

Abstract
Aims
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an ion channelopathy characterized by ventricular arrhythmia during exertion or stress. Mutations in RYR2-coded Ryanodine Receptor-2 (RyR2) and CASQ2-coded Calsequestrin-2 (CASQ2) genes underlie CPVT1 and CPVT2, respectively. However, prognostic markers are scarce. We sought to better characterize the phenotypic and genotypic spectrum of CPVT, and utilize molecular modelling to help account for clinical phenotypes.
Methods and results
This is a Pediatric and Congenital Electrophysiology Society multicentre, retrospective cohort study of CPVT patients diagnosed at <19 years of age and their first-degree relatives. Genetic testing was undertaken in 194 of 236 subjects (82%) during 3.5 (1.4–5.3) years of follow-up. The majority (60%) had RyR2-associated CPVT1. Variant locations were predicted based on a 3D structural model of RyR2. Specific residues appear to have key structural importance, supported by an association between cardiac arrest and mutations in the intersubunit interface of the N-terminus, and the S4–S5 linker and helices S5 and S6 of the RyR2 C-terminus. In approximately one quarter of symptomatic patients, cardiac events were precipitated by only normal wakeful activities.
Conclusion
This large, multicentre study identifies contemporary challenges related to the diagnosis and prognostication of CPVT patients. Structural modelling of RyR2 can improve our understanding severe CPVT phenotypes. Wakeful rest, rather than exertion, often precipitated life-threatening cardiac events.
Keywords: Catecholaminergic polymorphic , Ventricular tachycardia , RyR2 , Arrhythmia , Genetics , Sudden unexpected death
What’s new?
A three-dimensional model of human RyR2 can be reliably created based on newly elucidated crystal structures and cryo-electron microscopy structures of the ryanodine receptor and is useful in predicting the structural impact of catecholaminergic polymorphic ventricular tachycardia (CPVT)-related variants.
RYR2 mutations localizing to specific residues of key structural importance may underlie the most severe CPVT phenotypes, including cardiac arrest.
A substantial number of life-threatening cardiac events occurred during wakeful rest and normal daily activities, challenging the paradigm that CPVT arrhythmias are isolated to times of adrenergic stress.
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an uncommon but often lethal ion channelopathy1–3 characterized by bidirectional or polymorphic ventricular tachycardia (VT) during stress.1 In 2001, Priori et al.2 reported that mutations in RYR2-coded Ryanodine Receptor-2 (RyR2) underlie CPVT1. RyR2 is the largest known ion channel in the human genome and is responsible for calcium regulation in the cardiomyocyte.4 CPVT1 mutations usually cluster in one of four areas known as ‘hotspots’.4 Arrhythmias are generally related to a gain of function in RyR2, which are likely a consequence of excessive or untimely calcium release in the sarcoplasmic reticulum, leading to delayed after-depolarizations.4 Calsequestrin-2 (CASQ2) mutations account for autosomal recessive CPVT2,5 probably through abnormal attenuation of RyR2.4
Data on prognosis in CPVT are limited.2,3,6 Initially, males with CPVT1 were thought to have more severe phenotypes.2 However, more recent data has not reported this association.6 Furthermore, expressivity is variable in CPVT, making clinical diagnosis challenging.6 A higher incidence of non-sustained VT has been observed in patients with variants in the C-terminal channel-forming region of RyR2,6 however this has not changed treatment guidelines.7 We sought to better characterize the genotypic and phenotypic spectrum of CPVT in a large, multicentre registry cohort.
Methods
This is a retrospective, observational cohort study of CPVT that enrolled (1) children (<19 years of age) with CPVT and (2) their affected first-degree relatives (≥19 years of age). Adults (≥19 years) were only included if a paediatric proband (<19 years) from their family was also enrolled. Centres were solicited through the Pediatric and Congenital Electrophysiology Society and all obtained ethical approval locally. The study adheres to the Declaration of Helsinki. Treatment outcomes from this population have been reported previously.8
Data collection
Data were obtained from existing medical records, entered by each site into a data collection form and verified by the coordinating centre. Genetic testing was undertaken prior to the study period by the enrolling centre as part of routine care. REDCap9 electronic data capture tools hosted by the Child and Family Research Institute at British Columbia Children’s Hospital were used for data acquisition and storage.
Modelling
A 3D model of human RyR2 was created based on available crystal structures and cryo-electron microscopy (EM) structures. The program MODELLER (https://salilab.org/modeller/) was used to produce the homology-based fragments, using available structures of human RyR2, mouse RyR2 (97.4% sequence identity), and rabbit Ryanodine Receptor-1 (RyR1) (65.4% sequence identity). The high sequence identity implies basically unaltered domain folds, thus allowing for the construction of reliable models. The utilized crystal structure templates were as follows: human RyR2 N-terminal region (PDB: 4JKQ),10 with missing loops modelled based on the mouse RyR2 crystal structure (PDB: 3IM6); mouse RyR2 SPRY1 (PDB 5C33)11; rabbit RyR1 repeat12 (PDB 5C30)11; mouse RyR2 SPRY2 domain (PDB 4P9I)12; and mouse RyR2 Repeat34 domain (PDB 4ETV).13 All of the remaining regions were built based on the cryo-EM model of rabbit RyR1 (PDB: 3J8H).14 The best model for each was selected based on the MolPDF score. The final full-length human RyR2 model was created by superposing the structural models of individual domains onto the cryo-EM structural model of rabbit RyR1 using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/).
Definitions
A proband was defined as the index case of confirmed CPVT in a family. Pathogenicity was recorded from the original genetic report whenever possible, usually in the setting of commercial genetic testing. When no rare genetic variant [pathogenic mutation, probable pathogenic mutation or variant of unknown significance (VUS)] was identified despite molecular analysis, the case was classified as gene-elusive. Known benign variants were also classified into the gene-elusive group. Subjects who did not undergo genetic testing as part of routine care were not included in genetic comparisons. Definitions of RyR2 hotspots4 and regions15 are published elsewhere. RYR2 variants that did not localize to a known region and/or hotspot are termed ‘non-hotspot‘ and/or ‘non-region’. Ventricular arrhythmias (VA) were classified as mild (ventricular couplets, ventricular bigeminy, and/or frequent premature ventricular complexes) or severe (non-sustained VT, bidirectional sustained VT, and/or cardiac arrest) based on the highest grade of ectopy documented on any recording irrespective of drug therapy. Variable expressivity was defined by ≥1 asymptomatic subject and ≥1 severely affected subject (history of syncope and/or cardiac arrest) within a family. An arrhythmic syncope or cardiac arrest while on a beta-blocker was defined as beta-blocker failure.
Statistical analysis
Contingency tables were generated for all categorical data with the frequency (percentage) reported. Data are presented as the median (95% distribution-free confidence intervals). Dates of birth were collected as year and month only as per ethical considerations. Durations listed vary by ± 30 days. All statistical analyses were completed using SAS Statistical Software Version 9.4 (SAS Institute, Cary, NC).
Results
This study describes 236 CPVT subjects (52% female) including 171 probands (72%) and 65 relatives (28%) followed for 3.5 (95% CI 2.9–3.9) years, equivalent to 795 patient-years. Diagnosis occurred at a median age of 12.6 (11.9–13.2) years with a delay to diagnosis of 0.5 (0.3–0.9) years in those presenting symptomatically. Further detailed demographic data from this cohort are described in a previous publication.8
Clinical and genetic assessment
Cardiac symptoms were reported in 179 of 236 patients (76%) at presentation, including 24 patients (13%) with more than one presenting symptom. Presenting symptoms included 112 with syncope (54%), 58 with cardiac arrest (28%), 12 with palpitations (6%), 8 with seizures (4%), 6 with chest pain (3%), 3 with pre-syncope (1.5%), 3 with dyspnoea (1.5%), and 5 with other miscellaneous symptoms/incidental findings leading to diagnosis (2%). The remaining 57 patients (24%) presented with a family history of CPVT or of possible CPVT, such as unexplained sudden death in a young relative. Genetic testing was undertaken in 194 of 236 patients (82%) prior to the study period by the enrolling centre as part of routine care. Some subjects had more than one gene sequenced, resulting in a greater total number of genetic test results than study subjects. Variants were as follows: RYR2 in 117 of 194 (60%), CASQ2 in 9 (5%), KCNJ2 in 1 (1%), 17 patients (9%) who tested positive for more than one potential mutation, and 27 (14%) had a variant but the participating centre could not provide further details. There were 23 (12%) gene-elusive patients. Genetic testing occurred in private/commercial labs for 162 of 256 tests (63%), research labs for 9 tests (4%) and unknown/unreported for 85 (33%) tests. Ninety-six of 194 (49%) subjects had a known or probable disease-causing variant, 33 (17%) had a VUS, and 65 (34%) had no available prediction at the time of testing. A comparison between CPVT1 and gene-elusive CPVT subjects is summarized in Table 1. Demographic data by pathogenicity of RYR2 variant appears in Table 2. Data on RyR2 variants organized by hotspot and region are summarized in Table 3 and Figures 1 and 2.
Table 1.
Clinical comparisons between patients with CPVT1 and gene-elusive CPVT
| CPVT1a | Gene elusive | |
|---|---|---|
| n = 117 | n = 23 | |
| Male sex | 59/117 (50%) | 10/23 (43%) |
| Probands | 77/117 (66%) | 22/23 (96%) |
| Median age at diagnosis (years) | 11.7 (95% CI: 10.6–12.8) | 14.8 (95% CI: 12.3–17.2) |
| Median delay to diagnosis (years) | 0.6 (95% CI: 0.3–1.2) | 0.4 (95% CI: 0–2.2) |
| Syncope | 36/117 (31%) | 6/23 (26%) |
| Cardiac arrest | 43/117 (37%) | 9/23 (39%) |
| Atypical trigger for syncope | 8/33 (13%) | 3/9 (33%) |
| Atypical trigger for cardiac arrest | 10/37 (26%) | 4/10 (40%) |
| VA | 89/117 (76%) | 23/23 (100%) |
| Atrial arrhythmia | 26/117 (22%) | 5/23 (22%) |
| Beta-blocker failure | 15/117 (13%) | 5/23 (22%) |
| ICD use | 56/115 (49%) | 15/23 (65%) |
| Deaths | 4/117 (3%) | 0/23 (0%) |
Defined as carrying a rare variant in RYR2 classified as a pathogenic mutation, probable pathogenic mutation, or VUS at the time of genetic testing.
Table 2.
Clinical comparisons between patients with CPVT1 by pathogenicity of RYR2 variant
| Pathogenic /probable pathogenic RYR2 variant | RYR2 variant of undetermined /unknown significance | |
|---|---|---|
| n = 90 | n = 27 | |
| Male sex | 42 (47%) | 17 (63%) |
| Probands | 60 (67%) | 17 (63%) |
| Median age at diagnosis (years) | 11.7 (8.0–14.7) | 11.8 (9.8–14.9) |
| Median delay to diagnosis (years) | 0.5 (0.1–4.1) | 0.7 (0.1–4.1) |
| Syncope | 30 (33%) | 6 (22%) |
| Cardiac arrest | 33 (37%) | 10 (37%) |
| Atypical trigger for syncope | 10 (11%) | 3 (11%) |
| Atypical trigger for cardiac arrest | 6 (7%) | 2 (7%) |
| VA | 69 (77%) | 20 (74%) |
| Atrial arrhythmia | 20 (22%) | 6 (22%) |
| Beta-blocker failure | 12 (13%) | 3 (11%) |
| ICD use | 46 (51%) | 10 (37%) |
| Deaths | 4 (4%) | 0 (0%) |
Table 3.
Phenotypes of CPVT1 patients by RyR2 hotspot and region
| Mild VA | Severe VA | Syncopal events | Cardiac arrest events | |
|---|---|---|---|---|
| (n = 13) | (n = 76) | (n = 42) | (n = 52)a | |
| Hotspot I | 5 (38%) | 12 (16%) | 6 (14%) | 8 (16%) |
| Hotspot II | 4 30% | 16 (21%) | 10 (24%) | 11 (21%) |
| Hotspot III | 1 (8%) | 15 (20%) | 8 (19%) | 12 (23%) |
| Hotspot IV | 1 (8%) | 19 (25%) | 12 (29%) | 9 (17%) |
| Non-hotspotb | 1 (8%) | 11 (14%) | 5 (12%) | 8 (15%) |
| Unknownc | 1 (8%) | 3 (4%) | 1 (2%) | 4 (8%) |
| C-terminal region | 2 (15%) | 35 (46%) | 19 (45%) | 21 (42%) |
| Central region | 4 (31%) | 18 (24%) | 10 (24%) | 13 (26%) |
| N-terminal region | 6 (46%) | 12 (16%) | 6 (14%) | 8 (16%) |
| Non-regionb | 0 | 5 (6%) | 2 (5%) | 3 (6%) |
| Unknownc | 1 (8%) | 6 (8%) | 5 (12%) | 5 (10%) |
Number of cardiac arrest events reported for hotspots is n = 52, and the number reported for regions is n = 50.
Variant does not localize to a known hotspot and/or region in RyR2.
Insufficient data provided by participating centre to determine localization of RyR2 variant.
Figure 1.
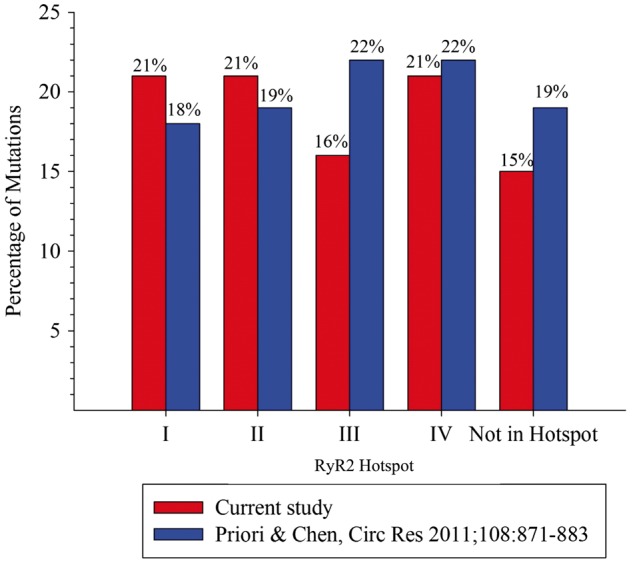
Plot of the percentage of variants found in hotspot areas on RyR2 thought to underlie most cases of CPVT4 in comparison to hotspot localization of RyR2 variants identified in the present study.
Figure 2.
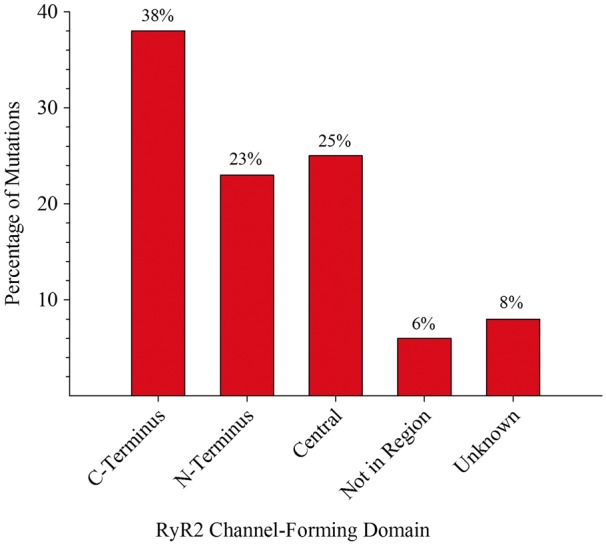
Plot of the percentage of variants by RyR2 region.
Triggers for life-threatening events
An atypical triggering event for syncope, defined by normal wakeful activity (e.g. playing a musical instrument, resting), occurred in 15 of 66 patients (23%) in whom the preceding circumstance was known. Cardiac arrest was atypically triggered in 12 of 45 (27%) patients. Six patients (3%) died as a result of confirmed/suspected CPVT-related arrhythmias including four with CPVT1. In three of the six deceased patients, an atypical trigger preceded death. An additional 2 decedents were excluded from mortality analysis as they were ascertained from a molecular autopsy. Both had variants in RYR2 and pre-mortem symptoms consistent with CPVT. Figure 3 summarizes the circumstances preceding life-threatening events.
Figure 3.
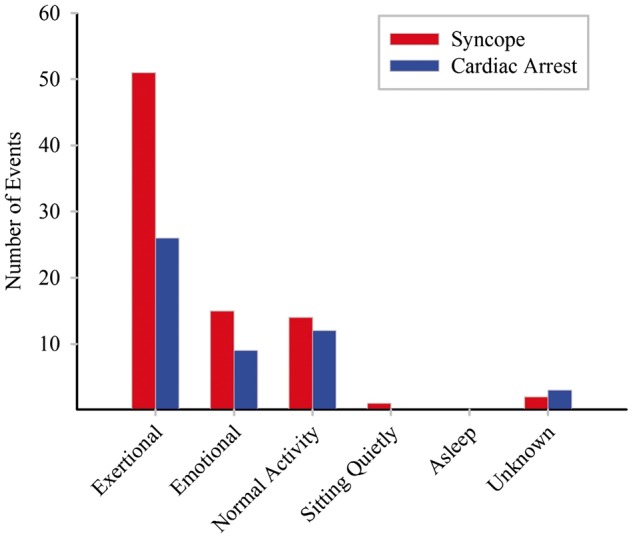
Circumstances immediately preceding all life-threatening events defined as syncope and/or cardiac arrest.
CPVT families
This cohort included 32 families comprising 79 patients (33%). Fifty-four patients from 23 families had CPVT1, while five patients from two families had CPVT2. In 13 of 32 families (41%), expressivity was variable among relatives. In 16 families (50%), there was ≥1 asymptomatic, genotype-positive case. A RYR2 variant at R420 (see Supplementary material online, Figure S1) was found in 14 of 117 CPVT1 patients (12%) (R420Q in 2 and R420W in 12). Nine of the RYR2-R420W subjects made up 4 families, all with variable expressivity.
CPVT2
There were 4 of 194 patients (2%) affected by homozygous CASQ2-associated CPVT2, and all had a history of life-threatening symptoms. The CASQ2 variants identified were as follows R251H, I270T, and Q245X. In addition, there was one family potentially affected by heterozygous CPVT2, which included three subjects with a heterozygous, probably pathogenic CASQ2 variant (D340stop). The proband experienced life-threatening symptoms, while her relatives had exercise-induced ventricular bigeminy.
Molecular modelling
A direct analysis of RYR2 variants on the overall 3D structure of our homology model e shows that: (1) most variants cluster close to the four-fold symmetry axis and virtually none are found towards the cytosolic corner regions, which are located more at the periphery (Figure 4A), (2) in hotspot 1, all seven variants found at the interface between neighbouring subunits are associated with cardiac arrest (see Supplementary material online, Figure S1), (3) all but one variant (I4867V) in the S5 and S6 helices, as well as the S4–S5 linker of the C-terminus, were implicated in cardiac arrest (Figure 4B).
Figure 4.
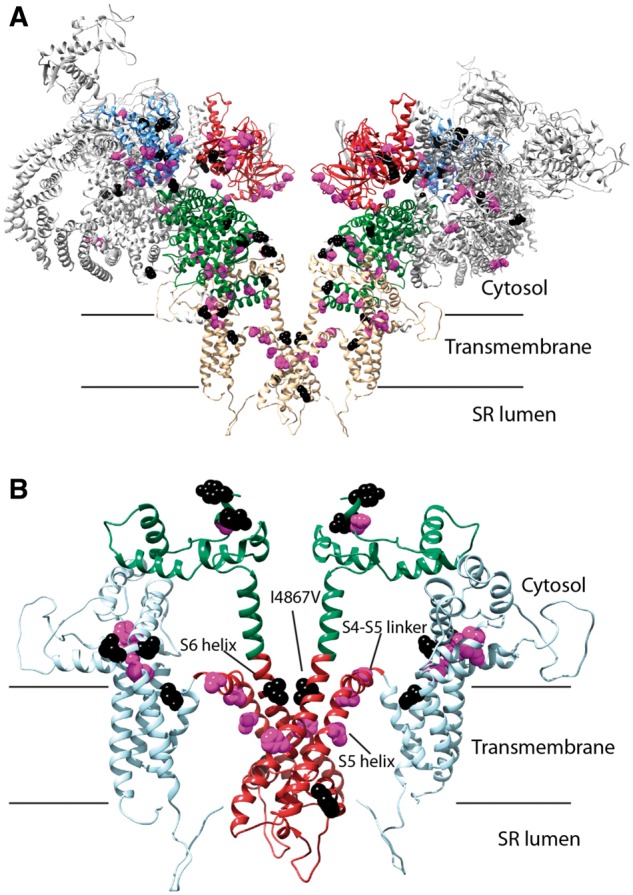
(A) Homology model of human RyR2. The protein is shown in cartoon form, with the disease hotspots highlighted in colours (hotspot 1: blue, hotspot 2: red; hotspot 3: green; hotspot 4: light blue). View is from the ‘side,’ parallel to the membrane. Only two out of four subunits are shown for clarity. Positions for CPVT-associated variants are highlighted, with atoms shown in Van der Waals representation (purple: associated with cardiac arrest, black: all others). (B) Close-up of the transmembrane region of the RyR2. This region forms the bulk of disease hotspot 4. The following subregions are highlighted: pore-forming region (red), additional transmembrane (cyan), and C-terminal cytosolic extension to the pore (green). Positions of CPVT-associated variants associated with cardiac arrest are highlighted in purple, and all others in black. The S5 and S6 helices and the S4–S5 linker are labelled.
Discussion
CPVT is a potentially lethal syndrome that almost entirely lacks prognostic data. Since the seminal description of RYR2-related CPVT more than 15 years ago, genotypic factors still do not inform prognosis or management. In some cohorts, CPVT1 patients appeared to be at higher arrhythmic risk,16 however this phenomenon has not been consistently observed.8 In 2012, van der Werf et al. showed that variant location in RyR2 could predict arrhythmia in several Dutch CPVT families,6 including a correlation between non-sustained VT on initial exercise stress test and C-terminus variants in RYR2. However, this potential prognosticator has not changed treatment standards.7 Instead, clinicians may make life-altering decisions based on anecdotal experience, patient sex, age, and/or family history despite a lack of reproducible evidence supporting any of these perceived risk factors. Herein, we describe the outcomes of multiple probands and families with CPVT1, and use molecular modelling to characterize the structural changes in RyR2 that underlie the most malignant CPVT phenotypes, in a large, heterogeneous registry population.
To better characterize the structural channel alterations in cases of CPVT1 complicated by life-threatening cardiac events, we modelled RYR2 variants from our cohort using a RyR2 homology model. The only available high-resolution 3D structure for human RyR2 is for the N-terminal region.10 However, reliable homology-based models can be built when templates are available with >50% sequence identity.17 The availability of multiple structures for mouse RyR2 domains, which displays >97% sequence identity with human RyR2, as well multiple crystal and cryo-EM structures of rabbit RyR1 (>65% identity)18 allowed for the building of a near complete homology-based model of RyR2 on which all variants could be mapped. Although portions of the cryo-EM study of RyR1 are at comparatively lower resolution, most variants mapped to the better-ordered regions and those for which high-resolution crystal structures are available. Figure 4B shows the location of the variants on our homology model of the RyR2 transmembrane region, which contains the C-terminus. This area forms the minimal ‘channel’ through which calcium ions permeate the sarcoplasmic reticulum membrane, whereas the cytosolic assembly mostly serves to allosterically modulate the gating properties of the channel region. Variants in the latter would thus be expected to be more permissive, whereas variants in the channel region would have a much higher impact on function. Within the transmembrane region, key structural elements are formed by the helices S5 and S6 of the pore-forming domain, and the linker between S4 and S5 has been suggested as an important allosteric coupling element between triggers and gating of the pore in several other ion channels.19 Accordingly, four of the five variants that map to these elements led to cardiac arrest in our cohort.
Our 3D model also confirms a previous study on the importance of the N-terminus in channel gating (see Supplementary material online, Figure S1): during channel opening, this area undergoes large conformational changes, whereby an interface between neighbouring subunits is thought to be disrupted.20 Variants that weaken this interface are thus, predicted to facilitate channel opening. All seven variants that mapped to this interface were associated with cardiac arrest, confirming a recent report that this interface is a crucial determinant of channel gating.20 These correlations within the C- and N-termini suggest that amino acid residues with key importance in RyR2 gating can give rise to the most severe disease phenotypes. In 2011, Priori and Chen4 also proposed that CPVT variants cluster in 1 of 4 ‘hotspots’ on RyR2. Hotspot 1 forms a gating ring at the cytosolic face,20 whereas hotspot 4 forms the transmembrane assembly.14 Hotspots 2 and 3 form a physical link with hotspots 1 and 4. Variant localizations by hotspot in our cohort mirror the data of Priori and Chen (Figure 1A), supporting the theory that alterations to these areas are most likely to underlie an arrhythmia phenotype.
A large number of CPVT1 families are reported in this cohort, probably due to utilization of cascade genetic screening. Half of families included ≥1 relative(s) with genetically confirmed, asymptomatic CPVT1. These cases of concealed CPVT1 are poorly understood. For example, in both the present study and existing data,6,21 the RYR2-R420W variant demonstrated incomplete penetrance and expressivity. R420 forms part of an extensive network of interactions between the domains in hotspot 1 (see Supplementary material online, Figure S1), coordinating a central chloride ion.22 Mutation of this residue leads to a marked disruption of the chloride binding site and allosteric changes within the hotspot in the setting of RYR2-R420W and R420Q.22,23 The growing number of concealed CPVT1 cases should be a topic of further study, including efforts to elucidate genetic modifiers that can account for phenotypic variability and to identify genotype-specific management strategies.
Traditionally, CPVT arrhythmias are described during periods of intense emotion or physical exertion, prompting experts to advise strict exercise restriction.1 However, a substantial proportion of events are triggered by wakeful rest in our cohort. Ventricular fibrillation unprovoked by exercise testing has been described in loss-of-function RYR2 mutation carriers, suggesting that other mechanisms could underlie arrhythmias in these patients.24 This cohort also includes a disproprortionately large number of children, who have elevated baseline heart rates compared to adult patients. This elevated heart rate at rest could potentially account for the higher number of arrhythmias precipitated by rest in this pediatric cohort.
Autosomal recessive CPVT2 is highly malignant.5 The present study includes four individuals with homozygous CPVT2, all of whom suffered from severe arrhythmias. We also report one small family with CPVT possibly related to a heterozygous variant in CASQ2, including one proband with life-threatening symptoms. Recently, the existence of heterozygous CPVT2 was documented using a whole exome sequencing approach,25 suggesting that the heterozygous CASQ2 variant observed in our family may also behave in an autosomal dominant fashion. Further molecular studies of CASQ2-related CPVT variants are needed.
Limitations
Our research was limited by selection bias, as those patients presenting with sudden death are rarely diagnosed or seen by a cardiologist, and minimally symptomatic or gene-elusive patients may have been excluded from enrolment. Adult subjects were under-represented as only paediatric centres participated, which may over-estimate disease severity. A diagnosis of CPVT was required for enrolment; however, we were unable to determine strength of diagnosis in all cases. A validated severity model for CPVT does not currently exist. The definition of mild vs. severe VA is based on clinical experience rather than robust data. Some centres did not have robust genotypic data beyond the amino acid sequence and pathogenicity as testing spanned a nearly 15 year period in this retrospective cohort. We could not determine disease-causation without linkage analysis. In some cases, it was not known whether one gene or multiple genes were analysed during the sequencing process. We attempted to verify genetic information with centres whenever possible. Future prospective studies are needed to assure that all patients undergo broad sequencing of all genes now known to underlie CPVT. For the RyR2 model, the portions for which crystal structures are available are the best defined, followed by the C-terminal region, for which the resolution of recent cryo-EM studies is the highest. As such, a direct analysis of variants in the N-terminal and C-terminal hotspots is the most reliable. For most other sections, a direct analysis of hydrogen bonds and ionic interactions of the variants is not yet possible, but there is no uncertainty regarding their general location in the 3D structure, since the overall fold is easily observed throughout current cryo-EM structures. Homology modelling is predictive in nature and cannot be independently used to determine pathogenicity.
Conclusions
This multicentre registry-based study of a large, heterogeneous, and extensively genotyped CPVT population describes contemporary challenges related to the diagnosis and prognostication of CPVT patients. Utilizing predictive homology modelling, areas of key structural importance in the RyR2 channel are elucidated and supported by clinical outcome data. Modelling RYR2 variants may be especially helpful for patients and families facing genetic uncertainty. Life-threatening events often occur during resting wakeful activities, highlighting the unpredictable nature of CPVT arrhythmias. Genetic expressivity of CPVT1 is also variable among relatives, which suggests that a family history of sudden death is an unreliable prognosticator. The present study moves us towards a better understanding of the interplay between RyR2 structure and clinical outcomes. Our findings also identify the challenges that still exist in CPVT, such as determining arrhythmic risk, and interpreting positive family histories and equivocal genetic results. Further functional and linkage analysis is needed to establish genetic causation in most cases of CPVT.
Supplementary material
Supplementary material is available at Europace online.
Supplementary Material
Acknowledgements
The authors thank the site coordinators and Taylor Cunningham for assisting with reference material.
Conflict of interest: Dr. Erickson is a paid consultant and lecturer for Medtronic.
Funding
This work was supported by the Rare Disease Foundation and BC Children’s Hospital Foundation, Vancouver, BC (T.M.R. and S.S.), National Institutes of Health–Grant no. HL108173, Bethesda, MD (P.J.K.), Canadian Institutes of Health Grant no. 125893, Ottawa, ON (F.V.P.), Caitlin Elizabeth Morris Fund of Appliance Canada (R.M.H.), MH CZ-DRO, University Hospital Motol, Prague, CZE 00064203 (P.K.).
References
- 1. Leenhardt A, Lucet V, Denjoy I, Grau F, Do Ngoc D, Coumel P.. Catecholaminergic polymorphic ventricular tachycardia in children: a 7-year follow-up of 21 patients. Circulation 1995;91:1512–9. [DOI] [PubMed] [Google Scholar]
- 2. Priori S, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R. et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196–200. [DOI] [PubMed] [Google Scholar]
- 3. Hayashi M, Denjoy I, Extramiana F, Maltret A, Roux-Buisson N, Lupoglazoff J. et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009;119:2426–34. [DOI] [PubMed] [Google Scholar]
- 4. Priori S, Chen S.. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res 2011;108:871–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O. et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 2001;69:1378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van der Werf C, Nederend I, Hofman N, van Geloven N, Ebink C, Frohn-Mulder I. et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2012;5:748–56. [DOI] [PubMed] [Google Scholar]
- 7. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C. et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013;15:1389–406. [DOI] [PubMed] [Google Scholar]
- 8. Roston T, Vinocur J, Maginot K, Mohammed S, Salerno J, Etheridge S. et al. Catecholaminergic polymorphic ventricular tachycardia in children: an analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol 2015;8:633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG.. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borko L, Bauerova-Hlinkova V, Hostinova E, Gasperik J, Beck K, Lai FA. et al. Structural insights into the human RyR2 N-terminal region involved in cardiac arrhythmias. Acta Crystallogr D Biol Crystallogr 2014;70(Pt 11):2897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuchi Z, Yuen SM, Lau K, Underhill AQ, Cornea RL, Fessenden JD. et al. Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nat Commun 2015;6:7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lau K, Van Petegem F.. Crystal structures of wild type and disease mutant forms of the ryanodine receptor SPRY2 domain. Nat Commun 2014;5:5397. [DOI] [PubMed] [Google Scholar]
- 13. Yuchi Z, Lau K, Van Petegem F.. Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure 2012;20:1201–11. [DOI] [PubMed] [Google Scholar]
- 14. Yan Z, Bai XC, Yan C, Wu J, Li Z, Xie T. et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 2015;517:50–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP. et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol 2009;54:2065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M. et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002;106:69–74. [DOI] [PubMed] [Google Scholar]
- 17. Sali A, Blundell TL.. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 18. Van Petegem F. Ryanodine receptors: allosteric ion channel giants. J Mol Biol 2015;427:31–53. [DOI] [PubMed] [Google Scholar]
- 19. Blunck R, Batulan Z.. Mechanism of electromechanical coupling in voltage-gated potassium channels. Front Pharmacol 2012;3:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kimlicka L, Lau K, Tung CC, Van Petegem F.. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat Commun 2013;4:1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N. et al. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J Am Coll Cardiol 2002;40:341–9. [DOI] [PubMed] [Google Scholar]
- 22. Kimlicka L, Tung CC, Carlsson AC, Lobo PA, Yuchi Z, Van Petegem F.. The cardiac ryanodine receptor N-terminal region contains an anion binding site that is targeted by disease mutations. Structure 2013;21:1440–9. [DOI] [PubMed] [Google Scholar]
- 23. Tung CC, Lobo PA, Kimlicka L, Van Petegem F.. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 2010;468:585–8. [DOI] [PubMed] [Google Scholar]
- 24. Jiang D, Chen W, Wang R, Zhang L, Chen SR.. Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci USA 2007;104:18309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gray B, Bagnall RD, Lam L, Ingles J, Turner C, Haan E. et al. A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2016;13:1652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.