim) positron emission tomography-computed tomography scan status is highly predictive of outcome in mantle cell lymphoma patients treated with R-HyperCVAD. Cancer. 2012;118(14):3565–3570. [DOI] [PubMed] [Google Scholar]
- 76. Bodet-Millin C, Touzeau C, Leux C, et al. Prognostic impact of 18F-fluoro-deoxyglucose positron emission tomography in untreated mantle cell lymphoma: A retrospective study from the GOELAMS group. Eur J Nucl Med Mol Imaging. 2010;37(9):1633–1642. [DOI] [PubMed] [Google Scholar]
- 77. Kedmi M, Avivi I, Ribakovsky E, et al. Is there a role for therapy response assessment with 2-[fluorine-18] fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography in mantle cell lymphoma? Leuk Lymp. 2014;55(11):2484–2489. [DOI] [PubMed] [Google Scholar]
- 78. Corradini P, Astolfi M, Cherasco C, et al. Molecular monitoring of minimal residual disease in follicular and mantle cell non-Hodgkin's lymphomas treated with high-dose chemotherapy and peripheral blood progenitor cell autografting. Blood. 1997;89(2):724–731. [PubMed] [Google Scholar]
- 79. Andersen NS, Donovan JW, Bonus JS, et al. Failure of immunologic purging in mantle cell lymphoma assessed by polymerase chain reaction detection of minimal residual disease. Blood. 1997;90(10):4212–4221. [PubMed] [Google Scholar]
- 80. Pott C, Hoster E, Delfau-Larue MH, et al. Molecular remission is an independent predictor of clinical outcome in patients with mantle cell lymphoma after combined immunochemotherapy: A European MCL intergroup study. Blood. 2010;115(16):3215–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pott C, Macintyre E, Delfau-Larue M, et al. MRD eradication should be the therapeutic goal in mantle cell lymphoma and may enable tailored treatment approaches: Results of the intergroup trials of the European MCL Network .Blood. 2014;124(21):147. [Google Scholar]
- 82. Callanan MB, Delfau MH, Macintyre E, et al. Predictive power of early, sequential MRD monitoring in peripheral blood and bone marrow in patients with mantle cell lymphoma following autologous stem cell transplantation with or without rituximab maintenance; interim results from the LyMa-MRD Project, conducted on behalf of the Lysa Group. Paper presented at the 57th meeting of the American Society for Hematology; December 2015; Orlando, FL.
- 83. Andersen NS, Pedersen LB, Laurell A, et al. Pre-emptive treatment with rituximab of molecular relapse after autologous stem cell transplantation in mantle cell lymphoma. J Clin Oncol. 2009;27(26):4365–4370. [DOI] [PubMed] [Google Scholar]
- 84. Bottcher S, Ritgen M, Buske S, et al. Minimal residual disease detection in mantle cell lymphoma: Methods and significance of four-color flow cytometry compared to consensus IGH-polymerase chain reaction at initial staging and for follow-up examinations. Haematologica. 2008;93(4):551–559. [DOI] [PubMed] [Google Scholar]
- 85. Ladetto M, Bruggerman M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leuk. 2014;28(6)1299–1307. [DOI] [PubMed] [Google Scholar]
- 86. Kalinova M, Fronkova E, Klener P, et al. The use of formalin-fixed, paraffin-embedded lymph node samples for the detection of minimal residual disease in mantle cell lymphoma. Br J Haematol. 2015;169(1):145–148. [DOI] [PubMed] [Google Scholar]
- 87. von Hohenstaufen KA, Conconi A, de Camos ZP, et al. Prognostic impact of monocyte count at presentation in mantle cell lymphoma. Br. J Haematol. 2013;162(4):465–473. [DOI] [PubMed] [Google Scholar]
- 88. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol. 2014;32(13):1338–1346. [DOI] [PubMed] [Google Scholar]
- 89. Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: Results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood. 2008;111(4):2385–2387. [DOI] [PubMed] [Google Scholar]
- 90. Dreyling M, Ferrero S, Vogt N, et al. New paradigms in mantle cell lymphoma: Is it time to risk-stratify treatment based on the proliferative signature? Clin Canc Res. 2014;20(20)5194–5206. [DOI] [PubMed] [Google Scholar]
- 91. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67, cytology, and growth pattern in mantle cell lymphoma: Results from randomized trials of the European MCL Network. J Clin Oncol. 2016;34(12):1386–1394. [DOI] [PubMed] [Google Scholar]
- 92. Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3(2):185–1897. [DOI] [PubMed] [Google Scholar]
- 93. Martinez N, Camacho FL, Algara P, et al. The molecular signature of mantle cell lymphoma reveals multiple signals favoring cell survival. Cancer Res. 2003;63(23):8226–8232. [PubMed] [Google Scholar]
- 94. Hartmann E, Fernandez V, Moreno V, et al. Five-gene model to predict survival in mantle-cell lymphoma using frozen or formalin-fixed, paraffin-embedded tissue. J Clin Oncol. 2008;26(30):4966–4972. [DOI] [PubMed] [Google Scholar]
- 95. Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123(8):1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nordstrom L, Sernbo S, Eden P, et al. SOX11 and TP53 add prognostic information to MIPI in a homogenously treated cohort of mantle cell lymphoma—a Nordic Lymphoma Group study. Br J Haematol. 2014;166(1):98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]